")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Organic anion transporter 3- and organic anion transporting polypeptides 1B1- and 1B3-mediated transport of catalposide
Authors Jeong H, Kwon M, Lee Y, Yoo JS, Shin DH, Song I, Lee HS
Received 4 October 2014
Accepted for publication 24 November 2014
Published 22 January 2015 Volume 2015:9 Pages 643—653
DOI https://doi.org/10.2147/DDDT.S75400
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Hyeon-Uk Jeong,1 Mihwa Kwon,2 Yongnam Lee,3 Ji Seok Yoo,3 Dae Hee Shin,3 Im-Sook Song,2 Hye Suk Lee1
1College of Pharmacy, The Catholic University of Korea, Bucheon 420-743, Korea; 2College of Pharmacy and Research Institute of Pharmaceutical Sciences, Kyungpook National University, Daegu 702-701, Korea; 3Central R&D Institute, Yungjin Pharm Co., Ltd., Suwon 443-270, Korea
Abstract: We investigated the in vitro transport characteristics of catalposide in HEK293 cells overexpressing organic anion transporter 1 (OAT1), OAT3, organic anion transporting polypeptide 1B1 (OATP1B1), OATP1B3, organic cation transporter 1 (OCT1), OCT2, P-glycoprotein (P-gp), and breast cancer resistance protein (BCRP). The transport mechanism of catalposide was investigated in HEK293 and LLC-PK1 cells overexpressing the relevant transporters. The uptake of catalposide was 319-, 13.6-, and 9.3-fold greater in HEK293 cells overexpressing OAT3, OATP1B1, and OATP1B3 transporters, respectively, than in HEK293 control cells. The increased uptake of catalposide via the OAT3, OATP1B1, and OATP1B3 transporters was decreased to basal levels in the presence of representative inhibitors such as probenecid, furosemide, and cimetidine (for OAT3) and cyclosporin A, gemfibrozil, and rifampin (for OATP1B1 and OATP1B3). The concentration-dependent OAT3-mediated uptake of catalposide revealed the following kinetic parameters: Michaelis constant (Km) =41.5 µM, maximum uptake rate (Vmax) =46.2 pmol/minute, and intrinsic clearance (CLint) =1.11 µL/minute. OATP1B1- and OATP1B3-mediated catalposide uptake also showed concentration dependency, with low CLint values of 0.035 and 0.034 µL/minute, respectively. However, the OCT1, OCT2, OAT1, P-gp, and BCRP transporters were apparently not involved in the uptake of catalposide into cells. In addition, catalposide inhibited the transport activities of OAT3, OATP1B1, and OATP1B3 with half-maximal inhibitory concentration values of 83, 200, and 235 µM, respectively. However, catalposide did not significantly inhibit the transport activities of OCT1, OCT2, OAT1, P-gp, or BCRP. In conclusion, OAT3, OATP1B1, and OATP1B3 are major transporters that may regulate the pharmacokinetic properties and may cause herb–drug interactions of catalposide, although their clinical relevance awaits further evaluation.
Keywords: catalposide, drug transporters, herb–drug interaction, OAT3, OATP1B1, OATP1B3
Introduction
Catalpa ovata has been used as traditional herbal medicines for the treatment of inflammation, itching, and scabies. It contains iridoid and naphthoquinones, and catalposide is a bioactive iridoid glucoside isolated from C. ovata (Figure 1).1,2 Recently, catalposide was reported to be a novel natural ligand of peroxisome proliferation-activated receptor α (PPAR-α), which regulates hepatic lipid metabolism.3 In addition, it showed inhibitory effects on tumor necrosis factor-α, interleukin 1β (IL-1β), and IL-6 production and nuclear factor-κB activation in lipopolysaccharide-activated RAW 264.7 macrophages, as well as cytoprotective effects against oxidative damage caused by the induction of heme oxygenase-1.4,5 The effective concentration of catalposide required for the suppression of cytokines, antioxidative effect, and PPAR-α activation has been reported in the range of 0.2–4 μM in an in vitro cell system.1,3,5,6 Catalposide attenuated the increased expression of intestinal epithelial proinflammatory gene and reduced the severity of colitis induced by trinitrobenzene sulfonic acid in mice at a dose of 0.5 mg/kg.1 Administration of higher-dose catalposide (1–2.5 mg/kg) resulted in a similar therapeutic effect without histologic toxicity.1
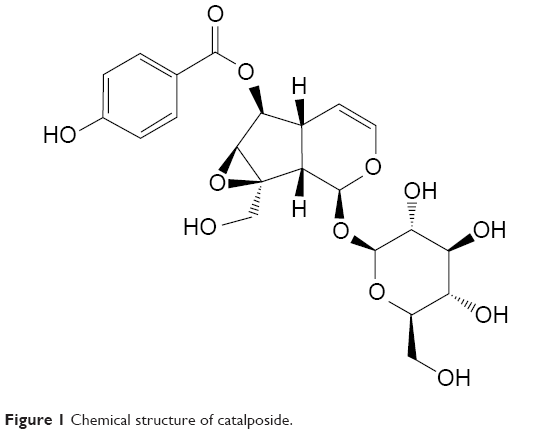 | Figure 1 Chemical structure of catalposide. |
Ji et al investigated the pharmacokinetics of catalposide in rats after intravenous administration.7 Plasma concentration of catalposide showed biphasic disposition with a terminal half-life of 19.3±9.5 minutes. It also showed a high distribution volume (2657.2±1396.9 mL/kg). In addition, systemic clearance of catalposide was 96.7±44.2 mL/minute/kg, and the renal and nonrenal clearance of catalposide was 8.47 and 88.2 mL/kg/minute, respectively. In the recovery of catalposide after intravenous administration (10 mg/kg), 9.9% was found in the urine. However, catalposide remained stable after a 3-hour incubation in rat and human plasma, as well as in the presence of NADPH in rat and human liver microsomes.7 These results, taken together, suggest catalposide is distributed rapidly into specific organs or the whole body and/or is subject to non-cytochrome P450 (non-CYP)-mediated metabolism, with subsequent excretion in the bile or urine. A significant amount of catalposide was excreted into the urine in its unchanged form (9.9% of the intravenous dose),7 suggesting a transport mechanism may be involved in its renal excretion. However, the metabolism and transport mechanism of catalposide and significance of drug metabolizing enzymes and transporters in the metabolism, distribution, and elimination need further investigation.
Recently, transporters have been suggested to be important in in vivo drug disposition, drug responses, and adverse drug reactions.8 In addition, information regarding drug transporters is increasing in drug labels and provides information for understanding the mechanisms of drug absorption, distribution, and elimination.8
A long and continuous history of dietary use has demonstrated the safety of many herbs, and some herb-derived drugs are important therapeutics.9 However, there is a growing trend for the concurrent administration of herbal ingredients with drugs, which can cause serious herb–drug interactions (HDIs). For example, hyperforin, contained in St John’s wort, significantly reduces plasma concentrations of cyclosporine, amitriptyline, digoxin, warfarin, phenprocoumon, midazolam, tacrolimus, indinavir, and theophylline.10,11 Common herbal medicines, including ginseng (Panax ginseng), ginkgo (Ginkgo biloba), garlic (Allium sativum), and ginger (Zingiber officinale), have been reported to interact with drugs such as digoxin, warfarin, midazolam, cyclosporine, tacrolimus, irinotecan, amitriptyline, and indinavir.10 Among the underlying mechanisms for the most commonly reported HDIs, modulation of drug transporters is an important mechanism.10,12,13
Thus, the purpose of this study was to evaluate the transport characteristics of catalposide and possible transporter-based catalposide–drug interactions, using HEK293 and LLC-PK1 cell systems overexpressing uptake and efflux transporters, such as organic anion transporter 1 (OAT1), OAT3, organic anion transporting polypeptide 1B1 (OATP1B1), OATP1B3, organic cation transporter 1 (OCT1), OCT2, P-glycoprotein (P-gp), and breast cancer resistance protein (BCRP).
Materials and methods
Materials
[3H]Para-aminohippuric acid (PAH; 0.13 TBq/mmol), [3H]estrone-3-sulfate (ES; 2.12 TBq/mmol), [3H]estradiol-17β-d-glucuronide (EG; 2.22 TBq/mmol),[3H]digoxin (1.103 TBq/mmol), and [3H]methyl-4-phenylpyridinium (MPP+; 2.9 TBq/mmol) were purchased from Perkin Elmer Inc. (Boston, MA, USA). Catalposide, tetraethylammonium chloride, probenecid, rifampin, cyclosporin A (CsA), 4-methylumbelliferone, and nonessential amino acids were obtained from Sigma-Aldrich (St Louis, MO, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), poly-d-lysine-coated 24-well plates, and Hank’s balanced salt solution (HBSS) were purchased from Corning-Gentest (Tewksbury, MA, USA). Methanol and acetonitrile (high-performance liquid chromatography-grade) were from Burdick & Jackson Inc. (Muskegon, MI, USA). Sodium butyrate and fumitrogen C (FTC) were obtained from Merck Millipore (Billerica, MA, USA). Other chemicals were of the highest quality available.
Evaluation of catalposide as a substrate of OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2, P-gp, and BCRP transporters
HEK293 cells overexpressing OAT1, OAT3, OATP1B1, OATP1B3, OCT1, and OCT2 transporters were maintained in a humidified atmosphere of 5% CO2at 37°C, in DMEM supplemented with 10% FBS, and 5 mM nonessential amino acids. For the experiments, 4×105 cells were seeded in poly-D-lysine-coated 24-well plates. After 24 hours, the growth medium was discarded and the attached cells were washed with HBSS and preincubated for 10 minutes in HBSS at 37°C.
The uptake of 10 μM catalposide was measured in the absence and presence of representative inhibitors (200 μM probenecid for OAT1 and OAT3, 100 μM rifampin for OATP1B1 and OATP1B3, and 10 mM tetraethylammonium for OCT1 and OCT2) for 10 minutes at 37°C, or 5 minutes at 37°C for OATP1B1 and OATP1B3. The cells were then washed three times with 100 μL ice-cold HBSS immediately after placing the plates on ice. Subsequently, 300 μL 80% methanol containing internal standard (4-methylumbelliferone 10 ng/mL) was added to each sample well, and the cell plates were shaken gently for 20 minutes at room temperature. One hundred microliters of each sample was added to 100 μL methanol and centrifuged (13,000× g, 4 minutes, 4°C), and the supernatant was recovered and evaporated. The residue was dissolved in 50 μL 15% methanol, and an aliquot (7 μL) was analyzed using the liquid chromatography–high-resolution mass spectrometry.
LLC-PK1 cells overexpressing P-gp and BCRP transporters were grown in tissue culture flasks in DMEM supplemented with 20% FBS, 4 mM L-glutamine, 1% nonessential amino acids, and 1% penicillin-streptomycin. The cells were seeded onto filter membranes at a density of 5×105 cells/well. Transepithelial electrical resistance values in the range of 300–650 O·cm2 were used in the transport experiment as a marker of the integrity of the cell monolayers. The apical-to-basal (A-to-B) transport of catalposide was measured by adding 0.5 mL HBSS containing 20 μM catalposide with or without 20 μM CsA for P-gp or 100 μM FTC for BCRP on the apical side and by adding 1.5 mL HBSS without catalposide on the basal side of the insert. The insert was transferred to a well containing fresh HBSS medium every 15 minutes for 1 hour. The basal-to-apical (B-to-A) transport of catalposide was measured by the addition of 1.5 mL of HBSS containing 20 μM catalposide with or without 20 μM CsA or 100 μM FTC on the basal side, and 0.5 mL of HBSS without catalposide on the apical side. The transport medium in the apical side was replaced with 0.4 mL fresh incubation medium every 15 minutes for 1 hour. One hundred fifty microliters B-to-A transport rate samples and 200 μL A-to-B transporter rate samples were added to 10 μL methanol containing the internal standard (4-methylumbelliferone 100 ng/mL) and 150 or 200 μL methanol, respectively. Subsequently, each sample was centrifuged (13,000× g, 4 minutes, 4°C), and the supernatant was recovered and evaporated. The residue was dissolved in 50 μL 30% methanol, and an aliquot (7 μL) was analyzed by liquid chromatography–high-resolution mass spectrometry.
To examine the concentration dependence of the uptake of catalposide in HEK293 cells overexpressing OAT3, OATP1B1, and OATP1B3 transporters, the cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in DMEM supplemented with 10% FBS and 5 mM nonessential amino acids. For the experiments, 4×105 cells were seeded in poly-D-lysine-coated 24-well plates. After 24 hours, the growth medium was discarded and the attached cells were washed with HBSS and preincubated for 10 minutes in HBSS at 37°C. To examine the concentration dependence of the uptake of catalposide via OAT3, OATP1B1, and OATP1B3, the uptake of various concentrations of catalposide (2–500 μM) was measured for 10 minutes at 37°C for OAT3, and for 5 minutes at 37°C for OATP1B1 and OATP1B3.
To evaluate the effects of representative inhibitors on the uptake of catalposide in HEK293 cells transiently expressing OAT3, OATP1B1, and OATP1B3 transporters, the cells were maintained and seeded as described previously, and the uptake of 10 μM catalposide was measured in the absence and presence of various concentrations of representative inhibitors (1, 10, and 200 μM probenecid and 10, 100, and 1,000 μM cimetidine and furosemide for OAT3, and 0.1, 1, and 10 μM CsA; 10, 100, and 1,000 μM gemfibrozil; and 1, 10, and 100 μM rifampin for OATP1B1 and OATP1B3) for 10 minutes at 37°C for OAT3 and for 5 minutes at 37°C for OATP1B1 and OATP1B3.
Analysis of catalposide
For the quantification of catalposide, an Exactive Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA, USA) coupled with an Accela ultraperformance liquid chromatography system was used. In the case of uptake transport studies, catalposide was eluted through a Halo C18 column (2.7 μm, 2.1 mm internal diameter ×50 mm; Advanced Materials Technology, Wilmington, DE, USA) with a mobile phase consisting of 5% methanol in ammonium formate (1 mM at pH 3.1; mobile phase A) and methanol (mobile phase B) at a flow rate of 0.5 mL/minute. The gradient mode was set as follows: 5% B for 2.7 minutes, 15% to 20% B in 4.3 minutes, 20% to 90% B in 0.5 minutes, 90% B for 3.0 minutes, 90% to 15% B in 0.5 minutes, and 15% B for 4 minutes.
In case of efflux transport studies and concentration dependency, quantification was performed using a Unison-C8 column (3.0 μm, 2.0 mm i.d. ×75 mm; Imtakt Corporation, Kyoto, Japan) with a gradient elution of 5% methanol in ammonium formate (1 mM at pH 3.1; mobile phase A) and methanol (mobile phase B) at a flow rate of 0.3 mL/minute: 30% B for 1.5 minutes, 30% to 95% B in 0.1 minutes, 95% B for 3.4 minutes, 95% to 30% B in 0.1 minutes, 30% B for 2.9 minutes at a flow rate of 0.4 mL/minute, 0.4 mL/minute to 0.3 mL/minute in 0.1 minutes, and 30% B for 0.4 minutes at a flow rate of 0.3 mL/minute. The column and autosampler temperatures were 40°C and 6°C, respectively. The mass spectrometer was equipped with an electrospray ionization (ESI) source and was operated in negative ion mode. The ESI source settings for the ionization of catalposide were as follows: capillary voltage, 3,000 V; vaporizer temperature, 400°C; capillary temperature, 330°C; sheath gas pressure, 50 psi; and auxiliary gas pressure, 15 psi. The accurate masses of the deprotonated molecular ions ([M-H]−) of catalposide and 4-methylumbelliferone were 481.13461 and 175.03952, respectively. Analytical data were processed using the Xcalibur software (Thermo Scientific).
Inhibitory effects of catalposide on transport activities of OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2, P-gp, and BCRP
HEK293 cells overexpressing OAT1, OAT3, OATP1B1, OATP1B3, OCT1, and OCT2 transporters were purchased from Corning-Gentest. The cells were maintained at 37°C in a humidified atmosphere of 5% CO2, in DMEM supplemented with 10% FBS, 5 mM nonessential amino acids, and 100 U/mL penicillin-streptomycin. For the experiments, 105 cells were seeded in 96-well plates. After 24 hours, the growth media were discarded and the attached cells were washed with HBSS and preincubated for 20 minutes in HBSS at 37°C. To examine the effects of catalposide on transporter activity, the uptake of 0.1 μM [3H]MPP+ for OCT1 and OCT2, 1 μM [3H]PAH for OAT1, 0.1 μM [3H]ES for OAT3 and OATP1B1, and 0.1 μM [3H]EG for OATP1B3 was measured in the presence of catalposide (0–100 μM for OAT1, OCT1, and OCT2 and 0–500 μM for OAT3, OATP1B1, and OATP1B3) for 10 minutes at 37°C. The cells were then washed three times with 100 μL ice-cold HBSS immediately after placing the plates on ice and lysed with 10% sodium dodecyl sulfate. The radioactivity of the probe substrate in the cells was measured using a liquid scintillation counter.
LLC-PK1-MDR1 (LLC-PK1 cells stably expressing P-gp; purchased from Corning-Gentest) and LLC-PK1-BCRP (LLC-PK1 cells stably expressing BCRP; obtained from Dr AH Schinkel, Netherlands Cancer Institute, Amsterdam, the Netherlands) cells were used for the comparison of the B-to-A transport rate of [3H]digoxin and [3H]ES in the absence and presence of catalposide. Briefly, the cells were seeded on filter inserts for 24-well Transwell plates at a density of 5×105 cells and grown for 5 days. Cell monolayers with transepithelial electrical resistance in the range of 300–850 Ω·cm2 were used in the transport experiment.14 B-to-A transport was initiated by adding 0.8 mL HBSS containing [3H]digoxin or [3H]ES (0.1 μM each) and catalposide (1, 10, 50, and 100 μM) on the basal side and adding 0.3 mL of fresh HBSS on the apical side. Every 15 minutes, 0.2 mL HBSS sample on the apical side was removed and replaced with 0.2 mL fresh HBSS for 1 hour. The radioactivity of [3H]digoxin or [3H]ES in the HBSS was measured using a liquid scintillation counter.
Data analysis
The percentages of inhibition were calculated by the ratio of the amounts of catalposide in the presence and absence of inhibitors, and half-maximal inhibitory concentration (IC50) values were calculated by using an inhibitory effect model [v=Emax(1-[I]/IC50+[I])], using WinNonlin (version 2.0; Pharsight, Mountain View, CA, USA).15,16 Here, Emax represents the maximum effect achieved by the system, [I] represents the concentration of an inhibitor, and IC50 represents the concentration of an inhibitor that is required for 50% inhibition. In the concentration-dependent uptake studies, the OAT3-, OATP1B1-, and OATP1B3-mediated transport of catalposide was calculated by subtracting the transport rates of the mock cells from those of the OAT3-, OATP1B1-, and OATP1B3-expressing HEK293 cells. Kinetic parameters for the OAT3-, OATP1B1-, and OATP1B3-mediated transport of catalposide were determined using the Hill equation [V=Vmax·Sn/(Kmn+Sn)] and the Enzyme Kinetics software (version 1.3; Systat Software Inc., San Jose, CA, USA). Here, Vmax represents the maximum uptake rate, S represents the concentration of a substrate, n is the Hill coefficient, and Km represents the concentration of a substrate that is required for 50% of Vmax. The statistical significance was assessed by Student’s t-test, using Statistical Package for the Social Sciences (SPSS Inc., Chicago, IL, USA).
Results
OAT3-, OATP1B1-, and OATP1B3-mediated transport of catalposide
We evaluated catalposide as a substrate of the OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2, P-gp, and BCRP transporters. The functionality of these transporters was confirmed by the significantly greater uptake rate of the substrates in HEK293 cells expressing various transporters than that in HEK293 mock cells (ie, 25.0-fold increase in PAH uptake for OAT1; 18.9-fold increase in ES uptake for OAT3; 17.3-fold increase in ES uptake for OATP1B1; 15.0-fold increase in EG uptake for OATP1B3; 13.2-fold increase in MPP+uptake for OCT1; and 17.8-fold increase in MPP+uptake for OCT2). Moreover, transport activity of P-gp and BCRP was also confirmed by the net efflux ratios of representative substrate in LLC-PK1 cells overexpressing P-gp and BCRP transporters. Net efflux ratio was calculated by comparing the efflux ratio of substrate in LLC-PK1 cells overexpressing P-gp and BCRP with that in LLC-PK1-mock cells. The net efflux ratio of digoxin and ES was 5.7 and 4.6 in LLC-PK1-MDR1 and LLC-PK1-BCRP compared with LLC-PK1-mock cells, respectively.
The uptake of catalposide was 319-, 13.6-, and 9.3-fold increased in HEK293 cells overexpressing the OAT3, OATP1B1, and OATP1B3 transporters, respectively, compared with that in HEK293 mock cells. Moreover, the increased uptake rate of catalposide mediated by OAT3, OATP1B1, and OATP1B3 was decreased to basal levels in the presence of representative inhibitors, such as probenecid and rifampin (Table 1). However, the OCT1, OCT2, and OAT1 transporters were apparently not involved in the uptake of catalposide into cells. In addition, the net efflux ratios of catalposide were 0.92 in LLC-PK1 cells overexpressing P-gp and BCRP transporters, and they were not changed by the presence of representative inhibitors of P-gp and BCRP (CsA and FTC, respectively) in LLC-PK1-MDR1 or LLC-PK1-BCRP cells (Table 2). On the basis of these results, it can also be concluded that catalposide is not a substrate of the MDR1 or BCRP transporters.
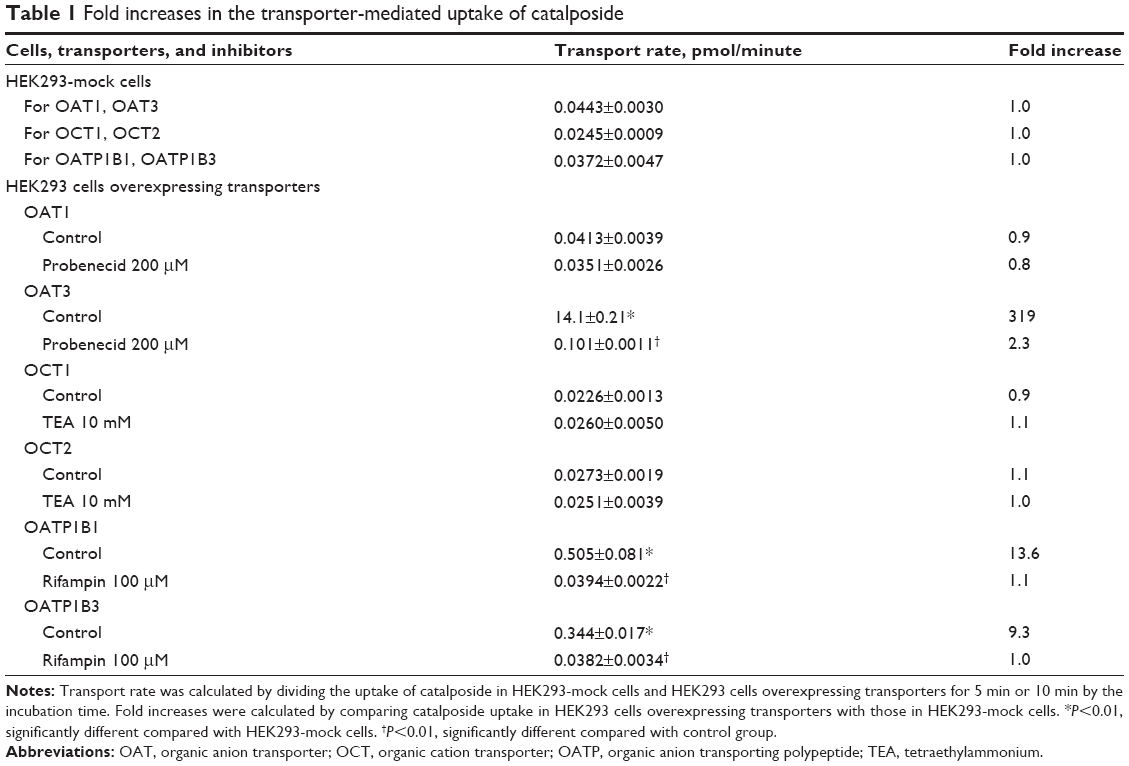 | Table 1 Fold increases in the transporter-mediated uptake of catalposide |
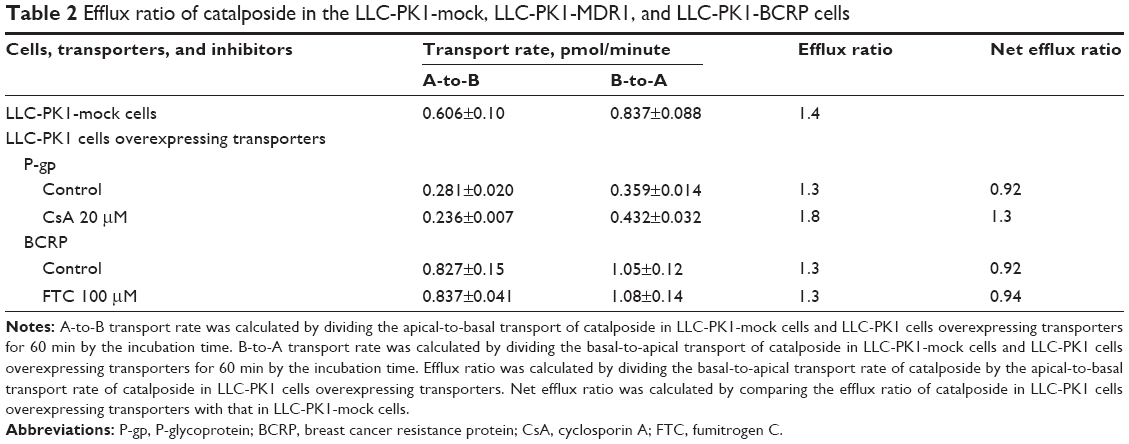 | Table 2 Efflux ratio of catalposide in the LLC-PK1-mock, LLC-PK1-MDR1, and LLC-PK1-BCRP cells |
We further evaluated OAT3-, OATP1B1-, and OATP1B3-mediated catalposide transport in the presence of representative inhibitors. Cimetidine, furosemide, and probenecid, representative inhibitors of OAT3,17,18 inhibited OAT3-mediated catalposide transport in a concentration-dependent manner (Figure 2A). Similarly, CsA, gemfibrozil, and rifampin, representative inhibitors of OATP1B1 and OATP1B3,19–21 also showed inhibitory effects on both OATP1B1- and OATP1B3-mediated catalposide transport in a concentration-dependent manner (Figure 2B and C). These results suggest that catalposide is actively taken up into cells via the OAT3, OATP1B1, and OATP1B3 transporters.
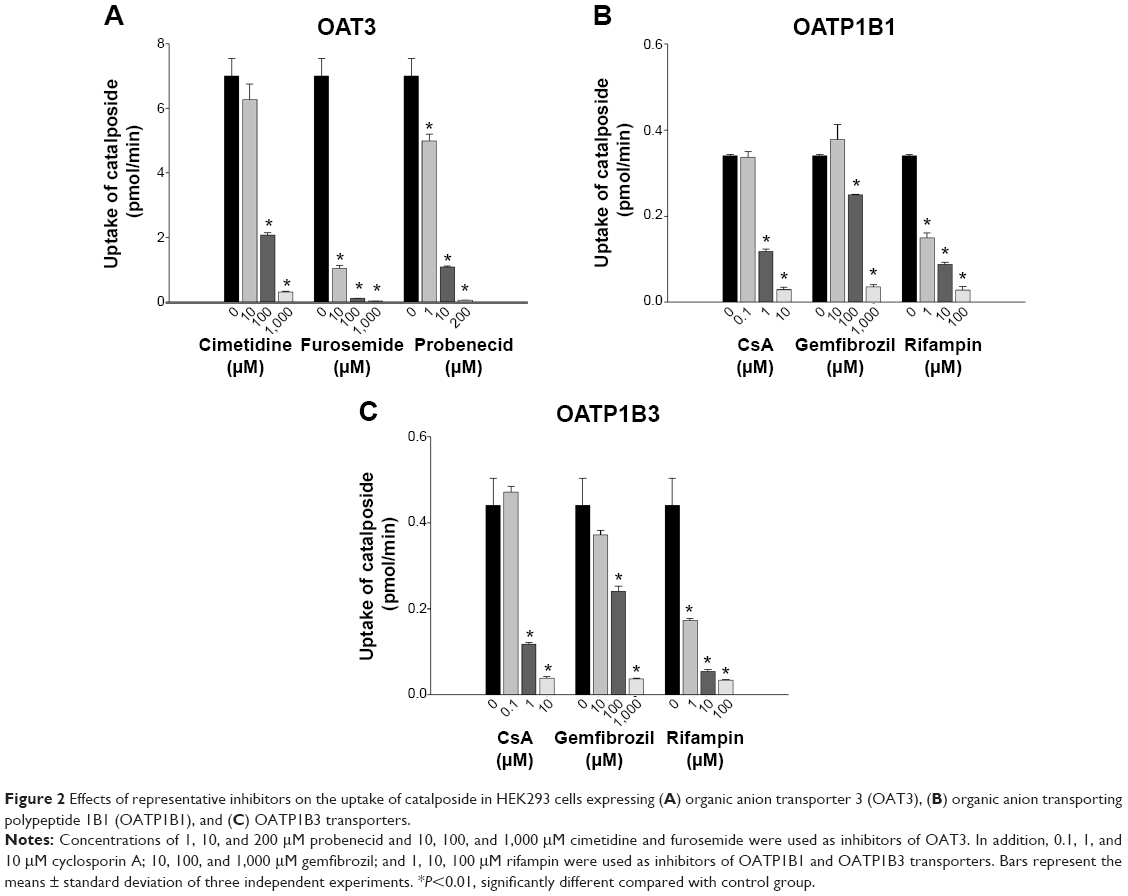 | Figure 2 Effects of representative inhibitors on the uptake of catalposide in HEK293 cells expressing (A) organic anion transporter 3 (OAT3), (B) organic anion transporting polypeptide 1B1 (OATP1B1), and (C) OATP1B3 transporters. |
To determine the kinetic parameters of concentration-dependent uptake of catalposide via OAT3, OATP1B1, and OATP1B3 transporters, the uptake of catalposide was measured in HEK293 cells overexpressing OAT3, OATP1B1, and OATP1B3 transporters with various concentrations of catalposide (2–500 μM; Figure 3). The Km, Vmax, and intrinsic clearance (Clint) values of catalposide uptake in OAT3 were 41.5 μM, 46.2 pmol/minute, and 1.11 μL/minute, respectively. In the case of OATP1B1, Km, Vmax, and Clint were 220.5 μM, 7.7 pmol/minute, and 0.035 μL/minute, respectively. For OATP1B3, Km, Vmax, and Clint were 502 μM, 16.9 pmol/minute, and 0.034 μL/minute, respectively.
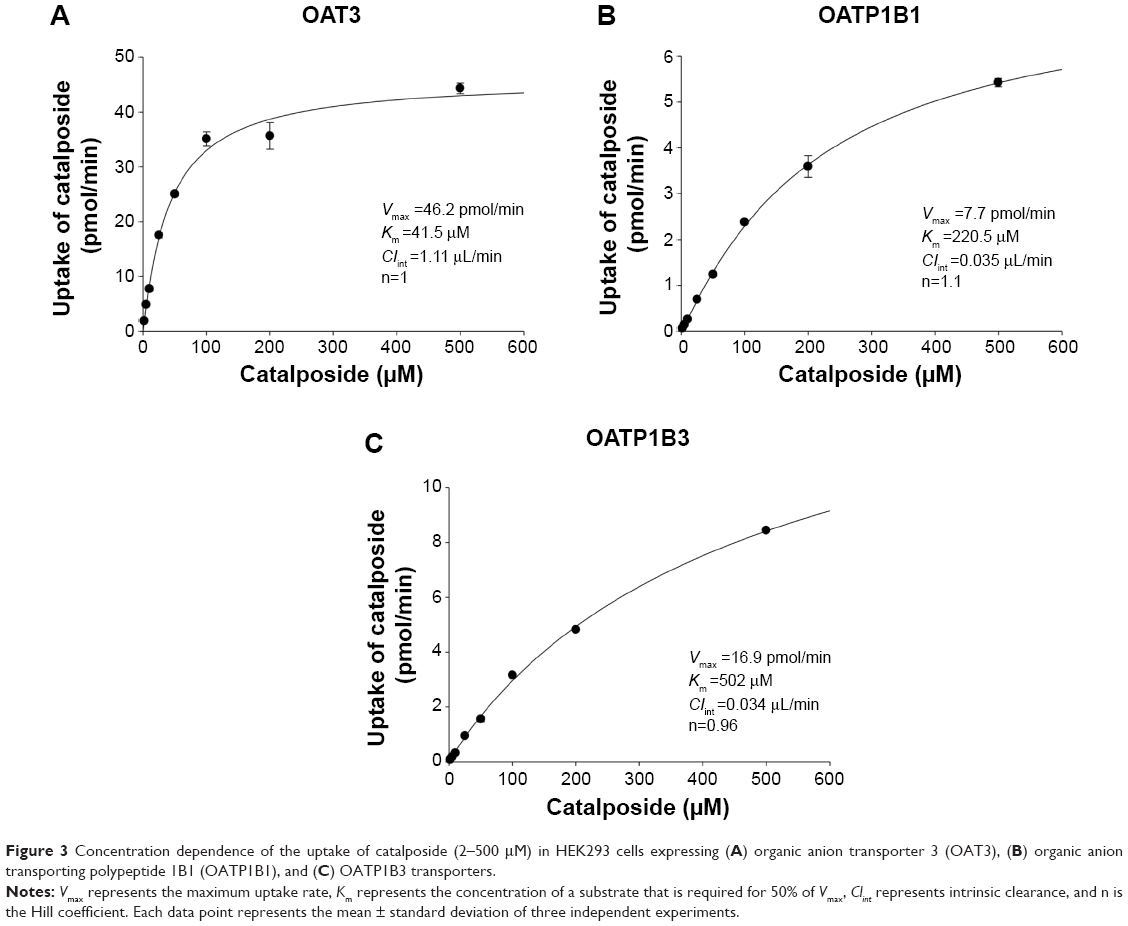 | Figure 3 Concentration dependence of the uptake of catalposide (2–500 μM) in HEK293 cells expressing (A) organic anion transporter 3 (OAT3), (B) organic anion transporting polypeptide 1B1 (OATP1B1), and (C) OATP1B3 transporters. |
Inhibitory effect of catalposide on OAT3, OATP1B1, and OATP1B3 transport activity
The inhibitory effects of catalposide on eight major transporters were evaluated using HEK293 and LLC-PK1 cell systems overexpressing OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2, P-gp, and BCRP transporters. Catalposide inhibited the transport activities of OAT3 with IC50 of 83 μM. Inhibitory effect of catalposide on the transport activities of OATP1B1 and OATP1B3 was also observed, as evidenced by high IC50 values of 200 and 235 μM, respectively (Figure 4). In contrast to these transporters, catalposide did not significantly inhibit transport activities of OCT1, OCT2, OAT1, P-gp, or BCRP in the concentration ranges tested (Figure 4).
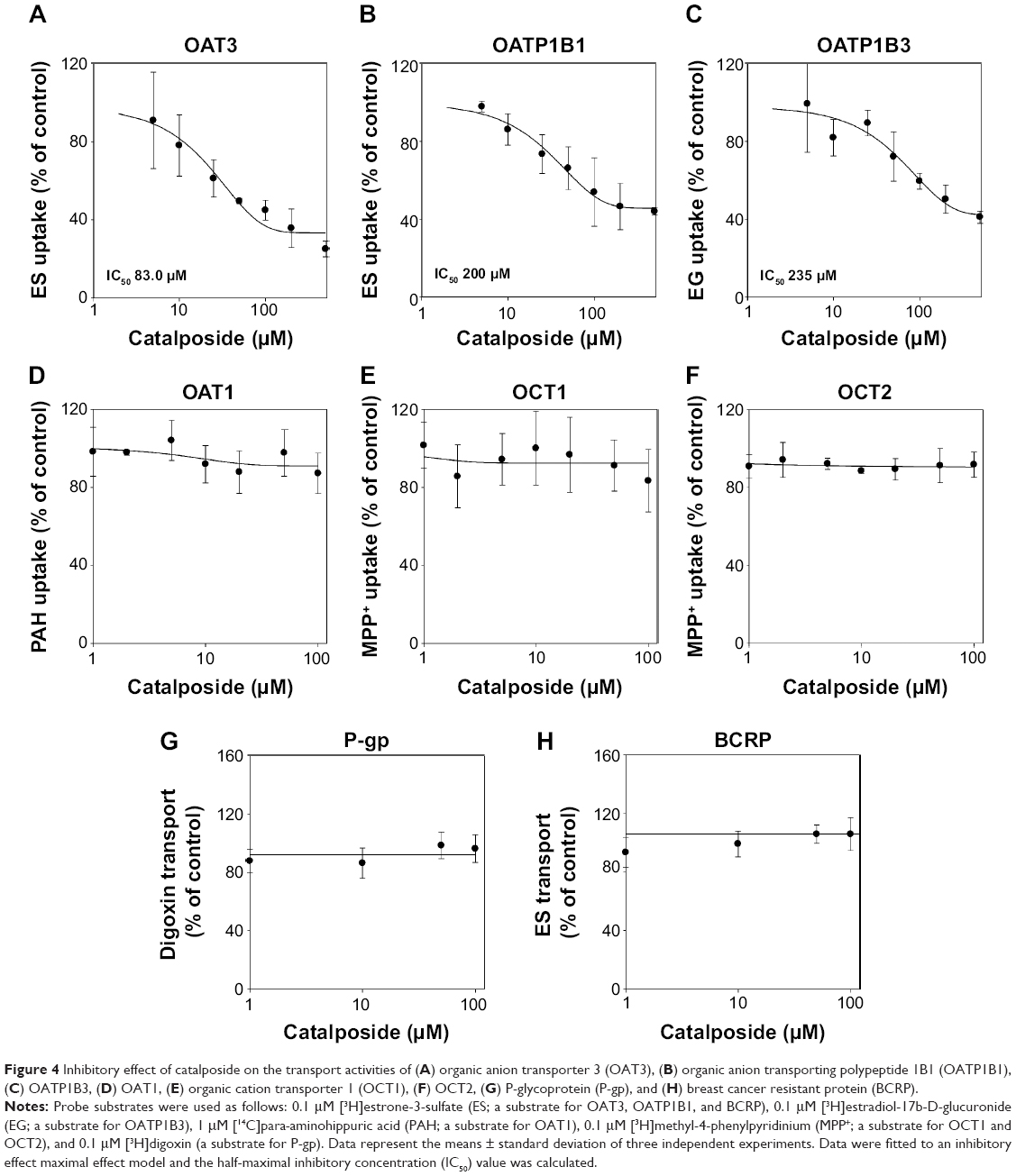 | Figure 4 Inhibitory effect of catalposide on the transport activities of (A) organic anion transporter 3 (OAT3), (B) organic anion transporting polypeptide 1B1 (OATP1B1), (C) OATP1B3, (D) OAT1, (E) organic cation transporter 1 (OCT1), (F) OCT2, (G) P-glycoprotein (P-gp), and (H) breast cancer resistant protein (BCRP). |
Discussion
In this study, we comprehensively assessed the substrate specificity of catalposide with regard to clinically important drug transporters such as OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2, P-gp, and BCRP, as well as the inhibitory effects of catalposide on these transporters. Among these eight transporters, cellular accumulation of catalposide was mediated by OAT3, OATP1B1, and OATP1B3 transporters, as evidenced by the 319-, 13.6-, and 9.3-fold increases in catalposide uptake in HEK293 cells overexpressing OAT3, OATP1B1, and OATP1B3, respectively, compared with that in HEK293 mock cells. We further confirmed the substrate specificity of catalposide by examining OAT3-, OATP1B1-, and OATP1B3-mediated catalposide uptake in the presence of representative inhibitors of these transporters. Cimetidine, furosemide, and probenecid were selected as inhibitors of OAT3, with IC50 values of 92.4±10.9, 51.1±10.5, and 4.93±27.9 μM, respectively.17,18 On the basis of these IC50 values, various concentrations of OAT3 inhibitors were applied to assess OAT3-mediated catalposide uptake. It was observed that 100 μM cimetidine and furosemide inhibited OAT3-mediated catalposide uptake by 70.4% and 98.3%, respectively, whereas 10 μM probenecid inhibited catalposide uptake by 84.5%. These differential inhibitory effects could be attributed to the different IC50 values of the OAT3 inhibitors, and collectively, the results show that OAT3 is involved in the catalposide uptake process. Similarly, CsA, gemfibrozil, and rifampin were selected as inhibitors of OATP1B1 with IC50 values of 1.88, 156.2, and 0.99 μM, respectively.19,20 CsA, gemfibrozil, and rifampin were also applied as inhibitors of OATP1B3, with IC50 values of 0.20, ~200, and 0.65 μM, respectively.19–21
Use of 1 μM CsA resulted in inhibition of OATP1B1- and OATP1B3-mediated catalposide uptake by 65.3% and 73.3%, respectively. Gemfibrozil and rifampin inhibited OATP1B1- and OATP1B3-mediated catalposide uptake in a similar manner. Consistent with these results, OATP1B1- and OATP1B3-mediated catalposide uptake showed concentration dependency (Figure 3). The Hill coefficients for OAT3, OATP1B1, and OATP1B3 were all close to 1, suggesting a reversible and competitive interaction between these transporters and catalposide, with 1:1 stoichiometry.22,23 The Clint value for OAT3 was 31.8-fold and 32.7-fold greater than those for OATP1B1 and OATP1B3, respectively, which seems to be mainly attributable to the low Km and high Vmax values compared with those for OATP1B1 and OATP1B3. Considering that OAT3 is expressed predominantly in the kidney and plays a role in the uptake of organic anionic drugs from the blood,24,25 OAT3 may contribute to the renal distribution and excretion of catalposide. Although the affinities of OATP1B1 and OATP1B3 were considerably lower than those of OAT3, OATP1B1, and OATP1B3, they may contribute to the hepatic distribution of this compound, based on their predominant expression in the liver.26
OAT3, OATP1B1, and OATP1B3 transporters mediated renal and hepatic uptake of catalposide, which would be excreted via renal and biliary routes with or without coupling non-CYP mediated metabolism. Therefore, we first explored the involvement of P-gp and BCRP. From the results of Table 2, we concluded that these transporters were not involved in the excretion of catalposide. In spite of the lack of involvement of P-gp and BCRP in the transport of catalposide (Table 2), we should note the limitation of the use of specific inhibitors. We used CsA (25 μM) and FTC (100 μM) as P-gp and BCRP inhibitors, respectively. CsA inhibits not only P-gp but also other transporters such as OATP family transporters, multidrug resistance-associated protein 2 (MRP2), bile salt efflux pump, and Na+-taurocholate cotransporting polypeptide.8 Therefore, the inhibition of other transporters by the CsA treatment can compromise the involvement of P-gp and other transporters. However, as we used LLC-PK1-MDR1 cells that overexpress P-gp, the inhibition of other transporters by CsA might play a minor role in the efflux of catalposide in LLC-PK1-MDR1 cells, if any. Similarly, a high concentration of FTC more than 10 μM was reported to inhibit P-gp function, as well as BCRP activity, in Caco-2 cells.27 However, BCRP inhibition by 100 μM FTC was performed in LLC-PK1-BCRP cells in this study; therefore, even though we measured a combined inhibitory effect of BCRP and P-gp on the catalposide efflux in our system, the contribution of P-gp might be negligible.
Because catalposide is an anionic weak acid, it can be speculated that MRP transporters may be involved in its membrane transport. MRP transporters are known to efflux organic anions as well as glucuronidated, sulfated, and glutathione-conjugated metabolites.28,29 Among the MRP isoforms, MRP2 is expressed at the canalicular membrane, whereas MRP3 and MRP4 are expressed at the sinusoidal membrane of hepatocytes.28 Because approximately 90% of catalposide was cleared by nonrenal clearance, it is possible that MRPs may facilitate biliary excretion of catalposide and/or its metabolites. MRP2 and MRP4 are also expressed at the apical membrane of renal proximal tubule epithelial cells,28 suggesting these transporters may also play a role in clearing catalposide28 via the renal route. Therefore, an investigation into MRP-mediated efflux transport of catalposide and its metabolites requires careful examination.
Because therapeutics are increasingly being used as “poly pills,” HDIs between coadministered herbs and drugs should be considered carefully, and the mechanisms underlying such HDIs have been investigated. Drug transporters, alone or in combination with drug-metabolizing enzymes, have drawn much attention in HDIs because they regulate the absorption, distribution, and elimination of drugs.8,16,30,31
It is recommended that clinically important transporters, such as OAT1, OAT3, OATP1B1, OATP1B3, OCT2, P-gp, and BCRP, be considered for screening during drug development.8 However, it should be noted that in vivo drug interactions can be predicted from in vitro inhibition studies, using the effective plasma concentration, unbound fraction, and IC50 values of inhibitors. Thus, it is important to determine whether the in vitro inhibition of OAT3, OATP1B1, and OATP1B3 transport activities by catalposide is in fact relevant through in vivo and, ultimately, clinical studies of the pharmacokinetics and efficacy of catalposide.8,16 As a result, IC50 values for the OAT3, OATP1B1, and OATP1B3 transporters were estimated to be 83, 200, and 253 μM, respectively (Figure 4). The effective concentration of catalposide required for anti-inflammatory effect and PPAR-α activation has been reported as about 0.2–4 μM in an in vitro cell system.1,3,5,6 However, the effective plasma concentration of catalposide has not been determined yet in in vivo animal and human studies, and the probability of in vivo drug–drug interactions between catalposide and substrates for OAT3, OATP1B1, and OATP1B3 should be carefully evaluated.
On this theme, previous research articles on the involvement of organic anion transporters in HDIs, and in particular OAT3, OATP1B1, and OATP1B3, were published.32–35 For example, bioactive flavonol, icariin, potentially inhibited OATP2B1 and OATP1B3 activities.33 Bicalein and wogonin (major components of Scutellaria baicalensis) significantly inhibited transport activities of OATP1B3, OAT1, and OAT3 and so on.32
In conclusion, a comprehensive evaluation of the transport mechanisms and transporter-mediated HDI of catalposide on eight major transporters was conducted across a wide range of catalposide concentrations using HEK293 and LLC-PK1 cell systems overexpressing the OCT1, OCT2, OAT1, OAT3, OATP1B1, OATP1B3, P-gp, and BCRP transporters (Table 3). Uptake of catalposide was increased greatly in HEK293 cells overexpressing OAT3, OATP1B1, and OATP1B3 transporters compared with that in mock cells. In addition, catalposide inhibited the transport activities of OAT3, OATP1B1, and OATP1B3, with IC50 values of 83, 200, and 253 μM, respectively. These findings suggest the OAT3, OATP1B1, and OATP1B3 transporters may be important in the pharmacokinetics and HDI of catalposide, although further evaluation is needed regarding their contribution to its pharmacokinetics in vivo.
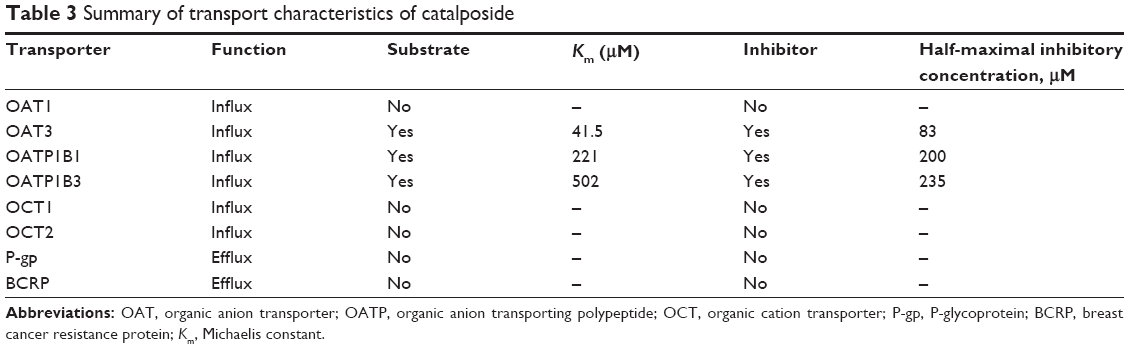 | Table 3 Summary of transport characteristics of catalposide |
Acknowledgments
This work was supported by the Industrial Strategic Technology Development Program (Global Botanical Drug Development for the US Food and Drug Administration approval of US clinical trials, 10039303) funded by the Ministry of Trade, Industry and Energy (MI, Korea) and The Catholic University of Korea, 2012 (M-2012-B0002-00024).
Disclosure
The authors report no conflicts of interest in this work.
References
Kim SW, Choi SC, Choi EY, et al. Catalposide, a compound isolated from catalpa ovata, attenuates induction of intestinal epithelial proinflammatory gene expression and reduces the severity of trinitrobenzene sulfonic Acid-induced colitis in mice. Inflamm Bowel Dis. 2004;10(5):564–572. | ||
Yang G, Choi CH, Lee K, Lee M, Ham I, Choi HY. Effects of Catalpa ovata stem bark on atopic dermatitis-like skin lesions in NC/Nga mice. J Ethnopharmacol. 2013;145(2):416–423. | ||
Lee JH, Jun HJ, Hoang MH, et al. Catalposide is a natural agonistic ligand of peroxisome proliferator-activated receptor-α. Biochem Biophys Res Commun. 2012;422(4):568–572. | ||
An SJ, Pae HO, Oh GS, et al. Inhibition of TNF-alpha, IL-1beta, and IL-6 productions and NF-kappa B activation in lipopolysaccharide-activated RAW 264.7 macrophages by catalposide, an iridoid glycoside isolated from Catalpa ovata G. Don (Bignoniaceae). Int Immunopharmacol. 2002;2(8):1173–1181. | ||
Moon MK, Choi BM, Oh GS, et al. Catalposide protects Neuro 2A cells from hydrogen peroxide-induced cytotoxicity via the expression of heme oxygenase-1. Toxicol Lett. 2003;145(1):46–54. | ||
Oh H, Pae HO, Oh GS, et al. Inhibition of inducible nitric oxide synthesis by catalposide from Catalpa ovata. Planta Med. 2002;68(8):685–689. | ||
Ji HY, Lee HW, Kim HH, Lee HI, Chung HT, Lee HS. Determination of catalposide in rat plasma by liquid chromatography-mass spectrometry. Anal Lett. 2003;36(14):2999–3009. | ||
Giacomini KM, Huang SM, Tweedie DJ, et al; International Transporter Consortium. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–236. | ||
Deng JW, Shon JH, Shin HJ, et al. Effect of silymarin supplement on the pharmacokinetics of rosuvastatin. Pharm Res. 2008;25(8): 1807–1814. | ||
Chen XW, Sneed KB, Pan SY, et al. Herb-drug interactions and mechanistic and clinical considerations. Curr Drug Metab. 2012;13(5):640–651. | ||
Mai I, Bauer S, Perloff ES, et al. Hyperforin content determines the magnitude of the St John’s wort-cyclosporine drug interaction. Clin Pharmacol Ther. 2004;76(4):330–340. | ||
Fasinu PS, Bouic PJ, Rosenkranz B. An overview of the evidence and mechanisms of herb-drug interactions. Front Pharmacol. 2012;3:69. | ||
Chen XW, Serag ES, Sneed KB, et al. Clinical herbal interactions with conventional drugs: from molecules to maladies. Curr Med Chem. 2011; 18(31):4836–4850. | ||
Choi MK, Song IS. Characterization of efflux transport of the PDE5 inhibitors, vardenafil and sildenafil. J Pharm Pharmacol. 2012;64(8): 1074–1083. | ||
Zhang S, Lovejoy KS, Shima JE, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66(17): 8847–8857. | ||
Choi MK, Kwon M, Ahn JH, Kim NJ, Bae MA, Song IS. Transport characteristics and transporter-based drug-drug interactions of TM-25659, a novel TAZ modulator. Biopharm Drug Dispos. 2014;35(3):183–194. | ||
Juhász V, Beéry E, Nagy Z, et al. Chlorothiazide is a substrate for the human uptake transporters OAT1 and OAT3. J Pharm Sci. 2013;102(5): 1683–1687. | ||
Khamdang S, Takeda M, Shimoda M, et al. Interactions of human- and rat-organic anion transporters with pravastatin and cimetidine. J Pharmacol Sci. 2004;94(2):197–202. | ||
Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 2009;158(3):693–705. | ||
Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology. 2002;36(1):164–172. | ||
Noé J, Portmann R, Brun ME, Funk C. Substrate-dependent drug-drug interactions between gemfibrozil, fluvastatin and other organic anion-transporting peptide (OATP) substrates on OATP1B1, OATP2B1, and OATP1B3. Drug Metab Dispos. 2007;35(8):1308–1314. | ||
Prinz H. Hill coefficients, dose-response curves and allosteric mechanisms. J Chem Biol. 2010;3(1):37–44. | ||
Weiss JN. The Hill equation revisited: uses and misuses. FASEB J. 1997;11(11):835–841. | ||
Sekine T, Miyazaki H, Endou H. Molecular physiology of renal organic anion transporters. Am J Physiol Renal Physiol. 2006;290(2): F251–F261. | ||
Jin L, Kikuchi R, Saji T, Kusuhara H, Sugiyama Y. Regulation of tissue-specific expression of renal organic anion transporters by hepatocyte nuclear factor 1 α/β and DNA methylation. J Pharmacol Exp Ther. 2012;340(3):648–655. | ||
Ho RH, Tirona RG, Leake BF, et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130(6):1793–1806. | ||
Mease K, Sane R, Podila L, Taub ME. Differential selectivity of efflux transporter inhibitors in Caco-2 and MDCK-MDR1 monolayers: a strategy to assess the interaction of a new chemical entity with P-gp, BCRP, and MRP2. J Pharm Sci. 2012;101(5):1888–1897. | ||
Dallas S, Miller DS, Bendayan R. Multidrug resistance-associated proteins: expression and function in the central nervous system. Pharmacol Rev. 2006;58(2):140–161. | ||
Keppler D. Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb Exp Pharmacol. 2011; (201):299–323. | ||
Choi MK, Jin QR, Ahn SH, Bae MA, Song IS. Sitagliptin attenuates metformin-mediated AMPK phosphorylation through inhibition of organic cation transporters. Xenobiotica. 2010;40(12):817–825. | ||
Chu X, Cai X, Cui D, et al. In vitro assessment of drug-drug interaction potential of boceprevir associated with drug metabolizing enzymes and transporters. Drug Metab Dispos. 2013;41(3):668–681. | ||
Xu F, Li Z, Zheng J, et al. The inhibitory effects of the bioactive components isolated from Scutellaria baicalensis on the cellular uptake mediated by the essential solute carrier transporters. J Pharm Sci. 2013;102(11):4205–4211. | ||
Li Z, Cheung FS, Zheng J, Chan T, Zhu L, Zhou F. Interaction of the bioactive flavonol, icariin, with the essential human solute carrier transporters. J Biochem Mol Toxicol. 2014;28(2):91–97. | ||
Li Z, Wang K, Zheng J, et al. Interactions of the active components of Punica granatum (pomegranate) with the essential renal and hepatic human Solute Carrier transporters. Pharm Biol. 2014;52(12): 1510–1517. | ||
Wang L, Pan X, Sweet DH. The anthraquinone drug rhein potently interferes with organic anion transporter-mediated renal elimination. Biochem Pharmacol. 2013;86(7):991–996. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.