")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Optimized nano-transfersomal films for enhanced sildenafil citrate transdermal delivery: ex vivo and in vivo evaluation
Authors Badr-Eldin S, Ahmed O
Received 24 December 2015
Accepted for publication 22 February 2016
Published 5 April 2016 Volume 2016:10 Pages 1323—1333
DOI https://doi.org/10.2147/DDDT.S103122
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Shaimaa M Badr-Eldin,1,2 Osamaa AA Ahmed1,3
1Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Kingdom of Saudi Arabia; 2Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University, Cairo, Egypt; 3Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Minia University, Minia, Egypt
Abstract: Sildenafil citrate (SLD) is a selective cyclic guanosine monophosphate-specific phosphodiesterase type 5 inhibitor used for the oral treatment of erectile dysfunction and, more recently, for other indications, including pulmonary hypertension. The challenges facing the oral administration of the drug include poor bioavailability and short duration of action that requires frequent administration. Thus, the objective of this work is to formulate optimized SLD nano-transfersomal transdermal films with enhanced and controlled permeation aiming at surmounting the previously mentioned challenges and hence improving the drug bioavailability. SLD nano-transfersomes were prepared using modified lipid hydration technique. Central composite design was applied for the optimization of SLD nano-transfersomes with minimized vesicular size. The independent variables studied were drug-to-phospholipid molar ratio, surfactant hydrophilic lipophilic balance, and hydration medium pH. The optimized SLD nano-transfersomes were developed and evaluated for vesicular size and morphology and then incorporated into hydroxypropyl methyl cellulose transdermal films. The optimized transfersomes were unilamellar and spherical in shape with vesicular size of 130 nm. The optimized SLD nano-transfersomal films exhibited enhanced ex vivo permeation parameters with controlled profile compared to SLD control films. Furthermore, enhanced bioavailability and extended absorption were demonstrated by SLD nano-transfersomal films as reflected by their significantly higher maximum plasma concentration (Cmax) and area under the curve and longer time to maximum plasma concentration (Tmax) compared to control films. These results highlighted the potentiality of optimized SLD nano-transfersomal films to enhance the transdermal permeation and the bioavailability of the drug with the possible consequence of reducing the dose and administration frequency.
Keywords: sildenafil citrate, central composite design, transfersomes, edge activator, permeation, transdermal, pharmacokinetics
Introduction
Sildenafil citrate (SLD), chemically known as 1-[[3-(6,7-dihydro-1-methyl-7-oxo-3- propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-ethoxyphenyl]sulfonyl]-4-methylpiperazine citrate, is a selective inhibitor of cyclic guanosine monophosphate-specific phosphodiesterase type 5.1 It was approved by the US Food and Drug Administration in 1998 for the oral treatment of erectile dysfunction.2 Recently, it has been used for the treatment of pulmonary arterial hypertension and the enhancement of uteroplacental perfusion in case of fetal growth retardation.3,4 However, the drug suffers from poor oral bioavailability (~40%) that could be attributed to its low water solubility (3.5 mg/mL) and extensive presystemic metabolism, in addition to absorption hindrance by food, especially fat-rich meals.5–9 To surpass this problem, researchers have been attracted to the investigation of SLD delivery via other routes of administration. Several SLD formulations avoiding peroral route have been developed, including orally disintegrating and orally dissolving films, intranasal microemulsions, and transdermal nanostructure lipid carriers and solid lipid nanoparticles.10–14
Transdermal delivery of drugs via the skin provides a leading alternative to peroral route due to bypassing the presystemic metabolism of drugs, prolonging their effect, and reducing inter- and intrasubject variability.15 A major challenge for transdermal drug delivery is the low permeation across the skin due to the natural barrier function of the stratum corneum (SC).16 Several approaches have been applied to improve skin penetration of drugs, including chemical approaches, that is, the use of penetration enhancers, in addition to physical approaches of iontophoresis and sonophoresis.17,18 Recently, attention has been focused on the use of lipid vesicular formulations for enhancing transdermal drug delivery.
Liposomes, the first generation of vesicular formulations, have been widely used as drug delivery system via several routes. Topical liposomal formulations have gained interest due to their safety, controlled release property, and enhanced clinical efficacy.19 However, higher tendency of conventional liposomes to deposit in the upper layers of the skin rather than to penetrate through the deeper layers to give systemic effect has been previously reported.20 New generations of liposomes have been developed to enhance transdermal drug delivery such as ethosomes and transfersomes.20,21 Transfersomes are flexible and ultradeformable vesicular systems, composed of phospholipids and a single-chain surfactant that acts as an edge activator. The edge activator destabilizes the lipid bilayers, thus imparting ultra-flexibility to the vesicular structure that enhances its squeezing and penetration through the skin.22,23 The utilization of transfersomes as a promising transdermal drug delivery system has been studied by several investigators.24–26 Due to the practical difficulty of applying vesicular systems on the skin, several studies have further incorporated them into gel bases and patch formulations.27–29
A screening study to investigate formulation and processing factors that affect the characteristics of SLD transfersomes has been previously conducted in our laboratory.30 Based on the findings of the aforementioned screening study, the aim of this work is to develop an optimized nanosized SLD transfersomal transdermal film with enhanced drug delivery through the skin to the systemic circulation and improved bioavailability. Central composite design (CCD) was applied for the optimization of SLD transfersomes. The optimized transdermal nano-transfersomal film was subjected to ex vivo permeation studies using abdominal rat skin. Furthermore, in vivo pharmacokinetic study was conducted in rats to assess the bioavailability of SLD from the films.
Materials
SLD, Span 80, Span 60, and hydroxypropyl methyl cellulose (HPMC), molecular weight (MW) 86,000 and viscosity 4,000 cP, were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). L-α-Phosphatidylcholine (soy-95%), MW 775.04, was purchased from Avanti Polar Lipids (Alabaster, AL, USA). High-performance liquid chromatography (HPLC) grade methanol and acetonitrile were obtained from Merck (Darmstadt, Germany). All other reagents and chemicals were of analytical grade. Double-distilled water was used throughout the experiments.
Methods
Preparation of SLD nano-transfersomes
Modified lipid film hydration technique was used to prepare SLD nano-transfersomes.30 Briefly, specified amount of the drug, L-α-phosphatidylcholine, and the edge activator (surfactant) were dissolved in methanol. The mixture was sonicated for 5 minutes in a water bath ultrasonicator (QS3, model F 0023902; Ultrawave Ltd, Cardiff, UK). Rotary evaporation of the solution was then conducted using a Buchi Rotavapor (R-200; BÜCHI Labortechnik AG, Flawil, Switzerland) at a temperature of 45°C under reduced pressure for the removal of excess methanol. The thin film, formed on the flask wall, was kept overnight in a vacuum oven (model 6505; Thermo Fisher Scientific, Waltham, MA, USA) for complete dryness. The dried film was then hydrated with 20 mL phosphate buffer saline (PBS) of specified pH with gentle shaking for 2 hours at room temperature.
Response surface methodology for the optimization of SLD nano-transferosomes
Based on the findings of the screening study previously conducted in our laboratory, response surface methodology was applied for the optimization of SLD nano-transfersomes with minimized vesicle size. Specifically, a three-factor three-level CCD was used. Drug-to-phospholipid molar ratio (X1), surfactant hydrophilic lipophilic balance (HLB) (X2), and hydration medium pH (X3) were considered as independent variables. The vesicle size (Y) was selected as dependent variable (response). All other formulation and processing variables were kept constant throughout the study. The variables and levels used in the design are presented in Table 1. Sixteen experimental runs were prepared including eight (23) fractional factorial points, six axial points to estimate curvature, along with two replicates in the center (Table 2). The axial points were located so that their distance from the center of the design space is equal to plus or minus alpha, |α|. The value of alpha (α) was set to 1.682 to achieve a rotatable design (to maintain rotatability, the value of α = [number of factorial runs]1/4 = [23]1/4 =1.682). Statistical analysis was performed using Design-Expert® Software version 7.0.0 (Stat-Ease Inc., Minneapolis, MN, USA) to assess the effect of the independent variables on the vesicle size (Y) as well as the interaction between these variables. To obtain the highest prediction power, three mathematical polynomial models, namely, linear (main effects only), two-factor interaction (effects and interactions), and quadratic models (effects, interactions, and quadratic terms) were evaluated. The equation was then determined according to the model selected to be either:
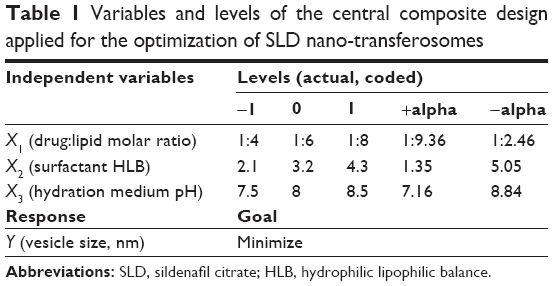 | Table 1 Variables and levels of the central composite design applied for the optimization of SLD nano-transferosomes |
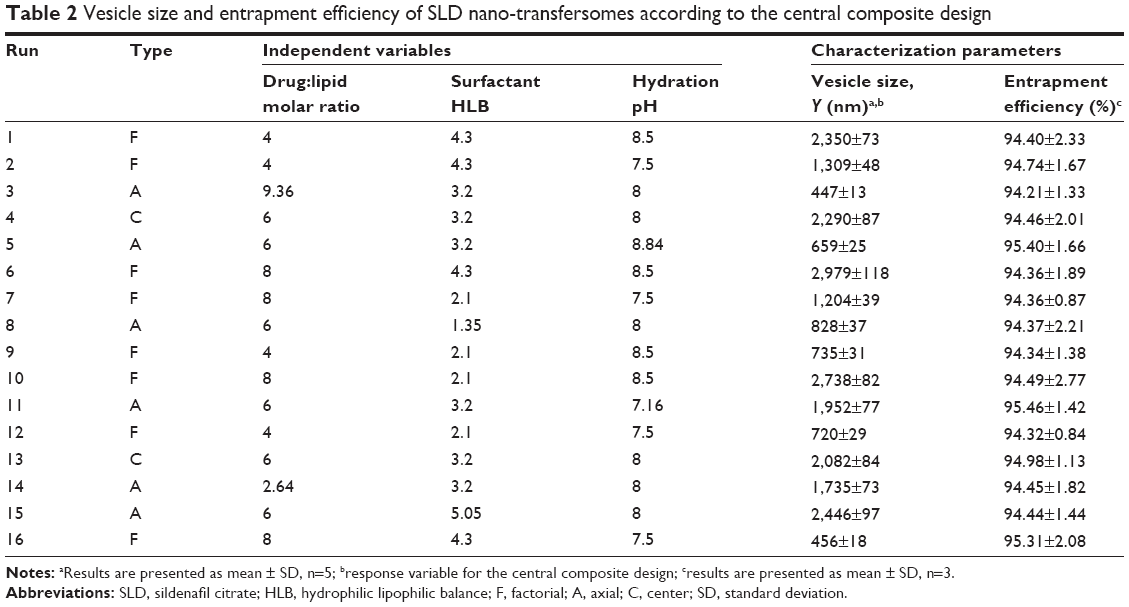 | Table 2 Vesicle size and entrapment efficiency of SLD nano-transfersomes according to the central composite design |
|
|
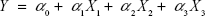
|
|
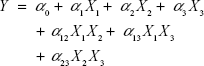
|
|
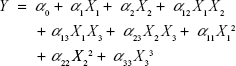
where Y is the measured response; α0 is a constant representing the arithmetic mean of the response of the 16 formulations; α1, α2, and α3 are the estimated linear coefficients of the factors X1, X2, and X3, respectively, that are related to the magnitude of the effect of these variables on the response; α12, α13, and α23 are the interaction coefficients between each two factors; and α11, α22, and α33 are their quadratic coefficients.26 A synergistic effect on the response is indicated by a positive sign of the coefficient, while an antagonistic effect is indicated by a negative sign.31 The level of statistical significance was set at P≤0.05. Numerical optimization method following desirability approach was applied to determine the composition of the optimized SLD nano-transfersomal formulation with minimized vesicular size. The observed and predicted values for the vesicle size of the optimized formulation were statistically compared.
Entrapment efficiency
The entrapment efficiency percent (EE%) of SLD nano-transfersomes was determined using indirect centrifugation method. The nano-transfersomal dispersions were centrifuged at 18,000 rpm for 1 hour at 4°C (Sigma Laboratory Centrifuge, Model 3K30, Osterode, Germany) to separate the free unentrapped drug from the vesicles. The supernatant was then filtered through a membrane filter (0.1 μm; EMD Millipore). Aliquots of the filtered supernatant were appropriately diluted, and the concentration of the unentrapped drug was determined using validated HPLC method of assay described later. The % SLD entrapped in the nano-transfersomes was calculated using the following equation:
|
|
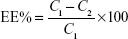
where C1 represents the initial amount of drug used and C2 represents the amount of free unentrapped drug in the supernatant.
HPLC analysis of SLD
Modified method of Sheu et al32 adapted to our laboratory was applied for the determination of SLD. Agilent 1200 series HPLC system consisting of quaternary pump (HP 1200; Agilent Technologies, Santa Clara, CA, USA) with a photodiode array detector (HP 1200; Agilent Technologies) was used. The analytical column was C18, 250 mm length ×4.6 mm internal diameter, and particle size 5 μm (Phenomenex, Torrance, CA, USA). The mobile phase was prepared by mixing 30 mM potassium dihydrogen phosphate and acetonitrile in the ratio of 55:45, and the pH was adjusted to 6 using 1N sodium hydroxide. In vitro calibration curve was constructed in the range of 10–1,000 ng/mL by using increasing aliquots of SLD stock solution in methanol (1 mg/mL). A 0.1 mL of propyl paraben solution in methanol as an internal standard stock solution (1 mg/mL) was added to the sample. The volume was adjusted with methanol to obtain the final desired concentration, and 20 μL aliquot of this solution was injected into the HPLC system. The flow rate was set at 0.8 mL/min at ambient temperature, and detection was carried out at 290 nm. The assay procedure was validated in terms of linearity, precision, and accuracy (correlation coefficient, R=0.998; lower limit of detection =10 ng/mL; lower limit of quantification =40 ng/mL; relative standard deviation for interday and intraday assay <5%, accuracy =96.60%). Concentrations of SLD in the withdrawn samples were calculated with reference to the calibration curve of drug/internal standard (IS) peak area ratio against the corresponding SLD concentration.
Vesicle size measurements
The mean vesicle size of SLD nano-transfersomes dispersions was determined by dynamic light scattering technique using Zetatrac (Microtrac Inc., Montgomeryville, PA, USA). Each measurement was done five times, and the mean vesicle size was calculated.
Transmission electron microscopy
The vesicular shape and morphology of the optimized SLD nano-transfersomes were studied using transmission electron microscope (100 CX-TEM; JEOL, Tokyo, Japan). A drop of diluted transfersomal dispersion was adsorbed onto a carbon-coated grid and then stained with 2% uranyl acid. The excess solution was removed, and the grid was allowed to dry thoroughly before visualization.
Preparation of SLD nano-transfersomal transdermal films
The optimized SLD nano-transfersomes were prepared, characterized for vesicular size, and then incorporated into HPMC transdermal films. Briefly, specified amounts of HPMC (matrix forming polymer) were dispersed in double-distilled water to obtain a final concentration of 2%. Propylene glycol was used as a plasticizer, and dimethyl sulfoxide was used as a penetration enhancer at a concentration of 1% for both. The films’ composition and the concentrations used were chosen based on preliminary studies for the film properties, performed according to the results of our previous study.33 Specified amount of optimized SLD nano-transfersomes was added to the casting solution with gentle stirring and then left for 24 hours at 4°C to obtain a clear solution. The formed gel was then poured into 9 cm petri dishes. Petri dishes were then left to dry in an oven at 40°C for complete evaporation of water. The films were then covered with backing membrane (CoTran™; 3M, St Paul, MN, USA) and cut into 1×1 cm square strips (1 cm2) of uniform thickness, each contained amount of transfersomes equivalent to 1 mg of drug. The strips were packed in aluminum foil and kept in a desiccator over CaCl2 at 25°C until further evaluation.33 Control films loaded with raw SLD powder were prepared using the same procedure and the same composition for comparison.
Ex vivo permeation of SLD from transdermal films
Permeation study
In vitro release and permeation of SLD from optimized SLD nano-transfersomal films and SLD films were studied using automated Franz diffusion cell apparatus (MicroettePlus; Hanson research, Chatsworth, CA, USA). Full thickness excised abdominal male Wistar rat skin (2.5×2.5 cm) was used as a membrane and carefully freed from any subcutaneous fats and examined using magnifier to assure skin integrity.34 The membrane was mounted between the two chambers of the diffusion cell with the SC facing the donor chamber and the dermis facing the receptor chamber. After equilibration in PBS for 15 minutes at 32°C, film strips were applied in the donor compartment through direct contact to the skin. PBS (pH 7.4) was used as a diffusion medium in the receptor chamber (effective diffusion area of 1.76 cm2 and volume of 7 mL). The temperature of the diffusion medium was maintained constant at 32°C±0.5°C, and the agitation rate was set to 400 rpm. Aliquots of 2 mL were automatically withdrawn at time intervals of 0.5, 1, 2, 4, 6, 8, 12, and 24 hours and replaced with fresh PBS to maintain constant volume.14 The withdrawn samples were analyzed using the previously described validated HPLC method.32 All experiments were performed in triplicate.
Permeation data analysis and kinetic modeling
Permeation parameters, namely, steady-state flux, permeability, and diffusion coefficients, were calculated. Steady-state flux, Jss (μg/cm2 H), was computed from the slope of the linear portion of the cumulative amount drug permeated per squared cm versus time plot. The permeability coefficient, Pc (cm/h), was determined by dividing the steady-state flux by the initial dug load.27 Diffusion coefficient (D) was obtained using the following equation:
|
|
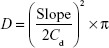
where Cd is the initial drug concentration in the donor compartment and the slope is that of the cumulative amount of drug permeated versus square root of time plot.35
The obtained results from the permeation study were fitted into equations of zero, first, Higuchi, and Korsmeyer–Peppas models to describe the kinetics and mechanism of drug permeation from the films.36–38 Linearity was determined by linear regression analysis, and the correlation coefficient (r) was used for evaluation of the goodness of fit to each model. The release exponent (n), calculated according to Korsmeyer–Peppas model, was used to elucidate the drug permeation mechanism.
In vivo evaluation of the optimized SLD nano-transfersomal transdermal film
Study design
In vivo evaluation of the optimized SLD nano-transfersomal transdermal films was performed in male Wistar rats weighing 200–250 g (n=36) in comparison to SLD transdermal films and SLD oral suspension. The study protocol was approved by the local Institutional Review Board for Preclinical & Clinical Research Ethics committee, Faculty of Pharmacy that ensured the care and use of animals according to the EU Directive 2010/63/EU on the protection of animals used for scientific purposes and Guiding Principle in Care and Use of Animals (DHEW publication NIH 80-23). Rats were housed at constant temperature and humidity, and were fasted overnight (with free access to water) prior to starting the experiment. The rats were randomly divided into three groups (A, B, and C), each of 12 rats. All the animals received SLD in a dose of 10 mg/kg,39 as follows:
- Group A (standard control): oral administration of SLD suspension
- Group B (positive control): transdermal application of SLD films (2.5 cm2)
- Group C (test): transdermal application of optimized SLD nano-transfersomal films (2.5 cm2).
The applied films were covered with plain adhesive patches. At predetermined intervals, blood specimens were withdrawn in a heparinized glass capillary tube from the tail vein at predetermined time intervals for 36 hours after drug administration. After centrifugation at 1,000 rpm for 8 minutes, plasma was transferred to sterile polypropylene tubes and stored at -20°C until analysis.
Preparation and analysis of plasma samples
Plasma samples preparation was performed by the extraction method adopted from Sheu et al.32 One milliliter of plasma sample was spiked with 0.1 mL of propyl paraben solution in methanol as an internal standard stock solution (1 mg/mL) and 0.1 mL of NaOH (1N). The sample was extracted with 3 mL ethyl acetate by vortex mixing for 5 minutes. The mixture was then centrifuged for 10 minutes at 3,000 rpm, and the supernatant was evaporated to dryness. The residue was dissolved in 1 mL of the mobile phase and 20 μL of this solution and injected into the HPLC for analysis using the previously described validated HPLC method.32 For construction of in vivo calibration curve (range of 10–1,000 ng/mL), increasing aliquots of SLD stock solution in methanol (1 mg/mL) were added to aliquots of 1 mL of drug-free plasma at the beginning of the procedure.
Pharmacokinetic and statistical analysis
Pharmacokinetic parameters, namely, elimination half-life (Ke), maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), and area under plasma concentration–time curve (AUC0–t and AUC0–∞) were calculated using Kinetica™ software (version 4; Thermo Fisher Scientific).
Pharmacokinetic data were statistically analyzed using SPSS® software, version 16 (SPSS Inc., Chicago, IL, USA). Two-way analysis of variance (ANOVA) followed by Tukey’s honest significant difference multiple comparisons test was performed on Cmax and AUC to assess the significance of the formulation and the subjects effects on the pharmacokinetic parameters. Differences are considered to be significant at P<0.05.
Results
Preparation and optimization of SLD nano-transfersomes
All SLD nano-transfersomes were prepared using modified lipid film hydration technique. Rotatable CCD was applied for the optimization of SLD nano-transfersomes with maximized entrapment efficiency and minimized vesicular size. Entrapment efficiency and vesicular size of all experimental runs were determined (Table 2). All prepared experimental trials showed SLD comparable high EE% ranging from 94.21%±1.33% to 95.46%±1.42%; accordingly, EE% was excluded as a response in the study. On the other hand, the experimental trials exhibited marked variations in the vesicular size ranging from 447±13 nm to 2,979±118 nm. Regression analysis of the measured vesicular size according to different polynomial models was performed (Table 3). The best fitting model for the data was found to be quadratic model. The validity of the model was confirmed by the residuals plot of the observed and predicted values of the vesicle size (Figure 1). ANOVA for the vesicle size response according to the quadratic model revealed the statistical significance of the model as depicted in Table 4 (P<0.05). The mathematical equation describing the model in terms of coded factors was generated as follows:
|
|
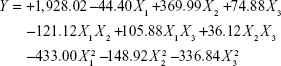
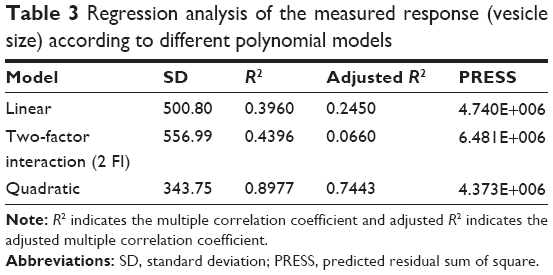 | Table 3 Regression analysis of the measured response (vesicle size) according to different polynomial models |
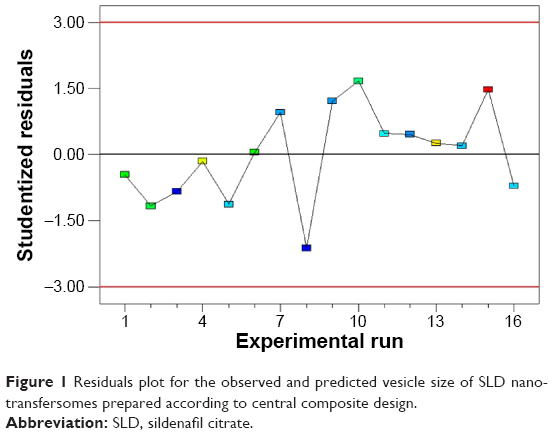 | Figure 1 Residuals plot for the observed and predicted vesicle size of SLD nano-transfersomes prepared according to central composite design. |
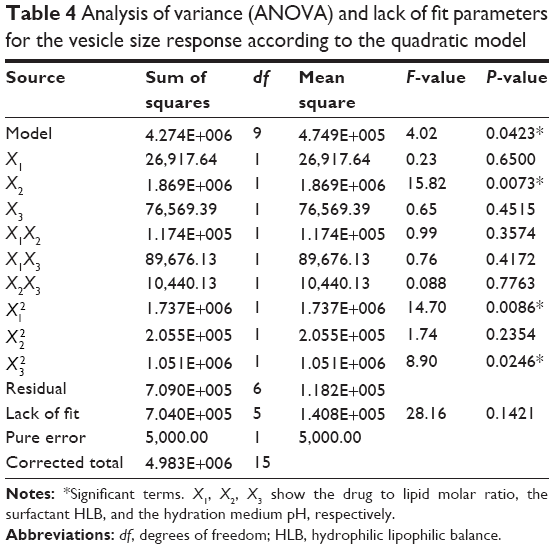 | Table 4 Analysis of variance (ANOVA) and lack of fit parameters for the vesicle size response according to the quadratic model |
ANOVA revealed significance of the linear term X2 corresponding to surfactant HLB (P=0.073) and the quadratic terms 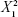 and
and 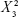 corresponding to drug:phospholipid molar ratio (P=0.0086) and hydration medium pH (P=0.0246), respectively. The effect of the independent variables on the vesicle size is illustrated in the three-dimensional surface plots and their corresponding contour plots (Figure 2). The composition of the optimized SLD nano-transfersomes with minimized vesicular size was generated using numerical optimization technique following desirability approach. The optimized formulation was prepared at X1, X2, and X3 levels of 1:2.74 (drug-to-phospholipid molar ratio), 2.08 (surfactant HLB), and 8.05 (hydration medium pH). The observed vesicle size of the optimized formulation (130 nm) was close to the predicted value (134.59 nm) showing no statistical significant difference (P<0.05) and % error of 3.41%, thus confirming the reliability of the optimization process.
corresponding to drug:phospholipid molar ratio (P=0.0086) and hydration medium pH (P=0.0246), respectively. The effect of the independent variables on the vesicle size is illustrated in the three-dimensional surface plots and their corresponding contour plots (Figure 2). The composition of the optimized SLD nano-transfersomes with minimized vesicular size was generated using numerical optimization technique following desirability approach. The optimized formulation was prepared at X1, X2, and X3 levels of 1:2.74 (drug-to-phospholipid molar ratio), 2.08 (surfactant HLB), and 8.05 (hydration medium pH). The observed vesicle size of the optimized formulation (130 nm) was close to the predicted value (134.59 nm) showing no statistical significant difference (P<0.05) and % error of 3.41%, thus confirming the reliability of the optimization process.
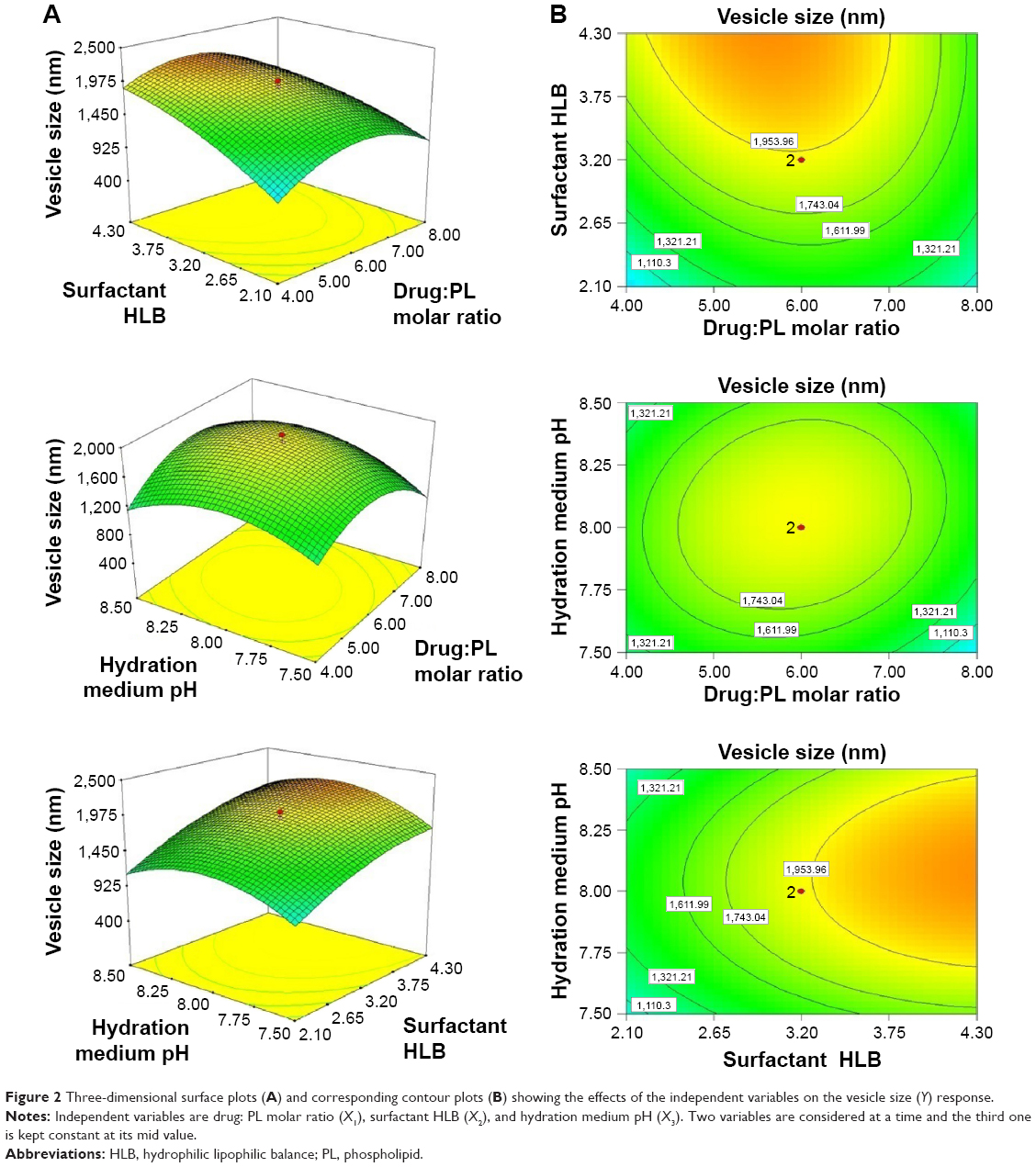 | Figure 2 Three-dimensional surface plots (A) and corresponding contour plots (B) showing the effects of the independent variables on the vesicle size (Y) response. |
Visualization of optimized SLD nano-transfersomes
Transmission electron microscopy has been utilized for evaluating shape and lamellarity of the optimized SLD nano-transfersomes. The representative photomicrograph (Figure 3) showed unilamellar vesicles with spherical shape.
 | Figure 3 TEM photomicrograph of optimized SLD nano-transfersomes (×10,000). |
Ex vivo permeation studies
Mean cumulative percent SLD permeated from both SLD nano-transfersomal films and SLD control films was determined (Figure 4). It was evident that the transfersomal films exhibited significantly higher cumulative percent drug permeated relative to the control films (P<0.05). The optimized transfersomal films showed a controlled gradual release over the study period reaching maximum amount of drug permeated of 1.54 folds compared with the control films. In addition, the computed permeability and diffusion coefficients for the optimized films were 1.25 and 1.57 folds higher compared with the control films, respectively. The permeation parameters of SLD from control and transfersomal films are compiled in Table 5.
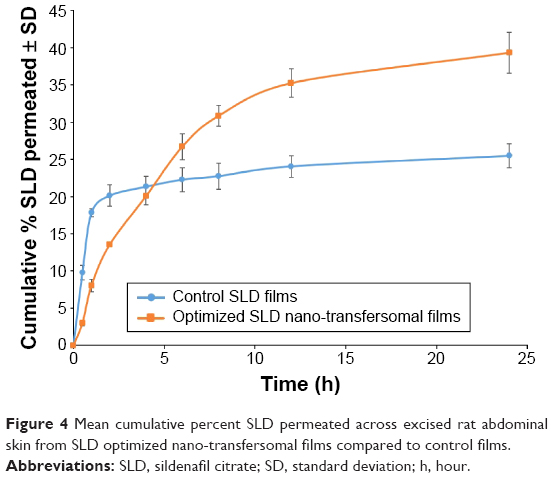 | Figure 4 Mean cumulative percent SLD permeated across excised rat abdominal skin from SLD optimized nano-transfersomal films compared to control films. |
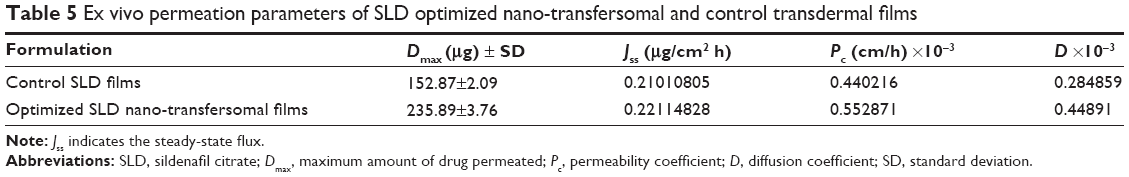 | Table 5 Ex vivo permeation parameters of SLD optimized nano-transfersomal and control transdermal films |
Kinetic analysis of the permeation data revealed that the permeation of SLD from both optimized SLD nano-transfersomal films and control SLD films followed Higuchi diffusion kinetics as evidenced by the highest correlation coefficient (R) of the model (Table 6). The (n) exponent obtained from the slope of plot of log fraction of drug released at time t (Mt/M∞) versus time indicates that the release mechanism from control SLD films follows Fickian diffusion (n<0.45), while that the release optimized SLD nano-transfersomal films follows non-Fickian diffusion, anamolous transport (0.45<n<0.89).
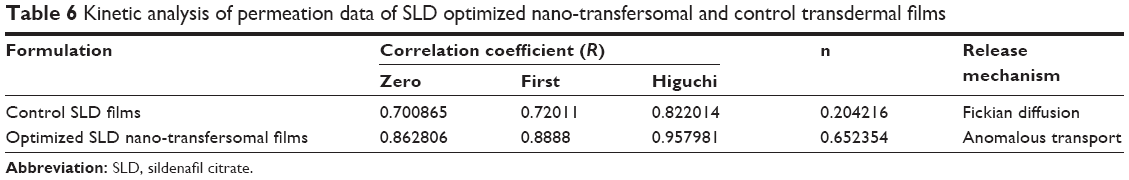 | Table 6 Kinetic analysis of permeation data of SLD optimized nano-transfersomal and control transdermal films |
In vivo pharmacokinetic studies
The concentration of SLD spiked in plasma was linearly correlated to the peak area ratios (SLD/internal standard) with a correlation coefficient of 0.996. The assay showed acceptable precision (coefficient of variation % of <5% and <8% for the intraday assay and the interday assay, respectively) and accuracy (96.00%). The extraction recovery of SLD from drug-fortified plasma samples ranged from 92.12% to 103.41%. Mean concentrations of SLD in rats’ plasma following oral administration of SLD suspension and transdermal application of control SLD films and optimized SLD nano-transfersomal films were determined (Figure 5). The optimized films showed significantly extended drug absorption compared to control films and suspension, reaching its peak plasma concentration after 15 hours. Both films exhibited significantly higher AUC (P<0.05) compared to oral suspension. The optimized SLD nano-transfersomal films exhibited significantly higher Cmax and almost doubled AUC (P<0.05) compared to control SLD films (Table 7). Although the oral suspension showed slightly higher Cmax than the optimized transfersomal film, yet this difference was statistically insignificant at the 95% confidence level.
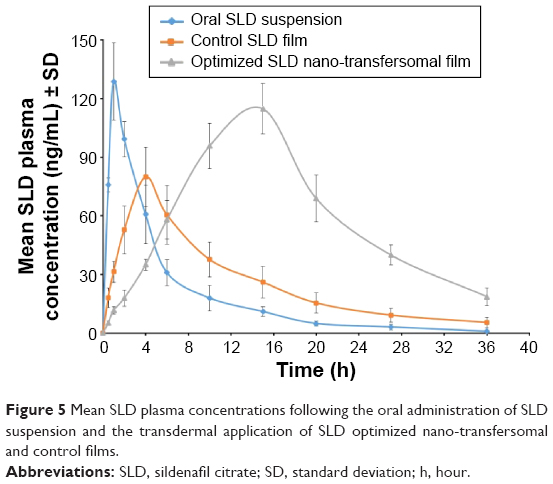 | Figure 5 Mean SLD plasma concentrations following the oral administration of SLD suspension and the transdermal application of SLD optimized nano-transfersomal and control films. |
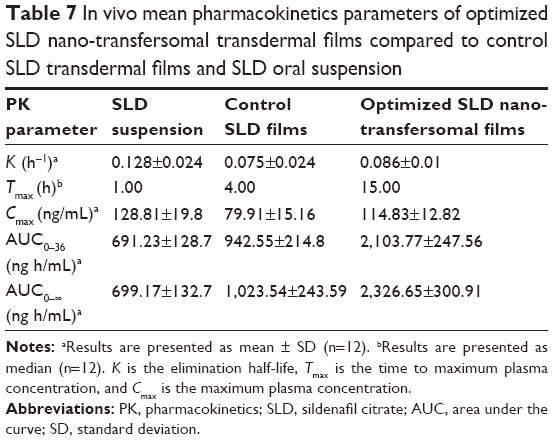 | Table 7 In vivo mean pharmacokinetics parameters of optimized SLD nano-transfersomal transdermal films compared to control SLD transdermal films and SLD oral suspension |
Discussion
Rotatable CCD was used to evaluate the relation among the independent variables, namely, drug-to-phospholipid molar ratio (X1), surfactant HLB (X2), and hydration medium pH (X3) and the vesicle size (Y) of the nano-transfersomes as a measured response. The observed high EE% could be explained based on the hydration medium pH range used and the solubility of the drug. SLD is an amphoteric drug that has two dissociation constants, pKa1 of 7.10 and pKa2 of 9.84 corresponding to the basic ionization of NH-piperazine and acidic ionization of NH-amide of pyrazolopyrimidine, respectively.40 Thus, SLD exhibits pH-dependent solubility in aqueous solutions with the least solubility at pH values between pKa1 and pKa2 owing to the saturation of the neutral species. In a study of the solubility of SLD over a pH range of 3–11, Wang et al41 reported the least solubility values for SLD at pH of 7–9 (0.025–0.040 mg/mL). Therefore, the high EE% of all trials could be attributed to the low solubility of the drug in the hydration medium (pH 7.5–8.5) that favors the entrapment of the drug into the vesicles. Since all trials showed comparable EE% with no marked difference among them, EE% was not considered as a response for analysis.
After fitting the measured vesicular sizes for individual trials into different polynomial equations, the quadratic model was selected for its largest multiple correlation coefficient (R2) and smallest predicted residual sum of squares compared to other models. The positive sign of the coefficient of the linear term X2 indicates a positive effect of the surfactant HLB on the response Y, that is, as the HLB of the surfactant moves toward hydrophobicity, a significant reduction in vesicle size is observed. The relation between surfactant HLB and vesicle size could be explained based on the reduced surface energy and the limited water uptake into vesicles core associated with increased hydrophobicity and low HLB values of the edge activators. Both effects could lead to the formation of smaller vesicles.42,43
Ex vivo permeation studies through excised rat abdominal skin were performed to offer an insight on the in vivo performance of the nano-transfersomal transdermal films in comparison with the control films. The enhanced permeation from transfersomal films could be explained based on the combined advantages of transfersomes and nanoparticulate systems. Based on the literature, transfersomes have potential ability to penetrate SC barrier and reach deeper dermal tissues into the systemic circulation owing to their flexibility and deformable structure. Moreover, permeation enhancement by transfersomes involves the presence of surfactants that act as edge activator, thus loosening or destabilizing the lipid bilayer of the membrane and consequently serving as a permeation enhancer.42,44 The nanosize of the vesicles contributes to the permeation enhancement effect since the small vesicular size implies a large surface area that increases the contact with the corneocytes and consequently increases the amount of encapsulated drug permeating the skin.14 The higher permeability and diffusion coefficients exhibited by the optimized nano-transfersomal films compared with control films could confirm their potentiality to enhance and control the permeation of SLD through skin with the possible consequence of improving its transdermal delivery, and thus, its bioavailability. The difference in permeation mechanism between the prepared films could suggest that the permeation of SLD from the control films was governed mainly by diffusion through the polymer matrix, while in case of transfersomal films, the permeation of the drug was controlled by coupled diffusion and erosion.37,38 It is worthy to note that the higher observed initial drug permeation from the control films could be attributed to the effect of the penetration enhancer on the free drug available on the film surface; however, a plateau was rapidly reached after 1 hour due to the slow diffusion of the drug from the film matrix.
Further in vivo pharmacokinetic studies were performed in rats to assess the effect of nano-transfersomal films on the bioavailability of SLD. The delayed Tmax exhibited by the optimized SLD nano-transfersomal films could be indicative for controlled and extended drug absorption. This result well correlates with the ex vivo permeation study that confirmed the ability of the nano-transfersomes to control drug permeation. The significantly higher extent of absorption of both films compared to oral suspension could be due to the avoidance of the first-pass effect by the transdermal route of administration. Moreover, the higher extent of absorption demonstrated by the optimized films compared to control films could be attributed to the enhanced permeation characteristics of SLD transfersomes owing to its ultra-flexibility and deformability that aid penetration of the vesicles through the skin layers and into systemic circulation.44 In addition, the small vesicle sizes in the nano range could potentially contribute to the permeation and the consequent bioavailability enhancement due to the increased surface area interacting on a fixed area of the SC.33
Conclusion
Nano-transfersomal films have been investigated as a possible transdermal drug delivery system for SLD. Response surface methodology, specifically CCD, was applied for the optimization of SLD nano-transfersomes with minimized vesicular size. Surfactant HLB was the most significant formulation factor affecting vesicular size. The optimized SLD nano-transfersomes (1:2.74 drug:PL molar ratio, surfactant HLB of 2.08, and hydration medium pH of 8.05) incorporated into HPMC-based transdermal films showed enhanced and controlled ex vivo permeation profile through excised rat abdominal skin compared to control SLD films. In vivo investigations confirmed the higher bioavailability and extended absorption of the drug from optimized films compared to oral suspensions and control films. Based on the previous results, optimized SLD nano-transfersomal transdermal films could be a potential promising drug delivery system for the drug. The enhanced and extended absorption of SLD from the optimized films provides insight on the possibility of the reduction of dose and frequency of administration of the drug.
Acknowledgments
This work was supported by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant number (166-659-D1435). The authors, therefore, gratefully acknowledge the DSR technical and financial support. The authors would also express their gratitude to Dr Tarek A Ahmed for his guidance.
Disclosure
The authors report no conflicts of interest in this work.
References
Badwan AA, Nabuls L, Al-Omari MM, Daraghmeh N, Ashour M. Sildenafil citrate. Anal Profiles Drug Subst Excipients. 2001;27:339–376. | ||
Kirby M, Creanga DL, Stecher VJ. Erectile function, erection hardness and tolerability in men treated with sildenafil 100 mg vs 50 mg for erectile dysfunction. Int J Clin Pract. 2013;67:1034–1039. | ||
Milger K, Felix JF, Voswinckel R, et al. Sildenafil versus nitric oxide for acute vasodilator testing in pulmonary arterial hypertension. Pulm Circ. 2015;5(2):305–312. | ||
Dastjerdi MV, Hosseini S, Bayani L. Sildenafil citrate and uteroplacental perfusion in fetal growth restriction. J Res Med Sci. 2012;17:632–636. | ||
Osman MA, El Maghraby GM, Hedaya MA. Intestinal absorption and presystemic disposition of sildenafil citrate in the rabbit: evidence for site-dependent absorptive clearance. Biopharm Drug Dispos. 2006;27:93–102. | ||
Zinner N. Do food and dose timing affect the efficacy of sildenafil? A randomized placebo-controlled study. J Sex Med. 2007;4:137–144. | ||
Hosny KM, Aljaeid BM. Sildenafil citrate as oral solid lipid nanoparticles: a novel formula with higher bioavailability and sustained action for treatment of erectile dysfunction. Expert Opin Drug Deliv. 2014;11:1015–1022. | ||
Sawatdee S, Phetmung H, Srichana T. Sildenafil citrate monohydrate–cyclodextrin nanosuspension complexes for use in metered-dose inhalers. Int J Pharm. 2013;455:248–258. | ||
Hosny KM, Mosly HA, Hassan AH. Soy polysaccharide as a novel superdisintegrant in sildenafil citrate sublingual tablets: preparation, characterization, and in vivo evaluation. Drug Des Devel Ther. 2015;9:465–472. | ||
Kim EY, Lee SY, Jeon JY, et al. Pharmacokinetics of a new orally soluble film formulation of sildenafil administered without water. Int J Clin Pharmacol Ther. 2014;52:437–445. | ||
Damle B, Duczynski G, Jeffers BW, Crownover P, Coupe A, LaBadie RR. Pharmacokinetics of a novel orodispersible tablet of sildenafil in healthy subjects. Clin Ther. 2014;36:236–244. | ||
Zayed R, Kamel A, Shukr M, El-Shamy A. An in vitro and in vivo comparative study of directly compressed solid dispersions and freeze dried sildenafil citrate sublingual tablets for management of pulmonary arterial hypertension. Acta Pharm. 2012;62:411–432. | ||
Elshafeey AH, Bendas ER, Mohamed OH. Intranasal microemulsion of sildenafil citrate: in vitro evaluation and in vivo pharmacokinetic study in rabbits. AAPS PharmSciTech. 2009;10:361–367. | ||
Elnaggar YS, El-Massik MA, Abdallah OY. Fabrication, appraisal, and transdermal permeation of sildenafil citrate loaded nanostructured lipid carriers versus solid lipid nanoparticles. Int J Nanomedicine. 2011;6:3195–3205. | ||
Barry B. Transdermal drug delivery. In: Aulton ME, editor. Pharmaceutics: The Science of Dosage Form Design. London: Churchill Livingstone; 2002:499–528. | ||
Paudel KS, Milewski M, Swadley CL, Brogden NK, Ghosh P, Stinchcomb AL. Challenges and opportunities in dermal/transdermal delivery. Ther Deliv. 2010;1:109–131. | ||
Mutalik S, Parekh HS, Davies NM, Udupa N. A combined approach of chemical enhancers and sonophoresis for the transdermal delivery of tizanidine hydrochloride. Drug Deliv. 2009;16:82–91. | ||
Schoellhammer CM, Blankschtein D, Langer R. Skin permeabilization for transdermal drug delivery: recent advances and future prospects. Expert Opin Drug Deliv. 2014;11:393–407. | ||
Maestrelli F, González-Rodríguez ML, Rabasco AM, Mura P. Effect of preparation technique on the properties of liposomes encapsulating ketoprofen-cyclodextrin complexes aimed for transdermal delivery. Int J Pharm. 2006;312:53–60. | ||
Elron-Gross I, Glucksam Y, Margalit R. Liposomal dexamethasone-diclofenac combinations for local osteoarthritis treatment. Int J Pharm. 2009;376:84–91. | ||
Jain S, Patel N, Madan P, Lin S. Quality by design approach for formulation, evaluation and statistical optimization of diclofenac-loaded ethosomes via transdermal route. Pharm Dev Technol. 2015;20(4):473–489. | ||
Benson HA. Elastic liposomes for topical and transdermal drug delivery. Curr Drug Deliv. 2009;6:217–226. | ||
Chen J, Lu WL, Gu W, Lu SS, Chen ZP, Cai BC. Skin permeation behavior of elastic liposomes: role of formulation ingredients. Expert Opin Drug Deliv. 2013;10:845–856. | ||
Bragagni M, Mennini N, Maestrelli F, Cirri M, Mura P. Comparative study of liposomes, transfersomes and ethosomes as carriers for improving topical delivery of celecoxib. Drug Deliv. 2012;19:354–361. | ||
Mahmood S, Taher M, Mandal UK. Experimental design and optimization of raloxifene hydrochloride loaded nanotransfersomes for transdermal application. Int J Nanomedicine. 2014;9:4331–4346. | ||
Duangjit S, Opanasopit P, Rojanarata T, Ngawhirunpat T. Evaluation of meloxicam-loaded cationic transfersomes as transdermal drug delivery carriers. AAPS PharmSciTech. 2013;14:133–140. | ||
El-Nabarawi MA, Bendas ER, El Rehem RT, Abary MY. Transdermal drug delivery of paroxetine through lipid-vesicular formulation to augment its bioavailability. Int J Pharm. 2013;443:307–317. | ||
Shamma RN, Elsayed I. Transfersomal lyophilized gel of buspirone HCl: formulation, evaluation and statistical optimization. J Liposome Res. 2013;23:244–254. | ||
Guan Y, Zuo T, Chang M, et al. Propranolol hydrochloride-loaded liposomal gel for transdermal delivery: characterization and in vivo evaluation. Int J Pharm. 2015;487:135–141. | ||
Ahmed TA. Preparation of transfersomes encapsulating sildenafil aimed for transdermal drug delivery: Plackett-Burman design and characterization. J Liposome Res. 2015;25:1–10. | ||
Fahmy RH, Badr-Eldin SM. Novel delivery approach for ketotifen fumarate: dissofilms formulation using 32 experimental design: in vitro/in vivo evaluation. Pharm Dev Technol. 2014;19:521–530. | ||
Sheu MT, Wu AB, Yeh GC, Hsia A, Ho HO. Development of a liquid chromatographic method for bioanalytical applications with sildenafil. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;791(1–2):255–262. | ||
El-Say KM, Ahmed TA, Badr-Eldin SM, Fahmy U, Aldawsari H, Ahmed OA. Enhanced permeation parameters of optimized nanostructured simvastatin transdermal films: ex vivo and in vivo evaluation. Pharm Dev Technol. 2015;20:919–926. | ||
Ahmed OAA, Afouna MI, El-Say KM, Abdel-Naim AB, Khedr A, Banjar ZM. Optimization of self nanoemulsifying systems for the enhancement of in vivo hypoglycemic efficacy of glimepiride transdermal patches. Expert Opin Drug Deliv. 2014;11:1005–1013. | ||
Ahmed O, Ahmed T, Abdel-Naim A, Khedr A, Banjar Z, Afouna M. Enhancement of in vitro skin transport and in vivo hypoglycemic efficacy of glimepiride transdermal patches. Trop J Pharm Res. 2014;13(8):1207–1213. | ||
Higuchi T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52:1145–1149. | ||
Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35. | ||
Peppas NA. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 1985;60:110–111. | ||
Ferrari F, Ottani A, Guiliani D. Influence of sildenafil on central dopamine-mediated behaviour in male rats. Life Sci. 2002;70(13):1501–1508. | ||
Al-Omari MM, Zughul MB, Davies JED, Badwan AA. Sildenafil/cyclodextrin complexation: stability constants, thermodynamics, and guest-host interactions probed by 1H NMR and molecular modeling studies. J Pharm Biomed Anal. 2006;41:857–865. | ||
Wang Y, Chow MS, Zuo Z. Mechanistic analysis of pH-dependent solubility and trans-membrane permeability of amphoteric compounds: application to sildenafil. Int J Pharm. 2008;352:217–224. | ||
El Zaafarany GM, Awad GAS, Holayel SM, Mortada ND. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int J Pharm. 2010;397:164–172. | ||
El-Laithy HM, Shoukry O, Mahran LG. Novel sugar esters proniosomes for transdermal delivery of vinpocetine: preclinical and clinical studies. Eur J Pharm Biopharm. 2011;77:43–55. | ||
Simões SI, Delgado TC, Lopes RM, et al. Developments in the rat adjuvant arthritis model and its use in therapeutic evaluation of novel non-invasive treatment by SOD in transfersomes. J Control Release. 2005;21:419–434. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.