")
Back to Journals » Journal of Experimental Pharmacology » Volume 15
OCE-205, a Selective V1a Partial Agonist, Reduces Portal Pressure in Rat Models of Portal Hypertension
Authors Bukofzer S , Harris G, Song S, Cable EE
Received 12 April 2023
Accepted for publication 4 July 2023
Published 13 July 2023 Volume 2023:15 Pages 279—290
DOI https://doi.org/10.2147/JEP.S416673
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Stan Bukofzer,1 Geoffrey Harris,2 Susan Song,2 Edward E Cable2
1Ocelot Bio, Inc., San Diego, CA, USA; 2Ferring Research Institute Inc., San Diego, CA, USA
Correspondence: Stan Bukofzer, Ocelot Bio, Inc., 12670 High Bluff Drive, San Diego, CA, 92130, USA, Tel +1 858-247-2272, Email [email protected]
Purpose: Management of decompensated cirrhosis may include the use of vasoconstrictors that can lead to serious adverse events. OCE-205 was designed as a highly selective V1a receptor partial agonist, intended to have a wider therapeutic window than full vasopressin agonists.
Methods: We aimed to characterize the activity of OCE-205 treatment in two rat models of portal hypertension (PHT). For both models, OCE-205 was administered as a subcutaneous bolus injection. Thirty male Wistar rats were fed a methionine/choline-deficient (MCD) diet to model PHT. Animals received OCE-205 (10, 25, 100, or 500 μg/kg) or intra-arterial terlipressin (100 μg/kg). In a more severe model of PHT, 11 male Sprague Dawley rats had the common bile duct surgically ligated (BDL) and received OCE-205. Portal pressure (PP) and mean arterial pressure (MAP) were measured.
Results: For PP in the MCD model, MAP increased while PP decreased in rats treated with OCE-205 or terlipressin; the peak changes to MAP were 14.7 and 33.5 mmHg, respectively. Changes in MAP began to plateau after 10 min in the OCE-205 groups, whereas in the terlipressin group, MAP rapidly increased and peaked after 20 min. Across all treatment groups in the BDL model, a dose-related decrease from baseline in PP was observed following OCE-205, plateauing as the dose increased. In all treatment groups, PP change remained negative throughout the 30-min testing period. In both PHT rat models, a reduction in PP was coupled to an increase in MAP, with both plateauing in dose–response curves.
Conclusion: Data support OCE-205 as a promising candidate for further development.
Institutional Protocol Number: Procedures were approved by the Ferring Research Institute (FRI) Institutional Animal Care and Use Committee on July 13, 2011, under protocol FRI-07-0002.
Keywords: mean arterial pressure, ascites, HRS-AKI, vasoconstriction, portal hypertension
Introduction
Decompensated cirrhosis and its serious hemodynamic complications are a critical area unmet need for patients. Systemic hemodynamic complications are typical of portal hypertension (PHT) and cirrhosis.1–4 PHT produces alterations in cardiovascular function and tone, with splanchnic arterial vasodilation, reduced systemic vascular resistance, and lower effective arterial blood volume along with arterial pressure reductions.5 Effective hypovolemia, low perfusion pressure, and reduced glomerular filtration rate can lead to vasoconstriction within the kidney with subsequent sodium and water retention, resulting in hepatorenal syndrome–acute kidney injury (HRS-AKI).5,6
HRS-AKI treatment relies on increasing blood volume with albumin supplementation and discontinuing diuretics with the hope of increasing renal perfusion to restore renal function. Often, short-duration therapy with systemic vasoconstrictors is needed to increase mean arterial pressure (MAP; 10–15 mmHg, which correlates with reversal of HRS-AKI7,8) to counteract renal dysfunction. Ultimately, the therapeutic goal is liver transplantation.9 Vasopressin agonists are among the best-characterized vasoconstrictors for the management of HRS-AKI. Arginine vasopressin (AVP), a key hormonal regulator of osmotic balance, is typically synthesized in the hypothalamus and released by the posterior pituitary gland.10 The V2 receptor, expressed in the distal tubules and collecting ducts of the kidney, principally regulates vasopressin and, once activated, causes water retention.10–12 Vasopressin activates the V1a receptor at elevated physiologic and pharmacologic concentrations; it then results in systemic vasoconstriction due to V1a expression on smooth muscle cells in the vasculature walls,10 including those in the splanchnic circulation.6 Vasopressin also activates the V1b receptor, expressed in the anterior pituitary; once activated, this stimulates corticotropin secretion, further increasing water retention.13
Current vasopressin agonists have a narrow therapeutic window. Underdosing does not achieve the desired clinical effect, and overdosing can produce excess vasoconstriction and potentially lead to life-threatening ischemic adverse events (AEs).10,12,14,15 All clinically utilized vasopressin agonists target the V2 agonists as well as the V1a receptor.16 The agonist activity at the V2 receptor contributes to the known AE profile, involving fluid overload and respiratory complications.12 The combination of fluid retention and risk of excess vasoconstriction currently limits the utilization of existing vasopressin agonists to relatively late in the development of HRS-AKI and other cirrhotic complications where the risk/benefit can be justified.
Historically, HRS-AKI treatment has utilized midodrine plus octreotide, norepinephrine,17,18 and, most recently, terlipressin. Norepinephrine and midodrine, as α-adrenergic agonists, can have limited therapeutic efficacy.19 Furthermore, norepinephrine administration requires a central venous line, typically given in an intensive care unit.9 Terlipressin was approved by the US Food and Drug Administration (FDA) in September 2022 to treat adults with HRS with rapid reduction in kidney function.20,21 Terlipressin (triglycyl-LVP) is a prodrug of lysine vasopressin (LVP, the porcine version of AVP) and shows activity at the V1a, V1b, and V2 receptors.22–25 Its systemic hemodynamic response has been associated with a decrease in portal pressure (PP) as well as lessening the hyperdynamic circulation without additional contraction of the arterial and central blood volume.26,27 The active metabolite of triglycyl-LVP is LVP, a full V1a agonist that is significantly more potent at the V2 receptor.22
Terlipressin is recommended for treatment of HRS-AKI,2,9 but can cause clinically significant AEs (eg, tissue hypoperfusion, ischemia) from its vasoconstrictive effects, likely due to LVP’s full agonism at the V1a receptor,14,28,29 as well as respiratory failure and fluid overload through V1a and V2 receptor activation, possibly related to sodium and water retention.23,30–33 An improved approach to targeting the V1a receptors with partial agonism (avoiding maximal stimulation), and no V2 receptor agonism, would lower the risks associated with vasopressin agonists14,28–30 and could offer therapeutic utility in conditions where a modest and capped increase in blood pressure is desirable.
To improve the safety profile of vasoconstriction through the vasopressin system in patients with HRS-AKI, a molecule with V1a receptor selectivity and minimal V1a receptor agonism is needed. A single molecule was proposed in 1996 that, by possessing both agonist and antagonist properties, would perform as a partial agonist.34
OCE-205 is a novel peptide drug designed to target the vasopressin receptor system as a mixed agonist/antagonist for the V1a receptor. Unlike terlipressin,22–25 OCE-205 is not a prodrug and does not require a liver first pass or other modifications in vivo to produce the desired pharmacological effects. The activity at human V1a receptors plateaus at ~50% maximum possible effect, with no activity at human V2 receptors at clinically relevant concentrations.35 OCE-205 has similar functional properties at rat and human vasopressin receptors; by examining the use of OCE-205 to reduce PHT and increase MAP in two rat models, we hope to pave the way for improvements in clinical safety and efficacy in human patients with HRS-AKI.
Materials and Methods
Animal Use
Housing conditions and animal care facilities were maintained in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Research Council. Procedures were approved by the Ferring Research Institute (FRI) Institutional Animal Care and Use Committee (IACUC) on July 13, 2011, under protocol FRI-07-0002.
Rat Models
Methionine/Choline-Deficient Model
The MCD model of PHT does not typically induce significant fibrosis nor development of cirrhosis, but leads to portal pressure (PP) elevation after 4 weeks of MCD.36–39 The MCD model of diet-induced PHT is clearly described elsewhere40 and is a considered a reliable model to induce PHT without being as severe and progressive as other interventions such as CCl4, or bile duct/portal vein ligation. The mechanism of inducing PHT is primarily from inducing a NAFLD/NASH-like phenotype with accumulation of intracellular lipid in hepatocytes. Presumably the increase in inflammation and congestion created by lipid accumulation increases hepatic resistance and therefore PP. Histological examination of the study animals were not performed as the livers were obviously steatotic upon macroscopic inspection.
Thirty adult male Wistar rats (Harlan, Indianapolis) were studied over multiple testing days and weighed 200–250 g at the start of the study. Animals were housed two per cage in a controlled environment with free access to an MCD diet (Harlan Teklad) and water. After ≥8 weeks on the MCD diet, the adult male Wistar rats underwent surgical placement of catheters in the portal vein and/or femoral artery. Animals were allowed to recover for ≥5 days. A saline vehicle was administered 5 to 15 min prior to experimental compounds to control for nonspecific responses. OCE-205 was bolus administered subcutaneously at doses of 500, 100, and 25 µg/kg to determine the degree of systemic vasoconstriction achieved for a given degree of reduction in portal pressure and whether these occurred at similar, identical, or disparate drug concentrations.
To avoid the need for a second surgery, terlipressin (100 µg/kg) was administered intra-arterially. Triglycyl-LVP (terlipressin) has been previously well characterized in both rats and dogs, at doses up to 50 µg/kg in combination with octreotide.41,42 Systolic blood pressure (SBP), diastolic blood pressure (DBP), and PP were measured by the fluidic transducers. Measurements were recorded continuously from time 0 (compound administration time) to 90 min following administration, unless technical difficulties (eg loss of catheter patency) resulted in cessation of the experiment prior to 90 min. To reduce the numbers of animals needed for analysis and as approved by the IACUC, 30 animals had repeat dosing with a minimum of 1 day of recovery between each administration. Altogether, 86 measurements with 1–8 repeat administrations per animal were used for this study.
Bile Duct Surgically Ligated Model
The BDL rat model typically causes larger increases in PHT than the MCD model, and significant renal dysfunction.36,43,44 Male Sprague Dawley rats had the common BDL to block the enterohepatic recirculation of bile acids. Eleven adult male Sprague Dawley rats (Harlan, Indianapolis) weighing 200–350 g were studied over multiple testing days. Animals were housed in a controlled environment with free access to food and water for ≥4 days after surgery and before experimentation.
A laparotomy was performed, and the portal vein exposed, following the procedure described by Gervaz et al.45 The bifurcation of the portal vein and the confluence of the gastric vein were identified. Proximal clamping for ≤5 min was performed to minimize blood loss and was well tolerated. Puncture of the anterior wall of the portal vein was performed with a 30-gauge needle just proximal to its bifurcation. The tip of the catheter was cut at 45°, and its sharp edge was introduced tangentially into the portal vein. Care was taken to avoid inserting the tip of the catheter too far into the vein lumen. The catheter was exteriorized through the abdominal wall, tunneled subcutaneously, and exteriorized in the intrascapular region. The catheter was locked with 50% glycerol/heparin. A jacket was positioned to hold the exterior portion of the catheter in place. Animals were allowed to recover for ≥5 days before receiving OCE-205.
To avoid the need for a second surgery, catheter placement and BDL were performed at the same time. The adult male Sprague Dawley rats had surgically placed portal vein catheters and BDL for an average of 25 days (range, 11–42 days). Vehicle (saline) was administered 5 to 15 min prior to experimental compound administration to control for nonspecific responses. No arterial measurements were taken due to bleeding issues in this more severe model of disease. OCE-205, administered as a subcutaneous bolus, was tested over a broader dose range (500, 100, 25, and 10 µg/kg) than in the MCD model, to establish a clearer dose–response relationship of changes in PP based on studies in healthy animals and evaluating changes in skin blood flow.
The PP was measured via the fluidic transducers and typically recorded continuously from time 0 (compound administration time) to 30 min following administration by NOTOCORD-hem™ data acquisition software, unless technical difficulties (eg, loss of catheter patency) resulted in cessation of experiment prior to 30 min. To reduce the number of animals needed for analysis and as approved by the IACUC, 11 animals had repeat dosing with a minimum of 16 h of recovery between each administration. A total of 38 measurements with 1–11 repeat administrations per animal were used for this study.
Test and Reference Compounds
OCE-205–TFA salt was the test compound in both studies. Terlipressin (triglycyl-LVP) was the reference compound in the MCD study.
Compound Formulation and Administration
OCE-205 was administered in a dose volume of 1 mL/kg subcutaneously by injection (OCE-205) into the animal’s lower back. Terlipressin (triglycyl-LVP) was administered intra-arterially via the femoral catheter used to measure pressure. The OCE-205 doses were 500, 100, 25, and 10 µg/kg. The terlipressin (triglycyl-LVP) dose was 100 µg/kg.
Subcutaneous Bolus Administration of Compounds
Overview
A minimum of four animals was used for testing each group to allow for the possibility of technical, and/or pharmacologic events. Most studied used each animal more than once. Animals were randomized to different testing stations (anesthesia stations and NOTOCORD™ software channels for automatic data acquisition). The investigators running the in-life portion of the studies were aware of the dosing solutions being administered.
On the day of experimental measurements for each study, animals were anesthetized, and catheters were flushed to maintain or restore patency. Catheters were then connected to fluidic pressure transducers linked to data acquisition stations. Prior to administration of compound, a stable baseline was obtained for the pressure readings, saline was administered, and an additional 5–15 min of pressure readings were taken to solidify the baseline readings and provide a per-animal vehicle control. Animals were excluded from the study if the study cannular was not patent or if stable baseline values could not be obtained.
Data Analysis
Data recorded by NOTOCORD-hem™ were transferred to Microsoft Excel for analysis. Data were evaluated as change (Δ) from baseline. MAP and PP were reported as the average pressure value (mmHg) recorded over 10s beginning at the first systole (PP and SBP) or diastole (DBP) following the time point of interest at nominal times of 0 (compound administration), 1, 2, 3, 5, 7, 10, 15, 20, and 30 min after administration in both studies, along with 60 and 90 min after administration in the MCD study. Data collected over multiple test days were compiled. Mean, SEM, and N were reported for PP, MAP, ΔPP, and ΔMAP for each compound dose and time point.
MAP was calculated from SBP and DBP:
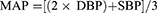
Change from baseline (delta Δ) for PP and MAP at each time point in each animal were calculated as follows:
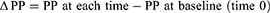
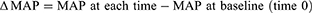
Statistical Analysis
The observed PP and MAP data were statistically analyzed using JMP software. Data from each animal at times 0 to 90 min (MCD study) and 0 to 30 min (BDL study) in the four treatment groups were compared using MANOVA with repeated measures. Each time course measurement was considered independent; animals were allowed to recover before any repeat administration. No corrections or extrapolations were performed if data were not collected for every time point.
The MANOVA outcome for treatment between subjects was not considered significant if p ≥ 0.05 (Prob>F); in the MCD study, subsequent contrasts were performed to compare treatment groups. Data from time 0 were statistically determined using one-way ANOVA to compare starting values in each treatment group. In cases when this ANOVA was significant (p < 0.05), Tukey–Kramer HSD post hoc analysis was used to compare treatment groups.
Results
OCE-205 Modulates Disease Physiology in an MCD Model of PHT
Mean Arterial Pressure
OCE-205 and terlipressin, when given to MCD-fed rats, resulted in increased MAP. For the 25 μg/kg (n = 13), 100 μg/kg (n = 23), and 500 μg/kg (n = 17) OCE-205 treatment groups, the highest observed absolute MAP values (mean ± SEM) were 79.2 ± 1.4, 86.8 ± 2.5, and 90.4 ± 3.7 mmHg, respectively (Table 1). There were greater increases in MAP resulting from the intra-arterial administration of terlipressin (100 μg/kg; n = 13) compared with the OCE-205 treatment groups, with the highest observed MAP of 116.6 ± 4.1 mmHg.
 |
Table 1 Change in MAP from Baseline After OCE-205 or Terlipressin Treatment in the MCD Model |
Observed MAP using MANOVA with repeated measures of the OCE-205 treatment groups showed a significant difference between the 25 and 100 μg/kg OCE-205 treatment groups (p = 0.03). There was no significant difference between the 100 and 500 μg/kg OCE-205 treatment groups (p = 0.63). MAP was significantly different between intra-arterial administration of terlipressin and subcutaneous administration of OCE-205 at 500 μg/kg (p < 0.01).
The maximum ΔMAP values (mean ± SEM) for OCE-205 were 5.3 ± 1.1, 11.3 ± 1.8, and 14.7 ± 2.6 mmHg in the 25, 100, and 500 μg/kg treatment groups, respectively (Figure 1). Following intra-arterial administration of terlipressin, the maximum ΔMAP (33.5 ± 2.6 mmHg) was greater than in any of the OCE-205 treatment groups.
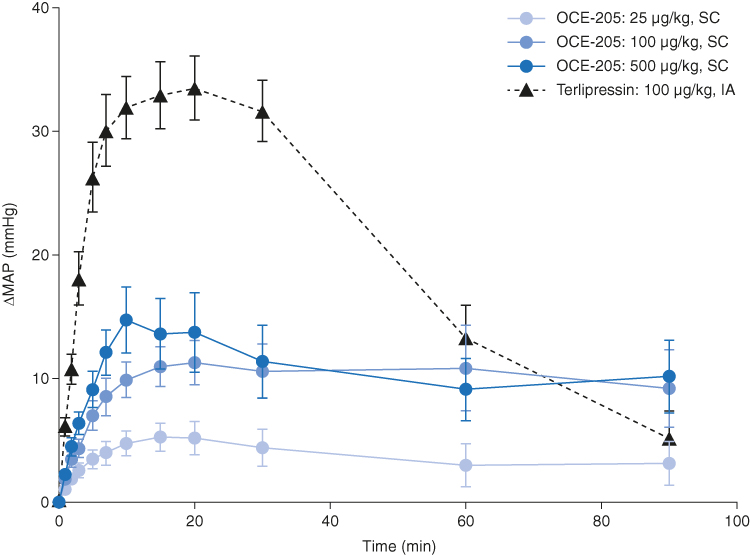 |
Figure 1 Change from baseline in mean arterial pressure following OCE-205 or terlipressin administration in a rat model of portal hypertension (methionine/choline-deficient diet). Abbreviations: IA, intra-arterial; SC, subcutaneous. |
Of note, the observed MAP baseline values (time 0) were statistically different for the terlipressin group versus each of the OCE-205 treatment groups. The values were determined by one-way ANOVA with Tukey–Kramer HSD post hoc analysis (p = 0.02 vs 25 μg/kg, p = 0.03 vs 100 µg/kg, p = 0.46 vs 500 μg/kg OCE-205).
Portal Pressure
There was a decrease in PP following treatment with OCE-205 or terlipressin. The observed PP values were not significantly different between any of the four treatment groups, as determined by MANOVA with repeated measures (Table 2). The maximum ΔPP (mean ± SEM) were −2.3 ± 0.2, −2.5 ± 0.3, and −3.9 ± 0.8 mmHg in the OCE-205 25, 100, and 500 μg/kg treatment groups, respectively (Figure 2). The change following intra-arterial administration of terlipressin was −2.8 ± 0.8 mmHg.
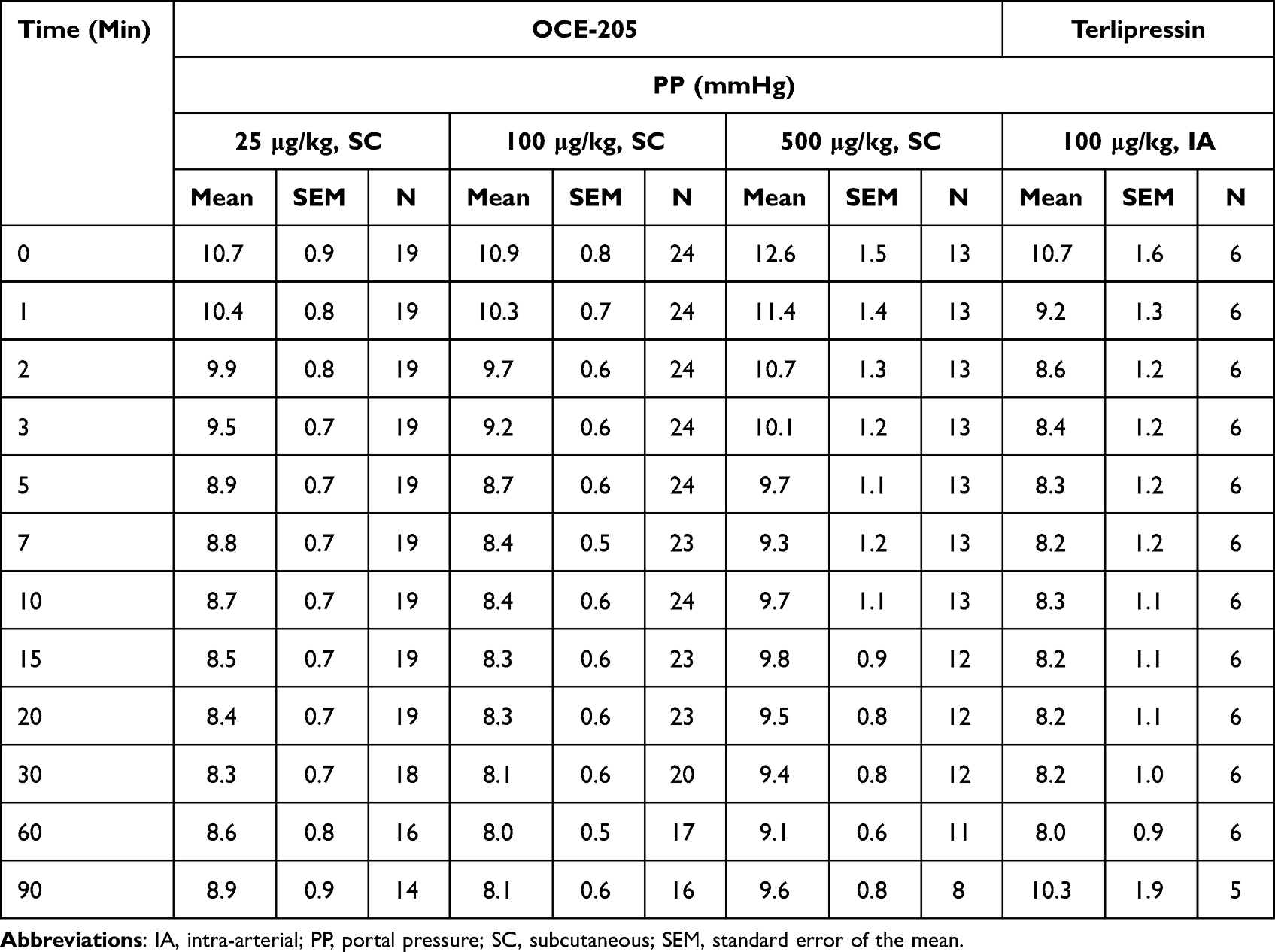 |
Table 2 Change in PP from Baseline After OCE-205 or Terlipressin Treatment in the MCD Model |
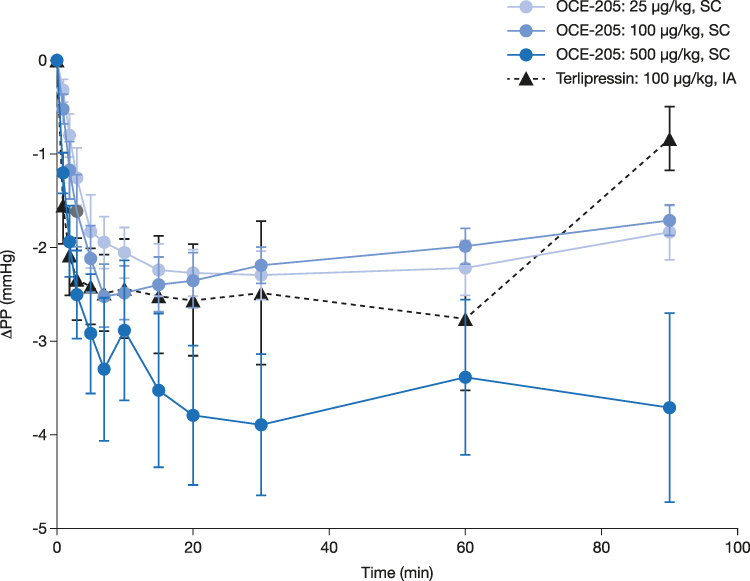 |
Figure 2 Change from baseline in portal pressure following OCE-205 or terlipressin administration in a rat model of portal hypertension (methionine/choline-deficient diet). Abbreviations: IA, intra-arterial; SC, subcutaneous. |
The observed PP values at baseline (time 0) were not statistically different between the treatment groups, as determined by one-way ANOVA.
Tolerability
No treatment-related deaths were observed after OCE-205 or terlipressin administration.
OCE-205 Modulates Pathophysiology in a BDL Rat Model of More Severe Disease
In the rats whose PHT was induced by BDL, OCE-205 treatment resulted in a decrease in PP from baseline. The observed PP values were not significantly different between any of the four treatment groups based on MANOVA with repeated measures (Table 3). Across all treatment groups, dose-related decreases from baseline in portal pressure (ΔPP) occurred following OCE-205 administration, with a plateau as the dose increased further (Figure 3).
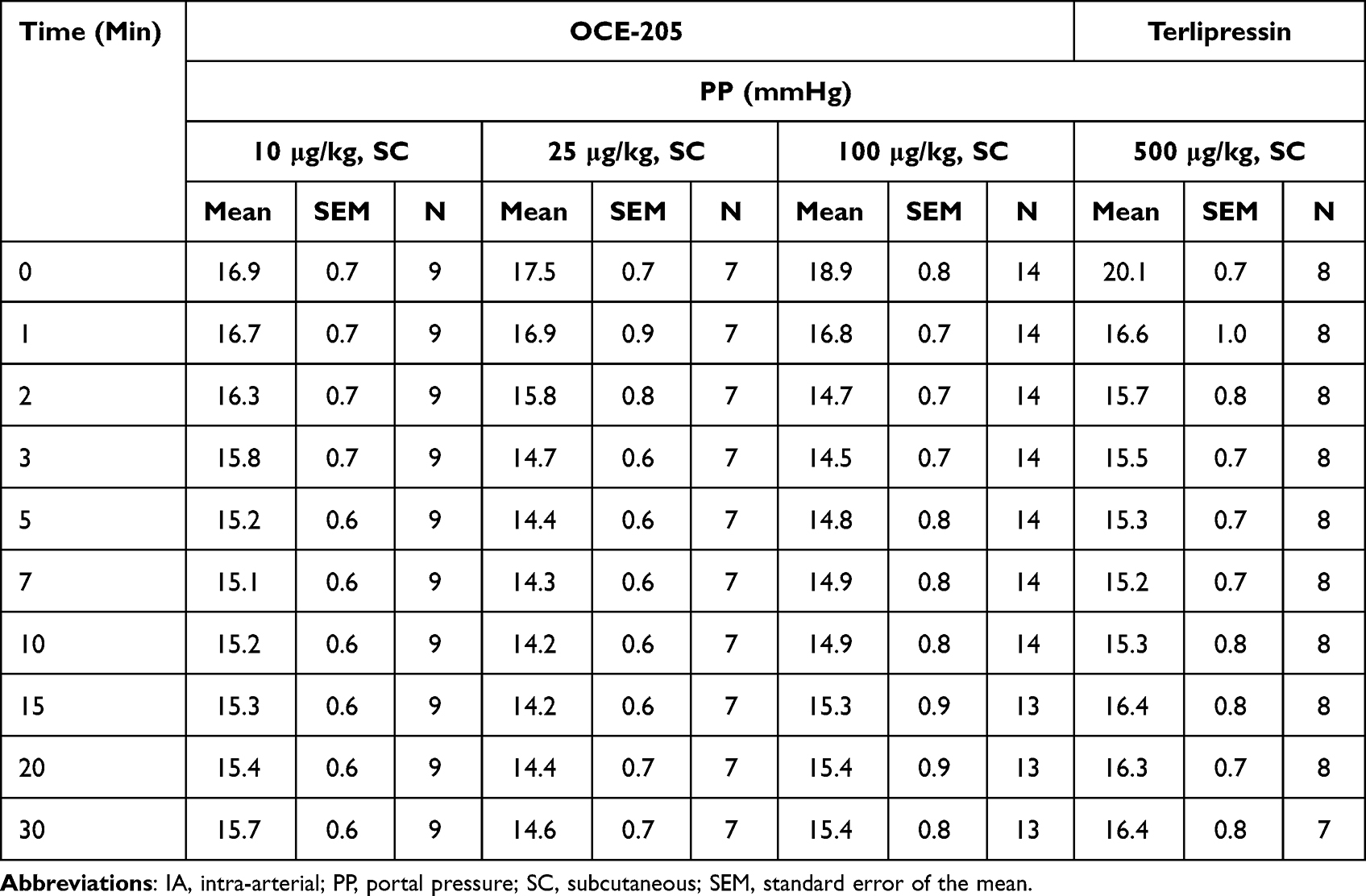 |
Table 3 Change in PP from Baseline After OCE-205 Treatment in the BDL Model |
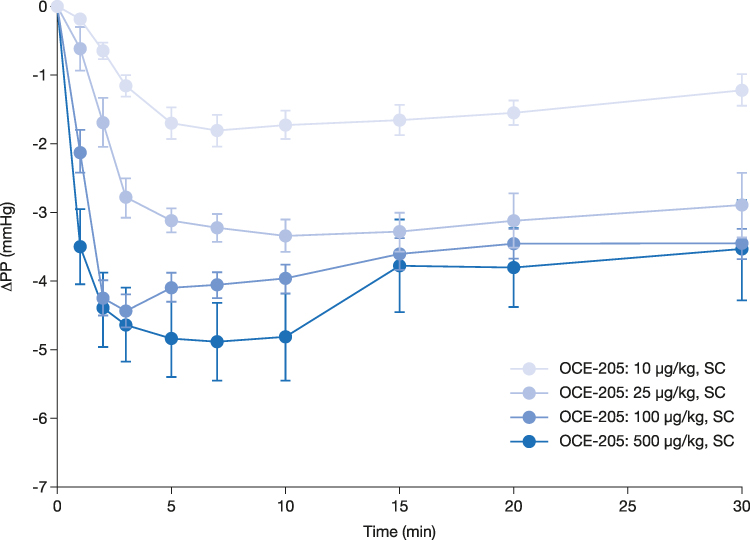 |
Figure 3 Change from baseline in portal pressure following subcutaneous (SC) OCE-205 administration in a rat model of bile duct ligation and portal hypertension. |
In the OCE-205 10 μg/kg (n = 9), 25 μg/kg (n = 7), 100 μg/kg (n = 14), and 500 μg/kg (n = 8) treatment groups, the maximum ΔPP (mean ± SEM) were −1.8 ± 0.2 mmHg at 7 min, −3.3 ± 0.2 mmHg at 10 min, −4.4 ± 0.2 mmHg at 3 min, and −4.9 ± 0.6 mmHg at 7 min, respectively. The decrease in PP remained present throughout the testing period (at 30 min) in all treatment groups: −1.2 ± 0.2, −2.9 ± 0.5, −3.4 ± 0.2, and −3.6 ± 0.7 mmHg, respectively.
When comparing results for ΔPP, the observed PP values at baseline (time 0) were statistically different between the 10 μg/kg (16.9 ± 0.7 mmHg) and 500 μg/kg (20.1 ± 0.7 mmHg) OCE-205 treatment groups. This was determined by one-way ANOVA with Tukey–Kramer HSD post hoc analysis (p = 0.045).
Tolerability
No treatment-related deaths were observed after OCE-205 administration.
Discussion
In two rat models of portal hypertension, OCE-205 achieved the therapeutic goal of decreasing PP without causing excessive vasoconstriction over a broad dose range. The study clearly shows that modest but significant decreases in PP can be obtained with OCE-205.
Studies in human blood vessels have demonstrated in vitro V1a selective activity and behavior akin to a partial agonist.35 Generating data on portal blood flow is much more challenging to accomplish in rodent models. Only two phenomena reduce PP: either splanchnic vasoconstriction restricts flow, or a change in hepatic resistance allows the same flow at decreased pressure. Vasopressin agonists exert their biological activity by increasing splanchnic resistance and reducing splanchnic blood flow. Here, we explored the potential therapeutic benefit of OCE-205 by demonstrating that it reduces PP in animals with PHT, and its safety by confirming a plateau effect where no further reductions in PP occur and there are no further increases in MAP compared with other known vasopressin analogs.
In the MCD model, which does not induce significant fibrosis nor development of cirrhosis but does lead to PP elevation,36,38,39,46 MAP increased within the desired treatment window, and plateaued over time and with increasing dose. This systemic increase was coupled with a significant reduction in PP. In contrast, administration of the full V1a receptor agonist terlipressin (triglycyl-LVP) resulted in increases in MAP that were beyond the desired treatment window of 10–15 mmHg. PP reductions in this group were similar to those seen with OCE-205. Although the recorded MAP baseline values were statistically different between the terlipressin (triglycyl-LVP) versus OCE-205 treatment groups, this observation could be attributed to inter-animal variability and smaller sample size for the terlipressin group (n = 6). The differences observed in baseline PP values in the 10 µg/kg versus 500 µg/kg groups could have been similarly affected. In the BDL rat model, which causes more profound PHT, and significant renal dysfunction,43,44 OCE-205 reduced PP with capped maximal vasoconstrictive activity that is consistent with a partial agonist-like effect.
The OCE-205 molecule was designed to widen the therapeutic window through an innovative combination of both a selective V1a agonist domain and a selective V1a antagonist domain that together achieve effective partial agonism. While this unique design allows the molecule to bind to the receptor in either orientation, any one molecule binds to one receptor at a time. Binding of the agonist domain to V1a receptors drives the desired vasoconstrictive effect, while binding of the antagonist domain in a competitive manner prevents maximal activation of the V1a system. Cell-based functional assays of OCE-205 support both its function as an effective partial agonist and its selectivity for the V1a receptor, with no activity at the human or rat V2 receptor at therapeutic concentrations.35 In healthy animals, this partial agonist mechanism was demonstrated, with a plateau effect achieving submaximal increases in MAP that were driven by similar increases in SBP and DBP (Bukofzer et al, “OCE-205 in Rats and Non-Human Primates: Pharmacokinetic and Pharmacodynamic Analysis”, submitted manuscript).
Without treatment, patients with HRS-AKI have a mortality of ~80% at 3 months, with a median survival of only 4 weeks.47 Furthermore, almost one-third of patients with HRS-AKI are readmitted to hospital within 30 days after initial hospitalization.48 While the current EASL guidelines recommend the use of terlipressin plus albumin,9 and the US FDA recently approved terlipressin for patients with HRS with a rapid reduction in kidney function,49 the observed side effects limit its use. Despite significantly improving renal function, terlipressin use is associated with serious AEs, including gastrointestinal disorders, and respiratory failure.50 LVP, the active metabolite of terlipressin (triglycyl-LVP), has activity at V1a, V1b, and V2 receptors22,23 and is both a full V1a and V2 receptor agonist.22 The known AE profile of terlipressin is likely related to the full V1a14,28,29 agonist activity of the primary metabolite LVP (which is responsible for the known pharmacology of terlipressin), leading to excessive vasoconstriction and significant V2 receptor activity which could contribute to further water retention.23,30 Because of its selective V1a functional partial agonist activity, OCE-205 could further benefit patients with HRS-AKI through increasing MAP by ~10–15 mmHg, within the desired treatment window, without the development of ischemic or pulmonary complications.
OCE-205 may have potential for more generalizable utility for other complications of decompensated cirrhosis, such as for resistant and refractory ascites and post-paracentesis–induced circulatory dysfunction, because of similar underlying mechanisms of renal dysfunction. Because of its innovative agonist/antagonist design, the selective binding of OCE-205 to V1a receptors should result in effective partial receptor agonism and limit maximal vasoconstriction. These properties could eliminate the excessive vasoconstriction and associated adverse effects observed with a full V1a receptor. Further studies utilizing OCE-205 in patients with refractory ascites are being planned.
Neither of these rat disease models fully replicates human liver disease, but rather reproduces the hemodynamic and fluid imbalances observed in patients with cirrhosis. Moreover, chronic liver diseases progress over many years in humans, whereas in rats the disease progression occurs over weeks or months. Rodent metabolism is much faster than human metabolism, which could lead to issues in replicating liver diseases related to metabolism (eg, NAFLD).51 Because animal models cannot completely predict the response in humans or replicate all the features of human liver disease, clinical trials in humans are needed to confirm the potential of OCE-205.
Conclusion
In our study, OCE-205, a novel peptide drug, led to predictable increases in MAP within the desired range for efficacy and safety while decreasing PP in both rat models. Its wide dose range, selective V1a receptor activation, and vasoconstrictive effects that elicit the target MAP increase suggest that OCE-205 is a promising candidate for further development to treat HRS-AKI. The first Phase 2 study is ongoing in patients with cirrhosis and ascites who develop HRS-AKI (NCT05309200).
Acknowledgments
Medical writing and editorial assistance was provided by Innovative Strategic Communications (Milford, PA) and by Richard Perry, PharmD, both funded by Ocelot Bio, Inc.
Funding
The original study was supported by Ferring Pharmaceuticals Inc., and publication of the data was supported by Ocelot Bio, Inc.
Disclosure
SB is the founding Chief Scientific and Medical Officer of Ocelot Bio, Inc. GH is a founder of and consultant to Ocelot Bio, Inc., and has multiple draft patents pending for V1a agonists. GH, SS, and EC were employees of Ferring Research Institute Inc., at the time of the study. The authors report no other conflicts of interest in this work.
References
1. Tellez L, Guerrero A. Management of liver decompensation in advanced liver disease (renal impairment, liver failure, adrenal insufficiency, cardiopulmonary complications). Clin Drug Investig. 2022;42(Suppl 1):15–23. doi:10.1007/s40261-022-01149-3
2. Biggins SW, Angeli P, Garcia-Tsao G, et al. Diagnosis, evaluation, and management of ascites, spontaneous bacterial peritonitis and hepatorenal syndrome: 2021 practice guidance by the American association for the study of liver diseases. Hepatology. 2021;74(2):1014–1048. doi:10.1002/hep.31884
3. Smith A, Baumgartner K, Bositis C. Cirrhosis: diagnosis and management. Am Fam Physician. 2019;100(12):759–770.
4. Moller S, Bendtsen F. The pathophysiology of arterial vasodilatation and hyperdynamic circulation in cirrhosis. Liver Int. 2018;38(4):570–580. doi:10.1111/liv.13589
5. Ginès P, Solà E, Angeli P, Wong F, Nadim MK, Kamath PS. Hepatorenal syndrome. Nat Rev Dis Primers. 2018;4(1):23. doi:10.1038/s41572-018-0022-7
6. Harper D, Chandler B. Splanchnic circulation. BJA Educ. 2015;16(2):66–71. doi:10.1093/bjaceaccp/mkv017
7. Velez JCQ. Hepatorenal syndrome type 1: from diagnosis ascertainment to goal-oriented pharmacologic therapy. Kidney360. 2022;3(2):382–395. doi:10.34067/KID.0006722021
8. Velez JC, Nietert PJ. Therapeutic response to vasoconstrictors in hepatorenal syndrome parallels increase in mean arterial pressure: a pooled analysis of clinical trials. Am J Kidney Dis. 2011;58(6):928–938. doi:10.1053/j.ajkd.2011.07.017
9. European Association for The Study of the Liver. EASL clinical practice guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69(2):406–460. doi:10.1016/j.jhep.2018.03.024
10. Treschan TA, Peters J, Warltier D. The vasopressin system: physiology and clinical strategies. Anesthesiology. 2006;105(3):599. doi:10.1097/00000542-200609000-00026
11. Barberis C, Mouillac B, Durroux T. Structural bases of vasopressin/oxytocin receptor function. J Endocrinol. 1998;156(2):223–229. doi:10.1677/joe.0.1560223
12. Koshimizu TA, Nakamura K, Egashira N, Hiroyama M, Nonoguchi H, Tanoue A. Vasopressin V1a and V1b receptors: from molecules to physiological systems. Physiol Rev. 2012;92(4):1813–1864. doi:10.1152/physrev.00035.2011
13. Albert RE, Smith WW, Eichna LW. Hemodynamic changes associated with fluid retention induced in noncardiac subjects by corticotropin (ACTH) and cortisone; comparison with the hemodynamic changes of congestive heart failure. Circulation. 1955;12(6):1047–1056. doi:10.1161/01.cir.12.6.1047
14. Donnellan F, Cullen G, Hegarty JE, McCormick PA. Ischaemic complications of glypressin in liver disease: a case series. Br J Clin Pharmacol. 2007;64(4):550–552. doi:10.1111/j.1365-2125.2007.02921.x
15. Gulberg V, Bilzer M, Gerbes AL. Long-term therapy and retreatment of hepatorenal syndrome type 1 with ornipressin and dopamine. Hepatology. 1999;30(4):870–875. doi:10.1002/hep.510300430
16. Glavas M, Gitlin-Domagalska A, Debowski D, Ptaszynska N, Legowska A, Rolka K. Vasopressin and its analogues: from natural hormones to multitasking peptides. Int J Mol Sci. 2022;23(6):3068. doi:10.3390/ijms23063068
17. Karwa R, Woodis CB. Midodrine and octreotide in treatment of cirrhosis-related hemodynamic complications. Ann Pharmacother. 2009;43(4):692–699. doi:10.1345/aph.1L373
18. Israelsen M, Krag A, Allegretti AS, et al. Terlipressin versus other vasoactive drugs for hepatorenal syndrome. Cochrane Database Syst Rev. 2017;9:CD011532. doi:10.1002/14651858.CD011532.pub2
19. Smythe CM, Nickel JF, Bradley SE. The effect of epinephrine (USP), L-epinephrine, and L -norepinephrine on glomerular filtration rate, renal plasma flow, and the urinary excretion of sodium, potassium, and water in normal man. J Clin Investig. 1952;31(5):499–506. doi:10.1172/JCI102634
20. Mallinckrodt Pharmaceuticals. TERLIVAZ [terlipressin] for injection. 2022; Available from: https://www.terlivaz.com/PI/.
21. U.S. Food & Drug Administration. FDA approves treatment to improve kidney function in adults with hepatorenal syndrome; 2022. Available from: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-treatment-improve-kidney-function-adults-hepatorenal-syndrome.
22. Jamil K, Pappas SC, Devarakonda KR. In vitro binding and receptor-mediated activity of terlipressin at vasopressin receptors V1 and V2. J Exp Pharmacol. 2018;10:1–7. doi:10.2147/JEP.S146034
23. Colson PH, Virsolvy A, Gaudard P, et al. Terlipressin, a vasoactive prodrug recommended in hepatorenal syndrome, is an agonist of human V1, V2 and V1B receptors: implications for its safety profile. Pharmacol Res. 2016;113(Pt A):257–264. doi:10.1016/j.phrs.2016.08.027
24. Ouattara A, Landi M, Le Manach Y, et al. Comparative cardiac effects of terlipressin, vasopressin, and norepinephrine on an isolated perfused rabbit heart. Anesthesiology. 2005;102(1):85–92. doi:10.1097/00000542-200501000-00016
25. Henderson KK, Byron KL. Vasopressin-induced vasoconstriction: two concentration-dependent signaling pathways. J Appl Physiol. 2007;102(4):1402–1409. doi:10.1152/japplphysiol.00825.2006
26. Moller S, Hansen EF, Becker U, Brinch K, Henriksen JH, Bendtsen F. Central and systemic haemodynamic effects of terlipressin in portal hypertensive patients. Liver. 2000;20(1):51–59. doi:10.1034/j.1600-0676.2000.020001051.x
27. Freeman JG, Barton JR, Record CO. Haemodynamic responses to 1.25 and 2 mg of terlipressin intravenously in man. Aliment Pharmacol Ther. 1988;2(4):361–367. doi:10.1111/j.1365-2036.1988.tb00709.x
28. Sarma P, Muktesh G, Singh RS, et al. Terlipressin-induced ischemic complications: a systematic review of published case reports. J Pharmacol Pharmacother. 2018;9(2):76–85. doi:10.4103/jpp.JPP_23_18
29. Boyer TD, Sanyal AJ, Wong F, et al. Terlipressin plus albumin is more effective than albumin alone in improving renal function in patients with cirrhosis and hepatorenal syndrome type 1. Gastroenterology. 2016;150(7):1579–1589 e2. doi:10.1053/j.gastro.2016.02.026
30. Krag A, Bendtsen F, Pedersen EB, Holstein-Rathlou NH, Moller S. Effects of terlipressin on the aquaretic system: evidence of antidiuretic effects. Am J Physiol Renal Physiol. 2008;295(5):F1295–F1300. doi:10.1152/ajprenal.90407.2008
31. Messmer AS, Zingg C, Müller M, Gerber JL, Schefold JC, Pfortmueller CA. Fluid overload and mortality in adult critical care patients-a systematic review and meta-analysis of observational studies. Crit Care Med. 2020;48(12):1862–1870. doi:10.1097/ccm.0000000000004617
32. Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351(2):159–169. doi:10.1056/NEJMra032401
33. Griffin BR, Liu KD, Teixeira JP. Critical care nephrology: core curriculum 2020. Am J Kidney Dis. 2020;75(3):435–452. doi:10.1053/j.ajkd.2019.10.010
34. Zhu BT. Rational design of receptor partial agonists and possible mechanisms of receptor partial activation: a theory. J Theor Biol. 1996;181(3):273–291. doi:10.1006/jtbi.1996.0130
35. Croston G, Cable E, Toy J, et al. Selective partial agonism of vasopressin 1a receptors in vitro by OCE-205. J Pharmacol Exp Ther. 2023;2023:1.
36. Konigshofer P, Brusilovskaya K, Schwabl P, Reiberger T. Animal models of portal hypertension. Biochim Biophys Acta Mol Basis Dis. 2019;1865(5):1019–1030. doi:10.1016/j.bbadis.2018.07.018
37. Bai Z, An Y, Guo X, et al. Role of terlipressin in cirrhotic patients with ascites and without hepatorenal syndrome: a systematic review of current evidence. Can J Gastroenterol Hepatol. 2020;2020:5106958. doi:10.1155/2020/5106958
38. Francque S, Wamutu S, Chatterjee S, et al. Non-alcoholic steatohepatitis induces non-fibrosis-related portal hypertension associated with splanchnic vasodilation and signs of a hyperdynamic circulation in vitro and in vivo in a rat model. Liver Int. 2010;30(3):365–375. doi:10.1111/j.1478-3231.2009.02136.x
39. Ryou M, Stylopoulos N, Baffy G. Nonalcoholic fatty liver disease and portal hypertension. Explor Med. 2020;1:149–169. doi:10.37349/emed.2020.00011
40. Rogers AE, Newberne PM. Animal model: fatty liver and cirrhosis in lipotrope-deficient male rats. Am J Pathol. 1973;73(3):817–820.
41. Oberti F, Veal N, Kaassis M, et al. Hemodynamic effects of terlipressin and octreotide administration alone or in combination in portal hypertensive rats. J Hepatol. 1998;29(1):103–111. doi:10.1016/s0168-8278(98)80184-0
42. Blei AT, Groszmann RJ, Gusberg R, Conn HO. Comparison of vasopressin and triglycyl-lysine vasopressin on splanchnic and systemic hemodynamics in dogs. Dig Dis Sci. 1980;25(9):688–694. doi:10.1007/BF01308328
43. Pereira RM, Dos Santos RA, Oliveira EA, et al. Development of hepatorenal syndrome in bile duct ligated rats. World J Gastroenterol. 2008;14(28):4505–4511. doi:10.3748/wjg.14.4505
44. Martinez-Prieto C, Ortiz MC, Fortepiani LA, Ruiz-Macia J, Atucha NM, Garcia-Estan J. Haemodynamic and renal evolution of the bile duct-ligated rat. Clin Sci. 2000;98(5):611–617. doi:10.1042/cs0980611
45. Gervaz P, Scholl B, Gillet M. Permanent access to the portal vein in rats: an experimental model. Eur Surg Res. 2000;32(3):203–206. doi:10.1159/000008764
46. Van der Graaff D, Kwanten WJ, Couturier FJ, et al. Severe steatosis induces portal hypertension by systemic arterial hyporeactivity and hepatic vasoconstrictor hyperreactivity in rats. Lab Invest. 2018;98(10):1263–1275. doi:10.1038/s41374-017-0018-z
47. Alessandria C, Ozdogan O, Guevara M, et al. MELD score and clinical type predict prognosis in hepatorenal syndrome: relevance to liver transplantation. Hepatology. 2005;41(6):1282–1289. doi:10.1002/hep.20687
48. Gilbertson EL, Krishnasamy R, Foote C, Kennard AL, Jardine MJ, Gray NA. Burden of care and quality of life among caregivers for adults receiving maintenance dialysis: a systematic review. Am J Kidney Dis. 2019;73(3):332–343. doi:10.1053/j.ajkd.2018.09.006
49. Terlivaz. US FDA prescribing information (terlipressin); 2022. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/022231s000lbl.pdf.
50. Wong F, Pappas SC, Curry MP, et al. Terlipressin plus albumin for the treatment of type 1 hepatorenal syndrome. N Engl J Med. 2021;384(9):818–828. doi:10.1056/NEJMoa2008290
51. Liu Y, Meyer C, Xu C, et al. Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol. 2013;304(5):G449–G468. doi:10.1152/ajpgi.00199.2012
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.