")
Back to Journals » Journal of Inflammation Research » Volume 14
Neuroinflammation and Microglial Activation at Rostral Ventrolateral Medulla Underpin Cadmium-Induced Cardiovascular Dysregulation in Rats
Authors Tsai CY , Fang C, Wu JCC, Wu CJ, Dai KY, Chen SM
Received 19 June 2021
Accepted for publication 28 July 2021
Published 11 August 2021 Volume 2021:14 Pages 3863—3877
DOI https://doi.org/10.2147/JIR.S325528
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Ching-Yi Tsai,1 Chi Fang,1 Jacqueline CC Wu,1 Chiung-Ju Wu,1 Kuang-Yu Dai,1 Shu-Mi Chen2,3
1Institute for Translational Research in Biomedicine, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan; 2Pharmacology and Toxicology, School of Medicine, Tzu Chi University, Hualien, Taiwan; 3Department of Pharmacy, Lotung Poh-Ai Hospital, Yilan, Taiwan
Correspondence: Ching-Yi Tsai
Institute for Translational Research in Biomedicine, Kaohsiung Chang Gung Memorial Hospital, 123 Dapi Rd, Kaohsiung 83301, Taiwan
Tel +886-7-7317123 ext.8598
Email [email protected]
Purpose: Cadmium is a heavy metal and environmental toxicant known to act on the central cardiovascular regulatory mechanisms, and one of its brain targets is the rostral ventrolateral medulla (RVLM), a brainstem site that maintains blood pressure and sympathetic vasomotor tone. The present study assessed the hypothesis that cadmium elicits cardiovascular dysregulation by inducing neuroinflammation and microglial activation, two potential cellular mechanisms, in RVLM.
Methods: Adult male Sprague–Dawley rats were used for measuring cardiovascular responses after intravenous administration of cadmium. We further conducted real-time PCR, immunofluorescence staining, in situ determination of mitochondrial superoxide, hematoxylin and eosin staining, and enzyme-linked immunosorbent assay (ELISA) to identify cytokine and chemokine mRNA expression, microglia activation, superoxide production, and necrotic and apoptotic cell death in RVLM.
Results: We found animals maintained under propofol anesthesia, intravenous administration of cadmium acetate (4 mg/kg) resulted in an increase, followed by a rebound and a secondary decrease in spontaneous baroreflex-mediated sympathetic vasomotor tone, a progressive reduction in mean arterial pressure and heart rate, alongside augmentation of pro-inflammatory cytokine and chemokine in RVLM. All those cardiovascular and neuroinflammatory events were reversed by pretreatment with an anti-inflammatory drug, pentoxifylline (50 mg/kg, i.p.). There were also concurrent microglial activation, reactive oxygen species production, hypoxia, reduced blood flow, and necrotic and apoptotic cell death in RVLM.
Conclusion: Based on these biochemical, pharmacological and morphological observations, we conclude that neuroinflammation and microglial activation at RVLM, and their downstream cellular mechanisms, causally underpin cadmium-induced cardiovascular dysregulation.
Keywords: reactive microglia, pro-inflammatory cytokine, RVLM, cardiovascular regulation, ROS
Introduction
Acute exposure to cadmium, a heavy metal and an environmental toxicant,1 induces cardiovascular toxicity that may be fatal.2 Cadmium is also capable of crossing the blood–brain barrier (BBB) and accumulates in the brain,3 leading to neurotoxicity.4 In the central nervous system, cadmium induces activation of various signaling pathways involved in inflammation, oxidative stress, and neuronal apoptosis.5–7
In vitro and in vivo studies showed that cadmium induces neuroinflammation via glial activation.5,8,9 As the key regulator of immune response in the brain, a logical glial target is the microglia.10,11 Microglia can respond quickly and undergo morphologic transformation in the event of infection and inflammation.11 These morphologically activated microglia play many functional roles, including phagocytosis of toxic products, release of cytokines, and promotion of repair and antigen-presenting.11,12
Neuroinflammation is reflected by an increase in transcription of pro-inflammatory cytokines and a decrease in transcription of anti-inflammatory cytokines.13 The accumulation of pro-inflammatory cytokines, such as interleukin-6 (IL-6), IL-1β, and tumor necrosis factor α (TNF-α), suppresses neuronal functions and induces neuronal death via oxidative stress.9,14 The transcription factor, nuclear factor-κB (NF-κB), in this regard, plays a significant role in inflammatory signaling that results in increased expression of pro-inflammatory cytokine genes.15
Cadmium is also known to act directly on central circulatory regulatory mechanisms and one potential brainstem target is the rostral ventrolateral medulla (RVLM).16 RVLM is a critical neural substrate in the baroreflex neural circuit and is intimately involved in the maintenance of stable blood pressure and sympathetic vasomotor tone.17 Myriad evidence from animal models of neurogenic hypertension,18 endotoxemia19 and organophosphate poisoning20 has confirmed that mitochondrial dysfunction-induced oxidative stress and apoptosis in RVLM is a major culprit of baroreflex dysregulation. Fu et al21 reported that systemic neuroinflammation enhances mitochondrial damage in the brain that leads to dysregulation of the baroreflex. Our previous study also revealed a significant increase in NF-κB activation and cytokine expression in RVLM 2 h after cadmium administration in rats.16
The causal relationship between neuroinflammation and microglial activation in RVLM, impairment of cardiovascular regulation and cadmium-induced sub-lethal cardiovascular toxicity remains to be established. The present study was therefore designed to assess the guiding hypothesis that neuroinflammation and microglial activation at RVLM causally underpin cadmium-induced cardiovascular dysregulation. Based on results from biochemical, pharmacological and morphological experiments, this hypothesis is validated.
Materials and Methods
Experimental Animals
Adult male Sprague–Dawley rats (254 to 330 g) purchased from BioLASCO, Taiwan, were used in this study and were housed in an AAALAC International-accredited facility maintained at 24–25°C and 12 h light/dark cycle (light on at 05:00) with freely accessible rat chow and water. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Kaohsiung Chang Gung Memorial Hospital (approval number 2017091402). The study was performed in accordance with the ARRIVE guidelines and was in compliance with the AAALAC-International Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals used as well as animal suffering during the experiment.
Recording of Cardiovascular Parameters
Animals were initially anesthetized with an induction dose of pentobarbital sodium (50 mg/kg, i.p.; SCI Pharmtech, Taoyuan, Taiwan) to carry out tracheotomy for intubation of the trachea and cannulation of a femoral artery and a femoral vein. As in our previous study,20 animals subsequently received intravenous infusion of propofol (Fresenius Kabi, Graz, Austria) at 25 mg/kg/h to provide satisfactory anesthetic maintenance while preserving the capacity of brainstem cardiovascular regulation.22 Arterial pressure (AP) recorded from the femoral artery was processed by an arterial blood pressure analyzer (APR31a; Notocord, Croissy Sur Seine, France), and the digitized signals were subjected to spectral analysis (SPA10; Notocord) to detect the low-frequency component (BLF; 0.25–0.8 Hz) of systolic blood pressure, which takes origin from RVLM23 and its power density reflects the prevalence of baroreflex-mediated sympathetic vasomotor tone.24 Heart rate (HR) was derived from the AP signals. The power density of the BLF component was displayed continuously during the experiment, alongside mean arterial pressure (MAP) and HR.
Administration of Cadmium and Test Agent
Cadmium solution (pH7.4) was freshly prepared by dissolving cadmium acetate (Sigma-Aldrich, St. Louis, MO, USA) in 0.9% normal saline. Intravenous administration of cadmium (4 mg/kg or 8 mg/kg) was delivered to the animals via the femoral vein. Injection of normal saline served as the vehicle and volume control. The volume of intravenous injection was 0.5 mL for each rat. These doses were selected from preliminary experiments and followed from our previous study.16
Animals were assigned randomly into three groups in initial experiments to measure cardiovascular responses: saline, cadmium (4 mg/kg) and cadmium (8 mg/kg). For mechanistic evaluations, we used only cadmium (4 mg/kg) group and sham-control group to identify cytokine and chemokine mRNA expression, microglia activation, superoxide production, necrotic and apoptotic cell death in RVLM.
Pretreatments with an anti-inflammatory drug, pentoxifylline (PTX; 50 mg/kg; Sigma-Aldrich), when employed, was intraperitoneally given 30 min before the administration of cadmium (at time zero in Figure 1). The dose used was determined in pilot experiments. Control infusion of sterile water served as the vehicle control. Animals in this series were assigned randomly into three groups for treatment: vehicle + saline, vehicle + cadmium and PTX + cadmium.
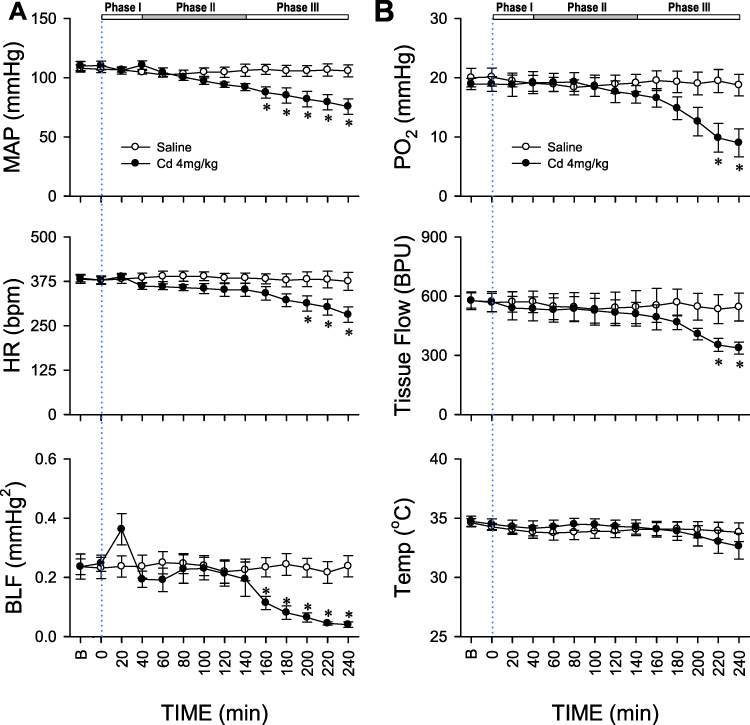 |
Figure 1 Cadmium induces triphasic cardiovascular responses in rats. (A) Temporal changes in mean arterial pressure (MAP), heart rate (HR), or power density of the low-frequency (BLF) component of systolic blood pressure spectrum in animals; (B) concurrent alterations in tissue oxygen tension (PO2), tissue perfusion (Tissue Flow) or tissue temperature (Temp) in rostral ventrolateral medulla (RVLM) of rats that received intravenous administration of cadmium (Cd; 4mg/kg) or saline. The three distinct phases based on augmented (Phase I), followed by a rebound (Phase II) and a secondary decrease (Phase III) power density of the BLF spectral component induced by cadmium are denoted on top of the figure. Values are mean ± SEM, n = 6 animals per experimental group. *P < 0.05 vs saline group at corresponding time points in the post hoc Tukey multiple-range test. Blue dotted line denotes time of cadmium injection. B denotes baseline values. |
Measurement of Tissue Oxygen Level, Blood Flow and Temperature in RVLM
As in our previous study,16 a combined oxygen/temperature/blood flow probe designed for simultaneous and continuous measurement of tissue oxygen, blood flow and temperature (Oxford Optronix, Abingdon, UK) was stereotaxically positioned into RVLM. The stereotaxic coordinates for RVLM were 4.5 to 5 mm posterior to lambda, 1.8 to 2.1 mm lateral to midline, and 8.1 to 8.4 mm below the dorsal surface of cerebellum.20 Instantaneous changes in local oxygen tension, compensated for fluctuations in tissue temperature, and real-time microvascular red blood cell perfusion in tissue were monitored. Blood flow was recorded in blood perfusion units (BPU), which is a relative unit defined against a controlled motility standard.
Collection of Tissue Samples from RVLM
Tissue samples were routinely collected at 20, 120 or 240 min after injection of cadmium. Animals were perfused with warm saline, and then a slice of medulla oblongata that contains RVLM (0.5 to 1.5 mm rostral to obex) was obtained. Tissues from both sides of the ventrolateral medulla that cover the anatomical boundaries of RVLM were subsequently collected and immediately stored in liquid nitrogen. Medullary tissues collected from anesthetized animals but without treatment served as the sham-controls.
RNA Extraction and Quantitative Real-Time PCR
RNA was extracted from RVLM tissue by a Total RNA mini kit (#RT300; Geneaid, Taipei, Taiwan) following the instructions protocol. Total RNA from each sample was transcribed into cDNA using a PrimeScript RT Reagent kit (#RR037A; Takara, Shiga, Japan). The cDNA product was diluted with RNase free water and stored at −20°C until used. Quantitative real-time PCR was carried out in duplicate using preoptimized primer/probe mixture (TaqMan Gene Expression Assays; #4331182; Applied Biosystems, Foster City, CA, USA) and TaqMan Fast Advanced Master Mix (#4444556; Applied Biosystems) on a StepOnePlus Real-time PCR system. The assay identification for each gene is IL-1β (#Rn00580432_m1), IL-6 (#Rn01410330_m1), monocyte chemoattractant protein 1 (MCP-1/Ccl2; #Rn00580555_m1), cytokine-induced neutrophil chemoattractant 1 (CINC-1/Cxcl1; #Rn00578225_m1), and GAPDH (#Rn01775763_g1). The levels of multiple cytokines and chemokines in a single sample were carried out using TaqMan Array Rat Cytokines & Chemokines 96-well plate (#RP47VT4; Applied Biosystems) according to the manufacturer’s instructions. Relative changes in mRNA expression were normalized to the levels of GAPDH and determined by the 2−ΔΔCt method.
Measurement of NF-κB Activation
Nuclear protein extracted from RVLM tissue was subjected to quantification of the transcription factor NF-κB activation by a DNA-binding ELISA kit (TransAM NF-κB p65; #40096; Active Motif; Carlsbad, CA, USA) according to the manufacturer’s protocol.
Determination of Apoptotic Cell Death
Cytoplasmic fraction of homogenized RVLM tissue was subjected to quantification of apoptotic cell death using a cell death detection kit (#11544675001; Roche, Mannheim, Germany) that measured the level of histone-associated DNA fragments following the instructions protocol.
Immunofluorescence Staining
As we reported previously,25 immunofluorescence staining was carried out using a rabbit polyclonal antiserum against Iba-1, a microglia-specific calcium-binding protein (#019-19741; FUJIFILM Wako Chemicals, Richmond, VA, USA), or Glial fibrillary acidic protein (GFAP), an astrocytes-specific marker (#Z0334; Agilent Technologies, Santa Clara, CA, USA), and a secondary goat anti-rabbit IgG conjugated with Alexa Fluor 488 (#A-11034; Invitrogen, Carlsbad, CA, USA). Slides were viewed under a confocal microscope (Olympus FV1000, Tokyo, Japan). ImageJ 2.1/1.53h program was used to quantify Iba-1 immunoreactivity and microglia morphology in RVLM according to the instructions described in the previous study.26
Detection of Mitochondrial Superoxide
For in situ determination of mitochondrial superoxide level, a highly selective fluorescent probe, MitoSOX (#M36008; Invitrogen) was used.20 Briefly, frozen sections of medulla oblongata were placed on glass slides. MitoSOX (2.5 μM) was applied to each section, and the slides were incubated in a light-protected humidified chamber at 37°C for 10 min. Images were viewed with a confocal microscope (Olympus).
Histological Examination
As reported previously,16 the brainstem was fixed with 4% paraformaldehyde for 24 h. The tissues were dehydrated, embedded in paraffin, sectioned at a thickness of 4 μm, and mounted for hematoxylin and eosin (H&E) staining for histopathological analysis. The stained brainstem sections were viewed and imaged using an automatic digital scanner (Pannoramic MIDI; 3DHISTECH, Budapest, Hungary).
Statistical Analysis
All values are expressed as mean ± SEM. Student’s t-test, one-way or two-way analysis of variance with repeated measures were used to assess group means, followed by Tukey’s test or Dunnett's multiple range test for post hoc assessment of individual means. P < 0.05 were considered statistically significant.
Results
Cadmium Induces Triphasic Responses in Baroreflex-Mediated Sympathetic Vasomotor Tone
Figure 1A shows that intravenous administration of cadmium (4 mg/kg) elicited an augmentation (Phase I), followed by a rebound (Phase II) and a secondary decrease (Phase III) in the power density of the BLF component of the systolic blood pressure spectrum, a valid experimental index for baroreflex-mediated sympathetic vasomotor tone. MAP and HR underwent a progressive reduction that became significant during Phase III. The cardiovascular parameters, however, were not affected by intravenous administration of saline (vehicle control). Intriguingly, cadmium (4 mg/kg) also induced significant reduction in tissue oxygen level and local blood flow in RVLM during the last 40 min of our 240-min observation period (Figure 1B), alongside insignificant changes in tissue temperature. Application of saline was again ineffective.
Administration of a higher-dose cadmium (8 mg/kg, i.v.) elicited high mortality. Fourteen of 15 animals died with 10 min post-cadmium, along with dramatic decrease in MAP, HR, BLF, and tissue oxygen and tissue blood flow in RVLM. The present study therefore focused on the effects of cadmium given at 4 mg/kg to induce sub-lethal cardiovascular depression.
Cadmium Induces Neuroinflammation in RVLM
Our transcriptional activity (Figure 2A) and quantitative real-time PCR (Figure 2B) results showed that, compared to sham-control, NF-κB activation and IL-1β, IL-6, CINC-1 or MCP-1 mRNA in RVLM exhibited a significant increase after cadmium (4 mg/kg) administration. We also found that most of the top 20 cytokine and chemokine genes in RVLM that underwent at least 8-fold increase at 240 min after delivery of cadmium (Table 1), were pro-inflammatory cytokine and chemokine genes.
 |
Table 1 Top 20 Cytokine and Chemokine Gene Changes in Rostral Ventrolateral Medulla (RVLM) of Rats |
 |
Figure 2 Cadmium induces neuroinflammation in RVLM. (A) Transcriptional activity of NF-κB or (B) mRNA level of IL-1β, IL-6, CINC-1 and MCP-1 in tissue collected from RVLM of rats 20, 120 or 240 min after cadmium (Cd; 4 mg/kg) administration or their sham-controls. Values are mean ± SEM, n = 3 animals per experimental group. *P < 0.05 vs sham-control (C) group in the Dunnett multiple-range test. |
Pentoxifylline Effectively Attenuates Cadmium-Induced Neuroinflammation in RVLM and Cardiovascular Depression
Our next series of experiments evaluated whether neuroinflammation in RVLM is causally related to cardiovascular dysregulation induced by cadmium. Pretreatment by the anti-inflammatory drug, pentoxifylline (PTX; 50 mg/kg, i.p.), given 30 min before the administration of cadmium, effectively retarded the augmented mRNA level of IL-1β, IL-6, CINC-1, and MCP-1 in RVLM induced by cadmium (Figure 3). Furthermore, PTX protected against the significant reduction in MAP and BLF power during Phase III (at 240 min) after administration of cadmium (Figure 4).
 |
Figure 3 Pentoxifylline attenuates cadmium-induced neuroinflammation in RVLM. mRNA level of IL-1β, IL-6, CINC-1 and MCP-1 in RVLM detected after intravenous administration of cadmium (Cd; 4 mg/kg) in rats that received pretreatment with an anti-inflammatory drug, pentoxifylline (PTX; 50 mg/kg, i.p.) or vehicle control (sterile water). Tissue samples were collected at 240 min after administration of cadmium. Values are mean ± SEM, n = 3 animals per experimental group. *P < 0.05 vs vehicle-control (VC) group in Student’s t-test. |
 |
Figure 4 Pentoxifylline attenuates cadmium-induced cardiovascular depression. Changes in MAP, HR, or BLF 240 min after intravenous administration of cadmium (Cd; 4mg/kg) or saline in animals that received pretreatment with an anti-inflammatory drug, pentoxifylline (PTX; 50 mg/kg, i.p.) or vehicle (Veh; sterile water). Values are mean ± SEM, n = 6 animals per experimental group. *P < 0.05 vs vehicle + saline (Veh + S) group in the Dunnett multiple-range test, + P < 0.05 vs vehicle + cadmium (Veh + Cd) group in the Tukey’s test. |
Cadmium Induces Microglia Activation and Increases the Number of Astrocytes in RVLM
Immunofluorescence staining (Figure 5) showed that the number of Iba-1 immunoreactive microglia in RVLM exhibited a significant increase after cadmium injection. In addition, morphological analysis (Figure 5) revealed that many of those microglia also underwent reduction of their endpoints and branch (process) length, suggesting that they were reactive microglia. We also observed a significant increase in the number of GFAP-immunoreactive astrocytes in RVLM after cadmium administration (Figure 6).
 |
Figure 5 Cadmium induces microglia activation in RVLM. (A) Representative photomicrographs of fluorescence images and their conversion to binary and skeletonized images showing low-power or high-power views of cells in ventrolateral medulla oblongata that were immunoreactive to the reactive microglia marker Iba-1 in rats treated with cadmium (4 mg/kg). (B) Quantitative and morphology analysis of Iba-1+ immunoreactivity in RVLM using ImageJ. These results are typical of 3 animals from each experimental group. *P < 0.05 vs sham-control (C) group in the Dunnett multiple-range test. Scale bar, 200 μm in low-power view or 20 μm in high-power view, binary and skeletonized images. Abbreviations: NA, nucleus ambiguus; RVLM, rostral ventrolateral medulla. |
 |
Figure 6 Cadmium increases the number of astrocytes in RVLM. (A) Representative photomicrographs of fluorescence images showing low-power or high-power views of cells in ventrolateral medulla oblongata that were immunoreactive to the reactive astrocyte marker GFAP in rats treated with cadmium (4 mg/kg). (B) Quantitative analysis of GFAP+ immunoreactivity in RVLM using ImageJ. These results are typical of 3 animals from each experimental group. *P < 0.05 vs sham-control (C) group in the Dunnett multiple-range test. Scale bar, 200 μm in low-power view or 20 μm in high-power view images. Abbreviations: NA, nucleus ambiguus; RVLM, rostral ventrolateral medulla. |
Production of Reactive Oxygen Species in RVLM by Cadmium
In situ detection of reactive oxygen species (ROS) using a mitochondrial superoxide indicator (MitoSOX; Figure 7) indicated that mitochondrial superoxide was significantly augmented in RVLM neurons after administration of cadmium.
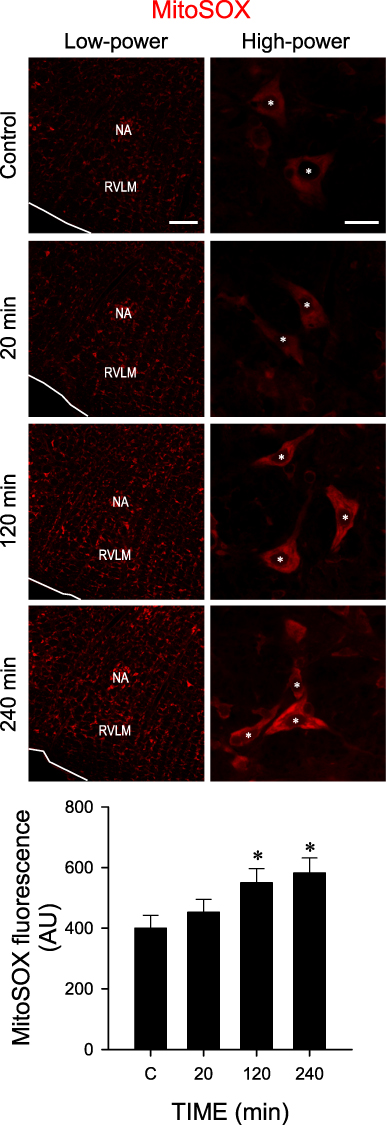 |
Figure 7 Cadmium induces ROS production in RVLM. Illustrative in situ detection of superoxide in RVLM using a mitochondrial superoxide indicator (MitoSOX) in sham-controls or 20, 120 or 240 min after intravenous administration of cadmium (Cd; 4 mg/kg). Values in quantification of mean intensity of MitoSOX fluorescence are mean ± SEM, n = 3 animals per experimental group. *P < 0.05 vs sham-control (C) group in the Dunnett multiple-range test. White asterisk denotes location of the nucleus in the corresponding RVLM neurons. Scale bar, 200 μm in low-power view or 20 μm in high-power view images. Abbreviation: NA, nucleus ambiguous. |
Both Necrotic and Apoptotic Cell Death in RVLM is Induced by Cadmium
Results from H&E staining (Figure 8A) revealed that animals that received intravenous administration of cadmium showed necrosis-appearing neurons in RVLM that were characterized by eosinophilicity (shrunken cell body, darkly stained red eosinophilic cytoplasm, and lacks discernible nucleolus), karyolysis (nuclear fading) or karyorrhexis (nuclear fragmentation). Quantitative analysis (Figure 8B) showed that 81.3 ± 2.5% of neurons in RVLM of sham-control rats exhibited clear hematoxylin-labeled nucleolus, intact nuclear membrane, nucleus and cytoplasm, as opposed to 70.6 ± 3.0%, 55.2 ± 2.8%, or 64.4 ± 4.6% at 20, 120 or 240 min after administration of cadmium (all P < 0.05 versus sham-control). At the same time, results based on the determination of histone-associated DNA fragments further showed that apoptotic cell death in RVLM from the cadmium group was also significantly increased (Figure 8C).
 |
Figure 8 Cadmium induces necrotic and apoptotic cell death in RVLM. (A) Representative photomicrographs of RVLM stained by hematoxylin and eosin showing nuclear karyolysis and eosinophilic neurons in rats 20, 120 or 240 min after receiving cadmium (Cd; 4 mg/kg, i.v.), or their sham-controls. (B) Stacked bar plots showing the average distribution of neurons in RVLM (in percentage) based on morphology from hematoxylin and eosin staining. These results are typical of 3 animals from each experimental group. In (A), NA, nucleus ambiguous; yellow arrow, eosinophilic neuron; black arrow, neuron exhibiting karyolysis. (C) changes in apoptotic cell death against sham-controls in tissues collected from RVLM of rats that received 20, 120 or 240 min after cadmium. Values are mean ± SEM, n = 4 animals per experimental group. *P < 0.05 vs sham-control (C) group in the Dunnett multiple-range test. |
Discussion
As a heavy metal and an environmental toxicant, cadmium can pass the BBB and accumulated in the brain to induce neurotoxicity and neuroinflammation4,5,8 The present study provided further biochemical, pharmacological and morphological evidence to support the notion that acute exposure to cadmium causally triggers neuroinflammation in RVLM, resulting in impaired baroreflex-mediated sympathetic vasomotor tone that leads to the decrease in blood pressure. We further demonstrated that the underlying cellular mechanisms in RVLM include activation of microglia, augmentation of astrocytes, generation of ROS, hypoxic responses and apoptotic and necrotic cell death.
Microglia represent 5–20% of the total glia cell population in rodents, and are key regulators of the immune response in the brain.10,11 It has been reported that during infection, inflammation, trauma, ischemia, and neurodegeneration, microglia can respond quickly and undergo morphological transformation from highly differentiated cells (ramified) to amoeboid cells (activated).27 There are two types of activation states, termed “M1” and “M2”.28 M1 phenotype microglia promote transcriptional activation of NF-κB and release pro-inflammatory cytokines or neurotoxic molecules to promote inflammation and cytotoxic reactions.11 In contrast, M2 phenotype microglia are associated with repair, regeneration and restoration of homeostasis by secreting anti-inflammatory cytokines and nutrient factors.12 Our immunofluorescence results showing morphologic transformation of microglia from ramified to activated phenotype, together with significant augmentation of pro-inflammatory cytokines and chemokines, therefore suggested that activation of M1 phenotype microglia underpins cadmium-induced neuroinflammation by promoting the release of pro-inflammatory cytokines and chemokines in RVLM.
We also observed a significant increase in the number of astrocytes in RVLM after cadmium administration. Cadmium has been shown to induce neuroinflammation via activation of astrocytes.8 Phuagkhaopong et al29 reported that astrocytes treated with cadmium elicit an upregulation and release of IL-6 and IL-8 via the mitogen-activated protein kinase (MAPK) and NF-κB pathways, possibly leading to neuroinflammation and neuronal death. It is therefore conceivable that astrocyte in RVLM may also play a role in the dysfunction of cardiovascular regulation after cadmium treatment.
Previous studies demonstrated that mitochondrial dysfunction, leading to the generation of ROS and necrotic or apoptotic cell death in RVLM, is a major culprit of baroreflex dysfunction.19,30,31 It thus is of interests that elevated mitochondrial superoxide in RVLM neurons, along with both necrotic and apoptotic cell death in RVLM, was found after cadmium treatment. Generation of ROS, in particular superoxide anion, may arise from a deficiency in mitochondrial electron transport chain.32 Induction of ROS production in mitochondria of cortical neuron7 via inhibiting the electron transfer chain33 is considered a possible pathogenic factor in cadmium-elicited cell death.4 Cadmium-induced mitochondrial dysfunction also leads to apoptosis or necrosis.34 It follows that mitochondrial dysfunction and ROS production in RVLM are potential action targets for cadmium toxicity against brainstem cardiovascular regulation.
We are cognizant that formation of peroxynitrite via a reaction between superoxide anion and inducible nitric oxide synthase (iNOS)-produced nitric oxide underpins endotoxin-induced apoptosis in RVLM that results in cardiovascular dysregulation;19 and transcriptional upregulation of iNOS in RVLM is induced by NF-κB.35 Our observed significant activation of NF-κB after cadmium administration therefore suggests that iNOS is a candidate target for NF-κB to mediate the secondary decrease of BLF power and MAP after cadmium treatment.
Our observation that hypoxia and reduced tissue perfusion in RVLM occurred simultaneously with decrease in MAP, HR and BLF power during Phase III is of interest. Hypoxia induces neuroinflammation and apoptosis via microglia activation.36 Acute hypoxia also initiates an immune response elicited by an increased expression of pro-inflammatory mediators in autonomic brain nuclei, including RVLM.37 An imbalance that favors activated M1 microglia through NF-κB signaling to increase the release of pro-inflammatory cytokines caused by acute hypoxia was shown in Alzheimer’s disease mice.38 It is therefore conceivable that hypoxia because of reduced blood flow in RVLM may be the common source for our observed activation of microglia and cell death promoted by cadmium.
Our previous work showed that cadmium induces apoptotic cell death in RVLM via activating caspase-3 pathway.16 Inhibitor of apoptosis proteins (IAPs) are a family of caspase inhibitors to prevent cell from apoptosis. The X-linked inhibitor of apoptosis protein (XIAP) is the most characterized member of IAPs to directly inhibit caspase-3 and caspase-7.39 Zhao et al reported that cadmium-induced mitochondrial ROS down-regulates expression of XIAP, leading to apoptosis in neuronal cells.40 It follows that mitochondrial dysfunction and ROS production in RVLM may inactivate XIAP pathway, leading to apoptotic cell death in our animal model.
Chronic cadmium exposure has been found to adversely affect various organs, including the brain, liver, kidney, lung, blood, and bone, leading to multiple organ toxicity.1 Recent reports showed that selenium is an essential mineral that possesses antioxidant and anti-inflammatory actions on multiple organ toxicity induced by cadmium in animals.41–43 Likewise, application of nutraceuticals such as myo-inositol or flavocoxid elicits protective effects in cadmium-induced organ injury.42,44 Combination of selenium with myo-inositol treatment showed greater protection against cadmium exposure in mice.42 Therefore, application of nutraceutical, antioxidant or anti-inflammatory compounds in combination with selenium may offer new directions on treatment strategies against cadmium intoxication.
We recognize that our study design did not include examination of how cadmium accumulates or activates microglia in RVLM. In vitro study in cerebrovascular endothelial cells indicated that cadmium stimulates intercellular adhesion molecule-1 (ICAM-1) expression via NF-κB activation, leading to BBB injury.45 It follows that cadmium may act directly or indirectly on microglia in RVLM to induce cardiovascular dysregulation. Future experiments are required to address these issues.
Conclusion
The present study provided biochemical, pharmacological and morphological evidence to support the notion that cadmium elicits sub-lethal circulatory depression by causally inducing neuroinflammation in RVLM that leads to cardiovascular dysregulation. We also demonstrated that the underlying cellular mechanisms (Figure 9) include activation of microglia, augmentation of astrocytes, generation of ROS, and cell death, all of which may be initiated by hypoxia because of reduced blood flow promoted by cadmium in RVLM. Our observed potency of the anti-inflammatory drug (PTX) to ameliorate the cadmium-induced dysfunction of the brainstem cardiovascular regulatory mechanism offers new insights into treatment strategies against cardiovascular toxicity induced by this heavy metal and environmental toxicant. In this regard, one potential therapeutic strategy is to exploit the synergy between nutraceuticals and anti-inflammatory or antioxidant compounds for the treatment of cadmium-induced toxicity.
 |
Figure 9 Proposed cellular mechanisms at RVLM that underpin cadmium-induced cardiovascular dysregulation. Activation of microglia and astrocytes, release of pro-inflammatory cytokines and chemokines, mitochondrial dysfunction and generation of ROS initiated by hypoxia because of reduced blood flow promoted by cadmium leads to cell death in RVLM, resulting in cardiovascular dysregulation. Also included are potential therapeutic targets against cadmium-induced toxicity. Note dotted line denotes that mitochondria may be a direct action target for cadmium. Abbreviations: Cd, cadmium; PTX, pentoxifylline; RVLM, rostral ventrolateral medulla. |
Acknowledgments
This study was supported by research grants from the Chang Gung Medical Foundation (CMRPG8G1401, CMRPG8G1402, and CMRPG8G1403 to CYT). All authors thank Professor Samuel H.H. Chan for providing critical comments on the manuscript. We also thank the Core Laboratory for Phenomics and Diagnostics, Department of Medical Research, Kaohsiung Chang Gung Memorial Hospital for providing the slide scanner (Pannoramic MIDI; 3DHISTECH) for histological examination.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. World Health Organization. Environmental Health Criteria 134, Cadmium.
2. Miura N, Yanagiba Y, Ohtani K, Mita M, Togawa M, Hasegawa T. Diurnal variation of cadmium-induced mortality in mice. J Toxicol Sci. 2012;37:191–196. doi:10.2131/jts.37.191
3. Shukla A, Shukla GS, Srimal RC. Cadmium-induced alterations in blood-brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Hum Exp Toxicol. 1996;15:400–405. doi:10.1177/096032719601500507
4. Wang B, Du Y. Cadmium and its neurotoxic effects. Oxid Med Cell Longev. 2013;2013:898034. doi:10.1155/2013/898034
5. Ashok A, Rai NK, Tripathi S, Bandyopadhyay S. Exposure to As-, Cd-, and Pb-mixture induces Aβ, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol Sci. 2015;143:64–80. doi:10.1093/toxsci/kfu208
6. Chen L, Liu L, Huang S. Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic Biol Med. 2008;45:1035–1044. doi:10.1016/j.freeradbiomed.2008.07.011
7. Xu C, Wang X, Zhu Y, et al. Rapamycin ameliorates cadmium-induced activation of MAPK pathway and neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A. Neuropharmacology. 2016;105:270–284. doi:10.1016/j.neuropharm.2016.01.030
8. Favorito R, Monaco A, Grimaldi MC, Ferrandino I. Effects of cadmium on the glial architecture in lizard brain. Eur J Histochem. 2017;61:2734.
9. Yang Z, Yang S, Qian SY, et al. Cadmium-induced toxicity in rat primary mid-brain neuroglia cultures: role of oxidative stress from microglia. Toxicol Sci. 2007;98:488–494. doi:10.1093/toxsci/kfm106
10. Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170.
11. Kawabori M, Yenari MA. The role of the microglia in acute CNS injury. Metab Brain Dis. 2015;30:381–392. doi:10.1007/s11011-014-9531-6
12. Choi JY, Kim JY, Kim JY, Park J, Lee WT, Lee JE. M2 phenotype microglia-derived cytokine stimulates proliferation and neuronal differentiation of endogenous stem cells in ischemic brain. Exp Neurobiol. 2017;26:33–41. doi:10.5607/en.2017.26.1.33
13. Menzel L, Kleber L, Friedrich C, et al. Progranulin protects against exaggerated axonal injury and astrogliosis following traumatic brain injury. Glia. 2017;65:278–292. doi:10.1002/glia.23091
14. Jung HY, Kwon HJ, Kim W, et al. Phosphoglycerate mutase 1 prevents neuronal death from ischemic damage by reducing neuroinflammation in the rabbit spinal cord. Int J Mol Sci. 2020;21:7425. doi:10.3390/ijms21197425
15. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi:10.1038/sigtrans.2017.23
16. Chen SM, Phuagkhaopong S, Fang C, et al. Dose-dependent acute circulatory fates elicited by cadmium are mediated by differential engagements of cardiovascular regulatory mechanisms in brain. Front Physiol. 2019;10:772. doi:10.3389/fphys.2019.00772
17. Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi:10.1038/nrn1902
18. Chan SHH, Wu KHL, Chang AYW, Tai MH, Chan JYH. Oxidative impairment of mitochondrial electron transport chain complexes in rostral ventrolateral medulla contributes to neurogenic hypertension. Hypertension. 2009;53:217–227. doi:10.1161/HYPERTENSIONAHA.108.116905
19. Chan SHH, Wu KLH, Wang LL, Chan JYH. Nitric oxide- and superoxide-dependent mitochondrial signaling in endotoxin-induced apoptosis in the rostral ventrolateral medulla of rats. Free Radic Biol Med. 2005;39:603–618. doi:10.1016/j.freeradbiomed.2005.04.012
20. Tsai CY, Wu CJ, Wu JCC, Fang C, Huang YH, Dai KY. Redox-active DJ-1 sustains brainstem cardiovascular regulation via maintenance of mitochondrial function during mevinphos intoxication. Neurochem Int. 2020;139:104791. doi:10.1016/j.neuint.2020.104791
21. Fu MH, Chen IC, Lee CH, et al. Anti-neuroinflammation ameliorates systemic inflammation-induced mitochondrial DNA impairment in the nucleus of the solitary tract and cardiovascular reflex dysfunction. J Neuroinflammation. 2019;16:224. doi:10.1186/s12974-019-1623-0
22. Yang CH, Shyr MH, Kuo TBJ, Tan PPC, Chan SHH. Effects of propofol on nociceptive response and power spectra of electroencephalographic and systemic arterial pressure signals in the rat: correlation with plasma concentration. J Pharmacol Exp Ther. 1995;275:1568–1574.
23. Kuo TBJ, Yang CC, Chan SHH. Selective activation of vasomotor component of SAP spectrum by nucleus reticularis ventrolateralis in rats. Am J Physiol. 1997;272:H485–H492.
24. Poon YY, Tsai CY, Cheng CD, Chang AYW, Chan SHH. Endogenous nitric oxide derived from NOS I or II in thoracic spinal cord exerts opposing tonic modulation on sympathetic vasomotor tone via disparate mechanisms in anesthetized rats. Am J Physiol Heart Circ Physiol. 2016;311:H555–H562. doi:10.1152/ajpheart.00246.2016
25. Tsai CY, Su CH, Chan JYH, Chan SHH. Nitrosative stress-induced disruption of baroreflex neural circuits in a rat model of hepatic encephalopathy: a DTI study. Sci Rep. 2017;7:40111. doi:10.1038/srep40111
26. Young K, Morrison H. Quantifying microglia morphology from photomicrographs of immunohistochemistry prepared tissue using ImageJ. J Vis Exp. 2018;136:57648.
27. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi:10.1016/0166-2236(96)10049-7
28. Tu H, Chu H, Guan S, et al. The role of the M1/M2 microglia in the process from cancer pain to morphine tolerance. Tissue Cell. 2021;68:101438. doi:10.1016/j.tice.2020.101438
29. Phuagkhaopong S, Ospondpant D, Kasemsuk T, et al. Cadmium-induced IL-6 and IL-8 expression and release from astrocytes are mediated by MAPK and NF-κB pathways. Neurotoxicology. 2017;60:82–91. doi:10.1016/j.neuro.2017.03.001
30. Chan JYH, Cheng HL, Chou JLJ, et al. Heat shock protein 60 or 70 activates NOS I- and inhibits NOS II-associated signaling, and depresses mitochondrial apoptotic cascade during brain stem death. J Biol Chem. 2007;282:4585–4600. doi:10.1074/jbc.M603394200
31. Chang AYW, Chan JYH, Chuang YC, Chan SHH, Kleinschnitz C. Brain stem death as the vital determinant for resumption of spontaneous circulation after cardiac arrest in rats. PLoS One. 2009;4:e7744. doi:10.1371/journal.pone.0007744
32. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi:10.1074/jbc.M304854200
33. Wang Y, Fang J, Leonard SS, Rao KM. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic Biol Med. 2004;36(11):1434–1443. doi:10.1016/j.freeradbiomed.2004.03.010
34. Messner B, Türkcan A, Ploner C, Laufer G, Bernhard D. Cadmium overkill: autophagy, apoptosis and necrosis signalling in endothelial cells exposed to cadmium. Cell Mol Life Sci. 2016;73:1699–1713. doi:10.1007/s00018-015-2094-9
35. Chan JYH, Ou CC, Wang LL, Chan SHH. Heat shock protein 70 confers cardiovascular protection during endotoxemia via Inhibition of nuclear factor-kB activation and inducible nitric oxide synthase expression in the rostral ventrolateral medulla. Circulation. 2004;110:3560–3566. doi:10.1161/01.CIR.0000143082.63063.33
36. Yang Z, Zhong L, Zhong S, Xian R, Yuan B. Hypoxia induces microglia autophagy and neural inflammation injury in focal cerebral ischemia model. Exp Mol Pathol. 2015;98:219–224. doi:10.1016/j.yexmp.2015.02.003
37. Silva TM, Chaar LJ, Silva RC, et al. Minocycline alters expression of inflammatory markers in autonomic brain areas and ventilatory responses induced by acute hypoxia. Exp Physiol. 2018;103:884–895. doi:10.1113/EP086780
38. Zhang F, Zhong R, Li S, et al. Acute hypoxia induced an imbalanced M1/M2 activation of microglia through NF-κB signaling in Alzheimer’s disease mice and wild-type littermates. Front Aging Neurosci. 2017;9:282. doi:10.3389/fnagi.2017.00282
39. Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi:10.1038/40901
40. Zhao R, Yu Q, Hou L, et al. Cadmium induces mitochondrial ROS inactivation of XIAP pathway leading to apoptosis in neuronal cells. Int J Biochem Cell Biol. 2020;121:105715. doi:10.1016/j.biocel.2020.105715
41. Tang KK, Li HQ, Qu KC, Fan RF. Selenium alleviates cadmium-induced inflammation and meat quality degradation via antioxidant and anti-inflammation in chicken breast muscles. Environ Sci Pollut Res Int. 2019;26:23453–23459. doi:10.1007/s11356-019-05675-0
42. Benvenga S, Marini HR, Micali A, et al. Protective effects of myo-inositol and selenium on cadmium-induced thyroid toxicity in mice. Nutrients. 2020;12:1222. doi:10.3390/nu12051222
43. Xue H, Cao H, Xing C, et al. Selenium triggers Nrf2-AMPK crosstalk to alleviate cadmium-induced autophagy in rabbit cerebrum. Toxicology. 2021;459:152855. doi:10.1016/j.tox.2021.152855
44. Micali A, Pallio G, Irrera N, et al. Flavocoxid, a natural antioxidant, protects mouse kidney from cadmium-induced toxicity. Oxid Med Cell Longev. 2018;2018:9162946. doi:10.1155/2018/9162946
45. Jeong EM, Moon CH, Kim CS, et al. Cadmium stimulates the expression of ICAM-1 via NF-κB activation in cerebrovascular endothelial cells. Biochem Biophys Res Commun. 2004;320:887–892. doi:10.1016/j.bbrc.2004.05.218
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.