")
Back to Journals » Substance Abuse and Rehabilitation » Volume 15
Nalmefene Hydrochloride: Potential Implications for Treating Alcohol and Opioid Use Disorder
Authors Green M, Veltri CA , Grundmann O
Received 5 December 2023
Accepted for publication 16 March 2024
Published 3 April 2024 Volume 2024:15 Pages 43—57
DOI https://doi.org/10.2147/SAR.S431270
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Rajendra D. Badgaiyan
MeShell Green,1 Charles A Veltri,1 Oliver Grundmann1,2
1College of Pharmacy, Department of Pharmaceutical Sciences, Midwestern University, Glendale, AZ, USA; 2College of Pharmacy, Department of Medicinal Chemistry, University of Florida, Gainesville, FL, USA
Correspondence: Oliver Grundmann, Department of Medicinal Chemistry, College of Pharmacy, University of Florida, 1345 Center Drive, Room P3-20, Gainesville, FL, 32611, USA, Tel +1-352-246-4994, Fax +1-352-392-9455, Email [email protected]
Abstract: Nalmefene hydrochloride was first discovered as an opioid antagonist derivative of naltrexone in 1975. It is among the most potent opioid antagonists currently on the market and is differentiated from naloxone and naltrexone by its partial agonist activity at the kappa-opioid receptor which may benefit in the treatment of alcohol use disorder. Oral nalmefene has been approved in the European Union for treatment of alcohol use disorder since 2013. As of 2023, nalmefene is available in the United States as an intranasal spray for reversal of opioid overdose but is not approved for alcohol or opioid use disorder as a maintenance treatment. The substantially longer half-life of nalmefene and 5-fold higher binding affinity to opioid receptors makes it a superior agent over naloxone in the reversal of high potency synthetic opioids like fentanyl and the emerging nitazenes. Nalmefene presents with a comparable side effect profile to other opioid antagonists and should be considered for further development as a maintenance treatment for opioid and other substance use disorders.
Keywords: opioid antagonist, withdrawal, substance use disorder, kappa opioid receptor
Introduction
Opioid use disorder (OUD) is characterized by an intense, reoccurring craving and uncontrollable desire to misuse opioids despite potential physical and psychological harm to the person using the substance.1 In 2019, approximately 1.6 million individuals aged 12 years and older in the United States were reported to suffer from an opioid dependency.2 Furthermore, an estimated 69,000 OUD overdose deaths were reported in 2020.2 OUD exerts a profound and adverse impact on one’s quality of life and increases potential risk factors for comorbidities, mental health disorders, and poor social and functional outcomes when left untreated.3 The global COVID-19 pandemic did worsen the OUD overdose rate primarily because of limited access to treatment facilities.4 In addition, a number of new psychoactive substances (NPS) have emerged in recent years, among them synthetic opioids that are more potent than fentanyl.5,6 The highest rate of fatality from NPS was from synthetic opioids, which may require many times the dose of naloxone available in emergency departments with limited resources.7,8 The numbers for OUD and opioid overdose deaths justify the term “opioid epidemic” used by both scientific and popular outlets and constitute a social and public health crisis that needs to be prioritized by implementing new prevention, treatment, and mitigation approaches, among them pharmacotherapy.
OUD medication-assisted treatment (MAT) can help reduce dependency and improve overall health. Current US Food and Drug Administration (FDA) therapies approved for OUD are buprenorphine, methadone, and naltrexone either alone or in combination.9 Methadone acts as a full agonist on the mu-opioid receptor (μOR), while buprenorphine functions as a partial agonist on the μOR. Both methadone and buprenorphine work to reduce cravings and alleviate withdrawal symptoms. Methadone was first approved for OUD MAT in 1972.10 Its effectiveness for OUD has been shown in several meta-analyses, while methadone also shows a lower potential for tolerance and dependence compared to other opioids likely based on its longer half-life and N-methyl-D-glutamate (NMDA) antagonist activity.11,12 Buprenorphine, alone or in combination with naloxone, was approved for OUD MAT in 2000 and can, similar to methadone, be associated with adverse effects if mis- or abused.13 Because it is a partial agonist, buprenorphine in theory may be limited in causing fatal respiratory depression because of its ceiling effect. It appears that buprenorphine resulted in lower recidivism compared to methadone despite a lower percentage of those using buprenorphine could be maintained on the MAT compared to methadone overall.14 It appears that OUD MAT should be personalized based on the patient’s individual characteristics and preferences. While some patients respond better to methadone as a full μOR agonist, others may prefer the longer duration and partial agonist activity of buprenorphine.14 Naltrexone is a pure μOR antagonist that counteracts the effects of opioid utilization.15 Although naltrexone has gained recognition for its role in blocking the euphoric effects of opioid consumption and suppressing cravings, it is less frequently used in the treatment of OUD when compared to methadone and buprenorphine, largely due to its sustained presence of withdrawal symptoms even after longer discontinuation, low patient acceptance, and non-adherence.16 Furthermore, low dose naltrexone (LDN) is receiving increasing attention for its potential in managing chronic pain and inflammation.17 Naloxone is a non-selective, competitive opioid receptor antagonist that is commonly paired intravenously with buprenorphine or when buprenorphine is given orally to counter the euphoric effects and potential abuse of buprenorphine.18 As monotherapy, naloxone is ineffective for the maintenance treatment of OUD due to its short half-life; however, it is the primary treatment for opioid overdose and, due to its safety profile, is now available as an over-the-counter opioid intoxication reversal agent.19 Buprenorphine, methadone, and naltrexone are clinically proven, effective therapies in the management of OUD; however, they can pose financial challenges as well. In 2021, the US Department of Defense estimated the annual cost per individual for treatment in a certified opioid treatment program (OTP) would be approximately $5980, $6552, and $14,112, respectively, excluding psychosocial support.20 Nevertheless, there is an ongoing need to expand research and develop additional therapy options for the individualized treatment of OUD.
Alcohol Use Disorder (AUD) remains among the most prevalent use disorders in the US, globally accounting for more than 14.5 million people with an AUD diagnosis and 95,000 deaths in the US in 2019.21 The most common pharmacotherapeutic treatment approaches involve the use of enzyme inhibitors (disulfiram) to prevent the degradation of acetaldehyde, the toxic metabolite of ethanol. Acamprosate serves to reduce withdrawal and cravings for alcohol by reducing inhibitory GABAA mediated signaling and increasing excitatory glutamate mediated signaling.22 In addition, naltrexone is commonly used to reduce withdrawal and cravings from alcohol as it mediates GABA release upon antagonism at opioid receptors. However, most pharmacotherapies present with limitations and a high recidivism rate of up to 75% at one year following treatment initiation.23
Nalmefene hydrochloride is a potent mu-opioid receptor antagonist and partial kappa-opioid receptor agonist, primarily employed for the treatment of AUD in the European Union, averting the compulsive urge to consume alcohol.24 Nalmefene has demonstrated efficacy in reversing opioid intoxication, in addition to curbing cocaine cravings.25,26 Additional potential application for nalmefene hydrochloride is its potential to provide relief in chronic pain and facilitate behavioral modifications.16,27 This review will evaluate the prospective effectiveness of nalmefene hydrochloride as an alternative, long-term therapy for the treatment of OUD and AUD.
Results
Pharmacodynamics
The opioid receptor system plays a crucial role in the modulation of pain, behavior, and antinociception. The mu (μ), kappa (κ), and delta (δ) opioid receptors are G-protein coupled receptors expressed in the central nervous system that affect analgesia, mood stability, and reward pathways which ultimately can lead to an OUD. The agonism of the μOR primarily operates in pain management, mood enhancement, and the stimulation of central dopamine reward pathways resulting in the sensation of euphoria.28 Additionally, the chronic stimulation of the μOR affects the respiratory center in the central nervous system, resulting in a depression in both the rate and depth of breathing, potentially reaching a dangerously low level. The δOR signaling pathway predominately serves the function of producing analgesia.28 The activation of κOR not only enhances the analgesic response but it also modulates various behaviors such as depression and reward. According to animal studies, the repeated activation of κOR was found to diminish the reward-potentiating impact of cocaine; however, the repeated stimulation was also associated with behavioral signs characteristic of depression-like responses.29,30
A preclinical study examined numerous compounds involved in opiate addiction at the μ, κ, and δ opioid receptors to quantify their binding affinity in efforts to evaluate potential opioid treatment therapies.31 Table 1 depicts the binding and inhibitory constants (Ki) of opioid receptor agonists and antagonists in Chinese hamster ovary (CHO) cells transfected with human opioid receptors. Morphine and fentanyl, opioid receptor agonists, exhibit high affinities primarily at the μOR. These drug therapies are frequently used for the relief of moderate-to-severe chronic pain, providing analgesic effects but also associated adverse events, including potential addiction and respiratory suppression when misused. Nalmefene and naltrexone, μOR antagonists, demonstrate greater affinities at the μ-opioid receptor compared to morphine, fentanyl, and its therapeutic counterparts and are utilized for the treatment of OUD. While naltrexone and naloxone are neutral antagonists that occupy but do not activate the opioid receptor, nalmefene acts as an inverse agonist, lowering the constitutive activity of opioid receptors below baseline.32 This suggests more efficient binding while reducing the likelihood of agonistic drug binding and receptor activation, thus, attenuating the effects of fentanyl and morphine. Antagonists for G-protein coupled receptors require approximately 60–90% target occupancy to elicit therapeutic effects in patients. An occupancy simulation in a PK/PD study demonstrated that a single administration of 20 mg nalmefene resulted in μOR occupancies of within or above 60–90% for up to 22–24 hours in 95% of the population.33 Several studies examined the μOR occupancy of various doses of naltrexone. Eight hours post-administration, 15 mg naltrexone occupied approximately 61% of the μ-opioid receptors, whereas 50 mg naltrexone achieved over 90% occupancy after about 49 hours.34,35 At the κ-opioid receptor, nalmefene acts as a high affinity partial agonist, potentially instigating aversion towards addictive behaviors, particularly in the treatment of AUD.36 The activation of κOR potentially leads to heightened activation of the hypothalamic-pituitary-adrenal (HPA) axis through increased adrenocorticotropic hormone and cortisol secretion.37 Elevated cortisol levels have been associated with mood disorders and depression. In contrast, naltrexone has an antagonistic effect on the κOR, potentially modulating dysphoria by alleviating symptoms of anxiety and depression in opiate dependent individuals.30,38,39 Another opioid-like receptor, the orphanin FQ/nociception receptor, is known to be antagonized by naltrexone, while it is not known what effect nalmefene administration has.40 The nociception receptor mediates spinal analgesia similar to the µOR.
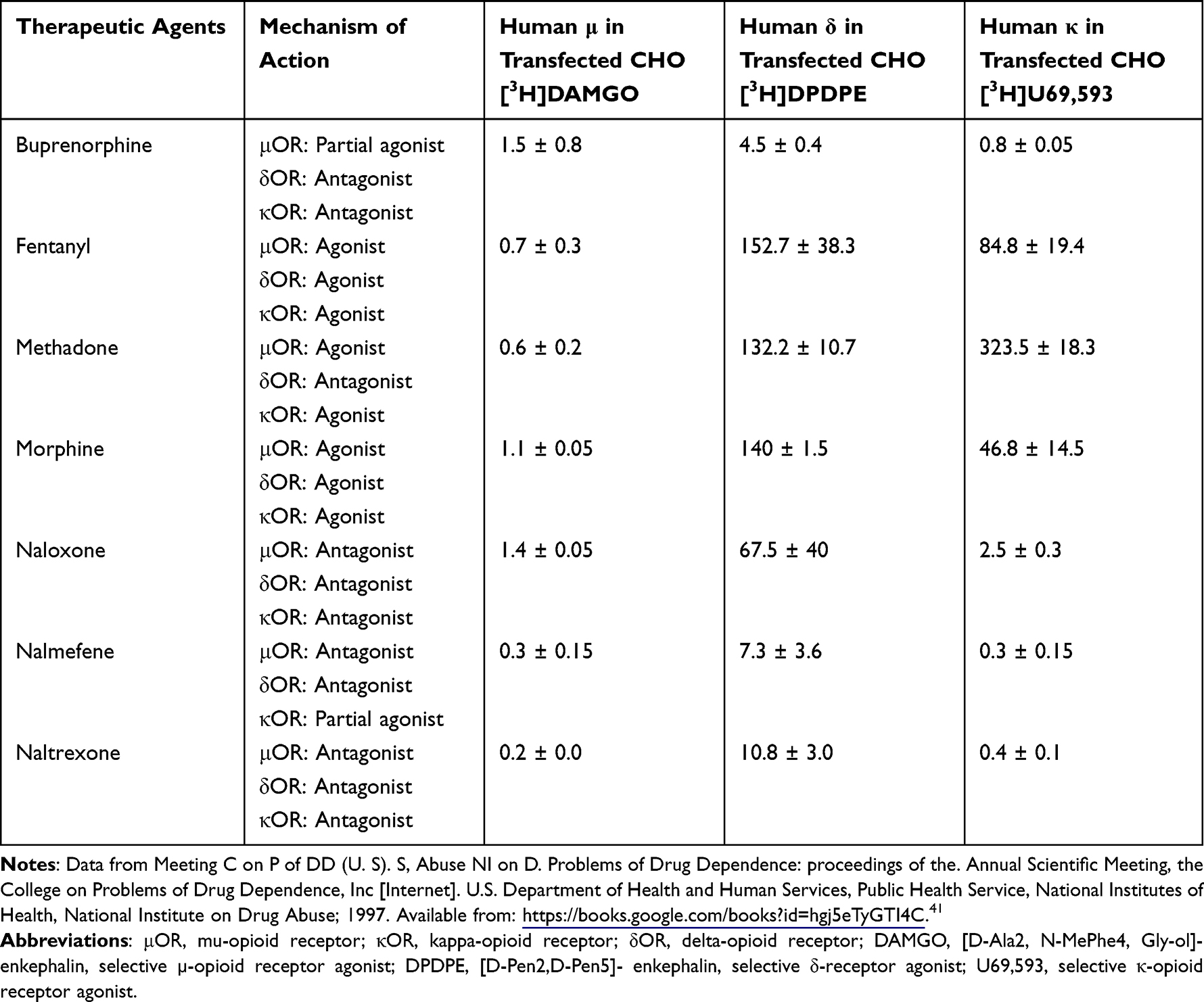 |
Table 1 Binding Affinities (Ki) in nM of Therapeutic Agents at the Humanized μ/κ/δ-Opioid Receptors on Transfected Chinese Hamster Ovarian Cells. |
A meta-analysis conducted to determine serious adverse events associated with the use of nalmefene in patients with substance abuse and impulse control disorders reported three long-term (24–48 weeks) randomized controlled trials that revealed psychiatric adverse events in 24 of the 1144 total participants (2.1%) across all three studies; however, no evidence of increased odds of overall psychiatric adverse events in the nalmefene group compared with the placebo group was present.42–45 Patients taking nalmefene were 3.22 times more likely to discontinue therapy due to transient side effects compared to placebo. Patients frequently encounter adverse effects such as nausea, dizziness, headache, and insomnia, contributing to early withdrawals from clinical studies and suboptimal adherence. Adverse events of nalmefene administration in opioid-dependent patients include mood changes, restlessness, nausea, vomiting, headache and dizziness.46 In clinical studies involving patients with OUD, reported side effects of naltrexone included nausea, vomiting, decreased appetite, low energy, and difficulty sleeping.47 Naltrexone has not been conclusively associated with cases of clinically apparent liver injury; however, close monitoring is recommended for patients with moderate-to-severe hepatic impairment.48 Interestingly, oral nalmefene does not exhibit dose-dependent hepatotoxicity, eliminating the need for dosing adjustments.49
Methadone serves as a full agonist at the μ and κ-opioid receptors and functions as an antagonist at the δ-opioid receptor. Methadone consists of both S and R-enantiomers, with R-methadone displaying high selectivity for the μ-opioid receptor and minimal affinity for the κ and δ-opioid receptors.50 In contrast to standard μOR agonists, methadone exhibits a diminished sense of euphoria upon oral administration, thereby reducing the intensity of cravings and withdrawal symptoms.51 Furthermore, the internalization and recycling of the μ-opioid receptor contributes to reduced opioid tolerance during long-term treatment.52 The non-opioid activities of methadone, such as N-methyl-D-aspartate (NMDA) receptor antagonism and the inhibition of serotonin and norepinephrine reuptake, have the potential to mitigate the development of desensitization associated with prolonged treatment but may not be of significant clinical impact at therapeutic concentrations.53,54 Chronic methadone exposure, leading to an excessive opioid receptor activity, can result in various toxic effects including respiratory suppression, cardiotoxicity characterized by bradycardia, QTc prolongation, and torsades de pointes (TdP), sensorineural hearing loss, and hypoglycemia.55
Buprenorphine is a partial agonist with a strong affinity for the μ-opioid receptor, provoking a stimulatory response.56 However, in the presence of a full agonist, buprenorphine acts as a functional antagonist, attenuating the effects generated by full agonists. Its low intrinsic activity at the μ-opioid receptor elicits a ceiling effect at higher doses, resembling a dose-dependent bell curve. This limits agonistic responses, decreasing the likelihood of abuse and the development of physical dependence. Buprenorphine exhibits a strong antagonistic affinity for the κOR, potentially mitigating dysphoric sensations akin to the effects observed with naltrexone.39,57 Several FDA approved formulations for OUD incorporate naloxone in conjunction with buprenorphine to reduce the risk of parenteral diversion and misuse of buprenorphine’s characteristic agonistic properties.56 In addition, there are several active metabolites of buprenorphine that interact with opioid receptors.58 Among them are norbuprenorphine and various glucuronides which also present with binding affinity at opioid receptors and the nociception receptor which is considered a target for opioids.
Naloxone, a high affinity μ-opioid receptor antagonist, quickly occupies the μOR and diminishes the stimulatory euphoric effects commonly induced by buprenorphine when administered intravenously. Yet, conflicting literature indicates that buprenorphine ultimately displaces naloxone resulting in a delayed euphoric experience, rendering the addition of naloxone ineffective.59,60 Severe complications associated with buprenorphine include central nervous system depression, hypotension, QTc prolongation, and lower seizure threshold. Less severe adverse events include nausea, dizziness, headache, and dry mouth.61 Furthermore, the active metabolite, norbuprenorphine, has been observed to penetrate the placental barrier in rats, consequently resulting in the reduction of myelinated axons, nerve growth factor, and the emergence of depression-like behavioral features in the exposed offspring, underscoring the importance for studying the impact of perinatal buprenorphine administration on humans.41,58
Naltrexone, buprenorphine, and nalmefene therapies can induce precipitated withdrawal when administered in the presence of a full μ-opioid agonist. Precipitated withdrawal occurs when the elevated binding affinities of buprenorphine, naltrexone, and nalmefene displace the full agonists, resulting in a rapid reduction in the μ-receptor activation from full to partial.62 It is characterized by the sudden onset of symptoms such as autonomic instability, seizures, body aches, nausea, and vomiting. Naltrexone therapies recommend that patients undergo a 7–10-day opioid wash-out period prior to initiation to prevent precipitated withdrawal.63 In contrast, before beginning buprenorphine therapy, an individual should wait until moderate withdrawal symptoms are present or at least 6–12 h after the last opioid administration. The amount of time from last use to moderate withdrawal varies depending on half-life of the opioid. When transitioning from methadone to buprenorphine, it is essential to gradually reduce the methadone dose to less than 30 mg. Additionally, the patient must wait at least 72 hours after the last methadone dose to prevent precipitated withdrawal. Individuals using naltrexone and nalmefene should wait until withdrawal symptoms are no longer present or receive smaller doses due to their characteristically high affinity for the μ-opioid receptor. However, nalmefene may present with distinct differences in precipitating withdrawal to naltrexone or naloxone given that it acts as an inverse agonist and may thus present with more severe adverse effects. There are currently no recommendations for the induction of nalmefene. During the induction period, it is crucial to closely monitor patients and titrate doses to suit the individual needs of the patient for all drug therapies.
Pharmacokinetics
Buprenorphine is a highly lipophilic drug available in numerous formulations including buccal film, transdermal films/patches, intramuscular (IM) injection, subcutaneous (SQ) injection, sublingual tablet, implant, and intravenous (IV) administration.64 The onset of buprenorphine varies from 15 minutes to 17 hours, depending on the dosage form and strength, with intravenous administration being most rapid and transdermal administration being the slowest.65,66 FDA- approved formulations for OUD encompass sublingual tablets, SQ injections, and the recently approved subdermal implant (Table 2). Transmucosal buprenorphine has a relatively high bioavailability (46–63%) reaching peak plasma concentrations within 40 minutes to 3.5 hours; however, the ingestion of liquids after administration reduces the bioavailability by 23–37%.67 The average half-life of buprenorphine spans from 31 to 36.5 hours following sublingual administration. Most recently, the subdermal implantable buprenorphine formulation provides long-term stable plasma concentrations for the duration of 6 months, eliminating the frequent dosing of transmucosal administration.68 Peak plasma concentrations are reached within 12–24 hours after implantation with a steady concentration attained in 3–8 weeks.68 Buprenorphine undergoes extensive metabolism in the liver by N-dealkylation via cytochrome P450 enzyme CYP3A4 to norbuprenorphine. Norbuprenorphine is an active metabolite that is present in peak plasma concentrations comparable to or exceeding those of buprenorphine, with a half-life that surpasses that of buprenorphine.69 In animal models, norbuprenorphine causes significantly marked respiratory depression (x10 fold) and has little analgesic effect (x0.02 fold) compared to buprenorphine.70,71 Furthermore, treatment should be monitored closely when taken in conjunction with CYP3A4 inducers and inhibitors, as they can potentially lead to hazardous decreases or increases in plasma concentrations of buprenorphine. Buprenorphine is additionally formulated with naloxone and available in transmucosal dosage forms (Table 2). When combined with naloxone, buprenorphine maintains it pharmacokinetic profile without undergoing any changes in its characteristics. Naloxone has a low oral bioavailability of approximately 10% and is largely ineffective in oral combination products. Given the extensive hepatic metabolism of buprenorphine, renal adjustment is not required; nevertheless, individuals with moderate-to-severe hepatic impairment should refrain from subcutaneous injections and the subdermal implant.72,73 Transmucosal dosage forms should be initiated at lower doses and slowly titrated up.67
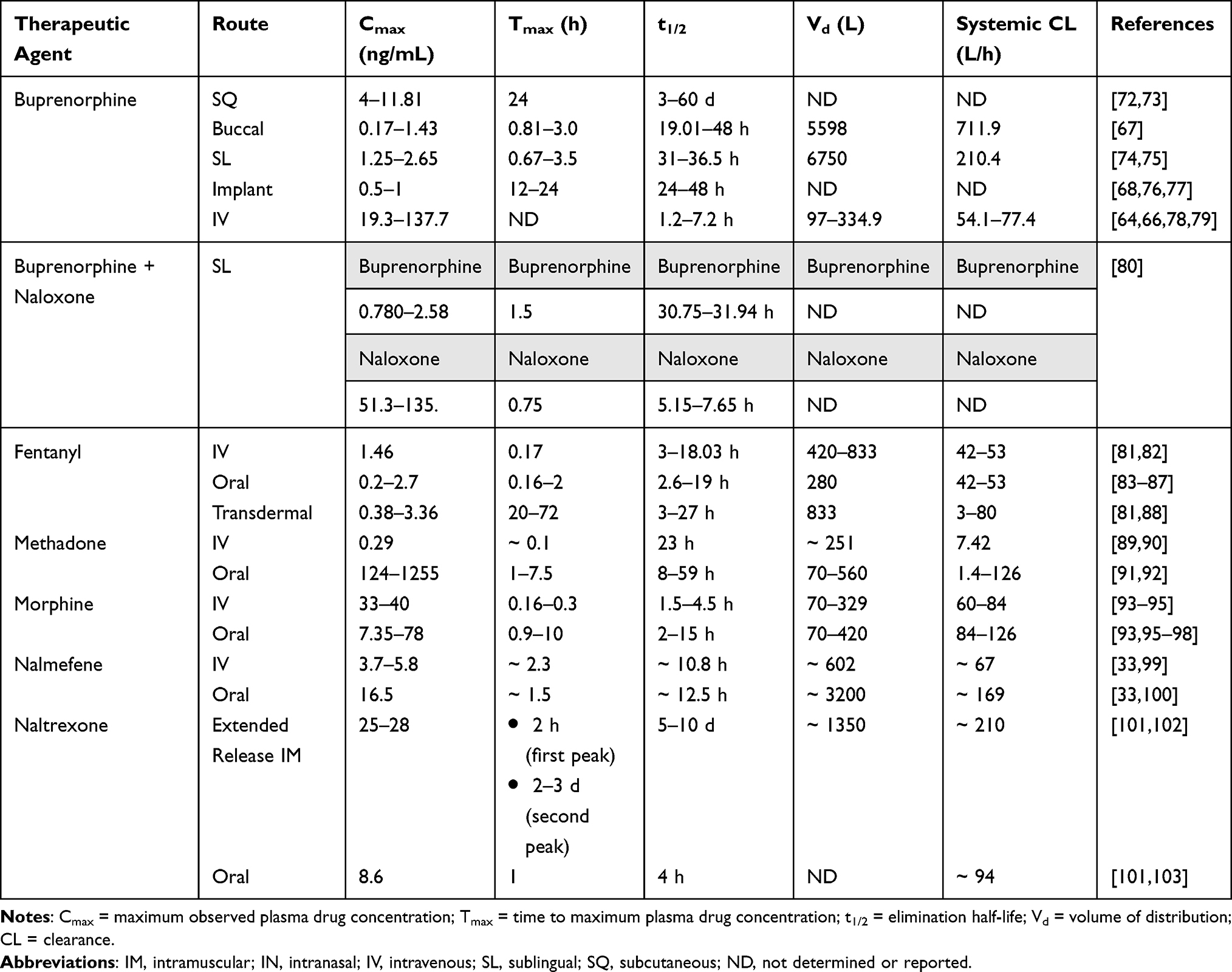 |
Table 2 Route, Cmax, Tmax, t1/2, Vd, and Systemic CL of Therapeutic Opioid Receptor Agents |
Methadone is available as a lipophilic hydrochloride salt in oral, IV, IM, SQ, epidural, and intrathecal formulation (Table 2). The onset of methadone ranges from approximately 15 minutes for intravenous formulations to 2 hours for oral formulations.104 For OUD, methadone is accessible in both oral liquid and tablet formulations, achieving peak plasma concentrations within 1 and 7.5 hours, respectively. Methadone has a high bioavailability of >80% and an extended half-life ranging from 8 to 59 hours.91,92,104 Methadone undergoes metabolism via N-demethylation mediated primarily by CYP2B6 and CYP3A4, producing the inactive metabolite 2-ethyl-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP).105 The CYP2B6 gene exhibits high variability; individuals carrying the CYP2B6*6 polymorphism metabolize methadone insufficiently, resulting in elevated plasma concentrations compared to those with the wild-type gene.106 Moreover, medications acting as CYP3A4 inducers and inhibitors pose potential risks and require close monitoring for interactions with methadone. Given that methadone undergoes hepatic metabolism, renal dosing adjustments are not generally required; however, caution should be exercised administering in patients with end-stage renal disease.
Naltrexone is available in oral and parenteral formulations. Extended-release IM naltrexone is currently the only formulation approved for the management of OUD (Table 2). Extended-release naltrexone (XR-NTX) requires monthly dosing with initial transient peak concentrations occurring within 2 hours following the gluteal injection, followed by a second peak observed approximately 2–3 days later.63 XR-NTX has an elimination half-life (t1/2) ranging from 5 to 10 days after administration. Oral naltrexone reaches peak concentrations in approximately 1 hour; however, it typically requires daily dosing due to a short half-ife (t1/2) of approximately 4 hours.101 Naltrexone undergoes extensive hepatic metabolism mediated by dihydrodiol dehydrogenase, a cytosolic family of enzymes, to the main active metabolite, 6-β-naltrexol. While a weaker antagonist than naltrexone, 6-β-naltrexol may play a role in the prolonged duration of action seen with naltrexone, given that plasma concentrations of the metabolite are consistently higher. A pharmacokinetic study revealed 1 hour following a 100 mg dose of oral naltrexone, the mean peak plasma concentrations of naltrexone and 6-β-naltrexol were 43.6 ± 29.9 and 87.2 ± 25.0 ng/L, respectively.107 However, animal studies have indicated that 6-β-naltrexol is 0.02 to 0.04 as potent as naltrexone as an active inhibitor, with minimal effects lasting 4 to 9 times longer. The mean elimination half-life (t1/2) for 6-β-naltrexol is approximately 13 hours. Administering intramuscularly circumvents first-pass metabolism, altering the exposure ratio of 6-β-naltrexol to naltrexone compared to oral administration, resulting in a similar mean elimination half-life as the parent compound (5–10 days).63 Dosing adjustments in patients with renal and hepatic impairment is not required for the administration in oral and IM naltrexone and patients should be monitored closely.108
Nalmefene is presently accessible in the US in intramuscular (IM) and intranasal (IN) formulations, while various countries offer oral dosage forms for the treatment of AUD. The oral formulation exhibits a bioavailability of 40–50% and a prolonged elimination half-life (t1/2) of 12.5 hours.100 The intravenous dosage form demonstrates a comparable half-life of approximately 10.8 hours.99 Nalmefene undergoes extensive liver metabolism, primarily through glucuronide conjugation, yielding inactive metabolites (Table 2). It demonstrates minimal pharmacokinetic variations between genders, across age groups, and among ethnicities. There does appear to be a minor influence of body size on the clearance of nalmefene. Additionally, clearance is reduced by approximately 28% in patients with mild-to-moderate hepatic dysfunction.99 Dosage adjustments are not required in individuals with moderate-to-severe renal insufficiency as well. A clinical investigation explored the pharmacokinetic profile in individuals with end-stage renal disease, revealing a reduction in systemic clearance and an increase in volume of distribution when compared to those with normal renal function.109 These two characteristics counterbalanced one another, leading to a plasma time concentration and clinical response that did not show significant differences when compared to healthy individuals. Nonetheless, close monitoring is recommended when using nalmefene.
The half-life of nalmefene following oral administration is approximately 3 times as long as naltrexone and double of naloxone. Given the longer half-life of fentanyl derivatives, nitazene opioids, and other new psychoactive substances that activate opioid receptors,5 nalmefene administration is less likely to result in rebound respiratory depression than the current use of naloxone.
Current Therapeutic Uses of Nalmefene
Alcohol Use Disorder
Preclinical studies on the application of opioid antagonists for the treatment of AUD began in the 1980s, revealing the potential indirect inhibition of opioid receptors by aliphatic alcohols, such as ethanol.110,111 In 1994, the FDA approved the use of oral naltrexone in the treatment of AUD, guided by the results of two randomized controlled trials that supported its efficacy and safety.112,113 Following that, in 2006, the FDA granted approval for a long-acting injectable formulation, eliminating the necessity for daily dosing.24 Simultaneously, as advancements were made with naltrexone, researchers sought to understand another selective opioid antagonist, nalmefene, for the treatment of AUD.
The administration of nalmefene utilizes its binding affinity for the μ, κ, and δ opioid receptors (ORs) to diminish the reinforcing effects of alcohol consumption.114 In pre-clinical animal trials, the antagonism of nalmefene on the μOR and δOR resulted in the suppression of alcohol self-administration in alcohol non-dependent and dependent rats. In comparison to naltrexone, which is currently approved in the US for AUD, nalmefene significantly attenuated alcohol self-administration to a greater extent in alcohol-dependent animals when administered at the same dose. The partial agonism of nalmefene on the κOR potentially contributed to suppressing self-administration in alcohol-dependent rats when compared to naltrexone.36 This is attributed to nalmefene’s, potential to enhance the aversive effects of the compensatory upregulated dynorphin/κ opioid receptor system on the μOR, resulting from prolonged alcohol intake.115,116 In contrast, naltrexone exhibits a comparable μOR affinity; however, it demonstrates a much lower affinity for κOR and δOR in both rats and humans. To better correlate nalmefene’s pharmacology at opioid receptors to its clinical relevance in AUD, a human study utilized positron emission tomography imaging that revealed μOR occupancy of 87% to 100% three hours after both single and multiple oral administrations of nalmefene hydrochloride, each at a clinically effective dose of 20 mg.117
Approved in the European Union (EU) in 2013, oral nalmefene hydrochloride (Selincro) is available for the as-needed use of alcohol consumption reduction in alcohol-dependent individuals who have a high drinking risk level consisting of a total pure ethanol consumption of >60 g/day in men or >40 g/day in women.118 Each tablet contains 21.9 mg of nalmefene hydrochloride dihydrate, equivalent to 18 mg nalmefene; the tablet is instructed for as-needed use, with a recommended limit of one tablet per day.100
The approval of oral nalmefene in the EU was based on three randomized, double-blind, multinational European trials. Two of the trials, ESENSE 1 and ESENSE 2, investigated the efficacy of oral nalmefene compared to placebo over a six-month period.43,44 The third clinical trial, SENSE, evaluated its safety and effectiveness over the duration of one year.119 In all three trials, as-needed oral nalmefene therapy was supplemented with motivational and adherence-enhancing interventions. Merging the findings from the ESENSE 1 and 2 trial for the specified target population (ie, patients with a high drinking risk level during selection and randomization) indicated that as-needed nalmefene treatment versus placebo led to a significant mean reduction of 3.2 heavy drinking days per month and a mean reduction of 14.3 g/day monthly alcohol consumption.45 In the SENSE trial, identical efficacy endpoints (reductions in number of heavy drinking days and total alcohol consumption) were assessed, alongside an evaluation of the safety and tolerability of nalmefene over the course of 13 months.45 Upon analysis of the target population (ie, patients who had at least a high drinking risk level), the use of as-needed nalmefene therapy resulted in a significant mean reduction of 3.6 heavy drinking days and a mean reduction of 17.3 g/day monthly alcohol consumption in comparison with the placebo.45 This affirmed the potential efficacy of oral nalmefene in reducing alcohol intake when compared to placebo in heavy drinking populations; nonetheless, significant criticism on its approval has occurred due to the limited supporting evidence of effectiveness when compared to active AUD medications (ie, naltrexone, disulfiram, acamprosate, etc.).120 Although the numbers needed to treat have not been established for nalmefene as they have been for naltrexone (NNT=20), the numbers needed to harm are lower for nalmefene (NNH=12 vs NNH=48 for naltrexone).22
Across all three clinical trials, frequent adverse events included nausea, insomnia, and dizziness, with symptoms ranging from mild to moderate in severity.43,44,119 Additionally, nalmefene was associated with more adverse events and trial dropouts in comparison to placebo. Throughout the trials, there were no clinically significant alterations or differences between nalmefene and placebo regarding vital signs, bodyweight, ECG recordings, or notable shifts in the occurrence of clinically significant laboratory findings, except for the reduction of ALT and γ-glutamyl transferase levels. Oral as needed, nalmefene for the reduction of alcohol intake is contraindicated in cases of severe renal or hepatic impairment, use of opioid analgesics, suspected recent use of opioids, current or recent opioid addiction, acute symptoms of opioid withdrawal, and recent history of acute alcohol withdrawal symptoms.
Opioid Overdose Treatment
Opioid-induced respiratory depression is driven by the overstimulation of μOR during an opioid overdose.121 Consequently, hypoxia resulting from respiratory depression has the potential to lead to fatalities or significant health complications in non-fatal overdoses. The extended potencies and half-lives of synthetic opioids have increased the complexity of reversing the symptoms of opioid overdose, enhancing the necessity for a more potent opioid antagonist with a longer half-life than the currently used antagonist, naloxone. A preclinical animal study indicated mice required a 10-fold higher dose of naloxone to reverse the respiratory depressant effects of fentanyl compared to morphine when administered at equipotent doses.122 A pre-clinical study compared the efficacy of nalmefene (9.4 −150 μg/kg, IM) and naloxone (150 μg/kg, IM) in reversing carfentanil-induced loss of righting reflex and respiratory depression in an overdose simulation involving rats.123 Upon intravenous administration of carfentanil (10 μg/kg), nalmefene doses ranging from 9.4 to 18.8 μg/kg exhibited a reduction in the duration of loss of righting reflex comparable to that achieved with naloxone 150 μg/kg. Furthermore, radioligand binding techniques utilizing μOR from non-human primate brain revealed nalmefene and naltrexone possess a 4.8 and 5.6-fold increased potency than naloxone, respectively.31 However, the findings from preclinical data have not yet translated into clinically significant therapeutic outcomes. A randomized, double blind multicenter study compared the efficacy of nalmefene and naloxone in 156 patients admitted to the Emergency Department suspected of an opioid overdose.124 Patients were administered a maximum of 4 doses of either nalmefene (1 mg or 2 mg) or naloxone (2 mg) intravenously every 5 minutes as needed; however, most patients required a single dose of the received treatment. Opioid positive patients (~43%) exhibited rapidly improved respiratory rates and no statistically significant differences in efficacy or withdrawal outcomes were observed between the treatment groups. It is important to highlight that the consumed opioids were unknown, and the study was conducted during a time when overdoses from synthetic opioids were not common. The primary attribute of nalmefene hydrochloride in countering opioid overdose is its extended half-life in comparison to naloxone. This feature has valuable implications, including the potential for reduced continuous patient surveillance and potentiality reducing re-administration due to its extensive antagonistic duration across synthetic opioid agonists with prolonged half-lives, like fentanyl. In 1995, nalmefene hydrochloride, marketed as Revex, gained its initial approval in the US market for parenteral administration. Revex was primarily aimed at managing the outcomes of opioid overdose in clinical settings; however, injectable nalmefene exhibits a slower onset of action, taking 5–15 minutes compared to naloxone’s 1–2 minutes, which may prove problematic if used as the first agent in emergency circumstances of acute severe respiratory depression (Table 3). In 2008, the 0.1 mg base/mL and 1.0 mg base/mL Revex formulations were removed from the market for reasons unrelated to safety or efficacy.125 On June 21, 2022, in response to the rise of overdose deaths, the demand drastically grew for an opioid overdose reversal agent capable of treating the effects of long-lasting synthetic opioids; one manufacturer announced the generic availability of nalmefene hydrochloride injection in 2 mg/2 mL vials for use by medical professionals.126
 |
Table 3 Onset, Tmax, t1/2, and Cmax for Nalmefene Intramuscular, Nalmefene Intranasal, Naloxone Intranasal, and Naltrexone + DDM Intranasal |
The critical development of nalmefene hydrochloride as an opioid overdose reversal agent occurred in May 2023 when the FDA authorized the first nalmefene hydrochloride nasal spray, known as Opvee.129 This intranasal (IN) spray delivers a single dose of 2.7 mg of nalmefene and is tailored for use by adults and pediatric patients aged 12 and older in emergency, community settings to reverse opioid overdose.130 When properly administered, Opvee holds the potential to mitigate or reverse respiratory depression, sedation, and hypotension. The vital difference between IN nalmefene and the injectable form is its quicker onset of action, facilitated by the addition of dodecyl maltoside (DDM), an absorption enhancer that works through widening the tight junctions between epithelial cells.130,131 Incorporating DDM into the IN formulation expedites the reversal of respiratory depression within a 2.5-to-5-minute timeframe.130 In a pharmacokinetic study, the addition of 0.25% DDM to IN 3 mg nalmefene resulted in a reduction of the time to peak concentration from 2 hours to 15 minutes and increased the peak plasma concentration by 2.2 fold when compared to IN 3 mg nalmefene alone (4.45 ng/mL vs 1.99 ng/mL).132 Additionally, IN nalmefene and DDM combination exhibited a peak plasma concentration x2.9 fold higher than IM 1.5 mg nalmefene (4.45 ng/mL vs 1.53 ng/mL). Prior to these discoveries, Krieter et al undertook a similar study investigating the incorporation of 0.25% DDM into 4 mg naltrexone. The combination of naltrexone with DDM exhibited a rapid time to peak concentration, like IN nalmefene + DDM, with an increased peak plasma concentration. The distinguishing characteristic between the two DDM therapies lies in the significantly prolonged half-life of nalmefene + DDM (Table 3).
Reported adverse effects were primarily nasal discomfort, headache, and nausea.130 Additionally, the use of Opvee in opioid dependent individuals may result in opioid withdrawal characterized by body aches, diarrhea, tachycardia, and fever.115 The faster onset of action and the extended half-life of IN nalmefene are vital when deployed in community settings to counter the symptoms of opioid overdose linked to synthetic opioids with prolonged half-lives.
Other Uses
The potent affinity of nalmefene hydrochloride for central opioid receptors has prompted its experimental application in the prevention of postoperative hyperalgesia, reversal of conscious opioid sedation, and reduction of reoccurring harmful behaviors. In a clinical study conducted by Jia et al, patients were intravenously administered either 0.20 μg/kg nalmefene, 0.50 μg/kg dexmedetomidine, 0.10 μg/kg nalmefene + 0.25 μg/kg dexmedetomidine, or placebo prior to receiving intraoperative 0.30 μg/kg/min remifentanil to investigate the efficacy of preoperative nalmefene administration in preventing remifentanil-induced hyperalgesia (RIH) during laparoscopic gynecological surgery.133 Nalmefene prevented RIH but did not reduce the onset of post-operative pain. However, the administration of pre-anesthetic nalmefene (0.10 μg/kg) in combination with dexmedetomidine (0.25 μg/kg) effectively prevented RIH, minimized the development of post-operative pain, and decreased the requirement for additional analgesic application. Simultaneously, nalmefene counteracted adverse reactions associated with the sole administration of dexmedetomidine, such as intraoperative bradycardia, and contributed to a more rapid post-anesthesia recovery. In a randomized, double-blind trial, the efficacy of nalmefene in counteracting opioid-induced sedation was evaluated in patients undergoing outpatient procedures such as incisions and drainage of soft tissue abscess (Barsan). Patients were administered a dose of meperidine (75–310 mg) for analgesia during the procedure, followed by a 1 mg dose of either nalmefene, naloxone, or placebo and responses were observed over the period of 4 hours. The results indicated that nalmefene significantly reduced patient opioid-induced sedation in comparison to naloxone within 60–150 minutes following antagonist administration. A multicenter, double-blind trial explored the efficacy of nalmefene for pathological gambling at 15 outpatient psychiatric treatment centers over a 16-week duration.134 Patients were administered oral doses of either 25 mg/day, 50 mg/day, 100 mg/day nalmefene, or a placebo. In the final assessment, clinicians rated 59% of patients allocated to 25 mg/day of nalmefene were rated as “much improved” or “very much improved”, in comparison to 34% of those who received the placebo. Additionally, a daily dose of either 25 mg or 50 mg nalmefene produced a significantly improved outcome score (Yale-Brown Obsessive Compulsive Scale Modified for Pathological Gambling total score) compared with the placebo group. However, data correspond with the adverse reactions (nausea, insomnia, dizziness) associated with nalmefene, leading to the withdrawal of two-thirds of the participants primarily due to challenges managing these reactions when higher doses of 50 mg/day and 100 mg/day were administered. While these studies warrant further investigation and confirmation to ascertain significance, they have paved the way for understanding the impact and potential additional uses of nalmefene hydrochloride.
Discussion
Buprenorphine has emerged as a popular option in clinical practice for treatment of OUD due to its patient acceptability, diminished risk of overdose (ceiling effect), and a low potential for abuse potential; nevertheless, it is essential to consider its various disadvantages.62 Daily dosing and long-term treatment are essential for effectively managing OUD with buprenorphine.135,136 A 2008 Cochrane systematic review and meta-analysis compared the treatment retention and suppression of illicit opiate use of buprenorphine and methadone.137 Buprenorphine did not have any advantages in terms of treatment retention and was less effective at suppressing illicit opioid use when administered at similar doses to methadone.138,139 The implantable formulation was designed for convenient administration, aiming to decrease potential diversion and ensure consistent dose concentrations. However, the insertion and removal of the implant require a trained professional, and the need for multiple implants (ranging from 1 to 5) to alleviate cravings and withdrawal symptoms may be uncomfortable and financially burdensome for the patient.140
Methadone is an FDA approved OUD therapy that necessitates daily dosing as well. Similar to buprenorphine, numerous states require daily treatment restricted to only specialized clinics, potentially negatively impacting adherence.137,138 Due to the preferred safety profile of buprenorphine, methadone is often reserved for individuals who do not respond to buprenorphine therapy. Methadone poses a higher risk of misuse and diversion and is more hazardous at high doses, leading to significantly increased effects of respiratory depression and euphoric sensations in comparison to buprenorphine. The coadministration of respiratory depressants, such as alcohol and benzodiazepines, can lead to severe respiratory suppression when using methadone.
Naltrexone is an FDA approved OUD therapy as an alternative for individuals who prefer not to use an opioid agonist, like buprenorphine and methadone. Due to naltrexone being an opioid antagonist, there is no risk for potential abuse or physical dependence and hence does not require tight regulation compared to buprenorphine and methadone.141 However, the only formulation available for OUD management is the long acting monthly injectable that, due to its high monthly cost, may pose a challenge for patient accessibility. The once-a-month dosing schedule of extended-release naltrexone (XR-NTX) allows for convenient administration.
Nalmefene potentially may be used as an as-needed oral maintenance alternative for the treatment of OUD or used daily in conjunction with buprenorphine. Coformulation of buprenorphine and nalmefene may potentially counteract the adverse events of one another leading to a superior formulation. Antagonism of the κ-opioid receptor leads to decreased spinal analgesia, dysphoria, miosis, and diuresis through inhibition of anti-diuretic hormone release.142 Additionally, the μ-antagonistic property of nalmefene diminishes the primary concern regarding buprenorphine diversion in transmucosal formulations. The prolonged half-life of naltrexone, in contrast to naloxone, has the potential to significantly diminish the risk of delayed euphoric effects when intravenously diverted. Furthermore, nalmefene lacks the analgesic properties that are provided by buprenorphine. This characteristic might enhance induction rates and adherence when compared to the use of nalmefene as monotherapy for OUD. The concurrent use of buprenorphine and nalmefene has the potential to substantially enhance the effectiveness of OUD treatment, leading to improved retention, negative urinalyses, and reduced dysphoria, mood symptoms, and cravings. It could represent an alternative strategy tailored to the specific requirements of individuals grappling with OUD, and there should be encouragement for both a systematic review of nalmefene for OUD and clinical studies involving the use of a nalmefene and buprenorphine formulation.
Conclusion
Nalmefene hydrochloride is a potent opioid receptor antagonist with unique pharmacological properties that distinguish it from other treatment options for OUD. Its partial agonist activity at κ-opioid receptors, high potency, and long half-life provide a differential effect profile that can benefit in the clinical treatment of substance use disorders, in particular if opioid agonist treatment is not feasible or cannot achieve the desired outcome alone. Its successful use in the treatment of AUD is indicative of benefits for OUD given the overlapping pharmacology at opioid and dopamine receptors. The introduction of nalmefene to reverse opioid overdoses comes at a critical time as fentanyl, nitazenes, and other potent illicit opioids require higher doses and longer-acting opioid antagonists to prevent an immediate or delayed fatality. With the current focus on naloxone in opioid overdose reversal, other options should be available not only for immediate mitigation but also in maintenance therapy given that a majority of patients on low to moderate dose buprenorphine maintenance continue to use illicit opioids.137 Nalmefene presents with a longer half-life and higher binding affinity at opioid receptors thus providing first responders and clinicians with a better option to reverse and stabilize a patient in acute overdose and at risk of a fatal outcome. Further clinical trials with nalmefene are warranted to establish its safety and efficacy in the maintenance treatment of opioid and other substance use disorders.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Dydyk AM, Jain NK, Gupta M. Opioid use disorder. In: StatPearls [Internet]. StatPearls Publishing; 2022.
2. Taylor JL, Samet JH. Opioid use disorder. Ann Intern Med. 2022;175:ITC1–16.
3. Webster LR. Risk factors for opioid-use disorder and overdose. Anesth Analg. 2017;125(5):1741–1748. doi:10.1213/ANE.0000000000002496
4. Haley DF, Saitz R. The opioid epidemic during the COVID-19 pandemic. JAMA. 2020;324:1615–1617. doi:10.1001/jama.2020.18543
5. Rinaldi R, Bersani G, Marinelli E, Zaami S. The rise of new psychoactive substances and psychiatric implications: a wide-ranging, multifaceted challenge that needs far‐reaching common legislative strategies. Hum Psychopharmacol Clin Exp. 2020;35:e2727.
6. Pichini S, Zaami S, Pacifici R, Tagliabracci A, Busardò FP. The challenge posed by new synthetic opioids: pharmacology and toxicology. Front Pharmacol. 2019;563. doi:10.3389/fphar.2019.00563
7. Lo Faro AF, Berardinelli D, Cassano T, et al. New psychoactive substances intoxications and fatalities during the COVID-19 epidemic. Biology. 2023;12:273. doi:10.3390/biology12020273
8. Amaducci A, Aldy K, Campleman SL, et al. Naloxone use in novel potent opioid and fentanyl overdoses in emergency department patients. JAMA Network Open. 2023;6(8):e2331264–e2331264. doi:10.1001/jamanetworkopen.2023.31264
9. Coffa D, Snyder H. Opioid use disorder: medical treatment options. Am Fam Physician. 2019;100:416–425.
10. Yarmolinsky A, Rettig RA. Federal regulation of methadone treatment; 1995.
11. Herman BH, Vocci F, Bridge P. The effects of NMDA receptor antagonists and nitric oxide synthase inhibitors on opioid tolerance and withdrawal. Neuropsychopharmacology. 1995;13:269–293. doi:10.1016/0893-133X(95)00140-9
12. Mattick RP, Breen C, Kimber J, Davoli M. Methadone maintenance therapy versus no opioid replacement therapy for opioid dependence. Cochrane Database Syst Rev. 2009. doi:10.1002/14651858.CD002209.pub2
13. Shulman M, Wai JM, Nunes EV. Buprenorphine treatment for opioid use disorder: an overview. CNS Drugs. 2019;33(6):567–580. doi:10.1007/s40263-019-00637-z
14. Pinto H, Maskrey V, Swift L, Rumball D, Wagle A, Holland R. The SUMMIT trial:: a field comparison of buprenorphine versus methadone maintenance treatment. J Subst Abuse Treat. 2010;39(4):340–352. doi:10.1016/j.jsat.2010.07.009
15. Alderks CE. Trends in the use of methadone, buprenorphine, and extended-release naltrexone at substance abuse treatment facilities: 2003–2015 (update); 2017.
16. Feng H, Elladki R, Jiang J, Wei G-W. Machine-learning analysis of opioid use disorder informed by MOR, DOR, KOR, NOR and ZOR-based interactome networks. Comput Biol Med. 2023;157:106745. doi:10.1016/j.compbiomed.2023.106745
17. Younger J, Noor N, McCue R, Mackey S. Low‐dose naltrexone for the treatment of fibromyalgia: findings of a small, randomized, double‐blind, placebo‐controlled, counterbalanced, crossover trial assessing daily pain levels. Arthritis Rheum. 2013;65:529–538. doi:10.1002/art.37734
18. Mammen K, Bell J. The clinical efficacy and abuse potential of combination buprenorphine–naloxone in the treatment of opioid dependence. Expert Opin Pharmacother. 2009;10(15):2537–2544. doi:10.1517/14656560903213405
19. Strang J, McDonald R, Campbell G, et al. Take-home naloxone for the emergency interim management of opioid overdose: the public health application of an emergency medicine. Drugs. 2019;79(13):1395–1418. doi:10.1007/s40265-019-01154-5
20. NIDA. How much does opioid treatment cost? 2023. Available from: https://nida.nih.gov/publications/research-reports/medications-to-treat-opioid-addiction/how-much-does-opioid-treatment-cost.
21. Goldfine CE, Tom JJ, Im DD, et al. The therapeutic use and efficacy of ketamine in alcohol use disorder and alcohol withdrawal syndrome: a scoping review. Front Psychiatry. 2023;14:1141836. doi:10.3389/fpsyt.2023.1141836
22. Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA. 2014;311(18):1889–1900. doi:10.1001/jama.2014.3628
23. Fan AZ, Chou SP, Zhang H, Jung J, Grant BF. Prevalence and correlates of past-year recovery from DSM-5 alcohol use disorder: results from national epidemiologic survey on alcohol and related conditions-III. Alcohol Clin Exp Res. 2019;43:2406–2420. doi:10.1111/acer.14192
24. Zindel LR, Kranzler HR. Pharmacotherapy of alcohol use disorders: seventy-five years of progress. J Stud Alcohol Drugs Suppl. 2014;79–88. doi:10.15288/jsads.2014.s17.79
25. Grosshans M, Mutschler J, Kiefer F. Treatment of cocaine craving with as-needed nalmefene, a partial κ opioid receptor agonist: first clinical experience. Int Clin Psychopharmacol. 2015;30(4):237–238. doi:10.1097/YIC.0000000000000069
26. Wang DS, Sternbach G, Varon J. Nalmefene: a long-acting opioid antagonist. Clinical applications in emergency medicine. J Emerg Med. 1998;16(3):471–475. doi:10.1016/S0736-4679(98)00019-5
27. Grant JE, Potenza MN, Hollander E, et al. Multicenter investigation of the opioid antagonist nalmefene in the treatment of pathological gambling. Am J Psychiatry. 2006;163(2):303–312. doi:10.1176/appi.ajp.163.2.303
28. Dhawan BN, Cesselin F, Raghubir R, et al. International Union of Pharmacology. XII. Classification of opioid receptors. Pharmacol Rev. 1996;48(4):567–592.
29. Potter DN, Damez-Werno D, Carlezon WA Jr, Cohen BM, Chartoff EH. Repeated exposure to the κ-opioid receptor agonist salvinorin A modulates extracellular signal-regulated kinase and reward sensitivity. Biol Psychiatry. 2011;70(8):744–753. doi:10.1016/j.biopsych.2011.05.021
30. Knoll AT, Carlezon WA
31. Woods JH, Medzihradsky F, Smith CB, Butelman ER, Winger G. Evaluation of new compounds for opioid activity (1996). Probl Drug Depend. 1997;396:1.
32. Wang D, Raehal KM, Bilsky EJ, Sadée W. Inverse agonists and neutral antagonists at µ opioid receptor (MOR): possible role of basal receptor signaling in narcotic dependence. J Neurochem. 2001;77:1590–1600. doi:10.1046/j.1471-4159.2001.00362.x
33. Kyhl LB, Li S, Faerch KU, Soegaard B, Larsen F, Areberg J. Population pharmacokinetics of nalmefene in healthy subjects and its relation to μ‐opioid receptor occupancy. Br J Clin Pharmacol. 2016;81:290–300. doi:10.1111/bcp.12805
34. Lee MC, Wagner HN, Tanada S, Frost JJ, Bice AN, Dannals RF. Duration of occupancy of opiate receptors by naltrexone. J Nucl Med. 1988;29:1207–1211.
35. Rabiner EA, Beaver J, Makwana A, et al. Pharmacological differentiation of opioid receptor antagonists by molecular and functional imaging of target occupancy and food reward-related brain activation in humans. Mol Psychiatry. 2011;16(8):826–835. doi:10.1038/mp.2011.29
36. Walker BM, Koob GF. Pharmacological evidence for a motivational role of κ-opioid systems in ethanol dependence. Neuropsychopharmacology. 2008;33(3):643–652. doi:10.1038/sj.npp.1301438
37. Schluger JH, Ho A, Borg L, et al. Nalmefene causes greater hypothalamic‐pituitary‐adrenal axis activation than naloxone in normal volunteers: implications for the treatment of alcoholism. Alcohol Clin Exp Res. 1998;22:1430–1436. doi:10.1111/j.1530-0277.1998.tb03931.x
38. Jacobson ML, Browne CA, Lucki I. Kappa opioid receptor antagonists as potential therapeutics for stress-related disorders. Annu Rev Pharmacol Toxicol. 2020;60(1):615–636. doi:10.1146/annurev-pharmtox-010919-023317
39. Benth JŠ, Solli KK, Opheim A, et al. Anxiety, depression, and insomnia among adults with opioid dependence treated with extended-release naltrexone vs buprenorphine-naloxone: a randomized clinical trial and follow-up study. JAMA Psychiatry. 2019;76:127–134. doi:10.1001/jamapsychiatry.2018.3537
40. Hao S, Ogawa H. Naltrexone, but not atropine or yohimbine, antagonizes suppression of formalin-induced spinal sensitization by intrathecal nociceptin. Life Sci. 1998;63(11):A167–73. doi:10.1016/S0024-3205(98)00359-2
41. Meeting C on P of DD (U. S). S, Abuse NI on D. Problems of Drug Dependence: proceedings of the. Annual Scientific Meeting, the College on Problems of Drug Dependence, Inc [Internet]. U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Institute on Drug Abuse; 1997. Available from: https://books.google.com/books?id=hgj5eTyGTI4C.
42. Johansen KGV, Tarp S, Astrup A, Lund H, Pagsberg AK, Christensen R. Harms associated with taking nalmefene for substance use and impulse control disorders: a systematic review and meta-analysis of randomised controlled trials. PLoS One. 2017;12:e0183821.
43. Gual A, He Y, Torup L, van den Brink W, Mann K; Group E 2 S. A randomised, double-blind, placebo-controlled, efficacy study of nalmefene, as-needed use, in patients with alcohol dependence. Eur Neuropsychopharmacol. 2013;23(11):1432–1442. doi:10.1016/j.euroneuro.2013.02.006
44. Mann K, Bladström A, Torup L, Gual A, van den Brink W. Extending the treatment options in alcohol dependence: a randomized controlled study of as-needed nalmefene. Biol Psychiatry. 2013;73(8):706–713. doi:10.1016/j.biopsych.2012.10.020
45. van den Brink W, Sørensen P, Torup L, Mann K, Gual A, Group SS. Long-term efficacy, tolerability and safety of nalmefene as-needed in patients with alcohol dependence: a 1-year, randomised controlled study. J Psychopharmacol. 2014;28:733–744. doi:10.1177/0269881114527362
46. Kaplan JL, Marx JA. Effectiveness and safety of intravenous nalmefene for emergency department patients with suspected narcotic overdose: a pilot study. Ann Emerg Med. 1993;22(2):187–190. doi:10.1016/S0196-0644(05)80200-8
47. Sarkar S, Addagadda SS, Bhatia G, Chadda RK. Adverse drug reactions with naltrexone: experience from an addiction treatment center. Indian J Psychiatry. 2021;63(2):206–207. doi:10.4103/psychiatry.IndianJPsychiatry_94_20
48. Hoofnagle JH. LiverTox: a website on drug-induced liver injury. In: Drug-induced liver disease. Elsevier. 2013;725–732.
49. Niciu MJ, Arias AJ. Targeted opioid receptor antagonists in the treatment of alcohol use disorders. CNS Drugs. 2013;27(10):777–787. doi:10.1007/s40263-013-0096-4
50. Kristensen K, Christensen CB, Christrup LL. The mu1, mu2, delta, kappa opioid receptor binding profiles of methadone stereoisomers and morphine. Life Sci. 1994;56(2):45–50. doi:10.1016/0024-3205(94)00937-6
51. Wang S. Historical review: opiate addiction and opioid receptors. Cell Transplant. 2019;28:233–238. doi:10.1177/0963689718811060
52. Walwyn WM, Miotto KA, Evans CJ. Opioid pharmaceuticals and addiction: the issues, and research directions seeking solutions. Drug Alcohol Depend. 2010;108(3):156–165. doi:10.1016/j.drugalcdep.2010.01.001
53. Gelfman LP, Chai EJ. Chapter 8 - What Special Considerations Should Guide the Safe Use of Methadone? In: Goldstein NE, Morrison RS, editors. Evidence-Based Practice in Palliative Medicine [Internet]. Philadelphia: W.B. Saunders; 2013:39–43.
54. Davis MP. Methadone does not block NMDA receptors. J Pain Symptom Manage. 2021;62(3):e7–8. doi:10.1016/j.jpainsymman.2021.05.014
55. Toce MS, Chai PR, Burns MM, Boyer EW. Pharmacologic treatment of opioid use disorder: a review of pharmacotherapy, adjuncts, and toxicity. J Med Toxicol. 2018;14(4):306–322. doi:10.1007/s13181-018-0685-1
56. Yokell M A, Zaller N D, Green T C, Rich J D. Buprenorphine and buprenorphine/naloxone diversion, misuse, and illicit use: an international review. Curr Drug Abuse Rev. 2011;4:28–41. doi:10.2174/1874473711104010028
57. Serafini G, Adavastro G, Canepa G, et al. The efficacy of buprenorphine in major depression, treatment-resistant depression and suicidal behavior: a systematic review. Int J Mol Sci. 2018;19(8):2410. doi:10.3390/ijms19082410
58. Pande LJ, Arnet RE, Piper BJ. An examination of the complex pharmacological properties of the non-selective opioid modulator buprenorphine. Pharmaceuticals. 2023;16(10):1397. doi:10.3390/ph16101397
59. Canestrelli C, Marie N, Noble F. Rewarding or aversive effects of buprenorphine/naloxone combination (Suboxone) depend on conditioning trial duration. Int J Neuropsychopharmacol. 2014;17:1367–1373. doi:10.1017/S146114571400025X
60. Comer SD, Sullivan MA, Vosburg SK, et al. Abuse liability of intravenous buprenorphine/naloxone and buprenorphine alone in buprenorphine‐maintained intravenous heroin abusers. Addiction. 2010;105:709–718. doi:10.1111/j.1360-0443.2009.02843.x
61. Rahimi‐Movaghar A, Gholami J, Amato L, Hoseinie L, Yousefi‐Nooraie R, Amin‐Esmaeili M. Pharmacological therapies for management of opium withdrawal. Cochrane Database Syst Rev. 2018. doi:10.1002/14651858.CD007522.pub2
62. Whelan PJ, Remski K. Buprenorphine vs methadone treatment: a review of evidence in both developed and developing worlds. J Neurosci Rural Pract. 2012;3:45–50. doi:10.4103/0976-3147.91934
63. VIVITROL XR [package insert]. Alkermes Inc. 2021.
64. Kumar R, Viswanath O, Saadabadi A. Buprenorphine; 2017.
65. BUTRANS [package insert]. Purdue Pharma LP; 2014.
66. BUPRENEX [package insert]. Indivior Inc.; 2019.
67. BELBUCA [package insert]. Ende Pharmaceuticals Inc.; 2015.
68. Kleppner SR, Patel R, Costantini LC, McDonough J. In-vitro and in-vivo characterization of a buprenorphine delivery system. J Pharm Pharmacol. 2006;58:295–302. doi:10.1211/jpp.58.3.0002
69. Moody DE, Slawson MH, Strain EC, Laycock JD, Spanbauer AC, Foltz RL. A liquid chromatographic-electrospray ionization-tandem mass spectrometric method for determination of buprenorphine, its metabolite, norbuprenorphine, and a coformulant, naloxone, that is suitable for in vivo and in vitro metabolism studies. Anal Biochem. 2002;306(1):31–39. doi:10.1006/abio.2002.5673
70. Brown SM, Holtzman M, Kim T, Kharasch ED. Buprenorphine metabolites, buprenorphine-3-glucuronide and norbuprenorphine-3-glucuronide, are biologically active. J Am Soc Anesthesiologists. 2011;115:1251–1260.
71. Ohtani M, Kotaki H, Sawada Y, Iga T. Comparative analysis of buprenorphine-and norbuprenorphine-induced analgesic effects based on pharmacokinetic-pharmacodynamic modeling. J Pharmacol Exp Ther. 1995;272:505–510.
72. SUBLOCADE [package insert]. Indivior Inc.; 2017.
73. BRIXADI [package insert]. Braeburn Inc.; 2023.
74. SUBUTEX [package insert]. Reckitt Benckiser Pharmaceuticals Inc; 2011.
75. BUPRENORPHINE SUBLINGUAL TABLETS [package insert]. Sun Pharmaceutical Industries Inc; 2023.
76. PROBUPHINE [package insert]. Braeburn Pharmaceuticals, Inc; 2016.
77. Barnwal P, Das S, Mondal S, Ramasamy A, Maiti T, Saha A. Probuphine®(buprenorphine implant): a promising candidate in opioid dependence. Ther Adv Psychopharmacol. 2017;7:119–134. doi:10.1177/2045125316681984
78. Bullingham RES, McQuay HJ, Moore A, Bennett MRD. Buprenorphine kinetics. Clin Pharmacol Ther. 1980;28:667–672. doi:10.1038/clpt.1980.219
79. Huestis MA, Cone EJ, Pirnay SO, Umbricht A, Preston KL. Intravenous buprenorphine and norbuprenorphine pharmacokinetics in humans. Drug Alcohol Depend. 2013;131(3):258–262. doi:10.1016/j.drugalcdep.2012.11.014
80. SUBOXONE [package insert]. Indivior Inc; 2022.
81. IONSYS [package insert]. Ortho McNeil Inc; 2006.
82. Vasisht N, Gever LN, Tagarro I, Finn AL. Single-dose pharmacokinetics of fentanyl buccal soluble film. Pain Med. 2010;11(7):1017–1023. doi:10.1111/j.1526-4637.2010.00875.x
83. Abstral [package insert]. Sentynl Therapeutics Inc; 2016.
84. ONSOLIS [package insert]. Meda Pharmaceuticals Inc; 2011.
85. FENTORA [package insert]. Cephalon Inc; 2011.
86. SUBSYS [package insert]. Insys Therapeutics Inc; 2016.
87. Actiq [package insert]. Abbott Laboratories; 1998.
88. DURAGESIC [package insert]. Janssen Pharmaceuticals Inc; 2021.
89. Inturrisi CE, Colburn WA, Kaiko RF, Houde RW, Foley KM. Pharmacokinetics and pharmacodynamics of methadone in patients with chronic pain. Clin Pharmacol Ther. 1987;41(4):392–401. doi:10.1038/clpt.1987.47
90. Horst J, Frei‐Jones M, Deych E, Shannon W, Kharasch ED. Pharmacokinetics and analgesic effects of methadone in children and adults with sickle cell disease. Pediatr Blood Cancer. 2016;63(12):2123–2130. doi:10.1002/pbc.26207
91. DOLOPHINE [package insert]. Roxane Laboratories; 2014.
92. METHADOSE [package insert]. Mallinckrodt Inc; 2016.
93. Hain RDW. Morphine and morphine‐6‐glucuronide in the plasma and cerebrospinal fluid of children. Br J Clin Pharmacol. 1999;48(1):37–42. doi:10.1046/j.1365-2125.1999.00948.x
94. DURAMORPH [package insert]. Baxter Healthcare Corporation; 2007.
95. MORPHINE SULFATE TABLETS [package insert]. West-Ward Pharmaceuticals Corp; 2021.
96. KADIAN [package insert]. Allergan USA Inc; 2016.
97. MS CONTIN [package insert]. Purdue Pharma LP; 2016.
98. Booij LHDJ, Vree TB, Koppers-Hoyset H, Lagerwerf AJ. Explorative pharmacokinetic study of preoperative administration of morphine 50mg sustained-release capsules (KapanolTM to surgical patients. Clin Drug Investig. 1999;18:125–132. doi:10.2165/00044011-199918020-00005
99. NALMEFENE HYDROCHLORIDE [package insert]. Purdue Pharma LP; 2022.
100. SELINCRO [package insert]. Elaiapharm; 2017.
101. REVIA [package insert]. Duramed Pharmaceuticals Inc; 2013.
102. Dunbar JL, Turncliff RZ, Dong Q, Silverman BL, Ehrich EW, Lasseter KC. Single-and multiple-dose pharmacokinetics of long‐acting injectable naltrexone. Alcohol Clin Exp Res. 2006;30:480–490. doi:10.1111/j.1530-0277.2006.00052.x
103. Meyer MC, Straughn AB, Lo MW, Schary WL, Whitney CC. Bioequivalence, dose-proportionality, and pharmacokinetics of naltrexone after oral administration. J Clin Psychiatry. 1984;45:15–19.
104. Dale OLA, Hoffer C, Sheffels P, Kharasch ED. Disposition of nasal, intravenous, and oral methadone in healthy volunteers. Clin Pharmacol Ther. 2002;72:536–545. doi:10.1067/mcp.2002.128386
105. Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of hepatic and intestinal cytochrome P450 3A and 2B6 in the metabolism, disposition, and miotic effects of methadone. Clin Pharmacol Ther. 2004;76:250–269. doi:10.1016/j.clpt.2004.05.003
106. Dennis BB, Bawor M, Thabane L, Sohani Z, Samaan Z, Zhang H. Impact of ABCB1 and CYP2B6 genetic polymorphisms on methadone metabolism, dose and treatment response in patients with opioid addiction: a systematic review and meta-analysis. PLoS One. 2014;9(1):e86114. doi:10.1371/journal.pone.0086114
107. Verebey K, Volavka J, Mule SJ, Resnick RB. Naltrexone: disposition, metabolism, and effects after acute and chronic dosing. Clin Pharmacol Ther. 1976;20(3):315–328. doi:10.1002/cpt1976203315
108. Mitchell MC Jr, Memisoglu A, Silverman BL. Hepatic safety of injectable extended-release naltrexone in patients with chronic hepatitis C and HIV infection. J Stud Alcohol Drugs. 2012;73(6):991–997. doi:10.15288/jsad.2012.73.991
109. Matzke GR, Frye RF, Alexander ACM, et al. The effect of renal insufficiency and hemodialysis on the pharmacokinetics of nalmefene. J Clin Pharmacol. 1996;36:144–151. doi:10.1002/j.1552-4604.1996.tb04179.x
110. Lucchi L, Bosio A, Spano PF, Trabucchi M. Action of ethanol and salsolinol on opiate receptor function. Brain Res. 1982;232:506–510. doi:10.1016/0006-8993(82)90297-9
111. Hiller JM, Angel LM, Simon EJ. Multiple opiate receptors: alcohol selectively inhibits binding to delta receptors. Science. 1981;214(4519):468–469. doi:10.1126/science.6270788
112. Volpicelli JR, Alterman AI, Hayashida M, O’Brien CP. Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry. 1992;49:876–880. doi:10.1001/archpsyc.1992.01820110040006
113. O’malley SS, Jaffe AJ, Chang G, Schottenfeld RS, Meyer RE, Rounsaville B. Naltrexone and coping skills therapy for alcohol dependence: a controlled study. Arch Gen Psychiatry. 1992;49:881–887. doi:10.1001/archpsyc.1992.01820110045007
114. Soyka M. Nalmefene for the treatment of alcohol use disorders: recent data and clinical potential. Expert Opin Pharmacother. 2016;17(4):619–626. doi:10.1517/14656566.2016.1146689
115. Kissler JL, Sirohi S, Reis DJ, et al. The one-two punch of alcoholism: role of central amygdala dynorphins/kappa-opioid receptors. Biol Psychiatry. 2014;75(10):774–782. doi:10.1016/j.biopsych.2013.03.014
116. Nealey KA, Smith AW, Davis SM, Smith DG, Walker BM. κ-opioid receptors are implicated in the increased potency of intra-accumbens nalmefene in ethanol-dependent rats. Neuropharmacology. 2011;61(1–2):35–42. doi:10.1016/j.neuropharm.2011.02.012
117. Ingman K, Hagelberg N, Aalto S, et al. Prolonged central μ-opioid receptor occupancy after single and repeated nalmefene dosing. Neuropsychopharmacology. 2005;30(12):2245–2253. doi:10.1038/sj.npp.1300790
118. Tadori Y. Pharmacological profile and clinical findings of nalmefene (Selincro®) for reducing alcohol consumption in patients with alcohol dependence. Nihon Yakurigaku Zasshi. 2020;155(2):113–119. doi:10.1254/fpj.19136
119. van den Brink W, Strang J, Gual A, Sørensen P, Jensen TJ, Mann K. Safety and tolerability of as-needed nalmefene in the treatment of alcohol dependence: results from the Phase III clinical programme. Expert Opin Drug Saf. 2015;14(4):495–504. doi:10.1517/14740338.2015.1011619
120. Naudet F, Fitzgerald N, Braillon A. Nalmefene for alcohol dependence: a NICE decision? Lancet Psychiatry. 2016;3(12):1104–1105. doi:10.1016/S2215-0366(16)30356-X
121. Bachmutsky I, Wei XP, Kish E, Yackle K. Opioids depress breathing through two small brainstem sites. Elife. 2020;9:e52694. doi:10.7554/eLife.52694
122. Hill R, Santhakumar R, Dewey W, Kelly E, Henderson G. Fentanyl depression of respiration: comparison with heroin and morphine. Br J Pharmacol. 2020;177(2):254–265. doi:10.1111/bph.14860
123. Yong Z, Gao X, Ma W, Dong H, Gong Z, Su R. Nalmefene reverses carfentanil-induced loss of righting reflex and respiratory depression in rats. Eur J Pharmacol. 2014;738:153–157. doi:10.1016/j.ejphar.2014.05.044
124. Kaplan JL, Marx JA, Calabro JJ, et al. Double-blind, randomized study of nalmefene and naloxone in emergency department patients with suspected narcotic overdose. Ann Emerg Med. 1999;34(1):42–50. doi:10.1016/S0196-0644(99)70270-2
125. Britch SC, Walsh SL. Treatment of opioid overdose: current approaches and recent advances. Psychopharmacology. 2022;239(7):2063–2081. doi:10.1007/s00213-022-06125-5
126. Purdue Pharma LP. Purdue pharma introduces nalmefene HCl injection, 2mg/2mL (1mg/1mL) in the U.S. for the treatment of known or suspected overdose with natural or synthetic opioids; 2022.
127. NARCAN [package insert]. Adapt Pharma Inc; 2015.
128. Krieter P, Chiang CN, Gyaw S, Skolnick P, Snyder R. Pharmacokinetic interaction between naloxone and naltrexone following intranasal administration to healthy subjects. Drug Metab Dispos. 2019;47(7):690–698. doi:10.1124/dmd.118.085977
129. United States Food and Drug Administration. FDA Approves Prescription Nasal Spray to Reverse Opioid Overdose. United States Food and Drug Administration; 2023.
130. OPVEE [package insert]. Opiant Pharm. 2023.
131. Crystal R, Ellison M, Purdon C, Skolnick P. Pharmacokinetic properties of an FDA‐approved intranasal nalmefene formulation for the treatment of opioid overdose. Clin Pharmacol Drug Dev. 2023;13(1):58–69. doi:10.1002/cpdd.1312
132. Krieter P, Gyaw S, Crystal R, Skolnick P. Fighting fire with fire: development of intranasal nalmefene to treat synthetic opioid overdose. J Pharmacol Exp Ther. 2019;371(2):409–415. doi:10.1124/jpet.118.256115
133. Jia Z, Chen Y, Gao T, et al. Nalmefene vs. dexmedetomidine for prevention of postoperative hyperalgesia in patients undergoing laparoscopic gynecological surgery with remifentanil infusion: a randomized double-blind controlled trial. Front Pharmacol. 2023;14:1131812. doi:10.3389/fphar.2023.1131812
134. Grant JE, Odlaug BL, Potenza MN, Hollander E, Kim SW. Nalmefene in the treatment of pathological gambling: multicentre, double-blind, placebo-controlled study. Br J Psychiatry. 2010;197(4):330–331. doi:10.1192/bjp.bp.110.078105
135. Weiss RD, Potter JS, Fiellin DA, et al. Adjunctive counseling during brief and extended buprenorphine-naloxone treatment for prescription opioid dependence: a 2-phase randomized controlled trial. Arch Gen Psychiatry. 2011;68:1238–1246. doi:10.1001/archgenpsychiatry.2011.121
136. Frost MC, Zhang L, Kim HM, Lin L. Use of and retention on video, telephone, and in-person buprenorphine treatment for opioid use disorder during the COVID-19 pandemic. JAMA Network Open. 2022;5(10):e2236298–e2236298. doi:10.1001/jamanetworkopen.2022.36298
137. Mattick RP, Breen C, Kimber J, Davoli M. Buprenorphine maintenance versus placebo or methadone maintenance for opioid dependence. Cochrane Database Syst Rev. 2014;2014(2). doi:10.1002/14651858.CD002207.pub4
138. Bell J, Trinh L, Butler B, Randall D, Rubin G. Comparing retention in treatment and mortality in people after initial entry to methadone and buprenorphine treatment. Addiction. 2009;104(7):1193–1200. doi:10.1111/j.1360-0443.2009.02627.x
139. Mégarbane B, Hreiche R, Pirnay S, Marie N, Baud FJ. Does high-dose buprenorphine cause respiratory depression? Possible mechanisms and therapeutic consequences. Toxicol Rev. 2006;25:79–85. doi:10.2165/00139709-200625020-00002
140. White J, Bell J, Saunders JB, et al. Open-label dose-finding trial of buprenorphine implants (Probuphine)® for treatment of heroin dependence. Drug Alcohol Depend. 2009;103(1–2):37–43. doi:10.1016/j.drugalcdep.2009.03.008
141. Jarvis BP, Holtyn AF, DeFulio A, et al. Effects of incentives for naltrexone adherence on opiate abstinence in heroin‐dependent adults. Addiction. 2017;112:830–837. doi:10.1111/add.13724
142. Jones HE, Johnson RE, Fudala PJ, Henningfield JE, Heishman SJ. Nalmefene: blockade of intravenous morphine challenge effects in opioid abusing humans. Drug Alcohol Depend. 2000;60(1):29–37. doi:10.1016/S0376-8716(00)80005-8
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.