")
Back to Journals » Drug Design, Development and Therapy » Volume 18
Molecular Modeling, Synthesis, and Antihyperglycemic Activity of the New Benzimidazole Derivatives – Imidazoline Receptor Agonists
Authors Martynov A , Farber B , Bomko T, Beckles DL , Kleyn I
Received 7 November 2023
Accepted for publication 28 February 2024
Published 3 April 2024 Volume 2024:18 Pages 1035—1052
DOI https://doi.org/10.2147/DDDT.S447289
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Artur Martynov,1 Boris Farber,2 Tatyana Bomko,1 Daniel L Beckles,3 Ilya Kleyn4
1Laboratory and Clinical department of Molecular Immunopharmacology, SI “ I. Mechnikov Institute of Microbiology and Immunology of National Academy of Medical Sciences of Ukraine, Kharkiv, Ukraine; 2R&D Department, Noigel LLC, New York, NY, USA; 3Baylor Scott & White Health, Round Rock, TX, USA; 4SUNY Downstate Medical Center / University Hospital of Brooklyn, New York, NY, USA
Correspondence: Artur Martynov, Email [email protected]
Introduction: The paper presents the results of a study on the first synthesized benzimidazole derivatives obtained from labile nature carboxylic acids. The synthesis conditions of these substances were studied, their structure was proved, and some components were found to have sugar-reducing activity on the model of alloxan diabetes in rats.
Methods: The study used molecular modeling methods such as docking based on the evolutionary model (igemdock), RP_HPLC method to monitor the synthesis reaction, and 1H NMR and 13C NMR, and other methods of organic chemistry to confirm the structures of synthesized substances.
Results & Discussion: The docking showed that the ursodeoxycholic acid benzimidazole derivatives have high tropics to all imidazoline receptor carriers (PDB ID: 2XCG, 2bk3, 3p0c, 1QH4). The ursodeoxycholic acid benzimidazole derivative and arginine and histidine benzimidazole derivatives showed the highest sugar-lowering activity in the experiment on alloxan-diabetic rats. For these derivatives, the difference in glucose levels of treated rats was significant against untreated control. Therefore, the new derivatives of benzimidazole and labile natural organic acids can be used to create new classes of imidazoline receptor inhibitors for the treatment of diabetes mellitus and hypertension.
Keywords: imidazoline receptors, benzimidazole derivatives, synthesis, antihyperglycemic activity
Introduction
Research interest in imidazoline receptor (IR) agonists has recently increased.
This is because the selective I1-imidazoline receptor (I1R) agonist moxonidine,1,2 the selective I2-imidazoline receptor (I2R) agonists diakamph3 and metformin4 have been found to have the ability to stimulate telomerase expression in cells and exhibit the ability to inhibit senescence.
In addition, the apparent ability of diakamph (V), moxonidine (VI), and metformin to increase tissue sensitivity to insulin has been shown. All three substances have been successfully used as medicines. Metformin is used as a treatment for diabetes mellitus, and moxonidine (VI) is a centrally-acting hypotensive drug (Figure 1). Particular attention should be paid to I3-imidazoline receptors (I3R), which are found in the pancreas and stimulated, leading to increased insulin production by beta-cells.5
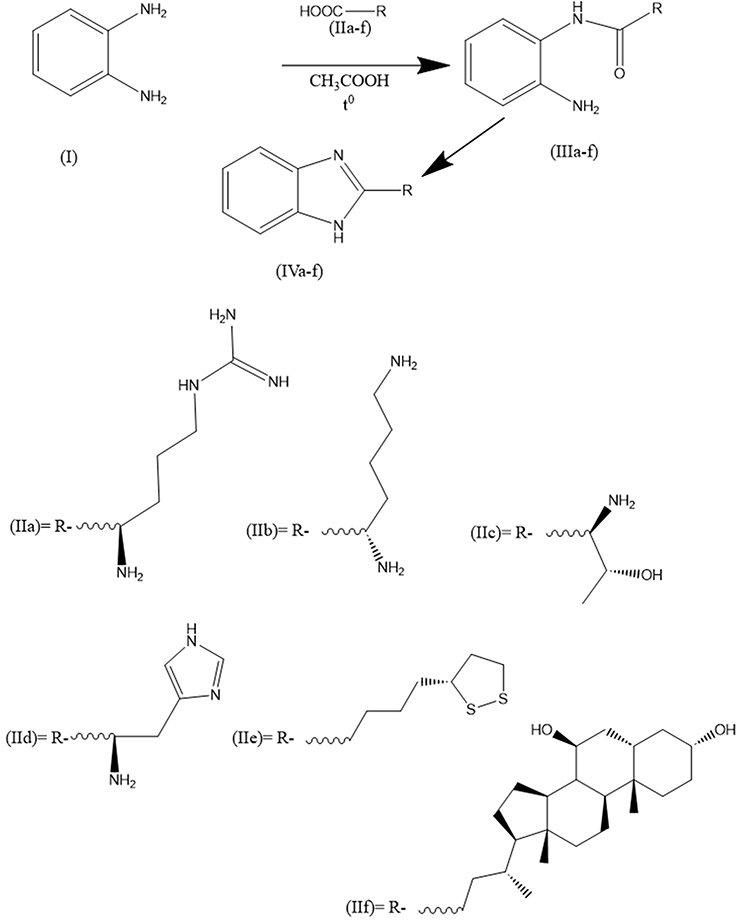 |
Figure 1 Synthesis of some benzimidazole derivatives (IVa-f). |
I1Rs are associated with phosphatidylcholine hydrolysis, the formation of arachidonic acid and eicosanoids, and are actively associated with catecholamine synthesis. Some studies indicate that I1Rs belong to neurocytokine receptors and are similar in action to interleukins.6–8 I1Rs are located in the brainstem and modulate blood pressure. In peripheral tissues, I1Rs are found in the kidneys and are associated with regulating sodium balance in the body.9 In the human brain, I1Rs are concentrated in the triatum, pale globe, and substantia nigra.7
The activation of I1Rs also negatively regulates the progression of fibrosis in hepatic stellate cells through a Nrf2-dependent pathway. This hepatic I1-dependent antifibrotic action was also observed in vivo in mice.10
Imidazoline Subtype 1 receptors have been shown to affect insulin and adiponectin positively. A study using insulin-secreting Min6 b-cells found that activation of I1Rs with a selective I1 receptor ligand, S43126, had a glucose-dependent insulinotropic action.11 This effect was prevented by efaroxan, a selective I1Rs antagonist. Similarly, it was also demonstrated that activating I1Rs on insulin-targeted tissues such as liver and adipose tissues improved insulin sensitivity.12 LNP599, a highly selective I1Rs compound, stimulates adiponectin secretion in 3T3-L1 adipocyte cultures.13
In human platelets, dexmedetomidine, an alpha-2 adrenergic agonist and I1Rs agonist suppressed ADP-induced platelet aggregation when alpha-2 adrenergic receptors were blocked by yohimbine. This specific activation of I1Rs led to an interesting antiaggregation effect on platelets.14
Selective I1 receptor agonists increased the expression of farnesoid X nuclear receptors in 3T3-L1 adipocyte cultures.15 This resulted in improved hepatic steatosis due to an increase in intracellular calcium and p38 phosphorylation. In vivo, an improvement in hepatic steatosis was observed in mice treated with rilmenidine.
I2Rs are present both in the human brain and in peripheral tissues. The highest number of I2Rs in the brain is found in the pineal gland.16,17 Decreased I2R density in the human brain is associated with Huntington’s chorea and Alzheimer’s disease, as well as in drug addiction.7 I2R was found most abundantly in mitochondrial membrane fractions derived from brain, kidney, heart, and liver.7,16 Some studies have found correlation of I2R with monoamine oxidase (MAO).12 In particular, purified I2R protein has the same amino acid sequence with MAO-A and MAO-B, and imidazoline-containing substances inhibit MAO activity.17
Over the years, Idazoxan has been used to identify I2Rs; however, it also binds to adrenoceptors. Because of this, medicinal chemistry has been actively developing selective I2Rs ligands with low I1 and a2-adrenergic receptor binding activities. This has led to the development of a large library of selective I2Rs ligands, and the effort continues today. For instance, a recent study describes a new family of (2-imidazolin-4-yl) phosphonates that have a high affinity for I2Rs.18 Several compounds have been used in previous studies to explore the pharmacology of I2Rs and have become valuable research tools. These include phenyzoline, 2-BFI, CR4056, BU224. Of note, two synthetic ligands have moved from preclinical research to human studies. One radiolabeled I2Rs ligand, 11C-BU99008, has been used in PET studies with human volunteers to demonstrate receptor distribution in the brain.19 In the future, the PET ligand 11C-BU99 could help expand our understanding of I2Rs in neurodegenerative disorders. This is because the density of I2Rs changes in several psychiatric conditions, especially where there is marked gliosis. Therefore, the ligand could be used to confirm and monitor neurodegenerative diseases as well as detect glioblastomas. The authors of the study have previously established that 11C-BU99 has low affinity for MAOB but high affinity and selectivity for I2Rs.20
I2Rs ligands have been studied extensively for their potential analgesic effects. These effects have been demonstrated by various compounds in multiple preclinical pain models. However, the research suggests that these compounds are not very effective in treating acute nociception in thermal stimulus-induced pain models. Previous studies have shown that I2Rs ligands, including tracizoline, LSL601012, BFI, and phenyzoline, do not produce significant antinociception in rodent models of acute nociception, such as radiant tail flick, and hot plate assays.21–23 Li’s group conducted recent studies that support these findings. Mild antinociception was observed with the use of 2-BFI and phenyzoline, while selective I2 receptor ligands such as BU224 and S22687 (5-[2methyl phenoxy methyl]-1,3-oxazolin-2-yl) amine) did not have any effect on the warm water tail flick assay.24,25 I2Rs agonists are generally effective in reducing pain caused by chemical stimuli. For instance, in a writhing test 2BFI, morphine and BU224 were found to have equal efficacy in reducing writhing response.26 During the formalin test, rodents exhibit nocifensive behaviors that consist of two phases: an early neurogenic Phase I, followed by a later inflammatory Phase II.27 During Phase II, CR4056, 2-BFI, and BU224 all demonstrated a dose-dependent reduction in the flinching response.28 In a different study, a new I2Rs agonist called CR4056 completely reversed capsaicin-induced neurogenic/inflammatory allodynia; the effect was dose-dependently blocked by idazoxan.29
Several studies have shown that both 2-BFI and phenyzoline can reduce mechanical allodynia in rats with chronic constriction injury (CCI) peripheral neuropathic pain.28 It has been confirmed that idazoxan attenuates the antiallodynic effects of 2-BFI and phenyzoline, which indicates that the mechanism is mediated by the I2 receptor.28,30 In multiple rat models of chronic pain, CR4056 has shown a strong antihyperalgesic effect. It has been found to reduce mechanical allodynia in rats treated with complete Freund’s adjuvant (CFA), attenuate mechanical hyperalgesia in diabetes-induced neuropathic pain, and reduce neuropathic pain induced by chronic treatment with the chemotherapeutic agent bortezomib.29,31 Additionally, CR4056 is effective in a rat model of fibromyalgia, reversing acidic saline-induced mechanical allodynia. In a postoperative pain model, CR4056 was fully effective in reversing mechanical allodynia, and this effect was blocked by idazoxan32 Interestingly, no sex difference was found for CR4056-induced antinociception. These studies show that I2Rs agonists can reduce chronic pain and may be a new form of painkillers with a wide range of effectiveness.
The I3Rs are believed to be a separate binding site from I1-2Rs. However, not much is known about their nature and function, despite previous research.33,34 Studies have shown that phentolamine and other imidazoline compounds can affect glycemic control in humans.35 In pancreatic islet tissue, protein bands consistent with I2Rs have been identified using an antiserum. However, the existence of I1Rs is disputed. Research has shown that some I2Rs agonists can regulate insulin secretion in pancreatic b-cells, while others cannot. An example of a potent potentiator of glucose-induced insulin secretion is 2-BFI. However, it requires higher concentrations than what is typical for interactions with I2Rs. Other ligands of I2Rs, such as idazoxan and RS45041-190, do not induce insulin release or antagonize the insulin secretory effect of other imidazoline compounds on b-cells.36,37 It seems that the presence of 2-BFI and its related compounds does not have a direct effect on b-cell insulin secretion through I2Rs. One possible explanation for how imidazoline compounds alter insulin secretion is that they bind to a site that is linked to the ATP-sensitive potassium (KATP) channel, which then lowers the rate of potassium efflux. This, in turn, increases the membrane potential and leads to membrane depolarization, ultimately triggering insulin secretion.34 The b-cell KATP channel is made up of two subunits, Kir6.2 and sulfonylurea receptor 1 (SUR1). Previous research has demonstrated that imidazolines can still function as blockers of KATP channels even in the absence of SUR1, when truncated Kir6.2 is present. This suggests that SUR1 is not necessary for imidazoline compounds to regulate the ionic conductance of Kir6.2. Additionally, the study indicates that the I3Rs are situated within Kir6.2.38,39 Further biochemical evidence supports this view.40 Therefore, I3Rs represent a binding site located on the Kir6.2 subunit of KATP channels. I3Rs were found in the pancreas by sensitivity to the imidazoline receptor agonist, efaroxan. Also, one of the imidazoline derivatives, diakamph (V), showed clear hypoglycemic activity and was positioned as a treatment for type 2 diabetes mellitus. In addition, it was found to have neuroprotective effects with oxidative cytotoxicity. Cytoprotective activity against cerebral ischemia in rats has been shown for other IR agonists.41 Agmatine, considered a natural agonist of IRb in experiment, showed antidepressant activity and exhibited anxiolytic, antinociceptive and anti-inflammatory properties.8,42–44
Among the identified receptors/enzymes for imidazoline derivatives, the following have been reliably confirmed: for I2R - monoamine oxidases A, B, and brain b-creatinine kinase,45 for I1R - nischarin.46 Another benzimidazole derivative –bendazol inhibits experimental myopia progression and decreases the ocular accumulation of HIF‐1α protein in young rabbits.47
Bendazole47 and diakamph48 are not pure imidazolines but only contain its fragment in the form of benzimidazole, while they exhibit more universal effects (hypotensive, sugar-reducing, adaptogenic, neuroprotective) all 3 types of imidazoline receptors, which gave us grounds to choose this group of compounds for modeling, synthesis and study of biological activity in relation to models of pathologies, in the treatment of which imidazoline receptor agonists have been successfully used.
Benzimidazole derivatives are crucial in many biologically active compounds, including antiparasitics, antimicrobials, antivirals, antifungals, and more. Drugs in various therapeutic lines have been developed by changing substituents around the core structure, such as albendazole, carbendazim, and omeprazole.49
The improved methodology for synthesizing benzimidazoles developed by us made it possible to obtain derivatives of thermolabile carboxylic acids, including those from amino acids and their derivatives, such as lipoic acid, in a more gentle regime.
The essence of the technique consists in boiling the reagents in glacial acetic acid with a reflux condenser and a Dean-Stark nozzle. After distillation of 2/3 of glacial acetic acid, the Dean-Stark nozzle is removed, and the reaction mixture continues to be heated for another 3–10 hours. This technique allows the use of hydrophilic acids and salts of amino acids as donors of carboxyl groups. Older methods involve using hydrophobic solvents such as benzene and toluene at higher boiling points. In this case, the product yield is minimal (up to 15%), some initial acids are decarboxylated, and some are simply not dissolved in toluene and benzene. This did not allow previously to obtain benzimidazoles from thermolabile or bifunctional compounds.
Materials and Methods
Experimental Section
The molecular docking method predicts how a protein binds to a ligand. Docking algorithms determine the best orientation with the lowest energy score.50
Computational Analysis
Ligand and Protein Preparation Ligands
As ligands for modeling, we used: benzimidazolyl-arginine (IVa), benzimidazolyl-lysine (IVb), benzimidazolyl-threonine (IVc), benzimidazolyl-histidine (IVd), benzimidazolyl-thioctic acid (IVe), benzimidazolyl-ursodeoxycholic acid (IVf), diakamph, moxonidine). As controls with known ability to stimulate all three types of imidazoline receptors, we used diakamph and moxonidine.
Molecular Docking
Diakamph (V) and moxonidine (VI) were obtained from PubChem database,51 the other substances were constructed using Yasara program,52 and their geometric optimization was performed using AMBER 15 IPQ method program,53 which includes geometric optimization in a water box with alternating sessions of molecular dynamics to select the most stable conformer with minimum energy. Protein obtained from PDB database (https://www.rcsb.org/): model monoamine oxidase-B (2BK3) in complex with the agonist farnesol, monoamine oxidase B in complex with the agonist (2-benzofuranyl)-2-imidazoline (2XCG), chicken brain creatinine kinase (1QH4) and human discharging PX-domain (3P0C).
Yasara software (http://www.yasara.org/) was used for docking after removing all water and ligand molecules from the protein preparation.
Previously, a comparative study of molecular docking methods was performed and showed that the methods’ results are comparable to each other and give statistically comparable results. Multiligand docking and the highest output parameters are given by the iGemdock program (http://gemdock.life.nctu.edu.tw/dock/igemdock.php). In addition, this program also takes pharmaceutical semi-empirical criteria such as PharmScore into account.
Molecular docking software used in iGemdock 2.1. iGemdock application, first, we have to select the protein binding site and ligands; virtual screening procedure of iGemdock consists of four main steps (Population size = 200, Generations = 70, Number of solutions = 2, and Default setting = Standard docking).54,55 iGemdock suite is an automated docking/screening tool used a generic evolutionary method for molecular docking and empirical scoring function. iGemdock useful tool for molecular recognition and used to systematically evaluate and improve docking scoring function.56,57 We used the PyMOL program (https://pymol.org/2/) to create a 3D model of the active centers with the ligands.58,59 Additionally, we utilized the LigPlus program (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/) to generate 2D projections of the complexes.60,61
Materials and Equipment
All chemicals and reagents used in the study were obtained from commercial vendors, including Merck (Germany) and Sigma Aldrich Sigma Chemical, Co. (USA), and were utilized without further purification. HPLC was performed using an HPLC microcolumn chromatograph (Millichrom A-02, RF)62,63 with a gradient of acetonitrile (5–100%)/0.1 M chloric acid and 0.5 M lithium perchlorate).
Melting points were determined using the open capillary method in an uncorrected Gallen Kamp MFB-59564,65 electric melting point instrument. UV spectra were obtained using a double-beam UV-Visible spectrophotometer GeneQuant 1300 (Model 2021, UK).66,67
The 1H-NMR, 13C-, NMR spectra were obtained using a Bruker Avance III 40068,69 at 400 and 100 MHz, with Tetramethylsilane (TMS) as an internal standard. The spectra were measured relative to TMS in DMSO-d6. The chemical shifts are represented as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets), and br s (broad singlet) in the appropriate solvent, along with coupling constants (J) in Hz (Hertz).
(R)-1-(4-Amino-4-(1H-Benzo[D]imidazol-2-Yl)butyl)guanidine (IVa)
L-arginine (base) (174 mg, 1.0 mmol) and benzene-1,2-diamine (108 mg, 1.0 mmol), dissolve in 3 mL anhydrous glacial acetic acid (AGAA) in a 10 mL round bottom flask, place in a system with a reflux condenser and a Dean-Stark nozzle. Boil with the reflux condenser and extract 2 mL of AGAA. Then, the Dean-Stark nozzle is disconnected, and the solution is boiled with reflux condenser for 5 hours. The solution is cooled, 6 mL of ethanol is added and placed in a −15 ° C freezer. The precipitate (IVa) is filtered off and recrystallized from a 1:3 mixture of AGAA and ethanol, and the precipitate is dried. (S)-1-(4-(4-amino-4-(1H-benzo[d]imidazol-2-yl)butyl) guanidine (IVa) is obtained as a gray powder. For analytical purposes, the product (IVa) is additionally was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm,70 mobile phase: [water (0.1%TClA) − ACN]: 20−55%, 12 min).1H-NMR (400 MHz, DMSO-d6): δ 1.50 (m, 2H), 1.81 (m, 2H), 2.52 (s, 1H), 3.33 (m, 2H),3.8 (s, 1H), 7.16 (d,1H), 7.52 (s, 1H), 8.76 (s, 2H), 6.63 (s, 2H), 7.82 (s, 1H), 12.16 (s, 1H). 13CNMR (126 MHz, CD3OD) δ 24.39, 25.70, 30.66, 43.07, 58.05, 58.66, 79.19, 100.37. HRMS (ESI+): m/z calcd for C12H18N6: 246.3212 [M + H]+; found: 246.33188 [M + H]+.
(S)-1-(1H-Benzo[D]imidazol-2-Yl)pentane-1,5-Diamine (IVb)
L-lysine (base) (146 mg, 1.0 mmol) and benzene-1,2-diamine (108 mg, 1.0 mmol), dissolve in 5 mL anhydrous glacial acetic acid (AGAA) in a 20 mL round bottom flask, place in a system with a reflux condenser and a Dean-Stark nozzle. Boil with the reflux condenser and extract 3 mL of AGAA. Then, the Dean-Stark nozzle is disconnected, and the solution is boiled with reflux condenser for 5 hours. The solution is cooled, 9 mL of ethanol is added and placed in a −15 ° C freezer. The precipitate (IVb) is filtered off and recrystallized from a 1:3 mixture of AGAA and ethanol, and the precipitate is dried. (S)-1-(1H-benzo[d]imidazol-2-yl)pentane-1,5-diamine (IVb) is obtained as a gray-yellow powder. For analytical purposes, the product (IVb) is additionally was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm, mobile phase: [water (0.1%TClA) − ACN]: 20−55%, 12 min).1H-NMR (400 MHz, DMSO-d6): δ 1.19 (m, 2H), 1.52 (m, 2H), 1.81 (m, 2H), 1.91 (m, 2H),2.74 (dd, 2H), 3.95 (br s,1H), 4.34 (m, 1H), 7.32 (2dd, 1H). 13CNMR (126 MHz, CD3OD) δ 22.09, 24.39, 27.69, 30.66, 41.90, 58.06, 58.66, 79.20, HRMS (ESI+): m/z calcd for C12H18N4: 218.3011 [M + H]+; found: 218.31149 [M + H]+.
(1R,2R)-1-Amino-1-(1H-Benzo[D]imidazol-2-Yl)propan-2-Ol (IVc)
L-threonine (119 mg, 1.0 mmol) and benzene-1,2-diamine (108 mg, 1.0 mmol), dissolve in 3 mL anhydrous glacial acetic acid (AGAA) in a 10 mL round bottom flask, place in a system with a reflux condenser and a Dean-Stark nozzle. Boil with the reflux condenser and extract 2 mL of AGAA. Then, the Dean-Stark nozzle is disconnected, and the solution is boiled with reflux condenser for 5 hours. The solution is cooled, 6 mL of methanol is added and placed in a −15 ° C freezer. The precipitate (IVc) is filtered off and recrystallized from a 1:3 mixture of AGAA and methanol, the precipitate is dried (1R,2R)-1-amino-1-(1H-benzo[d]imidazol-2-yl)propan-2-ol (IVc) is obtained as a light gray powder. For analytical purposes, the product (IVc) is additionally was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm, mobile phase: [water (0.1%TClA) − ACN]: 20−55%, 4 min).1H-NMR (400 MHz, DMSO-d6): δ 1.11 (m, 3H), 3.60 (br s, 2H), 3.81 (d, 1H), 4.00 (m, 1H), 7.30 (m, 1H), 7.35 (m, 1H), 3.95 (br s,1H). 13CNMR (126 MHz, CD3OD) δ 20.19, 24.39, 30.66, 41.90, 58.05, 58.66, 67.75, 79.20, HRMS (ESI+): m/z calcd for C10H13N3O: 191.2302 [M + H]+; found: 191.2050 [M + H]+.
1-(1H-Benzo[D]imidazol-2-Yl)-2-(1H-Imidazol-4-Yl)ethan-1-Amine (IVd)
Histidine (155 mg, 1.0 mmol) and benzene-1,2-diamine (108 mg, 1.0 mmol), dissolve in 5 mL anhydrous glacial acetic acid (AGAA) in a 20 mL round bottom flask, place in a system with a reflux condenser and a Dean-Stark nozzle. Boil with the reflux condenser and extract 3 mL of AGAA. Then, the Dean-Stark nozzle is disconnected, and the solution is boiled with reflux condenser for 7 hours. The solution is cooled, 6 mL of ethanol is added and placed in a −15 ° C freezer. The precipitate (IVd) is filtered off and recrystallized from a 1:3 mixture of AGAA and methanol, the precipitate is dried 1-(1H-benzo[d]imidazol-2-yl)-2-(1H-imidazol-4-yl)ethan-1-amine (IVd) is obtained as a blue powder. For analytical purposes, the product (IVd) is additionally was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm, mobile phase: [water (0.1%TClA) − ACN]: 20−55%, 4 min).1H-NMR (400 MHz, DMSO-d6): δ 2.61 (m, 2H), 2.86 (dd, 2H), 2.90 (d, 1H), 4.75 (d, 1H), 4.77 (d, 1H), 5.24 (m,1H), 7.33 (m, 1H). 13CNMR (126 MHz, CD3OD) δ 29.21, 58.05, 114.80, 115.90, 117.44, 122.45, 131.74, 135.74, 137.29, 152.89. HRMS (ESI+): m/z calcd for C10H13N3O: 227.2702 [M + H]+; found: 227.2212 [M + H]+.
(R)-2-(4-(1,2-Dithiolan-3-Yl)butyl)-1H-Benzo[D]imidazole (IVe)
Lipoic acid ((R)-5-(1,2-dithiolan-3-yl)pentanoic acid) (206 mg, 1.0 mmol) and benzene-1,2-diamine (108 mg, 1.0 mmol), dissolve in 5 mL anhydrous glacial acetic acid (AGAA) in a 20 mL round bottom flask, place in a system with a reflux condenser and a Dean-Stark nozzle. Boil with the reflux condenser and extract 3 mL of AGAA. Then, the Dean-Stark nozzle is disconnected, and the solution is boiled with a reflux condenser for 5 hours. The solution is cooled, 4 mL of anhydrous dioxan is added and placed in a −15 ° C freezer. The precipitate (IVe) is filtered off and recrystallized from a 1:2 mixture of AGAA and dioxan, and the precipitate is dried (R)-2-(4-(1,2-dithiolan-3-yl)butyl)-1H-benzo[d]imidazole (IVe) is obtained as a yellow powder. For analytical purposes, the product (IVe) is additionally was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm, mobile phase: [water (0.1%TClA) − ACN]: 20−55%, 17 min).1H-NMR (400 MHz, DMSO-d6): δ 1.34 (d, 2H), 1.36 (d, 2H), 1.38 (s, 2H), 1.41 (s, 1H), 1.48 (m, 2H), 1.70 (d,2H), 1.72 (d, 2H), 2.50 (m, 2H), 2.78 (s, 2H), 2.78 (s, 2H), 2.81 (d, 2H), 3.70 (m, 1H), 3.70 (m, 1H), 7.00 (m, 1H), 7.01 (m, 1H), 7.35 (m, 1H). 13CNMR (126 MHz, CD3OD) δ 26.03, 27.19, 27.73, 34.31, 38.33, 40.13, 56.21, 114.81, 115.91, 122.46, 122,54, 138.00, 155,71. HRMS (ESI+): m/z calcd for C14H18N2S2: 278.4305 [M + H]+; found: 278.4211 [M + H]+.
(3R,5S,7S,9S,10S,13R,14S,17R)-17-((R)-4-(1H-Benzo[D]imidazol-2-Yl)butan-2-Yl)-10,13-Dimethylhexadecahydro-1H-Cyclopenta[a]phenanthrene-3,7-Diol (IVf)
Ursodesoxycholic acid ((4R)-4-((3R,5S,7S,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoic acid) (392 mg, 1.0 mmol) and benzene-1,2-diamine (108 mg, 1.0 mmol), dissolve in 30 mL anhydrous glacial acetic acid (AGAA) in a 50 mL round bottom flask, place in a system with a reflux condenser and a Dean-Stark nozzle. Boil with the reflux condenser and extract 20 mL of AGAA. Then, the Dean-Stark nozzle is disconnected, and the solution is boiled with reflux condenser for 10 hours. The solution is cooled, 4 mL of methanol is added and placed in a −15 ° C freezer. The precipitate (IVf) is filtered off and recrystallized from a 1:3 mixture of AGAA and methanol, the precipitate is dried (3R,5S,7S,9S,10S,13R,14S,17R)-17-((R)-4-(1H-benzo[d]imidazol-2-yl)butan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol (IVf) is obtained as a white, yellow powder. For analytical purposes, the product (IVf) is additionally was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm, mobile phase: [water (0.1%TClA) − ACN]: 20−55%, 17 min). 1H-NMR (400 MHz, DMSO-d6): δ 0.70 (s, 3H), 0.95 (s, 3H), 1.32 (s, 2H), 1.47 (m, 2H), 1.58 (m, 2H), 1.61 (m,1H), 1.81 (m, 2H), 2.03 (m, 2H), 2.73 (d, 2H), 2.75 (d, 2H), 3.65 (br s, 1H), 3.74 (br s, 1H), 4.38 (br s, 1H), 7.02 (m, 1H), 7.34 (m, 1H). 13CNMR (126 MHz, CD3OD) δ 12.75, 18.89, 20.00, 22.40, 24.39, 24.89, 28.32, 28.84, 30.52, 30.66, 31.26, 34.19, 35.69, 35.83, 36.06, 36.39, 40.33, 40.40, 40.80, 42.63, 43.89, 51.80, 56.87, 58.66, 68.84, 71.30, 79.20. HRMS (ESI+): m/z calcd for C30H44N2O2: 464.3412 [M + H]+; found: 464.3600 [M + H]+.
All synthesized benzimidazole derivatives (IVa-f) have a characteristic double absorption band in the UV spectrum in the 260–280 nm (Figure 3).71,72 This double band indicates a benzimidazole fragment’s presence in the products’ structure (IVa-f).
 |
Figure 2 UV-specter of (IVd) benzimidazole derivative. |
The Study of Hypoglycemic Action of (IVa-f) Administered Orally to a Model of Alloxan diabetes
In this study, 70 male Wistar albino rats weighing 180–220 g were used. The rats were kept in standard vivarium conditions, including a controlled temperature (22–25°C), relative humidity (60–70%), dark/light cycle, and provided with a standard diet and unlimited access to water. All animal procedures were conducted according to the guidelines of the Animal Welfare Act and the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals. The rats were obtained from Biomodelservice in Kyiv, Ukraine, and housed in the vivarium at the Mechnikov Institute of Microbiology and Immunology. The animals were cared for by international guidelines, including the Principles of Good Laboratory Practice (GLP) 33,647–2015.),73 the international recommendations of the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (The European Convention, 1986).74 The protocol of the experimental study was approved by the Ethics Committee of Mechnikov Institute of Microbiology and Immunology of the National Academy of Medical Sciences (protocol № 4–2022 of 14.12.2022).
Alloxan monohydrate was used to induce diabetes in animals by a single intraperitoneal injection at a dose of 120 mg/kg freshly prepared from a 0.9% sodium chloride solution.75,76 The animals were fasted for 24 hours before the injection.77
After injecting the alloxan, rats developed diabetes within 72 hours. The rats chosen for the experiment had a fasting glucose content above 11.1 mmol/L. The level of glucose in their blood was measured by using a glucometer called “One Touch Ultra” (USA). Blood was taken from the tail vein.78
In the first set of experiments, without glucose, the animals were distributed into 14 groups of 5 animals per each:
1 – Intact control received saline;
2–7 – intact rats were injected with IVa-f
8 – diabetes control received saline;
9–14 animals with diabetes were injected with IVa-f
The rats were given food for 18 hours before and 3 hours after being given insulin and a placebo. IVa-f was used in a dosage of 50mg/kg and dissolved in a phosphate buffer solution. The control group was given saline solution in similar doses. The glucose level in the rats’ blood was measured before drug administration and after 0.5, 1, 2, 3, and 24 hours.
The animals were not given any food for 18 hours before the start of the experiment. After taking blood samples for a three-hour experiment, feeding was provided. The group (IVa-f) was given an injection of 50 mg/kg, while the animal control group received saline. After 15 minutes, rats were injected with glucose in a dose of 3 g/kg (40% solution, 0.75 mg/100 g) using the drug Glucose, which is a 40% injection solution in vials of 20 mL manufactured by JSC “Farmak” (Kiev, Ukraine). Glucose in blood serum was determined immediately before (IVa-f) injection and 1, 2, 3, and 24 hours after the glucose load. The research results are processed with the method of variational statistics using Student’s test, with a significance level Р ≤0,05.
The data are presented in Table 5.
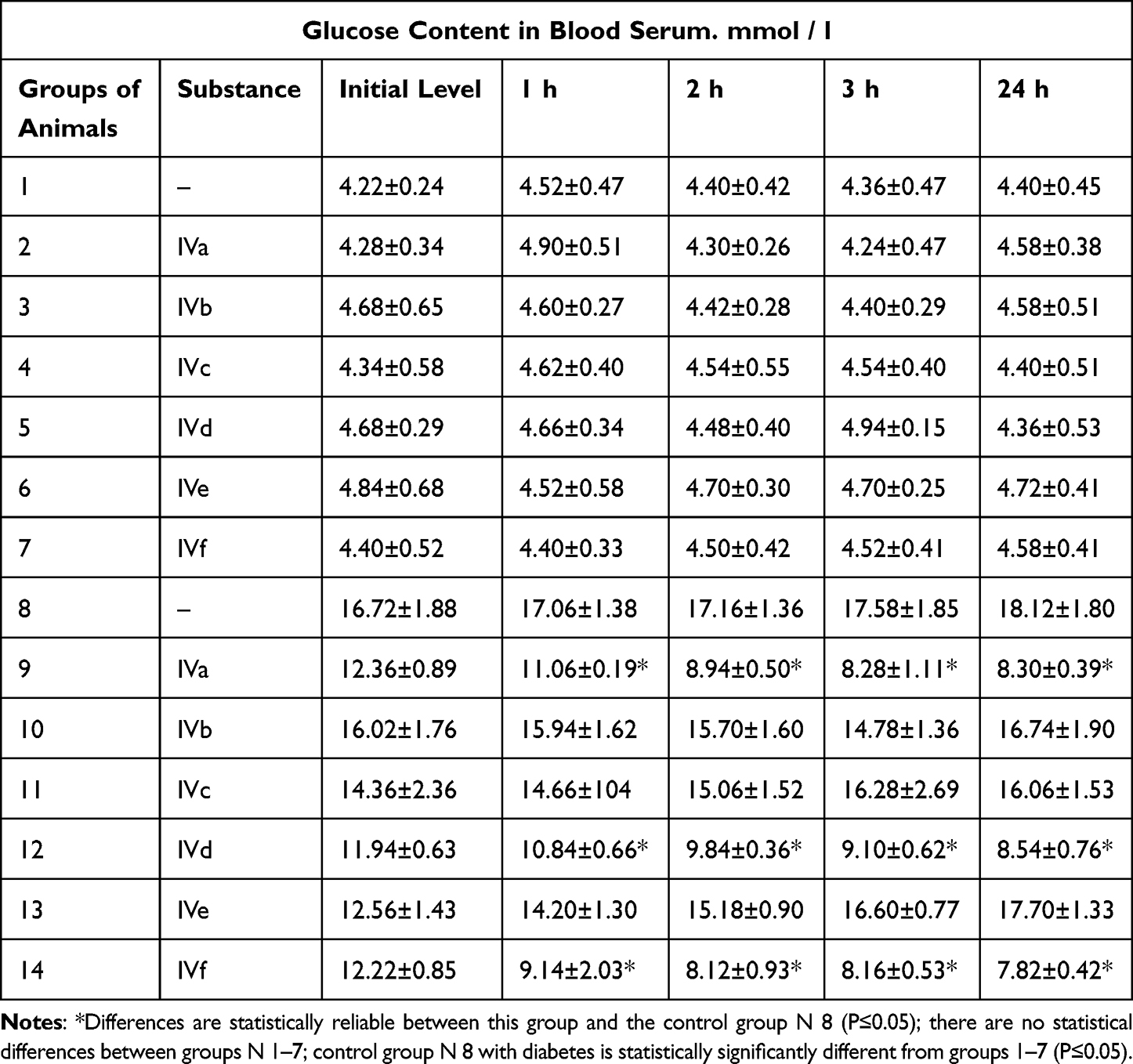 |
Table 1 Dynamics of Blood Glucose in Rats with Alloxan Diabetes After a Single Oral Administration of an Benzimidazoles IVa-f |
Results and Discussion
During the synthesis of each derivative (IVa-f), certain peculiarities were observed. The highly hydrophilic substances (IVa-d) caused the solution to turn green and dark blue during synthesis. No precipitates formed in the solutions after stopping the reaction and cooling the mixture. As a result, it was necessary to add ethanol or isopropanol and sometimes freeze the solution to obtain the residue. Recrystallization of substances (IIa-d) also involved adding ethanol or isopropanol to a solution of the derivative in glacial acetic acid, followed by freezing the solution.
After adding substances (IVe,f), the reaction mixture turned light blue and formed blue crystals upon settling. Based on the liquid chromatography data, the reaction time spanned from 2 to 8 hours, with peak 4 having the maximum area and peaks 1 and 3 decreasing in area. The figure displays the chromatogram of the reaction mixture during the product (IVd) after the solution acquired coloration. Band 1 is histidine, band 2 is probably the salts of o-phenylenediamine (I), histidine, and acetic acid, band 3 corresponds to the amide of histidine and o-phenylenediamine, and band 4 indicates the final product (IVd).
Preparative chromatographic purification gives one absorption band corresponding to the UV spectrum of benzimidazole with a split peak in the region 260–280 nm (Figure 2). A similar situation is observed in the synthesis of the derivative (IIc): bands 1 and 2 are mixtures of the starting components serine (IIc) and o-phenylenediamine (I), band two is associated with amides (IIIc), and band 4 is the final product (IVc).
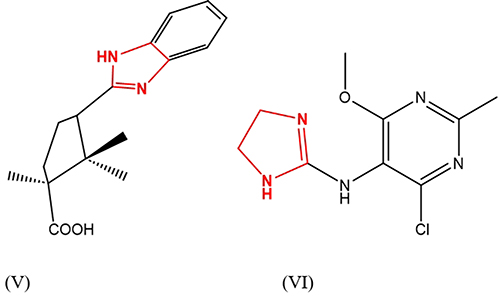 |
Figure 3 Diacamph structure (V), moxonidine structure (VI). |
The synthesis of diakamph, which was described in,79 was used as an example for comparison. Figures 4–6 show the results of RP-HPLC analysis of the reaction mixture. The mixture contains (I) and amides associated with band 2, as well as a mixture of diakamph isomers with bands 3 and 4. The spectral ratios for bands 4 in all the chromatograms exhibit a double.
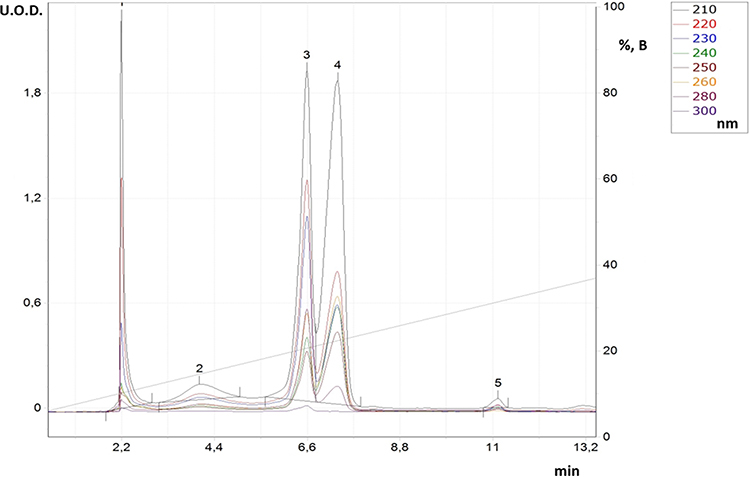 |
Figure 4 RP-HPLC for reaction solution in synthesis process for derivative (IVd). |
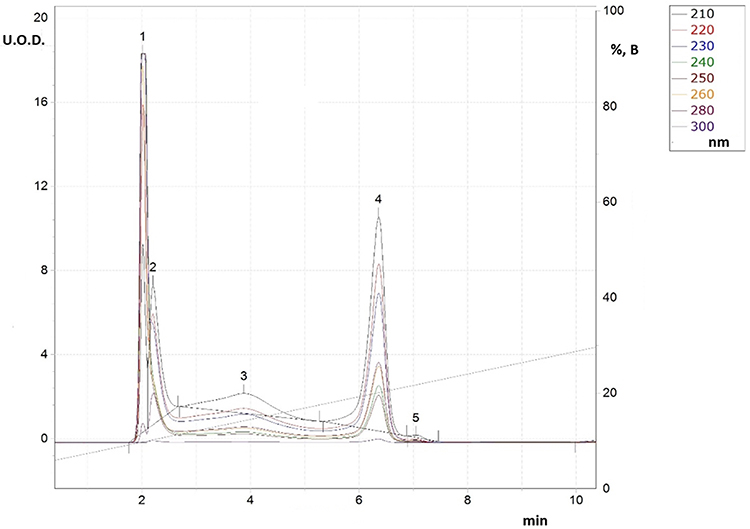 |
Figure 5 RP-HPLC for reaction solution in synthesis process for derivative (IVc). |
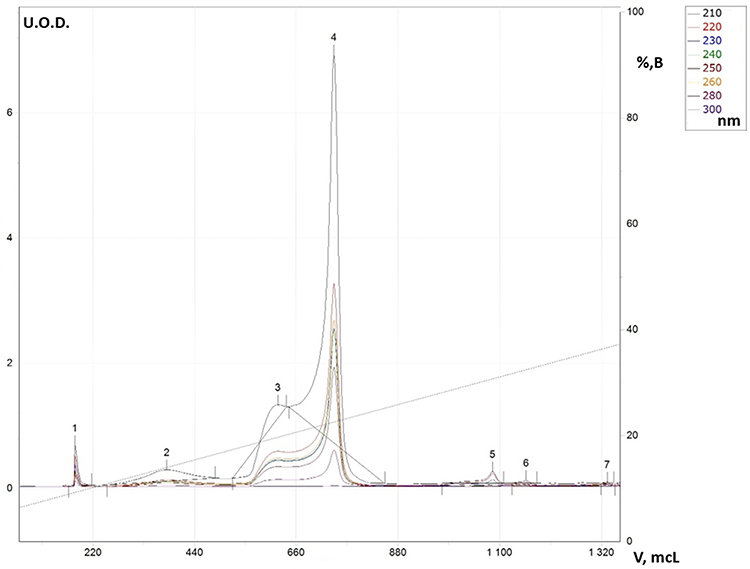 |
Figure 6 RP-HPLC for reaction solution in synthesis process for diacamph (V). |
Table 1 (Full data is shown in Supplementary Material) shows that giving derivatives (IVa-f) to animals did not affect their blood glucose levels. The average glucose level in groups 2–7 was the same as the control group (N 1). In the control group of animals with induced diabetes (N 8), glucose levels were between 16.7 and 18.1 mmol/l, which is typical for diabetes mellitus. A significant reduction in blood sugar levels (P ≤ 0.05) was observed for compounds (IVa), (IVd), and (IVf). Among these three compounds, the benzimidazole derivative and ursodeoxycholic acid (IVf) had the highest sugar-reducing effect and the lowest energy in docking.
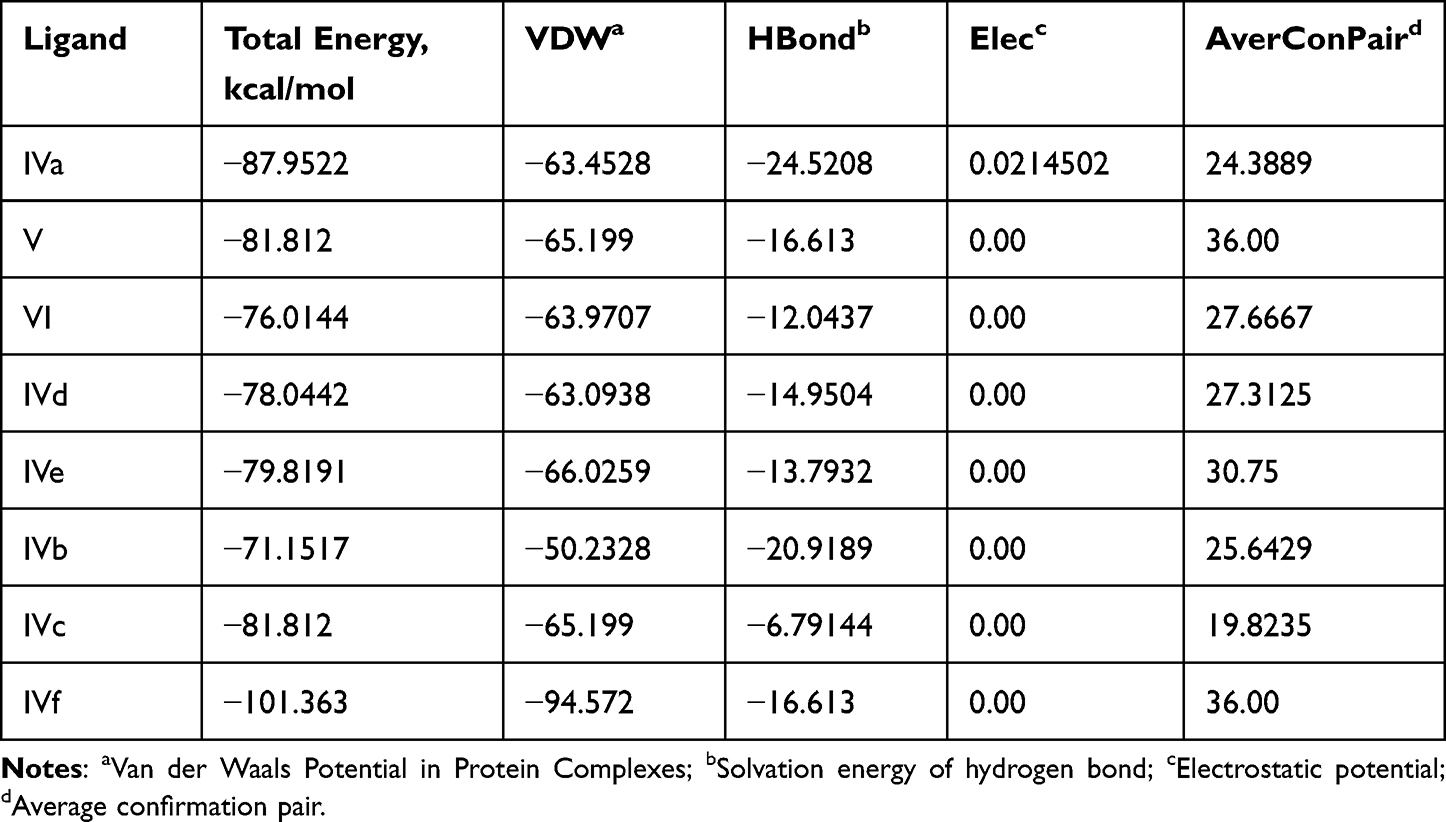 |
Table 2 Results of Molecular Docking Between Studied Substances and Chicken Brain-Type Creatine Kinase (PBPs) (PDB ID: 1QH4) |
The synthesized compounds (IVa-f) have minimum binding energy (Table 2) ranging from (−70 to −107 kcal/mol) with a good result achieved using compound 4 (−106.6 kcal/mol), which revealed Figure 3 3D (right) and 2D (left) representations of the binding interactions of (IVf) against Human monoamine oxidase b (PBPs) (PDB ID: 2XCG). Compound (IVf) interacts Arg-100 was involved in hydrogen bonding with -С=O of (IVf) and with NH2-group of Val-106. Hydrophobic/π-cation interactions were observed for Tyr-112 with the aromatic group, while Arg-120, Thr-479, Glu-483, Asn-116, Trp-119, Tyr-112 and Pro-105 showed Van der Waals residual interactions, respectively (Figure 7). The lowest energy of target-ligand complexes, except for compound (IVf), was observed in compounds (IVa) and the control substance I1-stimulant agonist moxonidine (VI). This suggests the presence of sugar-lowering activity in the synthesized substances.
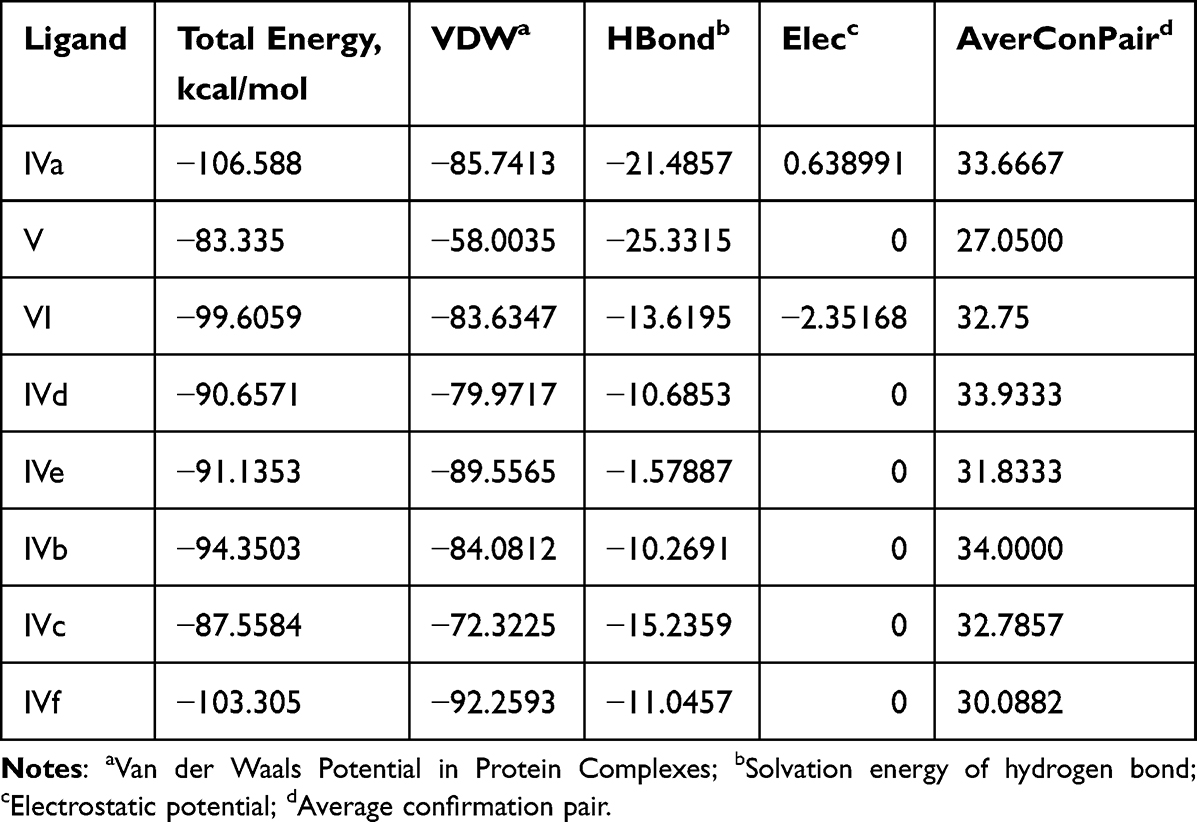 |
Table 3 Results of Molecular Docking Between Studied Substances and Human Monoamine Oxidase b (PBPs) (PDB ID: 2XCG) |
 |
Figure 7 3D (right) and 2D (left) representations of the binding interactions of (IVf) against Human monoamine oxidase b (PBPs) (PDB ID: 2XCG). |
Compound (IVa) interacts Asn-116 was involved in hydrogen bonding with -NH2 of (IVa) and with OH-group of Tyr112. Hydrophobic/π-cation interactions were observed for Phe-103 with the aromatic group, while Trp-119, Arg-127, Thr-479, Arg-120, Arg-484 and Glu-483 showed Van der Waals residual interactions, respectively (Figure 8). Among other molecules, (IVa) had one of the lowest complex energies with the 2bk3 target −94.5 kJ/mol (Table 3).
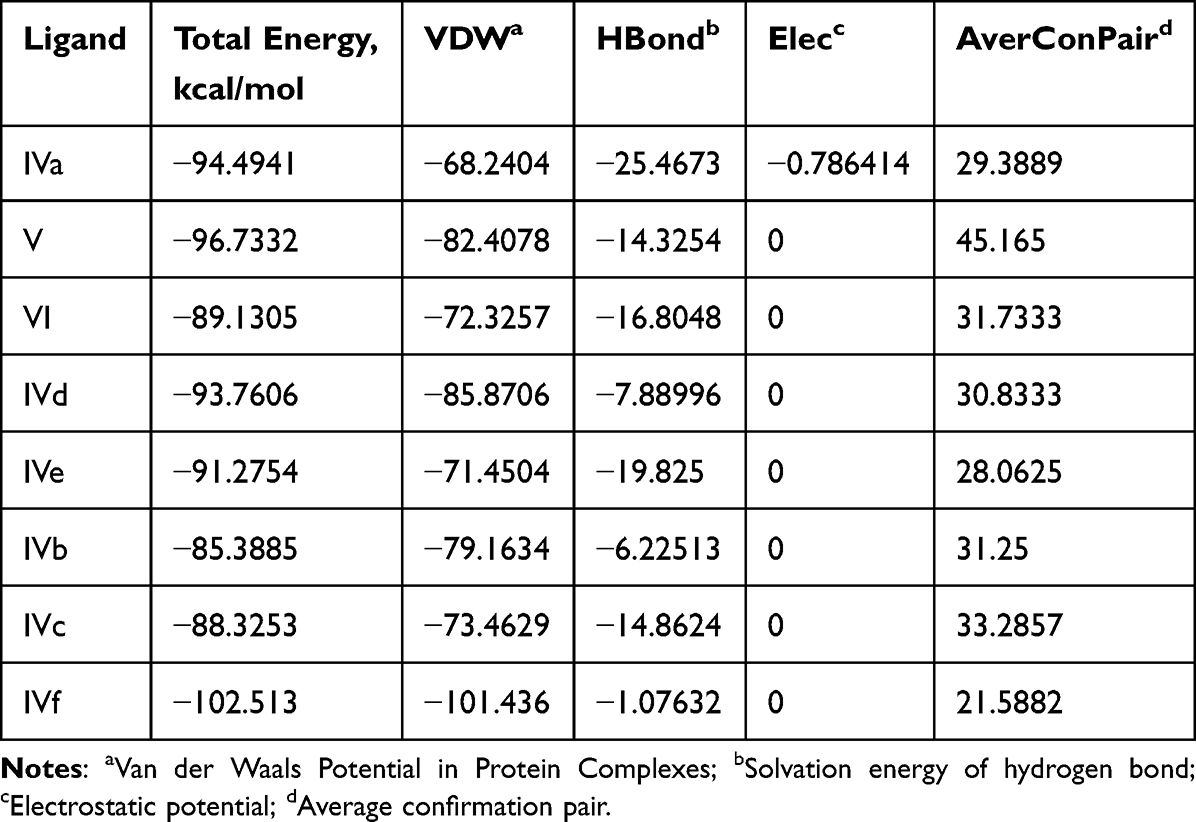 |
Table 4 Results of Molecular Docking Between Studied Substances and Human Monoamine Oxidase b (PBPs) (PDB ID: 2bk3) |
 |
Figure 8 3D (right) and 2D (left) representations of the binding interactions of (IVa) against Human monoamine oxidase b (PBPs) (PDB ID: 2bk3). |
Compound (IVf) interacts with Leu-26 through ionic bonds -CH2-OH of (IVf) and with -C=O-group of Val-22. An ionic bond is also formed between the -COO group in Glu-86 and the nitrogen of (IVf) imidazole fragment. Leu-84, Lys-87, Lys-90, Val-21, Arg-20 and Arg-82 showed Van der Waals residual interactions, respectively (Figure 9). The energy of the complex of Human Nischarin (PBPs) (PDB ID: 3P0C) with the derivative (IVf) was minimal −101.4 kJ/mol (Table 4). Of all compounds, the most promising 3P0C agonists can be considered compounds (IVa) and the comparison substance diakamph (V) with proven antagonism to nischarin.
 |
Table 5 Results of Molecular Docking Between Studied Substances and Nischarin (PBPs) (PDB ID: 3p0c) |
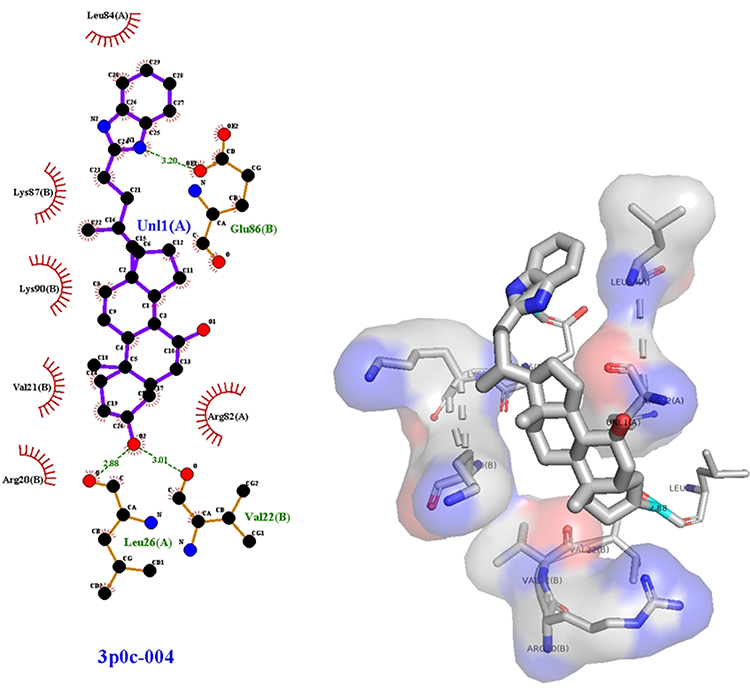 |
Figure 9 3D (right) and 2D (left) representations of the binding interactions of (IVf) against nischarin (PBPs) (PDB ID: 3p0c). |
Figure 10 3D (right) and 2D (left) representations of the binding interactions of (IVf) against chicken brain-type creatine kinase (PBPs) (PDB ID: 1QH4). Compound (IVf) interacts with Trp-211 through ionic bonds -C=O and -OH-02 in (IVf). Val-198, Pro-197, Lys-90, Leu-203, Phe-3, Arg-209, Ala-208, Leu-203, Asp-62, Lys-196, Pro-200, Pro-212 showed Van der Waals residual interactions, respectively (Figure 10). The energy of the complex of chicken brain-type creatine kinase (PBPs) (PDB ID: 1QH4) with the best pose-derivative (IVf) was minimal −101.4 kJ/mol (Table 5). Of all compounds, the most promising 1QH4 agonists can be considered compounds (IVa) and the comparison substance diakamph (V) with proven antagonism to nischarin.
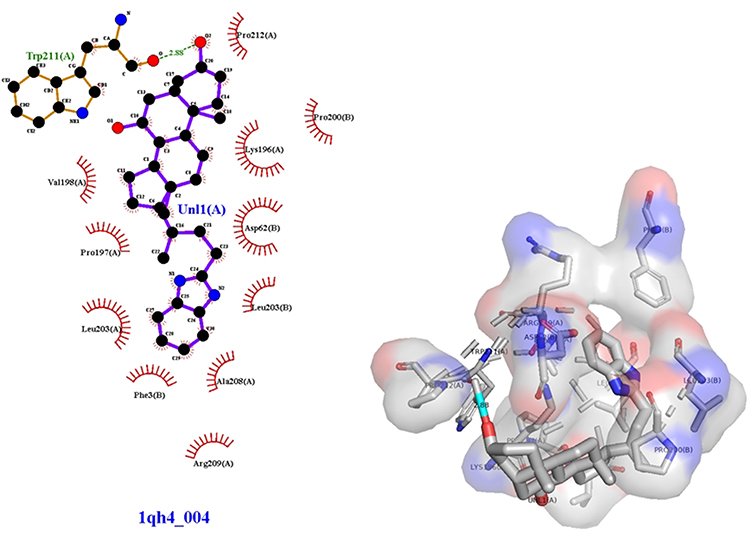 |
Figure 10 3D (right) and 2D (left) representations of the binding interactions of (IVf) against chicken brain-type creatine kinase (PBPs) (PDB ID: 1QH4). |
Conclusions
- Benzimidazole derivatives based on thermolabile amino acids (arginine, lysine, serine, histidine) and natural organic acids (ursodeoxycholic and lipoic acids) were synthesized.
- Molecular modeling was used to predict the highest affinity to imidazoline receptors for derivatives (IVa) and (IVf). Among these, the derivative (IVf) based on ursodeoxycholic acid displayed the lowest energies of complexes with imidazoline receptors of both type 1 and types 2 and 3. This derivative has the potential to be developed as a new antihyperglycemic agent for the treatment of severe forms of diabetes.
- The synthesized compounds were studied on the model of alloxan diabetes in rats. As a result, compounds (IVa), (IVd), and (IVf) showed a significant and statistically proven antihyperglycemic effect. Among these compounds, the ursodeoxycholic acid derivative (IVf) had the most pronounced effect, as predicted by the docking method.
- In the future, modeling is planned to provide molecular dynamic simulation data and thermodynamic stabilities of Ligand-Protein complex for the complex IVf–protein and Pharmacokinetic and bioavailability predictions.
- It is also planned to study the toxic properties of the most promising compounds (IVa) and (IVf).
Institutional Review Board Statement
The animals were cared for according to the international guidelines 33647-2015 (Principles of the Good Laboratory Practice, GLP), the international recommendations of the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (The European Convention, 1986). The protocol of the experimental study was approved by the Ethics Committee of Mechnikov Institute of Microbiology and Immunology of National Academy of Medical Sciences (protocol № 4-2022 of 14.12.2022). License for the software system Yasara # 179465823.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by National Academy of Medical Sciences of Ukraine, grant number 0122U000375.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Dudinskaya EN, Tkacheva ON, Bazaeva EV, et al. New possibilities of using moxonidin for blood pressure control in female patients with osteopenia. Kardiologiya. 2018;58:36–45. doi:10.18087/cardio.2508
2. Tkacheva O, Dudinskaya EN, Bazaeva EV. P3205 New features of use of moxonidine as telomerase activator. Eur Heart J. 2018;39(suppl_1):ehy563–P3205. doi:10.1093/eurheartj/ehy563.P3205
3. Sokoluk TV, Gorbenko NI, Podgayniy DG, Merzlikin SI. Experimental investigations of the influence of the pharmaceutical composition with the two antidiabetic agents on some manifestations of metabolic syndrome X. Farmatsevti Zhurnal. 2009;1:110–114.
4. Chen S, Gan D, Lin S, et al. Metformin in aging and aging-related diseases: clinical applications and relevant mechanisms. Theranostics. 2022;12(6):2722–2740. doi:10.7150/thno.71360
5. Cobos-Puc L, Aguayo-Morales H. cardiovascular effects mediated by imidazoline drugs: an update. Cardiov Haematol Disord Drug Targ. 2019;19(2):95–108. doi:10.2174/1871529X18666180629170336
6. Ernsberger P. The I1‐Imidazoline receptor and its cellular signaling pathways a. Ann NY Acad Sci. 1999;881(1):35–53. doi:10.1111/j.1749-6632.1999.tb09339.x
7. Holt A. Imidazoline binding sites on receptors and enzymes: emerging targets for novel antidepressant drugs? J Psychiatry Neurosci. 2003;28(6):409.
8. Takada K, Hayashi Y, Kamibayashi T, et al. The involvement of pertussis toxin‐sensitive G proteins in the post receptor mechanism of central I1‐imidazoline receptors. Br J Pharmacol. 1997;120(8):1575–1581. doi:10.1038/sj.bjp.0701090
9. Bousquet P, Hudson A, García-Sevilla JA, Li JX. Imidazoline receptor system: the past, the present, and the future. Pharmacol Rev 2020;721:50–79. doi:10.1124/pr.118.016311
10. Zhang W, Li X, Liu Y, Chen H, Gong J. Activation of imidazoline I1 receptor by moxonidine regulates the progression of liver fibrosis in the Nrf2-dependent pathway. Biomed Pharmacother. 2017;90:821–834. doi:10.1016/j.biopha.2017.04.025
11. Tesfai J, Crane L, Baziard-Mouysset G, Edwards LP. Novel I1-Imidazoline agonist S43126 augment insulin secretion in Min6 cells. J Diab Metabol. 2012;3(3). doi:10.4172/2155-6156.1000183
12. Weiss M, Bouchoucha S, Aiad F, et al. Imidazoline-like drugs improve insulin sensitivity through peripheral stimulation of adiponectin and AMPK pathways in a rat model of glucose intolerance. Am J Physiol Endocrinol Metab. 2015;309(2):E95–E104. doi:10.1152/ajpendo.00021.2015
13. Fellmann L, Nascimento AR, Tibiriça E, Bousquet P. Murine models for pharmacological studies of the metabolic syndrome. Pharmacol Ther. 2013;137(3):331–340. doi:10.1016/j.pharmthera.2012.11.004
14. Kawamoto S, Hirakata H, Sugita N, Fukuda K. Bidirectional effects of dexmedetomidine on human platelet functions in vitro. Eur J Pharmacol. 2015;766:122–128. doi:10.1016/j.ejphar.2015.09.049
15. Yang PS, Wu HT, Chung HH, et al. Rilmenidine improves hepatic steatosis through p38-dependent pathway to higher the expression of farnesoid X receptor. Naunyn-Schmiedeberg’s Arch Pharmacol. 2012;385:51–56. doi:10.1007/s00210-011-0691-1
16. Parini A, Moudanos CG, Pizzinat N, Lanier SM. The elusive family of imidazoline binding sites. Trends Pharmacol Sci. 1996;17(1):13–16. doi:10.1016/0165-6147(96)81564-1
17. Zomkowski AD, Hammes L, Lin J, Calixto JB, Santos ARS, Rodrigues ALS. Agmatine produces antidepressant-like effects in two models of depression in mice. Neuroreport. 2002;13(4):387–391. doi:10.1097/00001756-200203250-00005
18. Abás S, Erdozain AM, Keller B, et al. Neuroprotective effects of a structurally new family of high affinity imidazoline I2 receptor ligands. ACS Chem Neurosci. 2017;8(4):737–742. doi:10.1021/acschemneuro.6b00426
19. Tyacke RJ, Myers JF, Venkataraman A, et al. Evaluation of 11C-BU99008, a PET Ligand for the Imidazoline2 binding site in human brain. J Nucl Med. 2018;59(10):1597–1602. doi:10.2967/jnumed.118.208009
20. Parker CA, Nabulsi N, Holden D, et al. Evaluation of 11C-BU99008, a PET ligand for the imidazoline2 binding sites in rhesus brain. J Nucl Med. 2014;55(5):838–844. doi:10.2967/jnumed.113.131854
21. Boronat MA, Olmos G, García‐Sevilla JA. Attenuation of tolerance to opioid‐induced antinociception and protection against morphine‐induced decrease of neurofilament proteins by idazoxan and other I2‐imidazoline ligands. Br J Pharmacol. 1998;125(1):175–185. doi:10.1038/sj.bjp.0702031
22. Sánchez‐Blázquez P, Boronat MA, Olmos G, García‐Sevilla JA, Garzón J. Activation of I2‐imidazoline receptors enhances supraspinal morphine analgesia in mice: a model to detect agonist and antagonist activities at these receptors. Br J Pharmacol. 2000;130(1):146–152. doi:10.1038/sj.bjp.0703294
23. Gentili F, Cardinaletti C, Carrieri A, et al. Involvement of I2-imidazoline binding sites in positive and negative morphine analgesia modulatory effects. Eur J Pharmacol. 2006;553(1–3):73–81. doi:10.1016/j.ejphar.2006.09.031
24. Thorn DA, Zhang Y, Peng BW, Winter JC, Li JX. Effects of imidazoline I2 receptor ligands on morphine-and tramadol-induced antinociception in rats. Eur J Pharmacol. 2011;670(2–3):435–440. doi:10.1016/j.ejphar.2011.09.173
25. Sampson C, Zhang Y, Del Bello F, Li J-X. Effects of imidazoline I2 receptor ligands on acute nociception in rats. Neuroreport. 2012;23(2):73–77. doi:10.1097/WNR.0b013e32834e7db3
26. Li JX, Zhang Y. Imidazoline I2 receptors: target for new analgesics? Eur J Pharmacol. 2011;658(2–3):49–56. doi:10.1016/j.ejphar.2011.02.038
27. Hunskaar S, Hole K. The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain. 1987;30(1):103–114. doi:10.1016/0304-3959(87)90088-1
28. Thorn DA, Zhang Y, Li J. Effects of the imidazoline I2 receptor agonist 2‐BFI on the development of tolerance to and behavioural/physical dependence on morphine in rats. Br J Pharmacol. 2016;173(8):1363–1372. doi:10.1111/bph.13435
29. Ferrari F, Fiorentino S, Mennuni L, et al. Analgesic efficacy of CR4056, a novel imidazoline-2 receptor ligand, in rat models of inflammatory and neuropathic pain. J Pain Res;2011. 111–125. doi:10.2147/JPR.S18353
30. Siemian JN, Li J, Zhang Y, Li JX. Interactions between imidazoline I 2 receptor ligands and Acetaminophen in adult male rats: antinociception and schedule-controlled responding. Psychopharmacology. 2016;233:873–882. doi:10.1007/s00213-015-4166-9
31. Meregalli C, Ceresa C, Canta A, et al. CR4056, a new analgesic I2 ligand, is highly effective against bortezomib-induced painful neuropathy in rats. J Pain Res;2012. 151–167. doi:10.2147/JPR.S32122
32. Lanza M, Ferrari F, Menghetti I, Tremolada D, Caselli G. Modulation of imidazoline I 2 binding sites by CR 4056 relieves postoperative hyperalgesia in male and female rats. Br J Pharmacol. 2014;171(15):3693–3701. doi:10.1111/bph.12728
33. Eglen RM, Hudson AL, Kendall DA, et al. Seeing through a glass darkly’: casting light on imidazolineI’sites. Trends Pharmacol Sci. 1998;19(9):381–390. doi:10.1016/S0165-6147(98)01244-9
34. Morgan NG, Chan SL. Imidazoline binding sites in the endocrine pancreas: can they fulfil their potential as targets for the development of new insulin secretagogues? Curr Pharm Des. 2001;7(14):1413–1431. doi:10.2174/1381612013397366
35. Cerasi E, Effendic S, Luft R. Role of adrenergic receptors in glucose-induced insulin secretion in man. Lancet. 1969;294:7615):301–302. doi:10.1016/S0140-6736(69)90059-2
36. Chan SL, Brown CA, Scarpello KE, Morgan NG. The imidazoline site involved in control of insulin secretion: characteristics that distinguish it from I1‐and I2‐sites. Br J Pharmacol. 1994;112(4):1065–1070. doi:10.1111/j.1476-5381.1994.tb13191.x
37. Berdeu D, Gross R, Puech R, Loubatières-Mariani MM, Bertrand G. Evidence for two different imidazoline sites on pancreatic B cells and vascular bed in rat. Eur J Pharmacol. 1995;275(1):91–98. doi:10.1016/0014-2999(94)00757-X
38. Ishida‐Takahashi A, Horie M, Tsuura Y, Ishida H, Ai T, Sasayama S. Block of pancreatic ATP‐sensitive K+ channels and insulinotrophic action by the antiarrhythmic agent, cibenzoline. Br J Pharmacol. 1996;117(8):1749–1755. doi:10.1111/j.1476-5381.1996.tb15349.x
39. Proks P, Ashcroft FM. Phentolamine block of KATP channels is mediated by Kir6. 2. Proc Natl Acad Sci. 1997;94(21):11716–11720. doi:10.1073/pnas.94.21.11716
40. Monks LK, Cosgrove KE, Dunne MJ, Ramsden CA, Morgan NG, Chan SL. Affinity isolation of imidazoline binding proteins from rat brain using 5-amino-efaroxan as a ligand. FEBS Lett. 1999;447(1):61–64. doi:10.1016/S0014-5793(99)00264-1
41. Ferrer-Montiel AV, Merino JM, Planells-Cases R, Sun W, Montal M. Structural determinants of the blocker binding site in glutamate and NMDA receptor channels. Neuropharmacology. 1998;37(2):139–147. doi:10.1016/S0028-3908(98)00007-0
42. Roberg K, Johansson U, Öllinger K. Lysosomal release of cathepsin D precedes relocation of cytochrome c and loss of mitochondrial transmembrane potential during apoptosis induced by oxidative stress. Free Radic Biol Med. 1999;27(11–12):1228–1237. doi:10.1016/S0891-5849(99)00146-X
43. Ziegler D, Haxhiu MA, Kaan EC, Papp JG, Ernsberger P. Pharmacology of moxonidine, an I1-imidazoline receptor agonist. J Cardiov Pharmacol. 1996;27:S26–S37. doi:10.1097/00005344-199627003-00005
44. Yamakura T, Shimoji K. Subunit-and site-specific pharmacology of the NMDA receptor channel. Prog Neurobiol. 1999;59(3):279–298. doi:10.1016/S0301-0082(99)00007-6
45. Basile L, Pappalardo M, Guccione S, Milardi D, Ramsay RR. Computational comparison of imidazoline association with the I2 binding site in human monoamine oxidases. J Chem Inf Model. 2014;54(4):1200–1207. doi:10.1021/ci400346k
46. Nikolic K, Veljkovic N, Gemovic B, Srdic-Rajic T, Agbaba D. Imidazoline-1 receptor ligands as apoptotic agents: pharmacophore modeling and virtual docking study. Comb Chem High Through Screen. 2013;16(4):298–319. doi:10.2174/1386207311316040004
47. Tong L, Cui D, Zeng J. Topical bendazol inhibits experimental myopia progression and decreases the ocular accumulation of HIF‐1α protein in young rabbits. Ophthalmic Physiol Opt. 2020;40(5):567–576. doi:10.1111/opo.12717
48. Abramets II, Zayka TO, Evdokimov DV, Kuznetsov YV, Sidorova YV. Researches of the imidazole and indole derivatives cerebroprotective activity and its impact on effects of antidepressants. Pharmacokin Pharmacod. 2020;(1):18–24. doi:10.37489/2587-7836-2020-1-18-24
49. Brishty SR, MdJ H, Khandaker MU, Faruque MRI, Osman H, Rahman SMA. A comprehensive account on recent progress in pharmacological activities of benzimidazole derivatives. Front Pharmacol. 2021;12:762807. doi:10.3389/fphar.2021.762807
50. Atilgan E, Hu J. Improving protein docking using sustainable genetic algorithms. Int J Compu Inoform Sys Ind Manag App. 2011;3:248–255.
51. Štekláč M, Zajaček D, Bučinský L. 3CLpro and PLpro affinity, a docking study to fight COVID19 based on 900 compounds from PubChem and literature. Are there new drugs to be found? J Mol Struct. 2021;1245:130968. doi:10.1016/j.molstruc.2021.130968
52. Land H, Humble MS. YASARA: a tool to obtain structural guidance in biocatalytic investigations. Prot Engine. 2018;43–67. doi:10.1007/978-1-4939-7366-8_4
53. Belyaeva J, Zlobin A, Maslova V, Golovin A. Modern non-polarizable force fields diverge in modeling the enzyme–substrate complex of a canonical serine protease. Phys Chem Chem Phys. 2023;25(8):6352–6361. doi:10.1039/D2CP05502C
54. Hsu KC, Chen YF, Lin SR, Yang JM. iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinf. 2011;12(S1):S33. doi:10.1186/1471-2105-12-S1-S33
55. Azad I, Khan T, Maurya AK, Irfan Azad M, Mishra N, Alanazi AM. Identification of severe acute respiratory syndrome coronavirus‐2 inhibitors through in silico structure‐based virtual screening and molecular interaction studies. J Mol Recog. 2021;34(10):e2918. doi:10.1002/jmr.2918
56. Velavan S, Karnan R, Kanivalan N. A comparative study on In silico software’s in statistical relation to molecular docking scores. Asian J Innov Res. 2020;5(2):1–5.
57. Ahmad R. Steroidal glycoalkaloids from Solanum nigrum target cytoskeletal proteins: an in silico analysis. PeerJ. 2019;7:e6012. doi:10.7717/peerj.6012
58. Yuan S, Chan HS, Hu Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip Rev Comput Mol Sci. 2017;7(2):e1298. doi:10.1002/wcms.1298
59. Yuan S, Chan HS, Filipek S, Vogel H. PyMOL and Inkscape bridge the data and the data visualization. Structure. 2016;24(12):2041–2042. doi:10.1016/j.str.2016.11.012
60. Sandy S. In silico molecular docking of the antimalarial flavonoid compound macaranga (macaranga tanarius) against the PfDHFR Enzyme. Curr Trends Biotechnol Pharm. 2023;17(3):929–936.
61. Mahboobi M, Salmanian AH, Sedighian H, Bambai B. Molecular Modeling and Optimization of Type II E. coli l-Asparginase Activity by in silico Design and in vitro Site-directed Mutagenesis. Prot J. 2023;2023:1–11.
62. Kuvarina AE, Sukonnikov MA, Rogozhin EA, et al. Formation of various antimicrobial peptide emericellipsin isoforms in emericellopsos alkalina under different cultivation conditions. Appl Biochem Microbiol. 2023;59(2):160–167. doi:10.1134/S0003683823020060
63. Nishanth S, Chikunov AS, Thankappan S, Taran OP, Parmon VN, Uthandi S. Lignin derived aromatic monomers from birch wood by laccase (LccH) pretreatment and Ru/C catalyst: a two-pot approach for sustainable biorefineries. Biomass Convers Biorefin. 2022;2022:1–16.
64. Abdel-karim AM, Shahen S, Elsisi DM, Hyba AM, El-Shamy OA. Experimental and theoretical studies of corrosion resistance enhancement of carbon steel in 1 M HCl by quinoxalinosulfonamide hybrid-bearing theophylline moiety. J Bio- Tribo-Corros. 2022;8(3):70. doi:10.1007/s40735-022-00666-0
65. Ezzat A, Mohamed MBI, Mahmoud AM, Farag RS, El-Tabl AS, Ragab A. Synthesis, spectral characterization, antimicrobial evaluation and molecular docking studies of new Cu (II), Zn (II) thiosemicarbazone based on sulfonyl isatin. J Mol Struct. 2022;1251:132004. doi:10.1016/j.molstruc.2021.132004
66. Nasser M, Alyamani AA, Daou A, et al. Influence of the extraction solvent and of the altitude on the anticancer activity of Lebanese eucalyptus camaldulensis extract alone or in combination with low dose of cisplatin in A549 human lung adenocarcinoma cells. Processes. 2022;10(8):1461. doi:10.3390/pr10081461
67. Sarter M. Cooperative change in the internal dynamics of streptavidin caused by biotin binding. J Phys Chem A. 2023;127(14):3241–3247. doi:10.1021/acs.jpcb.3c00427
68. Yamada S, Kataoka M, Yoshida K, et al. Development of hydrogen-bonded dimer-type photoluminescent liquid crystals of fluorinated tolanecarboxylic acid. Crystals. 2022;13(1):25. doi:10.3390/cryst13010025
69. Vasil’ev SG, Panicheva KV. The self-di usion of 128-arm star-shaped polydimethylsiloxanes with a dendritic branching center. In: Magnetic Resonance and Its Applications. Spinus-2022; 2022:176–177.
70. Nekkalapudi AR, Veldi VG, Pippalla S. A novel RP-HPLC method for estimating fulvestrant, benzoyl alcohol, and benzyl benzoate in injection formulation. Am J Anal Chem. 2022;13(7):229–240. doi:10.4236/ajac.2022.137016
71. Slassi S, Aarjane M, Amine A. Synthesis, spectroscopic characterization (FT-IR, NMR, UV-Vis), DFT study, antibacterial and antioxidant in vitro investigations of 4, 6-bis ((E)-1-((3-(1H-imidazol-1-yl) propyl) imino) ethyl) benzene-1, 3-diol. J Mol Struct. 2022;1255:132457. doi:10.1016/j.molstruc.2022.132457
72. Ramirez H, Dominguez J, Fernandez-Moreira E, Rodrigues J, Rodriguez M, Charris JE. Synthesis of 4-Benzylsulfanyl and 4-benzylsulfonyl chalcones. biological evaluation as antimalarial agents. Vaccine. 2022;3:4.
73. Jena GB, Chavan S. Implementation of good laboratory practices (GLP) in basic scientific research: translating the concept beyond regulatory compliance. Regul Toxicol Pharmacol. 2017;89:20–25. doi:10.1016/j.yrtph.2017.07.010
74. Olsson IAS, da SSP, Townend D, Sandøe P. Protecting animals and enabling research in the European Union: an overview of development and implementation of directive 2010/63/EU. ILAR Jl. 2017;57(3):347–357. doi:10.1093/ilar/ilw029
75. Mustafakulov M, Kimsanova N, Hamdamova N, et al. Determination of antioxidant properties of l-cysteine in the liver of alloxan diabetes model rats. Internat J Contemp Sci Tech Res. 2023;2023:47–54.
76. Akoko S, Aleme BM, Uahomo PO. The effect of addition of extracts of Vernonia amygdalina and Moringa oleifera in the nutrition of alloxan-induced diabetic Wistar Rats. Inter J Phar Res Health Sci. 2022;10(4):3455–3462.
77. Etuk EU. Animals models for studying diabetes mellitus. Agric Biol J North Am. 2010;1(2):130–134.
78. Katz LB, Stewart L, Guthrie B, Cameron H. Patient satisfaction with a new, high accuracy blood glucose meter that provides personalized guidance, insight, and encouragement. J Diabet Sci Technol. 2020;14(2):318–323. doi:10.1177/1932296819867396
79. Maraš N, Kočevar M. Boric acid‐catalyzed direct condensation of carboxylic acids with benzene‐1, 2‐diamine into benzimidazoles. Helvetica Chim Acta. 2011;94(10):1860–1874. doi:10.1002/hlca.201100064
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.