")
Back to Journals » Journal of Inflammation Research » Volume 14
Microglial Activation Mediates Noradrenergic Locus Coeruleus Neurodegeneration via Complement Receptor 3 in a Rotenone-Induced Parkinson’s Disease Mouse Model
Authors Jing L, Hou L, Zhang D, Li S, Ruan Z, Zhang X, Hong JS, Wang Q
Received 4 January 2021
Accepted for publication 24 March 2021
Published 9 April 2021 Volume 2021:14 Pages 1341—1356
DOI https://doi.org/10.2147/JIR.S299927
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Lu Jing,1,2,* Liyan Hou,1,* Dongdong Zhang,1 Sheng Li,3 Zhengzheng Ruan,1 Xiaomeng Zhang,3 Jau-Shyong Hong,4 Qingshan Wang1,3
1Institute of Toxicology, School of Public Health, Dalian Medical University, Dalian, 116044, People’s Republic of China; 2Department of Neurology, The First Affiliated Hospital of Dalian Medical University, Dalian, 116011, People’s Republic of China; 3National-Local Joint Engineering Research Center for Drug-Research and Development (R & D) of Neurodegenerative Diseases, Dalian Medical University, Dalian, 116044, People’s Republic of China; 4Neurobiology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC, USA
*These authors contributed equally to this work
Correspondence: Qingshan Wang
Institute of Toxicology, School of Public Health, Dalian Medical University, No. 9 W. Lvshun South Road, Dalian, 116044, People’s Republic of China
Tel +86 138 4081 7365
Email [email protected]
Background: Chronic exposure to the insecticide rotenone can damage dopaminergic neurons and lead to an increased risk of Parkinson’s disease (PD). Whereas it is not clear whether rotenone induces neurodegeneration of noradrenergic locus coeruleus (LC/NE) neurons. Chronic neuroinflammation mediated by microglia has been involved in the pathogenesis of PD. Evidence shows that complement receptor 3 (CR3) is a crucial regulator of microglial activation and related neurodegeneration. However, it is not clear whether CR3 mediates rotenone-elicited degeneration of LC/NE neurons through microglia-mediated neuroinflammation.
Materials and Methods: Wild type (WT) and CR3 knockout (KO) mice were treated with rotenone. PLX3397 and minocycline were used to deplete or inactivate the microglia. Leukadherin-1 (LA-1) was used to modulate CR3. LC/NE neurodegeneration, microglial phenotype, and expression of CR3 were determined by using immunohistochemistry, Western blot and real-time polymerase chain reaction (PCR) techniques. The glutathione (GSH) and malondialdehyde (MDA) contents were measured by using commercial kits.
Results: Rotenone exposure led to dose- and time-dependent LC/NE neuronal loss and microglial activation in mice. Depletion of microglia by PLX3397 or inhibition of microglial activation by minocycline significantly reduced rotenone-induced LC/NE neurodegeneration. Mechanistic studies revealed that CR3 played an essential role in the rotenone-induced activation of microglia and neurodegeneration of LC/NE neurons. Rotenone elevated the expression of CR3, and genetic ablation of CR3 markedly reduced rotenone-induced microglial activation and M1 polarization. LA-1 also suppressed rotenone-induced toxic microglial M1 activation. Furthermore, lack of CR3 or treatment with LA-1 reduced oxidative stress in the brainstem of rotenone-intoxicated mice. Finally, we found that mice deficient in CR3 or treated with LA-1 were more resistant to rotenone-induced LC/NE neurodegeneration than WT or vehicle-treated mice, respectively.
Conclusion: Our results indicate that CR3-mediated microglial activation participates in rotenone-induced LC/NE neurodegeneration, providing novel insight into environmental toxin-induced neurotoxicity and related Parkinsonism.
Keywords: pesticide, noradrenergic neuron, Parkinson’s disease, neuroinflammation, CR3, locus coeruleus
Introduction
Rotenone is a widely used insecticide in home gardening and agriculture.1 Because of its high lipophilicity and ability to easily cross the blood-brain barrier, the neurotoxic damages of rotenone exposure to the central nervous system (CNS) have gained attention.1 Chronic exposure to rotenone has been shown to increase the risk of Parkinson’s disease (PD) in a variety of epidemiological studies.2 In experimental animals, rotenone causes dopaminergic neurodegeneration in the substantia nigra pars compacta (SNpc), a characteristic pathological change observed in PD.3–5 Clinical studies revealed that in addition to nigral dopaminergic neurodegeneration, the pathology of PD follows a caudo-rostral pattern in which loss of neurons in the locus coeruleus (LC) occurs earlier than that of neurons in the SNpc.6,7 Strong evidence suggests that LC noradrenergic (LC/NE) neurodegeneration is critical to producing most nonmotor symptoms and exacerbating the dopaminergic neurodegeneration in PD rodent models.8,9 Whereas, whether rotenone exposure could injure LC/NE neurons needs further study.
Chronic neuroinflammation mediated by microglia has been involved in the pathogenesis of PD.10,11 Activated microglia have been detected near the degenerating neurons in the SN and other brain regions such as the LC of PD patients.12 Consistent with these findings, microglial activation was present in the LC in experimental PD models generated by LPS or combined paraquat and maneb.13,14 Previous studies demonstrated that depletion of microglia in cocultures of mesencephalic neurons and glia significantly mitigated dopaminergic neurodegeneration induced by various toxins associated with PD, such as 2,5-hexanedione, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and paraquat.15–17 In addition, inhibition of microglial activation showed dopaminergic neuroprotection in vivo. Chen and colleagues found that transforming growth factor (TGF)-β inhibited microglial activation and thus protected dopaminergic neurons in a PD rat model generated by injecting 1-methyl-4-phenylpyridinium (MPP+) unilaterally into the striatum.18 Similarly, inhibition of the proinflammatory microglial phenotype by taurine also attenuated dopaminergic neuronal loss in the PD mouse model generated by paraquat and maneb,19 indicating that chronic microglial activation participates in dopaminergic neurodegeneration. There were activated microglia in the SN of rotenone-intoxicated experimental rodents as well.20,21 However, whether rotenone exposure induces microglial activation in the LC and subsequent NE neuronal degeneration has remained unclear.
Complement receptor 3 (CR3) belongs to the β2 integrin family and is expressed at high levels in microglia.22 Strong evidence shows that CR3 is a crucial regulator of microglial activation and related neurodegeneration. Das et al found that intranasal administration of CR3-targeting small interfering RNA (siRNA) significantly reduced proinflammatory microglial activation and the production of cytotoxic factors, and these effects were accompanied by mitigation of hypoxia-induced working memory deficits in mice.23 Knockout of CR3 also suppressed hypoxia, and LPS cotreatment induced microglial activation and inhibited long-term synaptic depression in hippocampal brain slice cultures.24 Elevated expression of CR3 was detected in postmortem PD patients and multiple experimental animal models.25,26 Genetic ablation or pharmacological inhibition of CR3 suppresses microglial activation elicited by α-synuclein, the major component of Lewy bodies in PD.27 We recently reported that blocking CR3 with an inhibitory antibody also mitigated microglia-mediated neuroinflammation and dopaminergic neurodegeneration in primary midbrain cultures treated with combined paraquat and maneb.28 However, it is not clear whether CR3 mediates rotenone-elicited degeneration of LC/NE neurons through microglia-mediated neuroinflammation.
The purpose of this research was to investigate whether microglial activation is associated with LC/NE neurodegeneration in rotenone-intoxicated mice by depleting or inactivating microglia with PLX3397, an antagonist of colony-stimulating factor receptor 1 (CSF1R)29 or the antibiotic minocycline, respectively. Furthermore, the effect of CR3 on rotenone-induced microglial activation and LC/NE neuronal degeneration was further explored by using CR3−/- mice and the CR3 modulator leukadherin-1 (LA-1). These results indicated that CR3 regulates microglial activation and M1 polarization to mediate degeneration of LC/NE neurons in rotenone-intoxicated mice.
Materials and Methods
Animal Dosing
Two-month-old adult male C57BL/6 mice were randomly divided into three groups, ie, a control group and two rotenone groups with different doses (n = 14 in each group). Rotenone was freshly prepared every day in a solution of 0.1% DMSO (diluted with PBS) and injected (i.p.) into mice at a dose of 0.75 or 1.5 mg/kg/day for 3 consecutive weeks as described previously.30–32 The mortality rate of mice treated with 0.75 or 1.5 mg/kg/day rotenone was 0% and 14.3%, respectively. Control mice received an equivalent volume of 0.1% DMSO (diluted with PBS). CR3−/- mice were also randomly divided into control and rotenone groups (n = 10 in each group). CR3−/- mice in the control and rotenone groups were treated (i.p.) with vehicle and 1.5 mg/kg/day rotenone, respectively, for 3 consecutive weeks. CR3−/- mice treated with rotenone did not show any mortality during experiments. After 1, 2, and 3 weeks of initial rotenone injection, mice (n = 4 in each time point) were sacrificed. Mice were anesthetized by intraperitoneal injection of 10% chloral hydrate. All animal procedures and their care were implemented in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Dalian Medical University.
PLX3397, Minocycline and LA-1 Treatment
PLX3397, a selective antagonist of CSF1R, significantly reduces microglial number in the brain.29 Mice were randomly divided into three groups, ie, control, rotenone and rotenone plus PLX3397 group (n = 12 in each group). PLX3397 were dissolved in 0.1% DMSO (diluted with PBS). To deplete brain microglia, mice received PLX3397 (40 mg/kg/day, S7818, Selleck, China) by gavage. After 1 week of PLX3397 administration, the mice were treated with PLX3397 followed by rotenone (i.p.) 30 min later until the end of the experiment. Other than a slight increase in body weight, no deleterious side effects were detected in the PLX3397-treated mice, which is in agreement with that of a previous study.33
Mice were randomly divided into three groups, ie, control, rotenone and rotenone plus minocycline group (n = 11 in each group). Minocycline was dissolved in PBS. Mice received (i.p.) minocycline (Sigma-Aldrich, St. Louis, MO, USA) before 2 days of rotenone treatment. Minocycline was injected daily at a dose of 50 mg/kg/day for 3 continuous weeks. The dose of minocycline was selected according to a previous report.34
Mice were randomly divided into four groups, ie, control, rotenone, rotenone plus LA-1 and LA-1 group (n = 12 in each group). LA-1 were dissolved in 0.1% DMSO (diluted with PBS). Mice received (i.p.) 500 μL of 50 μM LA-1 (S8306, Selleck, China) or vehicle 1 h after rotenone injection for 3 continuous weeks. The dose of LA-1 was selected according to a previous study.35
PLX3397, minocycline and LA-1 treatment reduced 1.5 mg/kg/day rotenone-induced mortality of mice and a similar mortality rate (7.1%) of mice treated with combined PLX3397, minocycline, LA-1 and rotenone (1.5 mg/kg/day) was observed.
Immunohistochemistry
Immunohistochemistry was performed based on a previous protocol.36,37 Briefly, mice were perfused transcardially with PBS and then with 4% paraformaldehyde (PFA). The brains were then postfixed with 4% PFA at 4 °C for 48 h. The brains were dehydrated with 30% sucrose in PBS for an additional 2 days. Free-floating sections (30 μm) were collected and incubated with 0.25% Triton/PBS containing 4% goat serum for 2 h. After that, a primary antibody against tyrosine hydroxylase (TH, 1:1000, EMD Millipore, Temecula, CA, USA) or ionized calcium-binding adaptor molecule-1 (Iba-1, 1:1000, Wako Chemicals, Richmond, VA, USA) was used to stain the sections at 4 °C overnight. On the next day, the sections were washed three times with PBS and then incubated with biotin-conjugated goat anti-rabbit IgG antibody at room temperature for 2 h. Antibody binding was visualized by using a Vectastain ABC Kit (Vector Laboratories, Inc, CA, USA) and 3,3ʹ-diaminobenzidine (DAB, Sigma, MO, USA) as substrate.
The number of TH immunoreactive (THir) neurons in the LC region was visually counted in both hemispheres of mice under a microscope (200×) as described previously.38 The boundary of LC was outlined under magnification of the 4× objective as per the atlas.39 Every three sections from the rostral of a series of 36 sections that cover the entire extent of LC were selected for immunostaining.13 The total number of THir neurons in the LC was counted manually by two individuals blinded to the treatments. Previous reports demonstrated that it’s rather consistency for quantification of THir neurons in mice between manual and stereology counts.14,40
ImageJ software was used to carry out the quantitative analysis of Iba-1 immunostaining as described previously.9 Briefly, the image was first converted into the grayscale picture, and the background was adjusted before the quantifying area was selected for the measurement of the total pixels. The relative density of the staining was compared based on the density of the total pixels of a certain brain region (total pixels/area).
Real-Time Polymerase Chain Reaction Analysis
For real-time polymerase chain reaction (PCR), mice were perfused transcardially with PBS to remove blood. The brains were collected and different brain regions were dissected on ice. All the samples were stored at −80 °C. Total RNA was extracted from the brainstem samples with TRIzol reagent. Then, the RNA was reverse transcribed with MuLV reverse transcriptase and oligo (dT) primers as described previously.36 Real-time PCR amplification was carried out with SYBR Premix Ex TaqTM II (Takara Bio Inc., Kusatsu, Shiga, Japan) and a Takara Thermal Cycler Dice™ Real Time System based on the manufacturer’s protocols. The sequences of primers used are listed in Table 1. Amplification was performed at 95 °C for 10 seconds, 55 °C for 30 seconds, and 72 °C for 30 seconds for 40 cycles. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as the internal control. All samples were normalized to GAPDH using the 2−ΔΔCt method.41 Fold changes for each treatment were normalized and are shown as percentages of the control.
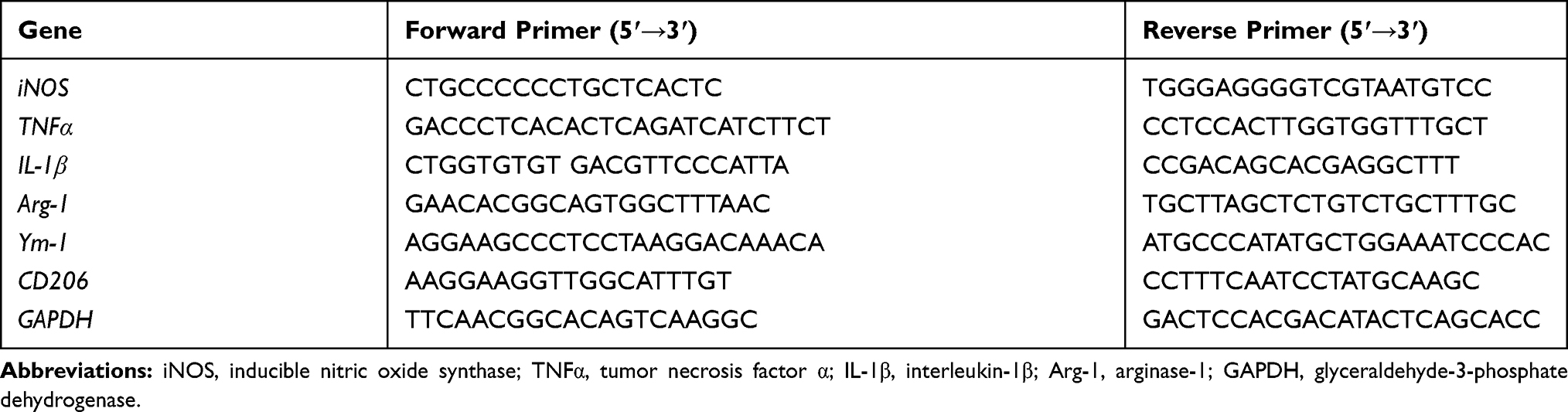 |
Table 1 Real-Time PCR Primer Sequences |
Glutathione and Malondialdehyde Measurement
Mice were perfused transcardially with PBS to remove blood. The brainstem samples were collected and were homogenized in ice-cold lysis buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, and protease inhibitor mixture. After 10 min of centrifugation at 10,000×g (4 °C), the contents of GSH and MDA in the collected supernatant were measured with commercial kits (GSH, S0052; MDA, S0131, Beyotime, Shanghai, China) according to the protocol provided by the manufacturer and previous studies.42–45 The rationale for the measurement of GSH based on that GSH reacts with chromogenic substrate DTNB to produce yellow products and the amount of GSH can be detected by measuring absorption at A412. While, MDA can react with thiobarbituric acid (TBA) in high temperature and acidic environment to form red MDA-TBA adduct. The MDA-TBA adduct has the maximum absorption at 535nm, which can be detected by colorimetric method.
Western Blot Analysis
For Western blot, mice were perfused transcardially with PBS to remove blood. The brains were collected and different brain regions were dissected on ice. All the samples were stored at −80 °C. The brainstem samples were homogenized in cold RIPA lysis buffer containing proteinase and phosphatase inhibitors.28,36 Equivalent amounts of protein from each group were added to a 4–12% Bis-Tris-polyacrylamide electrophoresis gel. Then, the proteins were transferred to polyvinylidene fluoride (PVDF) membranes. We blocked the membranes with 5% skim milk and then incubated them with primary antibodies against TH (1:1000, ab129991, Abcam, Cambridge, MA, USA), Iba-1 (1:1000, 019–19,741, Abcam, Cambridge, MA, USA), CR3 (1:1000, ab128797, Abcam, Cambridge, MA, USA) or GAPDH (1:5000, ab181602, Abcam, Cambridge, MA, USA) at 4 °C overnight. The membranes were then washed three times with PBST and incubated with horseradish peroxidase-linked anti-rabbit IgG (1:3000) antibody at room temperature for 2 h. The blot signals were detected by ECL reagents (Biological Industries, Cromwell, CT, USA). The densitometry of blots was quantified based on previous report.46 GAPDH was used as an internal control for each blotting. All blots were normalized to GAPDH. Fold changes for each treatment were normalized and are shown as percentages of the control.
Statistical Analysis
All data are shown as the mean ± SEM. The normally distributed and equal variance data were analyzed by Shapiro–Wilk and Bartlett’s tests, respectively. For normally distributed data, one-way (one parameter) or two-way (two or more independent parameters) ANOVA was used for comparisons among groups. Otherwise, the data were analyzed by a nonparametric test. Pairwise comparisons were done by Tukey’s post hoc test for variance homogeneity or Tamhane’s T2 test for variance. The value of p less than 0.05 was considered statistically significant. The software SPSS was used for statistical analysis.
Results
Rotenone Dose-Dependently Damages LC/NE Neurons in Mice
To investigate whether rotenone injures LC/NE neurons, mice were administered rotenone at a dose of 0.75 or 1.5 mg/kg/day for 3 consecutive weeks, and LC/NE neurons were immunostained with an antibody against TH. Our previous study showed high consistency for NE neuron quantitation by using TH+ and NeuN+ cell counts in mice.14 To employ an easier and more precise method, THir neurons counts were performed in this study. As shown in Figure 1A and B, compared with vehicle, rotenone dose-dependently damaged LC/NE neurons in mice, as shown by the gradual decrease in the number of TH+ cell number in the 0.75 and 1.5 mg/kg/day rotenone-treated mice. Dopaminergic neurodegeneration in the SNpc is an important pathological hallmark of PD, and nigral dopaminergic neurons were also quantified in the rotenone-intoxicated mice. Consistent with previous studies,30,31 a decreased number of dopaminergic neurons in the SNpc was noted in the rotenone-injected mice (Supplementary Figure 1A and B). Gait abnormality is one of the primary symptoms in PD.19 As seen in Supplementary Figure 2A–C, compared with vehicle, the mice exposed to rotenone performed shorter stride length and wider stride distance of limbs.
 |
Figure 1 Rotenone dose-dependently induces LC/NE neurodegeneration and microglial activation in mice. (A) Mice were injected with the indicated dose of rotenone for 3 weeks. Immunohistochemistry with an anti-TH antibody was performed to stain NE neurons in the LC of mice, and representative images are presented. (B) Quantification of the number of THir neurons. (C) Immunohistochemistry with an anti-Iba-1 antibody was performed to stain microglial cells in the LC of mice, and representative images are presented. (D) Quantification of the density of Iba-1 immunostaining. Results were mean ± SEM from six mice for each group and were analyzed by one-way ANOVA (THir neurons counts: F(2,15) = 50.221, P = 0.000; Iba-1 density: F(2,15) = 8.158, P = 0.004; post hoc analysis by Tukey’s multiple comparisons test). **P<0.01; Scale bar = 100 μm. |
Rotenone Time-Dependently Induces Microglial Activation and LC/NE Neurodegeneration in Mice
Iba-1 is one of the markers for microglia and can be upregulated upon microglial activation. To determine whether microglia participate in rotenone-induced LC/NE neurodegeneration, microglia were immunostained by using an antibody against Iba-1. As illustrated in Figure 1C, microglia in both 0.75 and 1.5 mg/kg/day rotenone-intoxicated mice showed morphological hypertrophy and enhanced Iba-1 staining, indicating microglial activation. We quantitatively analyzed the density of Iba-1 staining. The quantitative analysis results supported the morphological observations (Figure 1D). Consistent with our results, similar microglial activation with hypertrophy morphology and enhanced Iba-1 staining was observed in rotenone-induced rodent models of PD in previous studies.47–49
The temporal patterns of microglial activation and loss of LC/NE neurons were subsequently determined. After 1 week of rotenone injection, microglial activation in the LC was observed and remained until the end of the experiment (Figure 2A). Quantitative analysis showed that the density of Iba-1 immunostaining was increased after 1 week of rotenone treatment and continued to increase up until the study end-point (Figure 2B). In contrast, the number of LC/NE neurons decreased gradually during rotenone intoxication (Figure 2A and C). However, we did not detect significant difference in LC/NE neuron number between the control and rotenone group mice until 2 weeks after the initiation of rotenone treatment (Figure 2A and C). Compared with that of 2-week time point, a high degree of THir neuron loss was observed after 3 week of rotenone treatment; however, the difference was not statistical significant (Figure 2A and C).
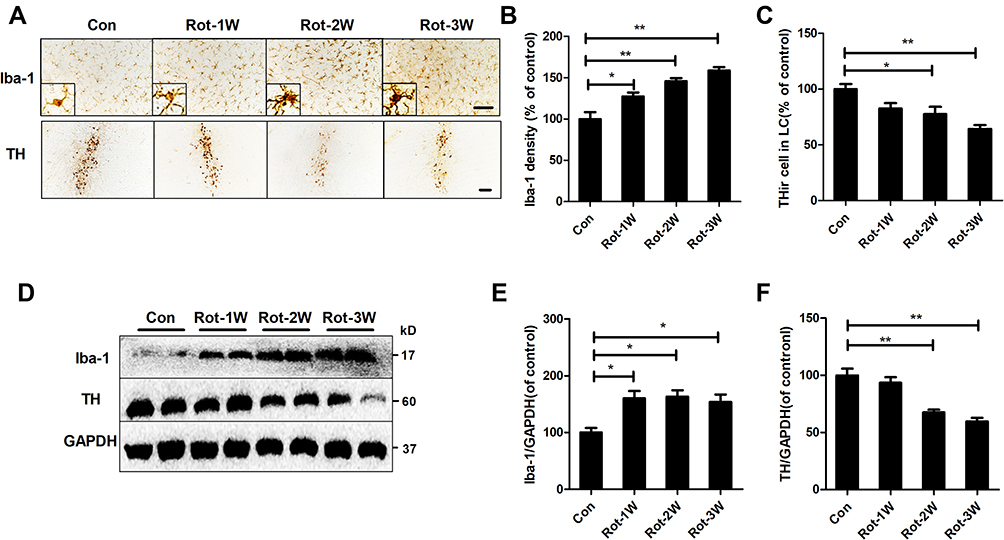 |
Figure 2 Rotenone time-dependently induces microglial activation and LC/NE neurodegeneration in mice. (A) Microglial cells and LC/NE neurons from mice at the indicated time points after rotenone treatment were immunostained with anti-Iba-1 and anti-TH antibodies, respectively, and representative images are shown. (B) Quantification of the density of Iba-1 immunostaining. (C) Quantification of the number of THir neurons. Results were mean ± SEM from four mice for each group and were analyzed by one-way ANOVA (Iba-1 density: F(3,12) = 21.464, P = 0.000; THir neurons counts: F(3,12) = 8.849, P = 0.002; post hoc analysis by Tukey’s multiple comparisons test). (D) The expression levels of Iba-1 and TH in the brainstem of mice were determined by Western blotting with specific antibodies, and representative blots are shown. (E and F) Quantification of the band densities of Iba-1 (E) and TH (F). Results were mean ± SEM from four mice for each group and were analyzed by one-way ANOVA (Iba-1: F(3,12) = 6.744, P = 0.006; TH: F(3,12) = 20.506, P = 0.000; post hoc analysis by Tukey’s multiple comparisons test). *P<0.05, **P<0.01; Scale bar = 100 μm. |
To confirm the activation of microglia and loss of LC/NE neurons in rotenone-injected mice, we determined the expression levels of Iba-1 and TH, markers of microglia and LC/NE neurons, respectively, at different time points after rotenone injection. Consistent with the previous results, Iba-1 expression in the brainstem of rotenone-intoxicated mice increased gradually beginning at 1 week after rotenone injection and remained distinctly higher than that of vehicle-treated control mice (Figure 2D and E). Compared with vehicle, rotenone decreased TH expression in the brainstem of mice at 2 weeks after the initial injection (Figure 2D and F).
PLX3397 and Minocycline Attenuate Rotenone-Induced LC/NE Neuronal Loss
To clarify whether the activation of microglia mediates the LC/NE neuronal loss in rotenone-intoxicated mice, we employed two methods to block microglia: pharmacological depletion of microglia by PLX3397 and inactivation of microglia by minocycline, a semisynthetic tetracycline antibiotic. Consistent with a previous report,29 PLX3397 treatment reduced Iba-1+ microglia by approximately 90% in the LC of mice (Figure 3A). The depletion of microglia by PLX3397 in the group cotreated with rotenone and PLX3397 was less severe (70% of control) (Figure 3A). Consistent with previous reports,50,51 microglia in mice cotreated with minocycline and rotenone displayed ramified morphology and decreased Iba-1 immunostaining density compared with those of the mice treated with rotenone alone (Figure 3B), indicating reduced microglial activation. The LC/NE neuronal loss was also detected in rotenone-intoxicated mice with or without PLX3397 or minocycline. In accordance with the results shown in Figures 1 and 2, the LC/NE neurons were lessened in number in the rotenone-intoxicated mice, and this reduction was significantly mitigated by PLX3397 and minocycline (Figure 3C and D).
 |
Figure 3 Microglial activation mediates rotenone-induced LC/NE neuronal loss in mice. (A) After 3 weeks of PLX3397 treatment, microglial cells in the LC of mice were immunostained with an anti-Iba-1 antibody, and the number of Iba-1+ microglia was quantified. (B) After 3 weeks of rotenone treatment with or without minocycline, microglial cells in the LC of mice were immunostained with an anti-Iba-1 antibody, and the density of Iba-1 in the brainstem was quantified. Results were mean ± SEM from three mice for each group and were analyzed by one-way ANOVA (Iba-1+ counts: F(3,8) = 251.733, P = 0.006; Iba-1 density: F(2,6) = 10.57, P = 0.011; post hoc analysis by Tukey’s multiple comparisons test). (C) Immunohistochemistry with an anti-TH antibody was performed to stain LC/NE neurons of rotenone-intoxicated mice with or without PLX3397 and minocycline, and representative images are shown. (D) Quantification of the number of THir neurons. Results were mean ± SEM from six mice for each group and were analyzed by one-way ANOVA (F(3,20) = 62.255, P = 0.000; post hoc analysis by Tukey’s multiple comparisons test). *P<0.05, **P<0.01; Scale bar = 100 μm. |
CR3 Contributes to Rotenone-Induced Microglial Activation
Subsequent experiments aimed to explore the potential mechanisms of rotenone-induced microglial activation. Integrin CR3 is expressed at high levels in microglia and is involved in the pathogenesis of multiple degenerative neurological disorders.11 To determine whether CR3 is essential for the rotenone-induced microglial activation, we detected the expression level of CR3 in rotenone-injected mice. Compared with vehicle-treated controls, Western blot analysis revealed increased CR3 expression in the brainstem of rotenone-treated mice (Figure 4A and B). Rotenone exposure also increased the mRNA levels of CR3 in mice (Figure 4C).
 |
Figure 4 CR3 contributes to microglial activation in rotenone-intoxicated mice. (A) The expression of CR3 in the brainstem was determined by Western blotting, and representative blots are shown. (B) Quantification of the band densities of the CR3 blots. Results were mean ± SEM from four mice for each group and were analyzed by t test (t = −9.098, V = 6, P = 0.000). (C) Real-time PCR was performed to detect the gene expression level of CR3 in the brainstem of mice. Results were mean ± SEM from six mice for each group and were analyzed by Wilcoxon (W = 22, Z = −2.722, P = 0.006). (D) Immunohistochemistry with an anti-Iba-1 antibody was performed to stain microglial cells in the LC of rotenone-intoxicated WT and CR3−/- mice, and representative images are shown. (E) Quantification of the density of Iba-1 immunostaining. Results were mean ± SEM from six mice for each group and were analyzed by two-way ANOVA (F(3,20) = 11.544, P = 0.000, post hoc analysis by Tukey’s multiple comparisons test). **P<0.01; Scale bar = 100 μm. |
The role of CR3 in rotenone-elicited activation of microglia was further studied by using CR3-deficient mice. Three weeks after the initial rotenone injection, microglia in the LC of wild type (WT) mice remained activated (Figure 4D and E). In contrast, microglia in rotenone-intoxicated CR3−/- mice showed ramified morphologies and reduced Iba-1 immunostaining, suggesting that CR3 is essential for rotenone-induced microglial activation (Figure 4D and E).
Activated microglia have two polarization states, namely neurotoxic (M1) and neuroprotective (M2) phenotypes. The effects of CR3 deficiency on rotenone-elicited microglial polarization were investigated. As shown in Figure 5A, rotenone treatment elevated the mRNA levels of M1 markers, including iNOS, TNFα and IL-1β, in WT mice. The upregulation of M1 marker genes was greatly mitigated in the CR3−/- mice (Figure 5A). Compared with vehicle, rotenone exposure decreased the mRNA levels of a number of M2 genes Arg-1 (Figure 5B). Although a decreased trend of CD206 and YM-1 genes in rotenone-treated WT mice was also observed, the difference was not statistical significant. Consistent with the previous results, the rotenone-induced decrease in M2 genes was attenuated in the CR3−/- mice (Figure 5B).
 |
Figure 5 Genetic deletion of CR3 reverses the rotenone-induced imbalance of microglial M1/M2 polarization in mice. (A) Real-time PCR was performed to detect the mRNA levels of iNOS, TNFα and IL-1β in the brainstem of WT and CR3−/- mice. Results were mean ± SEM from six mice for each group and were analyzed by two-way ANOVA (iNOS: F(3,20) = 20.362, P = 0.000; TNFα: F(3,20) = 25.809, P=0.000; IL-1β: F(3,20) = 20.25, P = 0.000; post hoc analysis by Tamhane’s T2 multiple comparisons test). (B) The mRNA levels of Arg-1, CD206 and YM-1 in the brainstem of WT and CR3−/- mice were determined by real-time PCR. Results were mean ± SEM from six mice for each group. Data of Arg-1 were analyzed by Kruskal Wallis H-test (H(3) = 12.767, P = 0.005) and results of CD206 and YM-1 were analyzed by two-way ANOVA (CD206: F(3,20) = 2.235, P = 0.116, YM-1: F(3,20) = 1.768, P = 0.186). *P<0.05, **P<0.01. |
LA-1, a Small Molecule Targeting CR3, Attenuates Rotenone-Induced Proinflammatory Microglial Activation
Our data above suggested that modulating CR3 could attenuate rotenone-induced proinflammatory microglial activation in mice. The small molecule leukadherins can enhance cell adhesion by binding to CR3.52 As we observed for the CR3-deficient mice, the mice treated with LA-1 were more resistant to rotenone-elicited microglial activation. As illustrated in Figure 6A, microglia with morphological hypertrophy as well as enhanced Iba-1 staining were present in the rotenone-injected mice but not in the LA-1 and rotenone-cotreated mice. Analysis of Iba-1 density also confirmed that LA-1 mitigated rotenone-induced microglial activation (Figure 6B). Consistent with these results, in contrast to treatment with rotenone alone, cotreatment with LA-1 and rotenone suppressed microglial M1 activation, as shown by reduced gene expression levels of iNOS, TNFα and IL-1β in the brainstem of mice (Figure 6C). The rotenone-induced reduction in microglial M2 genes including Arg-1 and YM-1 was also reversed by LA-1 (Figure 6D). The above findings suggested that CR3 disrupts the microglial M1/M2 balance in rotenone-treated mice.
 |
Figure 6 LA-1 abrogates rotenone-induced activation and M1 polarization of microglia in mice. (A) Immunohistochemistry with an anti-Iba-1 antibody was performed to stain microglial cells in the LC of rotenone-intoxicated mice with or without LA-1 treatment, and representative images are shown. (B) Quantification of the density of Iba-1 immunostaining. Results were mean ± SEM from five mice for each group and were analyzed by one-way ANOVA (F(3,16) = 10.109, P = 0.001). (C) The mRNA levels of iNOS, TNFα and IL-1β in the brainstem of rotenone-treated mice with or without LA-1 treatment were determined by real-time PCR. Results were mean ± SEM from five mice for each group and were analyzed by one-way ANOVA (iNOS: F(3,16) = 5.661, P = 0.008; TNFα: F(3,16) = 7.439, P = 0.002; IL-1β: F(3,16) = 3.589, P = 0.037; post hoc analysis by Tukey’s multiple comparisons test). (D) The mRNA levels of Arg-1, CD206 and YM-1 in the brainstem of rotenone-treated mice with or without LA-1 treatment were determined by real-time PCR. Results were mean ± SEM from five mice for each group and were analyzed by one-way ANOVA (Arg-1: F(3,16) = 12.575, P = 0.000, post hoc analysis by Tukey’s multiple comparisons test; CD206: F(3,16) = 6.956, P=0.003, post hoc analysis by Tamhane’s T2 multiple comparisons test; YM-1: F(3,16) = 10.109, P=0.001, post hoc analysis by Tukey’s multiple comparisons test). *P<0.05, **P<0.01; Scale bar = 100 μm. |
CR3 Deficiency and LA-1 Attenuate Rotenone-Induced Lipid Peroxidation in the Brainstem of Mice
Activated microglia, especially M1-polarized microglia, leads to oxidative stress by producing proinflammatory cytokines, chemokines and superoxide as well as other free radicals. The effects of CR3 knockout and LA-1 treatment on MDA content and the level of the antioxidant GSH, a product and a mediator of oxidative damage, respectively, were determined. Rotenone exposure resulted in increased MDA content and decreased GSH levels in WT mice (Figure 7A). In contrast, rotenone-elicited oxidative stress was significantly dampened in the CR3−/- mice, as shown by decreased MDA content and elevated GSH levels compared with those in the WT mice (Figure 7A). Consistent with our results, the rotenone-induced increase in MDA content and reduction in GSH levels in the brainstem of mice were also reversed by LA-1 (Figure 7B), suggesting that modulating CR3 mitigates rotenone-induced oxidative damage.
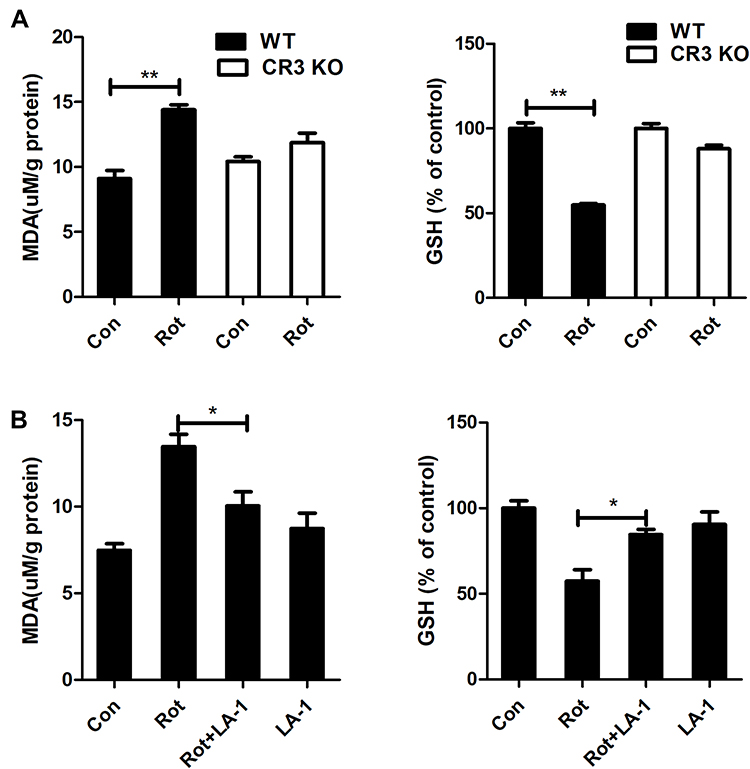 |
Figure 7 Genetic deletion of CR3 or LA-1 treatment mitigates oxidative stress in rotenone-intoxicated mice. (A) The MDA and GSH contents were determined in the brainstem of WT and CR3−/- mice after rotenone treatment by using commercial kits. Results were mean ± SEM from three mice for each group and were analyzed by two-way ANOVA (MDA: F(3,8) = 34.604, P = 0.000; GSH: F(3,8) = 37.129, P = 0.000; post hoc analysis by Tukey’s multiple comparisons test). (B) The MDA and GSH contents were determined in the brainstem of rotenone-treated mice with or without LA-1 treatment by using commercial kits. Results were mean ± SEM from four mice for each group and were analyzed by one-way ANOVA (MDA: F(3,12) = 12.753, P = 0.000; GSH: F(3,12) = 10.373, P = 0.001; post hoc analysis by Tukey’s multiple comparisons test). *P<0.05, **P<0.01. |
CR3 Deficiency and LA-1 Ameliorate Rotenone-Induced LC/NE Neuronal Loss
To explore whether CR3 mediates the rotenone-induced loss of LC/NE neurons, we counted the LC/NE neurons in rotenone-injected WT and CR3−/- mice. As shown in Figure 8A and B, rotenone induced a marked reduction in THir neurons in the LC of WT mice that was significantly attenuated in CR3−/- mice. Similarly, compared with that of rotenone-treated mice, we observed an increased number of THir neurons in the LC of LA-1 and rotenone-cotreated mice, indicating that LA-1 treatment attenuated rotenone-induced LC/NE neurodegeneration (Figure 8C and D). These results indicated that CR3 contributes to rotenone-induced toxicity in LC/NE neurons.
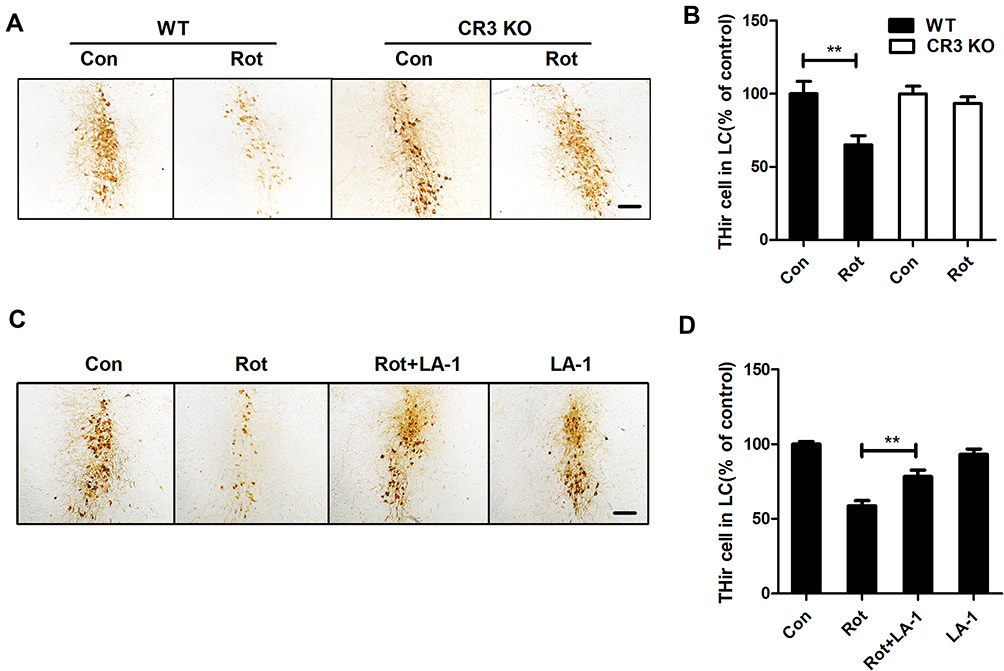 |
Figure 8 CR3 deficiency or LA-1 treatment alleviates rotenone-induced degeneration of LC/NE neurons in mice. (A) Immunohistochemistry with an anti-TH antibody was performed to stain LC/NE neurons of rotenone-intoxicated WT and CR3−/- mice, and representative images are shown. (B) Quantification of the number of THir neurons. Results were mean ± SEM from five to six mice for each group and were analyzed by two-way ANOVA (F(3,19) = 6.923, P = 0.002, post hoc analysis by Tukey’s multiple comparisons test). (C) Immunohistochemistry with an anti-TH antibody was performed to stain LC/NE neurons of rotenone-intoxicated mice with or without LA-1 treatment, and representative images are shown. (D) Quantification of the number of THir neurons. Results were mean ± SEM from five mice for each group and were analyzed by one-way ANOVA (F(3,16) = 27.527, P = 0.000, post hoc analysis by Tukey’s multiple comparisons test). **P<0.01; Scale bar = 100 μm. |
Discussion
In this study, we revealed an essential effect of CR3-dependent activation of microglia in rotenone-induced LC/NE neurodegeneration. Five salient features were found in this study: 1) Rotenone dose- and time-dependently induced LC/NE neuronal loss in mice; 2) Rotenone-induced activation of microglia occurred before LC/NE neurodegeneration; 3) Microglial depletion or inactivation attenuated rotenone-induced LC/NE neuronal loss in mice; 4) CR3 was critical for rotenone-induced microglial activation, M1 polarization and oxidative stress; and 5) Genetic ablation of CR3 or treatment with the CR3-modulating molecule LA-1 ameliorated LC/NE neurodegeneration in response to rotenone.
Microglial activation has long been recognized in a variety of neurological disorders. However, whether activated microglia are neurotoxic or neuroprotective remains controversial since findings so far have yielded mixed results. Minocycline, a semisynthetic tetracycline antibiotic, and PLX3397, a CSF1R antagonist, have been extensively applied to explore that how microglial activation contributes to neurodegenerative disease since minocycline and PLX3397 can efficiently block microglial activation and deplete brain microglia, respectively.53,54 Sriram et al reported that minocycline efficiently attenuates microglial activation in single MPTP- or methamphetamine-injected mice.55 However, minocycline fails to block dopaminergic neurotoxicity in the same model.55 In contrast, in a subchronic dopaminergic neurodegenerative rodent model, minocycline-inhibited microglial activation was accompanied by reduced dopaminergic neuron loss.56 Similarly, microglial depletion by PLX3397 failed to protect dopaminergic neurons against acute MPTP lesions but significantly alleviated amyloid accumulation, plaque deposition and pre-fibrillar oligomers formation in a chronic 5xFAD Alzheimer’s disease (AD) mouse model.57,58 In a multiple sclerosis animal model, microglial depletion also ameliorated demyelination, axonal pathology and neurodegeneration in the retinotectal system.33 These results suggest that blocking chronic and sustained microglial activation is neuroprotective. In this study, rotenone treatment resulted in sustained activation of microglia in the LC. Furthermore, inactivation of microglia by minocycline and microglial depletion by PLX3397 significantly ameliorated LC/NE neurodegeneration in rotenone-injected mice. Consistent with our findings, Gao et al found that rotenone was capable of directly stimulating microglial activation in vitro.59 Consistent with this result, activation of microglial occurred in the nigral regions of rotenone-treated mice.3 Moreover, Sun et al reported that minocycline attenuated rotenone-induced dopaminergic neurodegeneration in rats.51 We recently also found that the pesticides paraquat and maneb intoxication led to activation of microglia and LC/NE neuronal loss in mice,13 further supporting that microglial activation participates in LC/NE neurotoxicity. Two-month-old mice treated with PLX3397 for 21 days or 2 months showed no deficits in cognition, motor function, or behavior.29 A previous study showed that PLX3397 (40 mg/kg) and minocycline (50 mg/kg) had no effect on behavior, expression of pro-inflammatory cytokine or brain-derived neurotrophic factor levels in mesolimbic dopaminergic regions in WT mice.60 Du et al reported that eight-week-old mice administered minocycline (60, 90, or 120 mg/kg/day) did not show the loss of TH-positive neurons and striatal dopamine.61 We thought that PLX3397 and minocycline had no significant neuronal injury, so we did not set up PLX3397 and minocycline per se groups.
It is important to investigate the underlying mechanism of how rotenone-induced proinflammatory microglial activation drives progressive LC/NE neuronal loss. Here, we showed strong experimental evidence indicating that integrin CR3 is a critical mediator of the activation of microglia and the subsequent degeneration of LC/NE neurons induced by rotenone. First, rotenone elevated the expression and mRNA level of CR3 in the LC of mice. Second, CR3 deficiency and the CR3 modulator LA-1 mitigated rotenone-induced microglial activation. Third, mice deficient in CR3 or treated with LA-1 were more resistant to rotenone-induced LC/NE neurodegeneration than WT or vehicle-treated control mice, respectively. Consistent with our findings, Sherer62 and Shaikh63 reported increased CR3 immunoreactivity in rats and CHME-5 human microglial cells treated with rotenone. CR3−/- mice has been found to display greater resistance against β-amyloid (Aβ)-elicited synapse loss and neuronal damage than WT control mice in Hong et al’s report.64 Similarly, CR3 also mediates combined hypoxia and LPS-triggered neuroinflammation and long-term synaptic depression,24 an important form of synaptic plasticity related to cognition.65 These findings provide compelling evidence to indicate that CR3-mediated microglial activation could drive neurodegenerative processes in response to rotenone exposure.
Strong evidence suggests that microglial M1 polarization and the related production of cytotoxic factors are critical mediators of neuronal damage induced by microglia-mediated neuroinflammation.66 Yan et al reported that inhibition of microglial M1 activation by idebenone mitigated the production of proinflammatory factors in the SN of MPTP-treated mice and that this effect was associated with neuroprotection of dopaminergic neurons.67 Blocking microglial M1 polarization and the related production of reactive oxygen species (ROS) by taurine also attenuated neuronal damage in a paraquat- and maneb-generated PD mouse model.19,68 Additionally, suppression of microglial M1 polarization abrogated environmental toxin-induced neurotoxicity. In a 1-bromopropane-induced neurotoxic rat model, allyl sulfide counteracted 1-bromopropane-induced neuronal apoptosis by dampening microglial activation and oxidative stress.69 Here, we found that genetic ablation of CR3 or LA-1 treatment mitigated the rotenone-elicited microglial activation and generation of proinflammatory cytokines in mice. Furthermore, decreased MDA contents and elevated GSH levels were also observed in rotenone-intoxicated mice with LA-1 treatment or CR3 deficiency compared with vehicle-treated or WT mice, respectively. These results suggest that proinflammatory factors, ROS and related oxidative damage contribute to LC/NE neurodegeneration mediated by microglia via CR3 in response to rotenone.
However, at present, the mechanisms by which CR3 regulates rotenone-induced microglial activation and M1 polarization remain unclear. Although this point has not yet been explored, we could make a speculation. Src kinase is known to be the downstream signal of CR3 that regulates the inflammatory response.28 Activated Src kinase was found in rotenone-treated microglial cells.70 Furthermore, inhibition of Src kinase by SKI606 also efficiently prevented rotenone-induced accumulation of α-synuclein, and this effect was associated with attenuated neurotoxicity.70 These findings indicated that Src kinase might be an underlying mechanism responsible for the contribution of CR3 to activation and M1 polarization of microglia in response to rotenone. In addition, NADPH oxidase (NOX2), a superoxide-generating enzyme in microglia,71 might be the other downstream signal of CR3 to regulate inflammation induced by rotenone. Rotenone can stimulate NOX2 activation to produce superoxide, resulting in oxidative stress.72,73 Strong evidence indicates that CR3 can also regulate NOX2 activity. Gao et al reported that in microglial cultures, high-mobility group box 1 (HMGB1) binds to CR3, leading to an increase in NOX2 activity and subsequent microglial activation.74 Similar results were also reported in microglia treated with LPS, a ligand of CR3. Once activated, NOX2 produces extracellular and intracellular ROS that participated in the initiation and maintenance of chronic neuroinflammatory responses mediated by microglia. Our results showed that genetic deletion of CR3 or treatment with the CR3 modulator LA-1 inhibited rotenone-induced oxidative stress, supporting the contribution of NOX2 in mediating CR3-regulated microglial activation in response to rotenone.
Conclusion
In conclusion, our study revealed that microglia-mediated neuroinflammation played an important role in driving LC/NE neuronal damage in a rotenone-elicited PD mouse model. Further, integrin CR3 was identified as a principal factor in microglial activation as well as the subsequent loss of LC/NE neurons. Our results provide a new mechanistic view of the pathogenesis of LC/NE neurodegeneration elicited by environmental toxins and may result in the development of new tactics for combating this disease.
Abbreviations
AD, Alzheimer’s disease; Aβ, β-amyloid; CNS, central nervous system; CSF1R, colony-stimulating factor receptor 1; GSH, glutathione; HMGB1, high-mobility group box 1; iNOS, inducible nitric oxide synthase; IL-1β, interleukin-1β; LA-1, leukadherin-1; LC/NE, noradrenergic locus coeruleus; MDA, malondialdehyde; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MPP+, 1-methyl-4-phenylpyridinium; NOX2, NADPH oxidase; PD, Parkinson’s disease; ROS, reactive oxygen species; SNpc, substantia nigra pars compacta; TGF-β, transforming growth factor β; TNFα, tumor necrosis factor α.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Sciences Foundation of China (81973087), the NSFC-Liaoning Province United Foundation of China (U1908208), the National Natural Sciences Foundation of Liaoning Province (2019-MS-077; 2020-MS-264), the Liaoning Revitalization Talents Program (XLYC1907026; XLYC1808031) and the Liaoning BaiQianWan Talents Program (No. [2017]90).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Nistico R, Mehdawy B, Piccirilli S, et al. Paraquat- and rotenone-induced models of Parkinson’s disease. Int J Immunopathol Pharmacol. 2011;24(2):313–322. doi:10.1177/039463201102400205
2. Tanner CM, Kamel F, Ross GW, et al. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119(6):866–872. doi:10.1289/ehp.1002839
3. Zaitone SA, Ahmed E, Elsherbiny NM, et al. Caffeic acid improves locomotor activity and lessens inflammatory burden in a mouse model of rotenone-induced nigral neurodegeneration: relevance to Parkinson’s disease therapy. Pharmacol Rep. 2019;71(1):32–41. doi:10.1016/j.pharep.2018.08.004
4. Tapias V, McCoy JL, Greenamyre JT. Phenothiazine normalizes the NADH/NAD(+) ratio, maintains mitochondrial integrity and protects the nigrostriatal dopamine system in a chronic rotenone model of Parkinson’s disease. Redox Biol. 2019;24:101164. doi:10.1016/j.redox.2019.101164
5. Betarbet R, Sherer TB, MacKenzie G, et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3(12):1301–1306. doi:10.1038/81834
6. Braak H, Ghebremedhin E, Rüb U, et al. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318(1):121–134. doi:10.1007/s00441-004-0956-9
7. Zarow C, Lyness SA, Mortimer JA, et al. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60(3):337–341. doi:10.1001/archneur.60.3.337
8. Hou L, Sun F, Sun W, et al. Lesion of the locus coeruleus damages learning and memory performance in paraquat and maneb-induced mouse Parkinson’s disease model. Neuroscience. 2019;419:129–140. doi:10.1016/j.neuroscience.2019.09.006
9. Song S, Wang Q, Jiang L, et al. Noradrenergic dysfunction accelerates LPS-elicited inflammation-related ascending sequential neurodegeneration and deficits in non-motor/motor functions. Brain Behav Immun. 2019;81:374–387. doi:10.1016/j.bbi.2019.06.034
10. Subhramanyam CS, Wang C, Hu Q, et al. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol. 2019;94:112–120. doi:10.1016/j.semcdb.2019.05.004
11. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi:10.1038/nrn2038
12. O’Neill E, Harkin A. Targeting the noradrenergic system for anti-inflammatory and neuroprotective effects: implications for Parkinson’s disease. Neural Regen Res. 2018;13(8):1332–1337. doi:10.4103/1673-5374.235219
13. Hou L, Zhang C, Wang K, et al. Paraquat and maneb co-exposure induces noradrenergic locus coeruleus neurodegeneration through NADPH oxidase-mediated microglial activation. Toxicology. 2017;380:1–10. doi:10.1016/j.tox.2017.02.009
14. Song S, Jiang L, Oyarzabal EA, et al. Loss of brain norepinephrine elicits neuroinflammation-mediated oxidative injury and selective caudo-rostral neurodegeneration. Mol Neurobiol. 2019;56(4):2653–2669. doi:10.1007/s12035-018-1235-1
15. Zhang C, Hou L, Yang J, et al. 2,5-Hexanedione induces dopaminergic neurodegeneration through integrin α(M)β2/NADPH oxidase axis-mediated microglial activation. Cell Death Dis. 2018;9(2):60. doi:10.1038/s41419-017-0091-7
16. Gao HM, Liu B, Zhang W, et al. Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. FASEB J. 2003;17(13):1954–1956. doi:10.1096/fj.03-0109fje
17. Wu XF, Block ML, Zhang W, et al. The role of microglia in paraquat-induced dopaminergic neurotoxicity. Antioxid Redox Signal. 2005;7(5–6):654–661. doi:10.1089/ars.2005.7.654
18. Chen X, Liu Z, Cao BB, et al. TGF-β1 neuroprotection via inhibition of microglial activation in a rat model of Parkinson’s disease. J Neuroimmune Pharmacol. 2017;12(3):433–446. doi:10.1007/s11481-017-9732-y
19. Che Y, Hou L, Sun F, et al. Taurine protects dopaminergic neurons in a mouse Parkinson’s disease model through inhibition of microglial M1 polarization. Cell Death Dis. 2018;9(4):435. doi:10.1038/s41419-018-0468-2
20. De Miranda BR, Rocha EM, Bai Q, et al. Astrocyte-specific DJ-1 overexpression protects against rotenone-induced neurotoxicity in a rat model of Parkinson’s disease. Neurobiol Dis. 2018;115:101–114. doi:10.1016/j.nbd.2018.04.008
21. Zhou F, Wu JY, Sun XL, et al. Iptakalim alleviates rotenone-induced degeneration of dopaminergic neurons through inhibiting microglia-mediated neuroinflammation. Neuropsychopharmacology. 2007;32(12):2570–2580. doi:10.1038/sj.npp.1301381
22. Rosetti F, Mayadas TN. The many faces of Mac-1 in autoimmune disease. Immunol Rev. 2016;269(1):175–193. doi:10.1111/imr.12373
23. Das S, Mishra KP, Ganju L, et al. Intranasally delivered small interfering RNA-mediated suppression of scavenger receptor Mac-1 attenuates microglial phenotype switching and working memory impairment following hypoxia. Neuropharmacology. 2018;137:240–255. doi:10.1016/j.neuropharm.2018.05.002
24. Zhang J, Malik A, Choi HB, et al. Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron. 2014;82(1):195–207. doi:10.1016/j.neuron.2014.01.043
25. Liberatore GT, Jackson-Lewis V, Vukosavic S, et al. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5(12):1403–1409. doi:10.1038/70978
26. Wang Q, Qian L, Chen SH, et al. Post-treatment with an ultra-low dose of NADPH oxidase inhibitor diphenyleneiodonium attenuates disease progression in multiple Parkinson’s disease models. Brain. 2015;138(Pt 5):1247–1262. doi:10.1093/brain/awv034
27. Hou L, Bao X, Zang C, et al. Integrin CD11b mediates alpha-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018;14:600–608. doi:10.1016/j.redox.2017.11.010
28. Hou L, Wang K, Zhang C, et al. Complement receptor 3 mediates NADPH oxidase activation and dopaminergic neurodegeneration through a Src-Erk-dependent pathway. Redox Biol. 2018;14:250–260. doi:10.1016/j.redox.2017.09.017
29. Elmore MR, Najafi AR, Koike MA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–397. doi:10.1016/j.neuron.2014.02.040
30. Khatri DK, Juvekar AR. Neuroprotective effect of curcumin as evinced by abrogation of rotenone-induced motor deficits, oxidative and mitochondrial dysfunctions in mouse model of Parkinson’s disease. Pharmacol Biochem Behav. 2016;150–151:39–47. doi:10.1016/j.pbb.2016.09.002
31. Mitra S, Chakrabarti N, Dutta SS, et al. Gender-specific brain regional variation of neurons, endogenous estrogen, neuroinflammation and glial cells during rotenone-induced mouse model of Parkinson’s disease. Neuroscience. 2015;292:46–70. doi:10.1016/j.neuroscience.2014.12.052
32. Sharma N, Jamwal S, Kumar P. Beneficial effect of antidepressants against rotenone induced Parkinsonism like symptoms in rats. Pathophysiology. 2016;23(2):123–134. doi:10.1016/j.pathophys.2016.03.002
33. Groh J, Klein D, Berve K, et al. Targeting microglia attenuates neuroinflammation-related neural damage in mice carrying human PLP1 mutations. Glia. 2019;67(2):277–290. doi:10.1002/glia.23539
34. Manso Y, Holland PR, Kitamura A, et al. Minocycline reduces microgliosis and improves subcortical white matter function in a model of cerebral vascular disease. Glia. 2018;66(1):34–46. doi:10.1002/glia.23190
35. Czirr E, Castello NA, Mosher KI, et al. Microglial complement receptor 3 regulates brain Abeta levels through secreted proteolytic activity. J Exp Med. 2017;214(4):1081–1092. doi:10.1084/jem.20162011
36. Hou L, Sun F, Huang R, et al. Inhibition of NADPH oxidase by apocynin prevents learning and memory deficits in a mouse Parkinson’s disease model. Redox Biol. 2019;22:101134. doi:10.1016/j.redox.2019.101134
37. Hou L, Qu X, Qiu X, et al. Integrin CD11b mediates locus coeruleus noradrenergic neurodegeneration in a mouse Parkinson’s disease model. J Neuroinflammation. 2020;17(1):148. doi:10.1186/s12974-020-01823-3
38. Zhang W, Wang T, Qin L, et al. Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: role of NADPH oxidase. FASEB J. 2004;18(3):589–591. doi:10.1096/fj.03-0983fje
39. Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates.
40. Wang Q, Chu CH, Qian L, et al. Substance P exacerbates dopaminergic neurodegeneration through neurokinin-1 receptor-independent activation of microglial NADPH oxidase. J Neurosci. 2014;34(37):12490–12503. doi:10.1523/JNEUROSCI.2238-14.2014
41. Okubo H, Nakatsu Y, Sakoda H, et al. Mosapride citrate improves nonalcoholic steatohepatitis with increased fecal lactic acid bacteria and plasma glucagon-like peptide-1 level in a rodent model. Am J Physiol Gastrointest Liver Physiol. 2015;308(2):G151–G158. doi:10.1152/ajpgi.00198.2014
42. Hou L, Huang R, Sun F, et al. NADPH oxidase regulates paraquat and maneb-induced dopaminergic neurodegeneration through ferroptosis. Toxicology. 2019;417:64–73. doi:10.1016/j.tox.2019.02.011
43. Hou L, Yang J, Li S, et al. Glibenclamide attenuates 2,5-hexanedione-induced neurotoxicity in the spinal cord of rats through mitigation of NLRP3 inflammasome activation, neuroinflammation and oxidative stress. Toxicol Lett. 2020;331:152–158. doi:10.1016/j.toxlet.2020.06.002
44. Zhang D, Li S, Hou L, et al. Microglial activation contributes to cognitive impairments in rotenone-induced mouse Parkinson’s disease model. J Neuroinflammation. 2021;18(1):4. doi:10.1186/s12974-020-02065-z
45. Mao XY, Zhou HH, Jin WL. Ferroptosis induction in pentylenetetrazole kindling and pilocarpine-induced epileptic seizures in mice. Front Neurosci. 2019;13:721. doi:10.3389/fnins.2019.00721
46. Gassmann M, Grenacher B, Rohde B, et al. Quantifying western blots: pitfalls of densitometry. Electrophoresis. 2009;30(11):1845–1855. doi:10.1002/elps.200800720
47. Lee JM, Kim TW, Park SS, et al. Treadmill exercise improves motor function by suppressing purkinje cell loss in parkinson disease rats. Int Neurourol J. 2018;22(Suppl 3):S147–S155. doi:10.5213/inj.1836226.113
48. Javed H, Azimullah S, Abul Khair SB, et al. Neuroprotective effect of nerolidol against neuroinflammation and oxidative stress induced by rotenone. BMC Neurosci. 2016;17(1):58. doi:10.1186/s12868-016-0293-4
49. Ojha S, Javed H, Azimullah S, et al. Neuroprotective potential of ferulic acid in the rotenone model of Parkinson’s disease. Drug Des Devel Ther. 2015;9:5499–5510. doi:10.2147/DDDT.S90616
50. Verma DK, Singh DK, Gupta S, et al. Minocycline diminishes the rotenone induced neurotoxicity and glial activation via suppression of apoptosis, nitrite levels and oxidative stress. Neurotoxicology. 2018;65:9–21. doi:10.1016/j.neuro.2018.01.006
51. Sun C, Wang Y, Mo M, et al. Minocycline protects against rotenone-induced neurotoxicity correlating with upregulation of Nurr1 in a Parkinson’s disease rat model. Biomed Res Int. 2019;2019:6843265. doi:10.1155/2019/6843265
52. Maiguel D, Faridi MH, Wei C, et al. Small molecule-mediated activation of the integrin CD11b/CD18 reduces inflammatory disease. Sci Signal. 2011;4(189):ra57. doi:10.1126/scisignal.2001811
53. Budni J, Garcez ML, de Medeiros J, et al. The anti-inflammatory role of minocycline in Alzheimer s disease. Curr Alzheimer Res. 2016;13(12):1319–1329. doi:10.2174/1567205013666160819124206
54. Liang YJ, Feng SY, Qi YP, et al. Contribution of microglial reaction to increased nociceptive responses in high-fat-diet (HFD)-induced obesity in male mice. Brain Behav Immun. 2019;80:777–792. doi:10.1016/j.bbi.2019.05.026
55. Sriram K, Miller DB, O’Callaghan JP. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-alpha. J Neurochem. 2006;96(3):706–718. doi:10.1111/j.1471-4159.2005.03566.x
56. Kumar V, Singh BK, Chauhan AK, et al. Minocycline rescues from zinc-induced nigrostriatal dopaminergic neurodegeneration: biochemical and molecular interventions. Mol Neurobiol. 2016;53(5):2761–2777. doi:10.1007/s12035-015-9137-y
57. Yang X, Ren H, Wood K, et al. Depletion of microglia augments the dopaminergic neurotoxicity of MPTP. FASEB J. 2018;32(6):3336–3345. doi:10.1096/fj.201700833RR
58. Sosna J, Philipp S, Albay R
59. Gao F, Chen D, Hu Q, et al. Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS One. 2013;8(8):e72046. doi:10.1371/journal.pone.0072046
60. Wang J, Lai S, Li G, et al. Microglial activation contributes to depressive-like behavior in dopamine D3 receptor knockout mice. Brain Behav Immun. 2020;83:226–238. doi:10.1016/j.bbi.2019.10.016
61. Du Y, Ma Z, Lin S, et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98(25):14669–14674. doi:10.1073/pnas.251341998
62. Sherer TB, Betarbet R, Kim JH, et al. Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci Lett. 2003;341(2):87–90. doi:10.1016/S0304-3940(03)00172-1
63. Shaikh SB, Nicholson LF. Effects of chronic low dose rotenone treatment on human microglial cells. Mol Neurodegener. 2009;4(1):55. doi:10.1186/1750-1326-4-55
64. Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–716. doi:10.1126/science.aad8373
65. Van der Jeugd A, Ahmed T, Burnouf S, et al. Hippocampal tauopathy in tau transgenic mice coincides with impaired hippocampus-dependent learning and memory, and attenuated late-phase long-term depression of synaptic transmission. Neurobiol Learn Mem. 2011;95(3):296–304. doi:10.1016/j.nlm.2010.12.005
66. Subramaniam SR, Federoff HJ. Targeting microglial activation states as a therapeutic avenue in Parkinson’s disease. Front Aging Neurosci. 2017;9:176. doi:10.3389/fnagi.2017.00176
67. Yan A, Liu Z, Song L, et al. Idebenone alleviates neuroinflammation and modulates microglial Polarization in LPS-stimulated BV2 cells and MPTP-induced Parkinson’s disease mice. Front Cell Neurosci. 2018;12:529. doi:10.3389/fncel.2018.00529
68. Hou L, Che Y, Sun F, et al. Taurine protects noradrenergic locus coeruleus neurons in a mouse Parkinson’s disease model by inhibiting microglial M1 polarization. Amino Acids. 2018;50(5):547–556. doi:10.1007/s00726-018-2547-1
69. Suo J, Zhang C, Wang P, et al. Allyl sulfide counteracts 1-bromopropane-induced neurotoxicity by inhibiting neuroinflammation and oxidative stress. Toxicol Sci. 2019;167(2):397–407. doi:10.1093/toxsci/kfy240
70. Wang Y, Liu J, Chen M, et al. The novel mechanism of rotenone-induced alpha-synuclein phosphorylation via reduced protein phosphatase 2A activity. Int J Biochem Cell Biol. 2016;75:34–44. doi:10.1016/j.biocel.2016.03.007
71. Hou L, Zhang L, Hong JS, et al. Nicotinamide adenine dinucleotide phosphate oxidase and neurodegenerative diseases: mechanisms and therapy. Antioxid Redox Signal. 2020;33(5):374–393. doi:10.1089/ars.2019.8014
72. Pal R, Monroe TO, Palmieri M, et al. Rotenone induces neurotoxicity through Rac1-dependent activation of NADPH oxidase in SHSY-5Y cells. FEBS Lett. 2014;588(3):472–481. doi:10.1016/j.febslet.2013.12.011
73. Taetzsch T, Block ML. Pesticides, microglial NOX2, and Parkinson’s disease. J Biochem Mol Toxicol. 2013;27(2):137–149. doi:10.1002/jbt.21464
74. Gao HM, Zhou H, Zhang F, Wilson BC, Kam W, Hong JS. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci. 2011;31(3):1081–1092. doi:10.1523/JNEUROSCI.3732-10.2011
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.