")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Mechanism and pharmacological rescue of berberine-induced hERG channel deficiency
Authors Yan M, Zhang K, Shi Y, Feng L, Lv L, Li B
Received 1 July 2015
Accepted for publication 12 August 2015
Published 22 October 2015 Volume 2015:9 Pages 5737—5747
DOI https://doi.org/10.2147/DDDT.S91561
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Meng Yan,1 Kaiping Zhang,1 Yanhui Shi,1 Lifang Feng,1 Lin Lv,1 Baoxin Li1,2
1Department of Pharmacology, Harbin Medical University, 2State-Province Key Laboratory of Biopharmaceutical Engineering, Harbin, Heilongjiang, People’s Republic of China
Abstract: Berberine (BBR), an isoquinoline alkaloid mainly isolated from plants of Berberidaceae family, is extensively used to treat gastrointestinal infections in clinics. It has been reported that BBR can block human ether-a-go-go-related gene (hERG) potassium channel and inhibit its membrane expression. The hERG channel plays crucial role in cardiac repolarization and is the target of diverse proarrhythmic drugs. Dysfunction of hERG channel can cause long QT syndrome. However, the regulatory mechanisms of BBR effects on hERG at cell membrane level remain unknown. This study was designed to investigate in detail how BBR decreased hERG expression on cell surface and further explore its pharmacological rescue strategies. In this study, BBR decreases caveolin-1 expression in a concentration-dependent manner in human embryonic kidney 293 (HEK293) cells stably expressing hERG channel. Knocking down the basal expression of caveolin-1 alleviates BBR-induced hERG reduction. In addition, we found that aromatic tyrosine (Tyr652) and phenylalanine (Phe656) in S6 domain mediate the long-term effect of BBR on hERG by using mutation techniques. Considering both our previous and present work, we propose that BBR reduces hERG membrane stability with multiple mechanisms. Furthermore, we found that fexofenadine and resveratrol shorten action potential duration prolongated by BBR, thus having the potential effects of alleviating the cardiotoxicity of BBR.
Keywords: berberine, hERG, cavoline-1, cardiotoxicity, LQTS, pharmacological rescue
Corrigendum for this paper has been published.
Introduction
The human ether-a-go-go-related gene (hERG) potassium channel confers the rapid activating component of the cardiac delayed rectifier potassium current (IKr), which is crucial for repolarization of cardiac action potential. Dysfunction of the hERG channel could cause long QT syndrome (LQTS) characterized by delayed repolarization and prolonged QT interval, which increases the risk of ventricular arrhythmias and sudden death.1 However, diverse therapeutic drugs can induce LQTS via inhibition of hERG with a wide range of mechanisms. Dofetilide and cisapride acutely inhibit hERG current.2,3 Desipramine reduces hERG expression via inhibiting trafficking and promoting endocytosis and degradation.4 Pentamidine impedes hERG trafficking from endoplasmic reticulum (ER) to cell membrane.5 Arsenic trioxide simultaneously decreases channel synthesis and inhibits channel trafficking.6,7
Berberine (BBR), an alkaloid mainly extracted from plants belonging to Berberidaceae family, has been extensively used to treat gastrointestinal infections and has a variety of pharmacological effects including anti-hyperlipidemia, anti-inflammation, and anti-arrhythmia. Previous researches showed that BBR acutely blocks hERG channel in Xenopus oocytes by interacting with the two aromatic amino acid residues (tyrosine [Tyr652] and phenylalanine [Phe656], which are thought as the universal binding sites for various hERG channel blockers) located in S6 transmembrane helix.8 Drug binding to special residues may induce conformational changes of the hERG channel.9 In addition, we recently reported that BBR reduces hERG plasma membrane expression, through disrupting trafficking after long-term exposure.10
However, the regulatory mechanisms at cell membrane level and the binding sites mediating hERG expression reduction after BBR incubation have not been documented. Caveolin-1 (Cav-1), a major protein constituent of caveolae, co-localizes with hERG protein on cell surface.11 The expression level of cav-1 is closely associated with cholesterol on membrane.12 Coincidently, BBR has been identified to possess cholesterol-lowering effect.13 Hence, we speculate that cav-1 involves in BBR induced reduction of the hERG channel. Previous report illustrated that BBR acutely block hERG channel by interacting with specific residues (Tyr652 and Phe656).8 However, there was no report to elucidate that the binding sites mediate hERG reduction triggered by BBR incubation. This study uses site-directed mutagenesis to identify the binding sites that mediate long-term effect. Moreover, we try to search for rescue strategies for BBR-triggered hERG channel deficiency on account of its high proarrhythmic potential. Various reagents have been found to effectively rescue hERG deficiency. In this report, we tested three drugs for their ability to rescue BBR-induced hERG inhibition.
Materials and methods
Reagents
BBR (Sigma-Aldrich Co., St Louis, MO, USA), astemizole (Sigma-Aldrich Co.), and fexofenadine (Sigma-Aldrich Co.) were dissolved in DMSO as a stock (10 μM, 5 μM, and 5 μM, respectively). Resveratrol (Aladdin Industrial Corporation, Shanghai, People’s Republic of China) was dissolved in ethanol as a stock (10 μM). Drug solutions were diluted in cultured medium before use. Cells were incubated with drugs for 24 hours.
Cell culture
Human embryonic kidney 293 (HEK293) cells and HEK293 cells stably expressing the wild-type (WT) hERG gene (hERG-HEK293, provided by Professor Zhiguo Wang, Harbin Medical University) were cultured at 37°C and 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM; Hyclone, Logan, Utah, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; Bioind, Israel). For hERG-HEK293 cells, 400 μg·mL−1 gentamycin (G418; Thermo Fisher Scientific, Waltham, MA,USA) was added to the medium.
Neonatal rat ventricular myocytes (NRVM) were isolated from dissected hearts of 1- to 2-day-old neonatal Sprague Dawley rat (The Experimental Animal Center of The Second Affiliated Hospital of Harbin Medical University, Harbin, People’s Republic of China). Ventricular parts were excised and minced on ice, treated with an enzyme solution containing 0.1% trypsin (Beyotime, Shanghai, People’s Republic of China) at 37°C for 10 minutes. After discarding the first digestion supernatants, three repeated digestions were performed. The supernatants were stored in DMEM containing 10% FBS and 1% penicillin–streptomycin (100 U·mL−1 and 100 μg·mL−1, respectively) and then centrifuged for 5 minutes at 1500 g·min−1. The cell pellets were incubated to obtain cardiomyocytes by differential adhesion for 1.5 hours. The supernatant was then plated onto new dishes with DMEM containing 10% FBS and 1% penicillin–streptomycin. The proliferation of the remaining fibroblasts was inhibited by the addition of 10 μM 5-bromodeoxyuridine (Sigma-Aldrich Co.). Cardiomyocytes were cultured for 2 days before use.
Guinea pigs (250–300 g, either sexes) were bought from The Experimental Animal Center of The Second Affiliated Hospital of Harbin Medical University. The procedure of myocyte isolation was done as described previously.14 Briefly, the guinea pigs were killed by cervical dislocation and their hearts were rapidly retrogradely perfused on a modified Langendorff perfusion apparatus with Tyrode’s solution (136 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 0.33 mM NaH2PO4, 10 mM glucose, and 10 mM HEPES, with pH 7.4) at 37°C for a few minutes until the effluent is without blood. Then, the heart was perfused with Ca2+-free Tyrode’s solution till the heart stopped beating, followed by the perfusion in the same solution containing 150 kU·L−1 type II collagenase (Worthington Biochemical Corporation, Lakewood, New Jersey, USA) and 1% bovine serum albumin (Huashun Biotechnology, Wuhan, Hubei Province, People’s Republic of China). Cells were obtained by pipetting the softened, minced tissue gently in KB solution (70 mM glutamic acid, 15 mM taurine, 30 mM KCl, 10 mM KH2PO4, 0.5 mM MgCl2, 0.5 mM ethylene glycol tetraacetic acid, 10 mM HEPES, 10 mM glucose, and 1% bovine serum albumin, with pH 7.4) and stored at 4°C for analysis. All solutions were oxygenated at 37°C for 1 hour. Guinea pigs were housed with up to two other animals of the same sex at a room temperature of 19°C–25°C with a relative humidity of 30%–70%. All experiments were approved by the institutional animal care and ethics committee of Harbin Medical University.
Western blot analysis
The cells were scraped into tubes after adding RIPA (Beyotime, Shanghai, People’s Republic of China) and PMSF (100 mM; Beyotime, People’s Republic of China). Cell lysis was performed by sonicating three times and then centrifuging at 13,500 rpm for 15 minutes. The supernatants were collected and protein concentration was determined by Bradford’s method. The proteins (100 μg per sample) were separated by SDS-PAGE and then transferred to a polyvinylidene difluoride membrane. Subsequently, the membrane was probed with antibodies against hERG (Santa Cruz, USA), cav-1 (Cell Signaling Technology, USA), and actin (Zhongshanjinqiao, Beijing, People’s Republic of China), respectively. After that, membranes were incubated with horseradish peroxidase–conjugated secondary antibodies (Li-CoR). The bands were detected with the Odyssey instrument (American Gene Corporation). The blots were analyzed and quantified with a Scion Image.
Transfection
Cav-1-siRNA (Santa Cruz Biotechnology Inc., Dallas, TX, USA) was used to knockdown the basal expression level of cav-1. The hERG cDNA mutations, Y652A (tyrosine to alanine) and F656V (phenylalanine to valine), were kindly provided by Professor Zhiguo Wang. Ctl-siRNA or Cav-1-siRNA was transfected into hERG-HEK293 cells and WT, F656V or Y652A cDNA, was transfected into HEK293 cells. In brief, cells were plated in 25 mm dishes at the required density and were incubated overnight. Approximately 3–4 hours prior to transfection, the media in the dishes were changed with 4.5 mL DMEM. Then, 250 μL opti-MEM and 12.5 μL lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) were added to the tube labeled A and 250 μL opti-DMEM and 2 μg siRNA or 4 μg cDNA were added to the tube labeled B, then the solutions in A and B were mixed. After incubation at room temperature for 20 minutes, the mixture was added to cells in the dishes. The cells were cultured at 37°C in a humidified CO2 incubator. The transfection took place 48 hours prior to analysis.
Patch-clamp recording techniques
hERG current and action potential recordings were performed using whole cell patch-clamp techniques. Cells were harvested after trypsinization, centrifugation, and then stored at 4°C in bath solution (136 mM NaCl, 5.4 mM KCl, 5 mM HEPES, 1 mM MgCl2, 1 mM CaCl2, and 10 mM glucose, with pH 7.4). The cell suspensions were seeded in a recording chamber using a microscope (Olympus IX-70; Olympus Corporation, Tokyo, Japan) and were superfused with the bath solution at a rate of 1.0 mL·min−1. The whole-cell configuration was formed by applying a pipette with a tip resistance of 1–3 MΩ. The cell membrane was sucked to form a whole-cell configuration after offsetting the junction resistance. The current was recorded with an Axonpatch-200B amplifier (Axon Instruments Inc, Union City, CA, USA). The action potentials in guinea pig cells and NRVM were recorded after overnight culture in M199 medium (Thermo Fisher Scientific, Waltham, MA, USA). The recordings were performed with a myocyte pipette solution (20 mM KCl, 1 mM MgCl2, 10 mM HEPES, 5 mM ATP, 5 mM EGTA, and 110 mM aspartate with pH 7.3). pClamp 9.2 (Axon Instruments) was used to control the protocols.
Data analysis
The data are expressed as the mean ± standard error of the mean (SEM). Differences between the means were obtained using an unpaired two-tailed Student’s t test. P-values <0.05 were considered statistically significant.
Results
BBR inhibited hERG channel on membrane via cav-1 interference
To find out whether the regulatory mechanisms at the cell membrane level take part in BBR-induced hERG channel deficiency, we tested the effect of BBR on cav-1. Cav-1, whose expression level is closely associated with cholesterol on the membrane, is reported to co-localize with hERG protein on the cell surface.11 Moreover, BBR is able to lower the cholesterol levels via the LDLR pathway.13 Therefore, we hypothesize that cav-1 involves in BBR-induced reduction of hERG channel. As shown in Figure 1A, after incubation with BBR for 24 hours, cav-1 in hERG-HEK293 cells was decreased to 87.37%±4.50% (1 μM) and 56.94%±2.14% (10 μM), respectively. Then, we transfected hERG-HEK293 cells with cav-1-specific siRNA to further test whether cav-1 participates in BBR-induced hERG stability defect at the cell surface (cav-1 was successfully inhibited to 75.59%±1.64% in Figure 1B). The inhibition ratio of 155 kDa hERG protein by 10 μM BBR was found to reduce from 66.03%±7.05% (Ctl-siRNA) to 39.04%±8.38% (Cav-1-siRNA) (Figure 1C). Together, these results suggested that BBR could reduce hERG expression on membrane by interfering with cav-1.
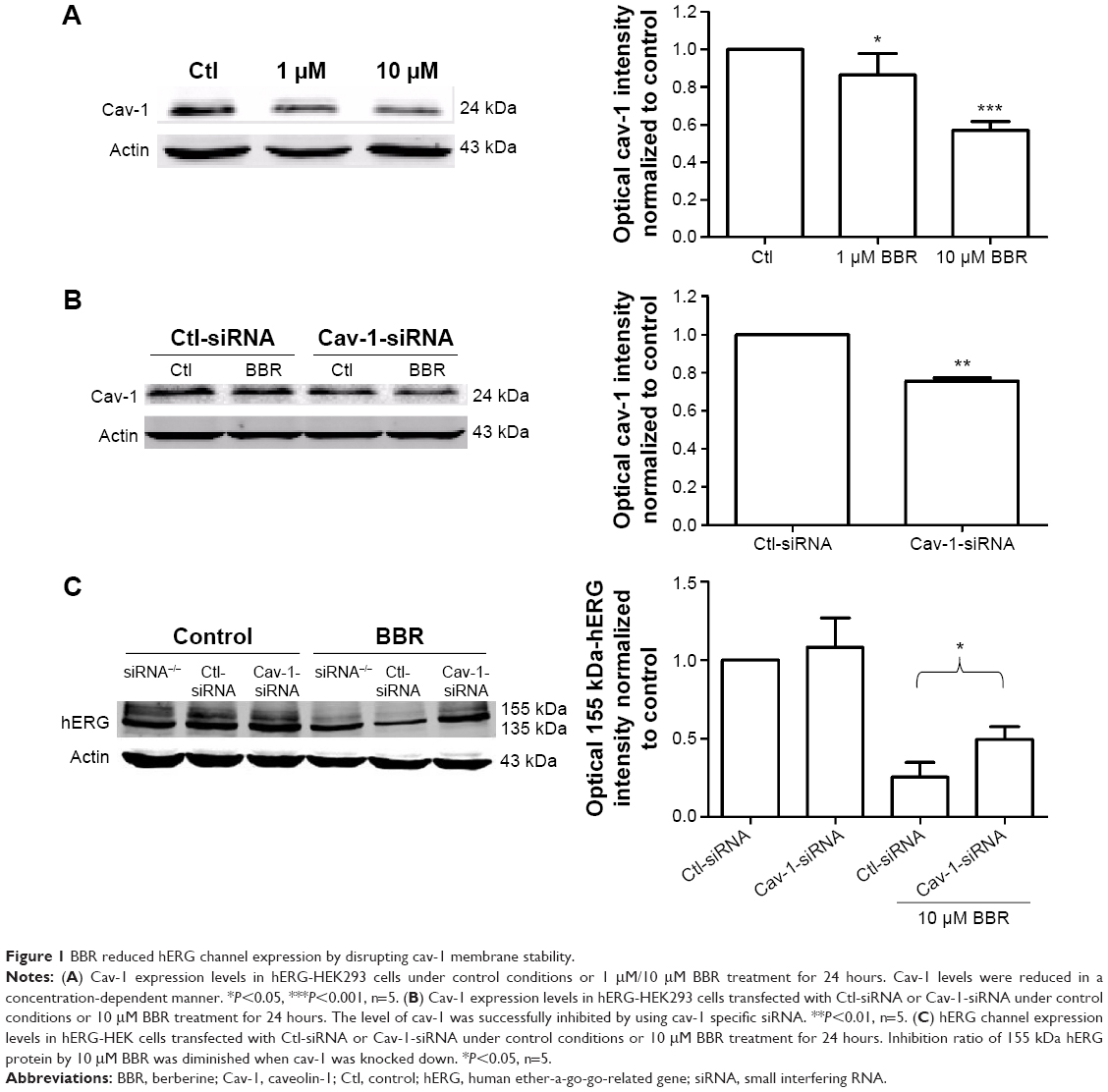 | Figure 1 BBR reduced hERG channel expression by disrupting cav-1 membrane stability. |
Phe656 and Tyr652 binding accounts for BBR-induced hERG channel deficiency
To clarify whether hERG channel deficiency caused by BBR incubation was also on account of Phe656 and Tyr652 binding similar to acute application of BBR.8 We studied the effects of BBR on mutant channels by transfecting HEK293 cells with WT-hERG, Y652A-hERG, or F656V-hERG. Figure 2A shows Western blot analysis and statistics. The expression of mature 155-kDa hERG protein was strongly inhibited by 10 μM BBR, at an inhibition ratio of 29.20%±2.73%. While BBR shows no obvious effect on that of F656V-hERG or Y652A-hERG. The electrophysiological recordings were consistent with western blots, where we measured WT-hERG tail current was significantly inhibited by 10 μM BBR after incubation for 24 hours (the inhibition ratio under 40 mV is 81.40%) and F656V-hERG or Y652A-hERG tail current was not affected (Figure 2B–D). The hERG currents were elicited by a 3-second depolarizing step in 10 mV increments from −60 mV to 40 mV, from a holding potential of −80 mV, followed by a 3-second step to −50 mV record tail current. These results suggest that BBR-induced hERG channel deficiency was on account of Phe656 and Tyr652 binding.
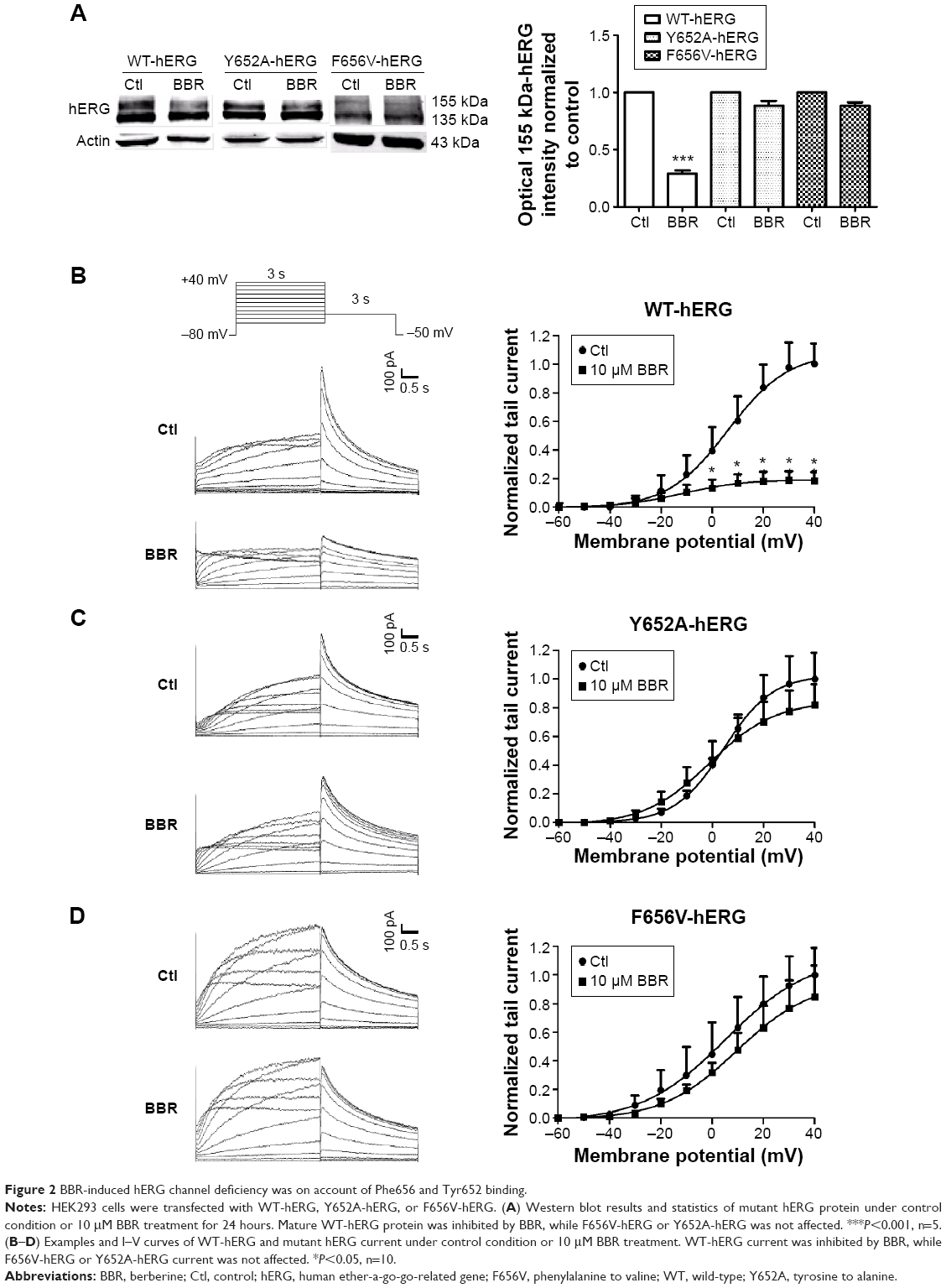 | Figure 2 BBR-induced hERG channel deficiency was on account of Phe656 and Tyr652 binding. |
Pharmacological rescue of BBR-induced hERG channel deficiency
To seek rescue strategies for BBR-induced hERG channel deficiency, we chose three drugs (resveratrol, astemizole, and fexofenadine, which were previously used to correct trafficking of hERG channel) to test whether the misprocessed hERG channel by BBR could be transported to the cell surface. Resveratrol is able to rescue the trafficking inhibition of hERG and relieve the ER stress triggered by arsenic trioxide.14 Astemizole promotes forward trafficking from ER to cell surface inhibited by pentamidine in a competitive way.5 Fexofenadine is often used to rescue trafficking defect of hERG because of the advantage that rescue without blocking the channel.15 hERG-HEK293 cells were incubated with 10 μM BBR, followed by 10 μM resveratrol (Figure 3A), 5 μM astemizole (Figure 3B,) or 1 μM fexofenadine (Figure 3C) for 24 hours before immunoblotting. As shown in Figure 3, the fully glycosylated 155 kDa hERG protein inhibited by 10 μM BBR was successfully restored by all these drugs and there was no distinct difference among their ability to rescue hERG trafficking.
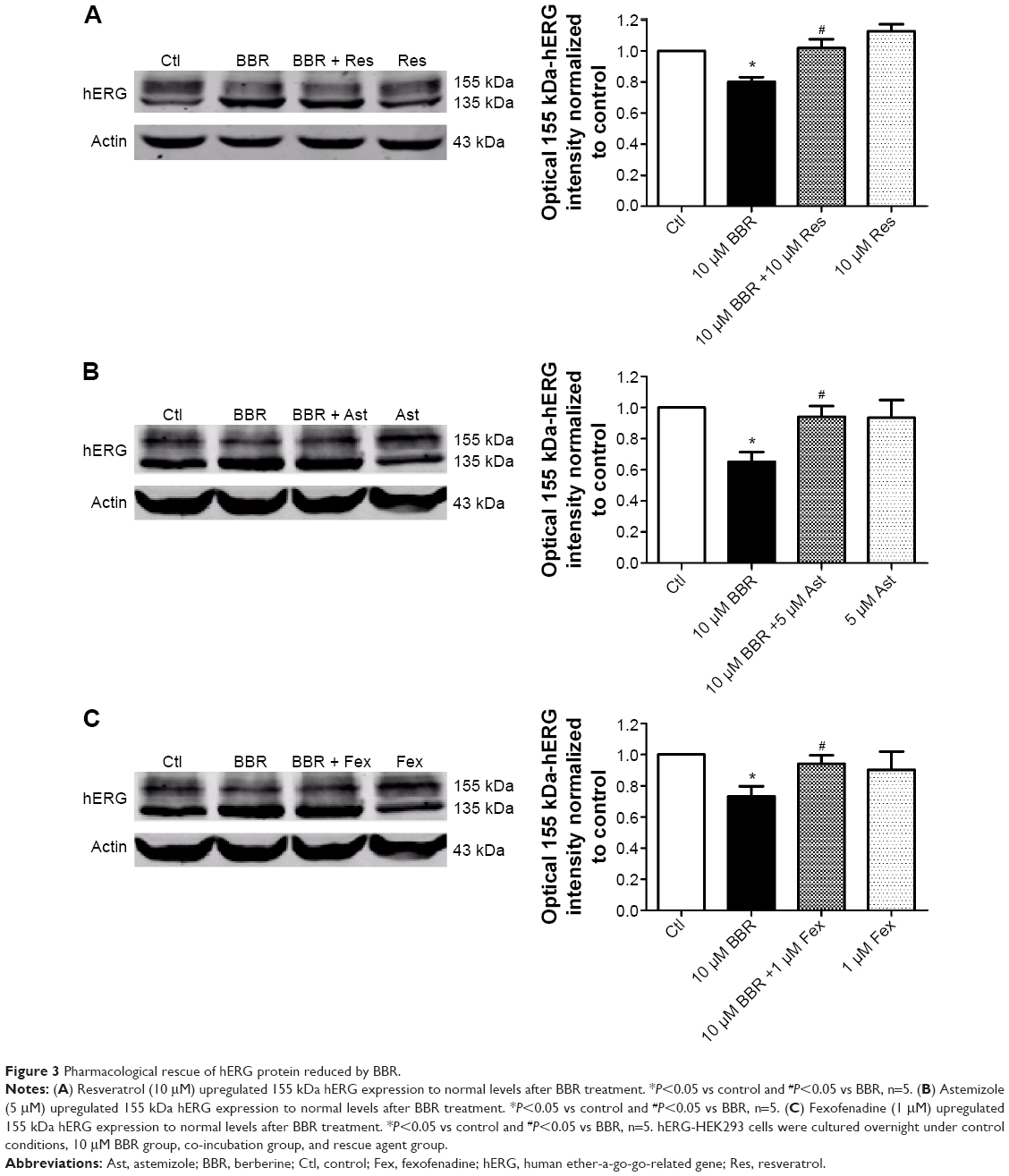 | Figure 3 Pharmacological rescue of hERG protein reduced by BBR. |
To further investigate whether the rescued mature hERG proteins were functional, currents recorded from hERG-HEK293 cells under the same conditions were analyzed. The rescue ratio was calculated by the function (Ico-incubation−IBBR)/Icontrol. Before recording, astemizole was washed out for 1 hour because of its high-affinity block on hERG. In Figure 4A and C, application of 10 μM resveratrol and 1 μM fexofenadine rescued hERG tail current reduction in the presence of 10 μM BBR. The rescue ratios of resveratrol and fexofenadine were 16.38% and 14.00% at +40 mV, respectively. However, astemizole showed no rescue effect on inhibited hERG current by BBR (Figure 4B), which was not coincident with the result from the Western blot.
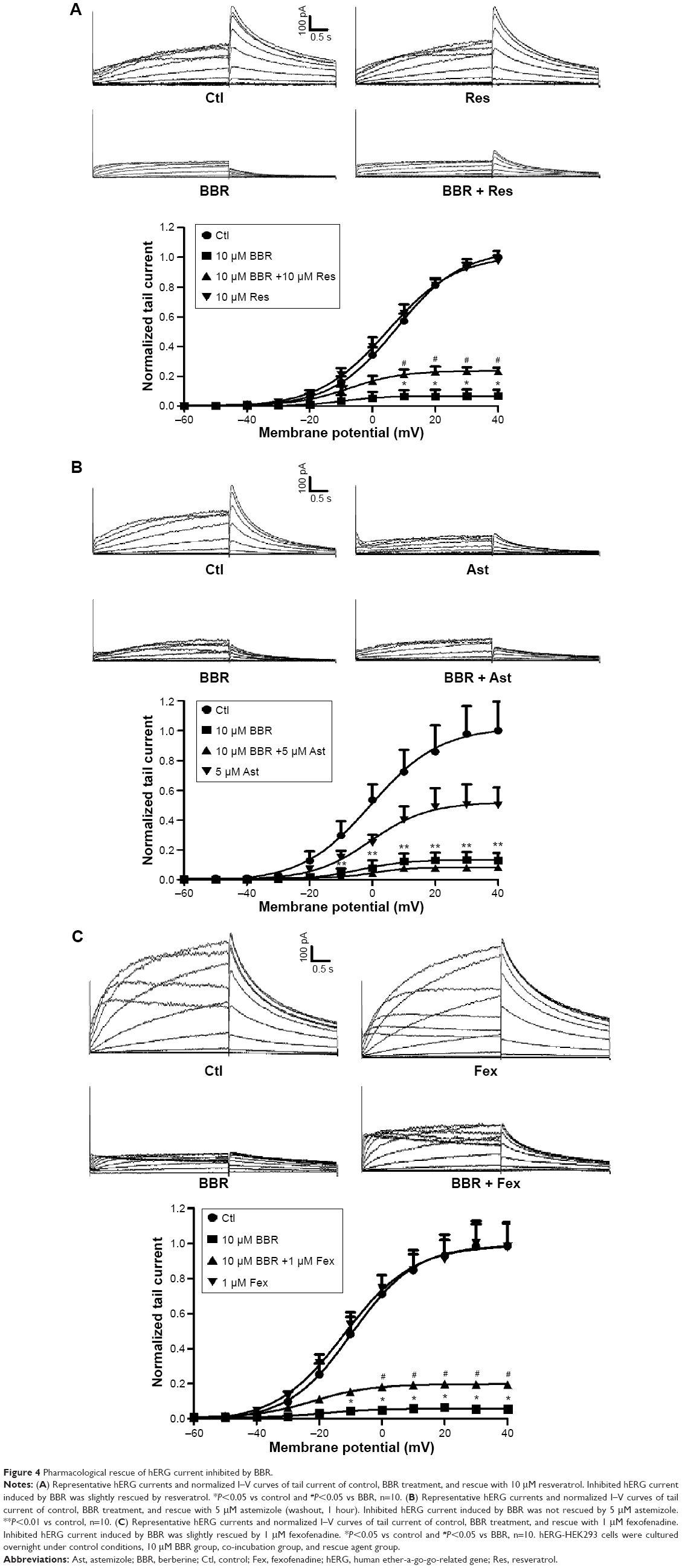 | Figure 4 Pharmacological rescue of hERG current inhibited by BBR. |
Together, these data illustrate that resveratrol and fexofenadine showed functional rescue of hERG channel deficiency triggered by BBR, while astemizole only recovered protein expression.
Reverse of BBR-induced action potential duration prolongation
To find whether fexofenadine and resveratrol could also rescue BBR-induced action potential duration (APD) prolongation,10 we studied the effects of BBR and fexofenadine or resveratrol on action potential recorded in NRVM. Myocytes were incubated for 24 hours under control conditions, with 10 μM BBR, 10 μM BBR +10 μM resveratrol or 1 μM fexofenadine. Figure 5A shows that APD of NRVM was significantly prolonged by 10 μM BBR (blue line). The APD50 and APD90 were prolonged from 5.80±0.49 ms and 25.47±1.28 ms in control to 27.32±1.74 ms and 135.25±11.76 ms, respectively. Resveratrol (10 μM) shortened the prolonged APD by BBR (green line). The APD50 and APD90 were shortened to 10.98±1.80 ms and 46.87±7.27 ms, respectively. Fexofenadine (1 μM) also shortened prolonged APD of NRVM by BBR (red line), the APD50 and APD90 were shortened to 7.75±0.63 ms and 37.78±2.72 ms, respectively.
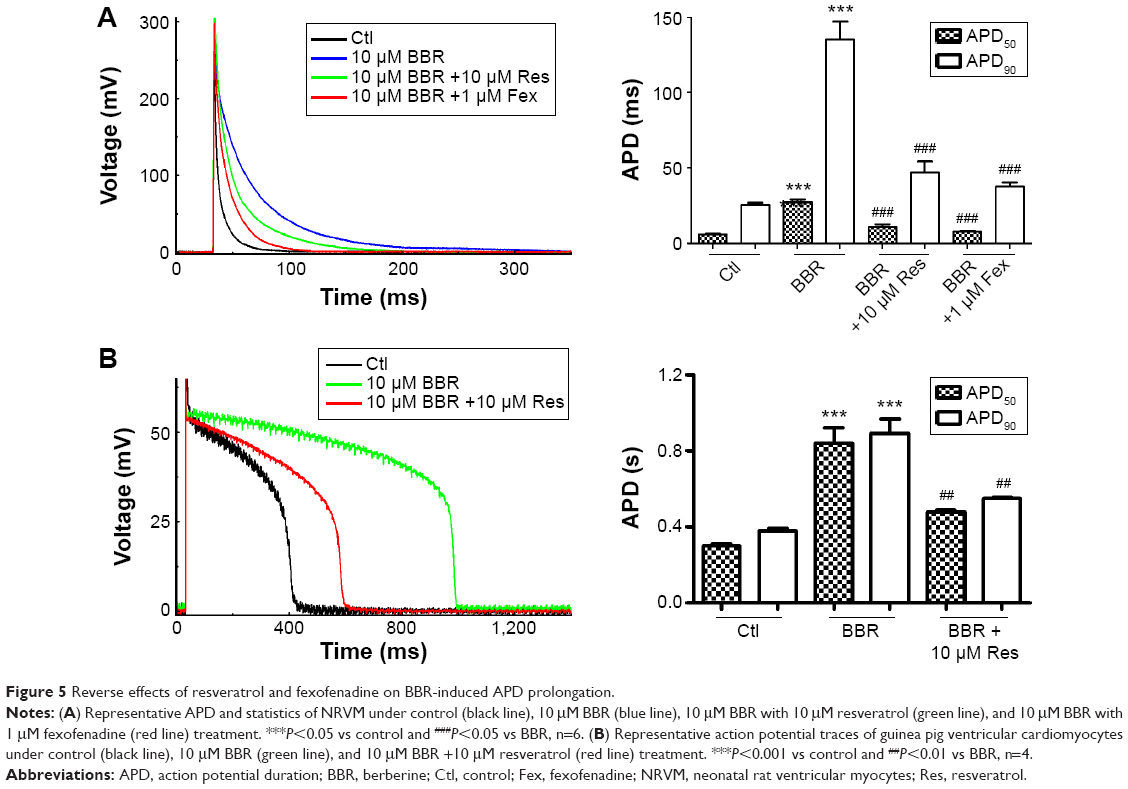 | Figure 5 Reverse effects of resveratrol and fexofenadine on BBR-induced APD prolongation. |
Moreover, we applied 10 μM resveratrol to detect whether prolonged APD in guinea pigs by BBR10 could also be shortened. The freshly isolated guinea pig cardiomyocytes were incubated overnight under control conditions, with 10 μM BBR and 10 μM BBR +10 μM resveratrol. Figure 5B shows that APD50 and APD90 of guinea pigs cardiomyocytes were prolonged from 298.60±12.23 ms and 380.10±10.63 ms of control (black line) to 841.68±77.43 ms and 891.95±75.86 ms, respectively, by 10 μM BBR (green line). Resveratrol (10 μM) (red line) shortened the APD50 and APD90 of guinea pig cardiomyocytes to 478.60±10.09 ms and 549.33±7.30 ms, respectively.
Together, these results suggest that both resveratrol and fexofenadine could be used to rescue BBR-induced APD prolongation of cardiomyocytes.
Discussion
In this study, we demonstrated that 1) cav-1 participates in BBR-induced hERG deficiency on cell surface; 2) residues Tyr652 and Phe656, essential components of hERG binding sites, mediate hERG surface expression inhibition by BBR incubation; and 3) fexofenadine and resveratrol can shorten APD of ventricular cardiomyocytes prolonged by BBR.
BBR is an isoquinoline, which is extensively used to treat gastrointestinal infections in clinics for many years. BBR has been reported previously to block hERG currents expressed in HEK-293 cells and Xenopus oocytes after acute application.8 Recently, we found that long-term application of BBR induced hERG channel reduction by disrupting channel trafficking.10 In addition to acutely blocking hERG channel and indirectly inhibiting hERG protein trafficking to cell membrane, there are clues inferring direct effect of BBR on the membrane. Overall comprehension of the mechanisms that caused hERG dysfunction will help to discover more rescue approaches. Cav-1, an integral membrane protein known to modulate numerous proteins at the plasma membrane including ion channels, has been reported to interact with hERG channel and negatively regulate hERG expression.11 The expression level of caveolin is closely associated with the level of cellular cholesterol.12 Otherwise, BBR was reported to be a cholesterol-lowering drug working through elevating LDLR expression.13 Thus, we proposed the hypothesis that BBR could affect hERG expression via cav-1. The results of our study confirmed this hypothesis. BBR decreased the expression of cav-1 (Figure 1A) and knocking down the basal expression of cav-1 alleviated BBR-induced hERG decrease on plasma membrane (Figure 1B and C). Our data are consistent with the report that probucol, also a cholesterol-lowering drug, decreases hERG expression through accelerating cav-1 degradation.16 Hence, we conclude that BBR reduces surface hERG protein through promoting hERG internalization due to its co-localization with cav-1 on the membrane.
The mechanisms of drug-induced hERG channel deficiency mainly include two types: 1) directly blocking hERG channel and 2) indirectly disrupting channel protein synthesis, trafficking, or degradation. The direct mechanism proposed is binding to the amino acid residues (Tyr652 and Phe656) in the S6 transmembrane helix of hERG channel subunit. High-affinity dofetilide binding to hERG expressed in Xenopus oocytes involves Phe656 residue.17 Quinidine, a class IA antiarrhythmic drug, blocks hERG channel, which is influenced by Tyr652 residue.18 However, the binding sites mediate drug-induced indirect mechanism that is less elucidated. Fluoxetine and norfluoxetine have been found to reduce IhERG by blocking hERG channel and disrupting trafficking; however, the two effects were not mediated by a single binding site. The direct block was mediated by drug binding to S6 domain of the hERG subunit, while trafficking deficiency was mediated by binding to a different site or a related protein.19 In this study, we used site-directed mutagenesis to test whether Y652A or F656V mutation alter the BBR sensitivity of hERG after long-term treatment. In Figure 2, BBR showed no remarkable inhibition of Y652A-hERG and F656V-hERG protein or current after overnight incubation. The result surprisingly manifested that binding to the two residues accounts for BBR-induced hERG defect. For this reason, we propose that BBR binds to both immature and mature conformation of hERG channel, the former leads to conformation defect of hERG, thus abnormal trafficking, and the latter results in direct block.
On account of the high proarrhythmic potential of BBR, we strived to search for rescue approaches of drug-induced hERG dysfunction. hERG channel blockers are widely used to rescue hERG dysfunction. One of the reasons is that many high-affinity hERG channel-blocking drugs, such as E4031 and terfenadine, can result in pharmacological rescue.9,15 However, a great majority of rescued hERG channels are dysfunctional because the binding sites of those drugs that have reverse effects are often the same with the sites as drugs that block the channels. Therefore, we should look forward to more effective and appropriate rescue strategies. In this study, we tested three drugs that have previously been reported to rescue trafficking-deficient hERG channel for restoration of BBR-induced hERG defect. We first chose the cardioprotective drug, resveratrol, which was used to correct trafficking defect caused by arsenic trioxide via acceleration of interaction between hERG and chaperones, and alleviation of ER stress.14 In addition, resveratrol shortened APD50 and APD90 recorded in freshly isolated guinea pig ventricular myocytes by inhibition of Ica-L and restoration of Ikr when co-incubated with arsenic trioxide. We also used astemizole (an antihistamine drug), a high-affinity blocker of hERG channel, which is thought to interact with the amino acid residue Tyr652.20 Astemizole is able to restore not only trafficking of hERG N470D and G601S mutants,9,21 but also trafficking inhibition induced by pentamidine in which astemizole acts as a conformation-sensitive chaperone and competes with pentamidine for overlapping binding sites.5 The third agent is another antihistamine drug called fexofenadine, a weak blocker of hERG, which is thought to be devoid of QT interval prolongation and other serious arrhythmia at clinical relevant concentration.22,23 Fexofenadine also corrects trafficking of hERG N470D, which is different from astemizole that the RC50 of fexofenadine is approximately 300-fold lower than IC50, which indicates that pharmacological rescue can occur without channel block.15 However, the mechanisms of this profitable manner remain unknown. In Figures 3 and 4, we observed that all the three drugs rescued hERG expression on cell surface in the presence of 10 μM BBR. However, proteins rescued by astemizole were nonconducting, the reason of which might be that recovered channels were blocked by astemizole due to its high affinity and washout of the drug is extremely difficult. Nevertheless, the mechanisms of rescue mediated by these drugs still need further exploration.
In summary, we clarified that disruption of cav-1 and binding to the residues, Tyr652 and Phe656, are responsible for BBR-induced hERG reduction on the membrane, which contribute to better understanding of the mechanisms underlying drug-induced LQTS. Moreover, we first investigated the pharmacological rescue of BBR-triggered hERG defect and provided reference for clinical safety of BBR.
Acknowledgment
This work is supported by grants from the Major Program of National Natural Science Foundation of Heilongjiang (number ZD2015015), the National Natural Science Foundation of China (number 31173050), and the Research Fund for the Doctoral Program of Higher Education of China (number 20122307110007).
Disclosure
The authors report no conflicts of interest in this work.
References
Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440(7083):463–469. | ||
Snyders DJ, Chaudhary A. High affinity open channel block by dofetilide of HERG expressed in a human cell line. Mol Pharmacol. 1996;49(6):949–955. | ||
Walker BD, Singleton CB, Bursill JA, et al. Inhibition of the human ether-a-go-go-related gene (HERG) potassium channel by cisapride: affinity for open and inactivated states. Br J Pharmacol. 1999;128(2):444–450. | ||
Dennis AT, Nassal D, Deschenes I, Thomas D, Ficker E. Antidepressant-induced ubiquitination and degradation of the cardiac potassium channel hERG. J Biol Chem. 2011;286(39):34413–34425. | ||
Dennis AT, Wang L, Wan H, Nassal D, Deschenes I, Ficker E. Molecular determinants of pentamidine-induced hERG trafficking inhibition. Mol Pharmacol. 2012;81(2):198–209. | ||
Zhang Y, Dong Z, Jin L, et al. Arsenic trioxide-induced hERG K(+) channel deficiency can be rescued by matrine and oxymatrine through up-regulating transcription factor Sp1 expression. Biochem Pharmacol. 2013;85(1):59–68. | ||
Ficker E, Kuryshev YA, Dennis AT, et al. Mechanisms of arsenic-induced prolongation of cardiac repolarization. Mol Pharmacol. 2004;66(1):33–44. | ||
Rodriguez-Menchaca A, Ferrer-Villada T, Lara J, Fernandez D, Navarro-Polanco RA, Sanchez-Chapula JA. Block of HERG channels by berberine: mechanisms of voltage- and state-dependence probed with site-directed mutant channels. J Cardiovasc Pharmacol. 2006;47(1):21–29. | ||
Ficker E, Obejero-Paz CA, Zhao S, Brown AM. The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether-a-gogo-related gene (HERG) mutations. J Biol Chem. 2002;277(7):4989–4998. | ||
Zhang K, Zhi D, Huang T, et al. Berberine induces hERG channel deficiency through trafficking inhibition. Cell Physiol Biochem. 2014;34(3):691–702. | ||
Lin J, Lin S, Choy PC, et al. The regulation of the cardiac potassium channel (HERG) by caveolin-1. Biochem Cell Biol. 2008;86(5):405–415. | ||
Hailstones D, Sleer LS, Parton RG, Stanley KK. Regulation of caveolin and caveolae by cholesterol in MDCK cells. J Lipid Res. 1998;39(2):369–379. | ||
Kong W, Wei J, Abidi P, et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med. 2004;10(12):1344–1351. | ||
Zhao X, Zhang KP, Huang T, et al. The rescuable function and mechanism of resveratrol on As2O3-induced hERG K(+) channel deficiency. Naunyn Schmiedebergs Arch Pharmacol. 2014;387(11):1079–1089. | ||
Rajamani S, Anderson CL, Anson BD, January CT. Pharmacological rescue of human K(+) channel long-QT2 mutations: human ether-a-go-go-related gene rescue without block. Circulation. 2002;105(24):2830–2835. | ||
Guo J, Li X, Shallow H, et al. Involvement of caveolin in probucol-induced reduction in hERG plasma-membrane expression. Mol Pharmacol. 2011;79(5):806–813. | ||
Lees-Miller JP, Duan Y, Teng GQ, Duff HJ. Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol Pharmacol. 2000;57(2):367–374. | ||
Sanchez-Chapula JA, Ferrer T, Navarro-Polanco RA, Sanguinetti MC. Voltage-dependent profile of human ether-a-go-go-related gene channel block is influenced by a single residue in the S6 transmembrane domain. Mol Pharmacol. 2003;63(5):1051–1058. | ||
Rajamani S, Eckhardt LL, Valdivia CR, et al. Drug-induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br J Pharmacol. 2006;149(5):481–489. | ||
Tu DN, Liao YH, Zhou AR, Xiao H, Wang XP, Li L. Biophysical properties and molecular mechanism of HERG channels blockade by astemizole. Shandong Med J. 2008;48(29):3. | ||
Zhou Z, Gong Q, January CT. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J Biol Chem. 1999;274(44):31123–31126. | ||
Taglialatela M, Castaldo P, Pannaccione A, et al. Cardiac ion channels and antihistamines: possible mechanisms of cardiotoxicity. Clin Exp Allergy. 1999;29(suppl 3):182–189. | ||
Grzelewska-Rzymowska I, Pietrzkowicz M, Gorska M. The effect of second generation histamine antagonists on the heart. Pneumonol Alergol Pol. 2001;69(3–4):217–226. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.