")
Back to Journals » Biologics: Targets and Therapy » Volume 18
MDMX in Cancer: A Partner of p53 and a p53-Independent Effector
Authors Lin W, Yan Y, Huang Q, Zheng D
Received 15 September 2023
Accepted for publication 8 December 2023
Published 31 January 2024 Volume 2024:18 Pages 61—78
DOI https://doi.org/10.2147/BTT.S436629
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Doris Benbrook
Wu Lin,1,* Yuxiang Yan,2,* Qingling Huang,1 Dali Zheng2
1Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Fujian Medical University, Fuzhou, People’s Republic of China; 2Fujian Key Laboratory of Oral Diseases, School and Hospital of Stomatology, Fujian Medical University, Fuzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dali Zheng, Fujian Key Laboratory of Oral Diseases, School and Hospital of Stomatology, Fujian Medical University, 88 Jiao Tong Road, Fuzhou, 350004, People’s Republic of China, Email [email protected]
Abstract: The p53 tumor suppressor protein plays an important role in physiological and pathological processes. MDM2 and its homolog MDMX are the most important negative regulators of p53. Many studies have shown that MDMX promotes the growth of cancer cells by influencing the regulation of the downstream target gene of tumor suppressor p53. Studies have found that inhibiting the MDMX-p53 interaction can effectively restore the tumor suppressor activity of p53. MDMX has growth-promoting activities without p53 or in the presence of mutant p53. Therefore, it is extremely important to study the function of MDMX in tumorigenesis, progression and prognosis. This article mainly reviews the current research progress and mechanism on MDMX function, summarizes known MDMX inhibitors and provides new ideas for the development of more specific and effective MDMX inhibitors for cancer treatment.
Keywords: MDMX, P53, cancer, inhibitors
Introduction
The transcription factor p53 is a powerful tumor suppressor1 that plays an important role in cell growth and development. The TP53 gene, which encodes p53, is mutated or deleted in more than 50% of human cancers, causing p53 to lose its function as a tumor suppressor.2 p53 plays a key role in regulating many cellular processes; thus, the level and activity of p53 are strictly controlled. MDM2 is the main regulator and inhibitor of p53. In cells containing wild-type p53, p53 can bind to MDM2-P2 promoter and regulate MDM2 transcription, leading to an increase in MDM2 mRNA and protein.3 MDM2 protein directly binds to p53 through its amino terminus, which can inhibit the function of p53 by blocking its binding to target DNA and promoting its degradation.4,5
MDMX, also known as MDM4, is a binding protein of p53, which has a high degree of homology with the sequence of MDM2 and is thus named MDMX.6 MDM2 and MDMX have similar functionality, and both can bind to p53 N-terminus and inhibit p53 transcriptional activity.7 MDM2 is an E3 ubiquitin ligase, which can degrade p53. MDMX can promote the E3 ubiquitin ligase activity of MDM2. However, MDMX itself is not an E3 ubiquitin ligase,8,9 and MDMX and MDM2 can directly interact to enhance MDM2 activity and stabilize MDM2 protein.10 Therefore, MDMX is gradually being considered an important component of the p53-MDM2/MDMX pathway.11,12 In the past 30 years, scientists have conducted extensive studies on the p53-MDM2 pathway and found that it is closely related to the progress of cancer.13–15 Since MDMX participates in the regulation of the p53-MDM2 pathway, whether MDMX can participate in human cancer has also become a research direction. Many studies have shown that MDMX is highly expressed in many different cancers and has an impact on the prognosis of tumor patients. Therefore, MDMX may be a very meaningful tumor therapy target. In this review, we will discuss the mechanism of MDMX participating in the occurrence and development of different cancers and the existing MDMX inhibitors.
The Structure of MDMX and Its Effect on p53 Activity
As Figure 1 shows, the MDMX protein is consisted of 490 amino acids and includes four main conserved large domains, namely, the amino-terminal hydrophobic p53 binding domain, the carboxy-terminal RING domain, the zinc finger domain and the central acidic domain.11 Consistent with MDM2, MDMX also interacts with p53 through the N-terminal p53 binding domain to play a role in inhibiting p53 transcription.16 In general, MDMX and MDMX cooperatives inhibit the activation of p53 through the 367th amino acid. Therefore, this domain is crucial for the combination of MDMX and MDM2.17 The mutation of serine to alanine at position 367 enhances the synergistic inhibitory effect of MDM2 and MDMX on the transcriptional activity of p53. The same amino acids are necessary for the interaction between the TAD domain of p53 and MDM2 and MDMX. However, the binding domains of p53 to MDM2 and MDMX are different, resulting in different binding properties of small molecule inhibitors of MDM2 and MDMX.18
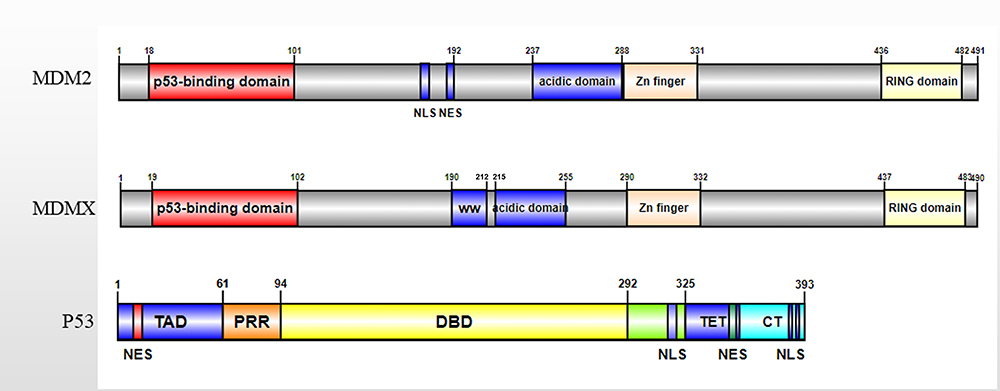 |
Figure 1 Diagram of the MDM2, MDMX, and p53. The structure of MDMX is similar to that of MDM2. Both MDMX and MDM2 contain four domains, including the amino-terminal hydrophobic p53 binding domain, the carboxy-terminal RING domain, the zinc finger domain and the central acidic domain. MDM2 has a unique NLS and NES signal sequence, which is related to the location of MDM2. |
In addition to the p53 binding domain, the acid domain of MDMX can also be combined with p53. The latest research shows that the acid domain of MDMX can inhibit the DNA binding activity of p53 through CK1α and assist MDM2 in controlling the level of p53.19 In addition, the acidic domain of MDMX can disrupt the p53-MDMX interaction.20 The zinc finger domain of MDMX is independent of p53, studies have found that the zinc finger domain interacts with retinoblastoma protein (Rb).21 In addition, there is an MDMX self-inhibitory sequence motif called the WW domain, which is located in the tryptophan-rich fragment and centered on the 200th and 201st tyrosine residues of MDMX. The WW domain of MDMX binds to the N-terminal domain of MDMX and prevents it from interacting with p53.22 The RING domain of MDMX and MDM2 is essential for the dimerization of MDM2 and MDMX proteins.23 This function is related to the interaction between the seven C-terminal amino acid residues (485–491) in the RING domain.24 The RING domain of MDM2 has E3 ubiquitin ligase activity, while the RING domain of MDMX does not.23 Unlike MDM2, the RING domain of MDMX does not have E3 ubiquitin ligase activity, so MDMX does not have the ability to degrade p53. MDMX promotes the ubiquitination degradation of p53 by MDM2 through interaction with MDM2. The C-terminal domain of MDMX can replace the C-terminal of MDM2 in the MDMX-MDM2 complex, thus restoring the E3 ubiquitin ligase activity of MDM2.25 Therefore, the RING domain of MDMX is crucial for the combination of MDMX and MDM2. The deletion or mutation of MDMX will affect the formation of MDMX-MDM2 complex and the regulation of MDM2 on p53.26 This phenomenon indicates that MDMX regulates the E3 ligase activity of mutant MDM2. The interaction of MDM2 and MDMX increases MDM2 protein stability and also enhances the activity of its E3 ligase.25 The MDM2-MDMX heterocyclic complex has stronger E3 ligase activity than the MDM2 homocyclic complex, indicating that the MDM2-MDMX heterodimer has a critical role in the control of p53 activity. The regulation of p53-related MDMX function is summarized in Figure 2.
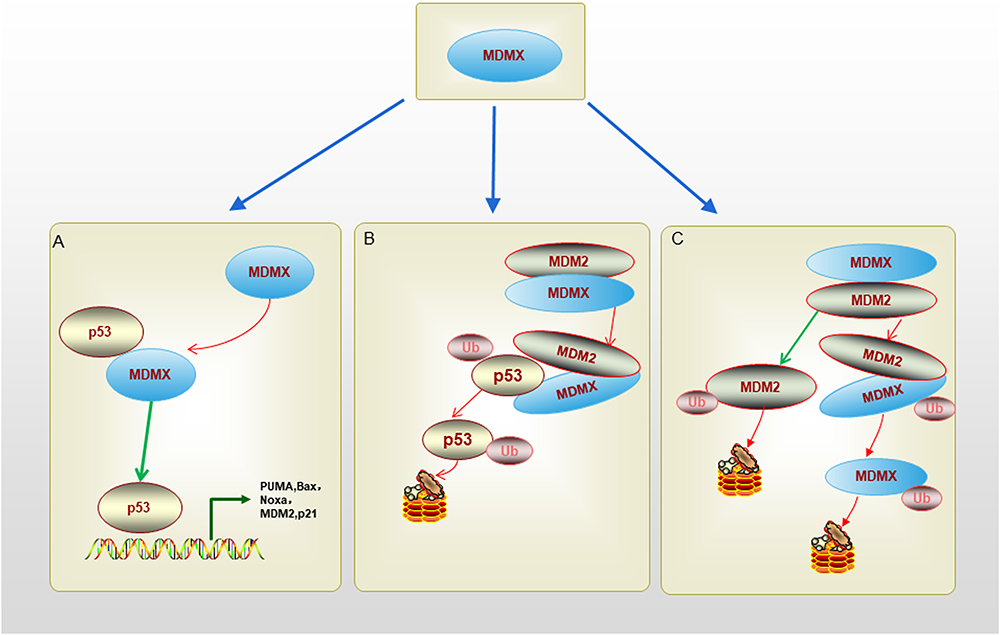 |
Figure 2 The schematic diagram of regulation of p53-related MDMX function. (A). N terminus p53-binding domain of MDMX can bind to p53 transactivation domain directly and inhibit p53 transactivation activity without promoting p53 degradation. (B). MDMX-MDM2 heterodimer is formed after the RING domains of them bind together, the heterodimer can promote MDM2-mediated p53 ubiquitination and degradation. (C). MDMX-MDM2 heterodimer can inhibit MDM2 ubiquitination, and increase MDM2 stability. MDM2 can promote MDMX ubiquitination and degradation too. |
The binding and interaction of p53-MDM2/X have been analyzed by X-ray crystallography, which demonstrates that p53 maintains an α-helical conformation that binds to MDM2 or MDMX. The p53 amino acid residues, including Phe19, Trp23, and Leu26, mediate the MDM2/MDMX/P53 interaction, and the π-π interaction between Phe19 and Trp23 is important for maintaining the complex structural stability.26 The p53 binding sites of MDM2 and MDMX both consist of 14 amino acid residues, four of which are different.27 The spatial orientation of Phe19 and Trp23 residues in the MDM2/MDMX/p53 complex has high similarity, in contrast, the orientation of Leu26 is different.28 The Met53 and Tyr99 residues on the p53 binding domain of MDMX play an important role in maintaining the shape of MDMX, the residues protrude into the hydrophobic crack of MDMX, so the shape of the binding capsule of MDMX is inconsistent with the shape of MDM2.29 The tryptophan-rich sequence located in the central acidic domain of MDMX is important in regulating the interaction between MDMX and p53. Solution-state NMR spectroscopy studies show that the tryptophan-rich central acidic domain has a propensity for adopting an extended structure.30
The Role of MDMX in p53-MDM2 Interaction
The interaction between p53 and MDM2 has been extensively studied.1,31,32 MDM2, a transcription target and negative regulator of p53, can promote tumor formation by targeting p53 for proteasome degradation or inhibiting p53 transcription.33 In the p53-MDM2 regulatory pathway, p53 can promote the transcription of MDM2, thereby regulating the expression of p53 itself.34 MDMX can regulate the protein expression of MDM2 and p53 respectively. In the p53/MDMX/MDM2 loop, MDMX can inhibit the function of p53 by enhancing the E3 ubiquitin ligase activity of MDM2 and inhibiting the transcription activity of p53, but MDMX will not affect the stability of p53.35 The phosphorylation of MDMX caused by casein kinase 1α (CK1α) play an important role in regulation of MDMX-p53 interaction and MDMX-mediated p53 inactivation.36 MDMX-MDM2 heterodimer is the basis for the formation of the p53-MDM2/MDMX loop. The RING domain of MDMX can enhance the E3 ubiquitin ligase activity of MDM2 and promote the p53 ubiquitination mediated by MDM2.23 However, under stress, the spatial conformation of MDMX will change, thereby inhibiting the degradation of p53 by proteasome. Therefore, the regulatory mechanism of MDMX on p53 and MDM2 is very complex, and more research is needed to explain the role of MDMX.37 Jing et al found that the seven amino acids at the C-terminal of MDMX are crucial for the binding of UbcH5c.38 These seven amino acids were grafted onto MDM2, which could make MDM2 interact with E2 ligase UbcH5c. However, the seven amino acids grafted onto other proteins did not interact with UbcH5c. Sequences other than the last seven residues in MDMX seem to be involved in the binding of UbcH5c. Therefore, the author believes that MDMX may recruit UbcH5c through the C-terminal domain to enable UbcH5c to interact with MDM2. This shows that MDMX promotes the transfer of ubiquitin from UbcH5c to p53 by MDM2 by bringing UbcH5c to the adjacent MDM2 RING domain, thereby enhancing the activity of the MDM2 E3 ligase. In addition, the binding of MDM2 to MDMX may cause a conformational change in the RING domain of MDM2, which allows MDM2 to interact with UbcH5c and activate MDM2 E3 ligase activity. This conformational change has been confirmed in some other E3 ligases, such as cIAP1-SMAC and DIABLO.39
How Post-Translational Modifications (PTMs) of MDMX Affect Its Function
PTMs play a key role in protein function regulation because they regulate the protein activity, location, and its interactions with other cellular molecules such as other proteins, nucleic acids and cofactors. After PTMs occur, the physicochemical properties and conformation of proteins will be significantly changed, thus directly changing the binding capacity and function of proteins. Therefore, even if the expression level of the protein does not alter, the function of the protein will also change significantly if the status of post-translational modification changes. PTMs also play regulation roles in MDMX function, such as ubiquitination, phosphorylation and sumoylation, and much more. Here, we summarize the various post-translational modifications involved in MDMX and the effects on the function of MDMX.
Ubiquitination
MDMX can promote the ubiquitination of MDM2, which in turn leads to the ubiquitination of MDMX, which is related to the function of MDM2 in regulating its expression (Figure 2C).40 In addition, Peli1 is another E3 ligase too. It can bind MDMX and polyubiquitinate it, promote MDMX translocation to the cytoplasm and activate p53, which is critical for tumorigenesis.41
SUMOylation
Similar to p53 and MDM2, MDMX can be modified in vivo and in vitro by binding to SUMO-1. Double mutation of two lysine residues K254 and K379 of MDMX abrogated MDMX SUMOylation in vivo. However, the localization, ubiquitination, and degradation of MDMX protein were not affected in SUMOylation-deficient MDMX mutant, and SUMOylation is not required for several activities of MDMX.42
Phosphorylation
MDMX has multiple phosphorylation sites, which can be activated by other protein kinases under external stimulation. After DNA damage, the stress-activated kinase c-Abl will phosphorylate the phosphorylation sites on the amino-terminus of MDMX, leading to the phosphorylation of MDMX and reducing the binding affinity of MDMX and p53.43–45 It has been shown that the MDMX Ser289 site is phosphorylated by kinase CK1α, which stimulates MDMX interaction with p53 and inhibits p53 activities.46,47 At present, there is no clear conclusion on the phosphorylation regulation of MDMX in the case of p53 deletion or mutation. Some studies have proved that the tyrosine kinase receptor AXL may promote the phosphorylation of MDMX in p53 mutant cell lines. Phosphorylation results in the nuclear localization of MDMX and increased affinity between MDMX and MDM2.48 Tumor cell-associated stress also induces down-regulation of p53 in peri-tumor cells through phosphorylation of MDMX-Ser314. As a result, an immunosuppressive TME was generated, reflected in reduced immune cell infiltration into the tumor and impaired M1 polarization of macrophages.49 The nuclear localization of MDMX is related to the phosphorylation of S367. MDMX will interact with several 14-3-3 family proteins, leading to MDMX entering the nucleus and being degraded.17
The Function of MDMX in Human Cancer
MDMX has many cancer-related functions, and the role of MDMX overexpression in tumorigenesis has yielded different results in mouse models. The MDMX-overexpression model showed increased tumorigenesis, mainly in sarcoma.50 However, different model studies have reported increased embryonic mortality in homozygous mice, while tumorigenesis did not increase in heterozygous mice.51 There may be several explanations for this difference, including differences in expression levels. Therefore, the role of MDMX in tumorigenesis remains to be studied.
MDMX Affects the Occurrence and Development of Tumors
Breast Cancer
MDM2 and MDMX are involved in the metastasis of triple-negative breast cancer (TNBC). A decrease in MDM2 leads to an increase in MDMX, but a decrease in MDMX does not lead to an increase in MDM2.52 Studies have shown that, in the context of British cells expressing mTP53, interference with the stability and/or function of the MDMX protein may be beneficial to the survival of patients.53 In breast cancer, MDMX promotes tumor metastasis to the lung and is also related to the upregulation of the G protein-coupled receptor C-X-C chemokine receptor 4 (CXCR4), which promotes metastasis. In addition, MDM2 and MDMX affect the occurrence and development of breast cancer at least to a certain extent, and they depend on the status of estrogen receptor α (ERα).54 MDM4 and MDM2 are elevated in primary human luminal A/B subtype breast cancer independent of p53 mutation. Research shows that the expression of MDMX is increased in primary human cavity type A/B breast cancer, which is related to ERα. ERα can increase the expression of MDMX independently of p53 function, and these effects can be blocked by fulvestrant and tamoxifen. In cells, MDMX inhibits ERα expression, indicating that there is a negative feedback regulation between them. In breast cancer cell lines, MDMX will form protein complexes with ER, which indicates that ER may participate in the regulation of p53 MDM2/MDMX in breast cancer.50 Therefore, the next research should focus on the role of ER in the MDM2/MDMX complex, and prove that this effect may be related to the formation and development of breast cancer.
Gastrointestinal Cancer
MDMX expression affects the prognosis of gastric cancer, as the high expression of MDMX is associated with lymph node metastasis of gastric adenocarcinoma.51 In colorectal cancer, MDMX expression is induced by insulin-like growth factor 1 (IGF1) and activated by the phosphorylation of extracellular signal-associated kinase (ERK),55 suggesting that mitotic signaling regulation may promote p53 inactivation. Amplification of the MDMX gene in colorectal cancer and TP53 mutation is not exclusive. Paradoxically, the level of MDMX protein decreases in cases of gene amplification.56 Therefore, the functional significance of MDMX is still unclear and needs further study. Unlike in hepatocellular carcinoma, TP53 mutation is not a common oncogenic driver in fibrolamellar hepatocellular carcinoma. In tumors lacking p53 mutations, the tumor suppressor activity of p53 is dysregulated in fibrolamellar hepatocellular carcinoma, which is attributed to MDMX overexpression. MDMX is a negative regulator of p53, inhibiting the ability of p53 to induce apoptosis and promote DNA repair. MDMX transcription levels are elevated in most tumor samples, and nuclear MDMX levels are significantly elevated in tumor tissue compared with nonneoplastic liver tissue.57 It is suggested that the expression of MDMX and the increase in nuclear localization may be the potential mechanism of abnormal regulation of p53.
Melanoma
MDMX is overexpressed in about 65% of human melanoma, and mouse experiments have also confirmed that overexpression of MDMX can promote the proliferation of melanoma.58 p53 has proapoptotic activity by regulating the expression of apoptosis related gene. MDMX can promote the proliferation of human metastatic melanoma by inhibiting the proapoptotic activity of p53. In malignant melanoma cells, MDMX will bind to p53 through p53 binding domain and inhibit p53 transcription activity. In addition, MDMX will also degrade p53 by forming the MDMX-MDM2 complex.6 Notably, inhibition of the interaction of MDMX-p53 can restore the activity of p53 in melanoma cells, thereby increasing sensitivity to chemotherapy and mutant BRAF (V600E) inhibitors. Therefore, MDMX is a key factor in the tumorigenesis and development of human melanoma and has been designated as a promising target for anti-melanoma combination clinical therapy.
Prostate Cancer
Although no research has confirmed that MDMX plays a role in the progression of prostate cancer, it is known that the expression of MDM2 is related to advanced prostate cancer. At the same time, TCGA database analysis shows that MDM2 and MDMX will be amplified simultaneously in most prostate cancer cells, and the amplification rate of MDMX in CRPC is higher than MDM2. At the same time, with the increase of tumor malignancy, the proportion of MDM2 and MDMX co-amplified/ co-overexpressed cells will also increase. MDM2 and MDMX will jointly inhibit the progress of CRPC. In addition to the p53 MDM2/MDMX feedback loop, MDM2 and MDMX will inhibit the degradation of androgen receptors, while inhibiting the expression of MDM2 and MDMX can inhibit AR signal, resulting in more potent growth inhibition of prostate cancer cells compared to treatments that activate p53 alone. Therefore, MDM2 and MDMX may regulate the p53 and AR signaling pathways to promote CRPC progression.59
Acute Myeloid Leukemia
It has been found that overexpression of MDMX can affect the progression of AML. Overexpression of MDMX increases the number and competitive advantage of preleukemia stem cells (pre-LSCs). The results of animal experiments show that MDMX can affect the progression of pre-leukemia status to dominant AML in different mice, and MDMX plays this role by activating Wnt/β-Catenin signaling in pre-LSCs. Mechanistically, MDMX binds to CK1α and causes β-Catenin to accumulate in a p53-independent manner. The Wnt/β-Catenin inhibitor reversed the MDMX-induced increasement of β-Catenin in pre-LSCs and synergized with the MDMX-p53 inhibitor.60
Other Tumors
In Burkitt’s lymphoma, increased MDMX cannot regulate the TP53 pathway in the absence of TP53 or MDM2 abnormalities. The protein and mRNA expression levels of MDM2 were not associated with TP53 protein expression; however, detectable TP53 protein expression was inversely associated with MDMX copy number increases and mRNA expression. The TP53 pathway is maladjusted in some BL cases, and increased MDMX expression may be the main mechanism.61 For retinoblastoma, MDMX gene amplification is reported to occur in 65% of tumors and is unrelated to TP53 gene changes.62 Retinoblastoma retains wild-type TP53, and MDMX expansion is thought to block the p53 pathway, leading to tumorigenesis.63 In addition, the mRNA expression of MDMX in non-small-cell lung cancer tissues was significantly higher than that in the corresponding nontumor tissues. High MDMX expression is related to poor differentiation of tumor cells, advanced TNM, and the occurrence of lymph node metastasis.64 MDMX is also found to be overexpressed in acute lymphoblastic leukemia (more than 80%) and head and neck squamous carcinomas (more than 50%).65,66 MDMX is involved in p53 signaling in anaplastic large cell lymphoma (ALCL).67 In wt-p53 ALCL cells, MDMX pharmacological inhibition or siRNA-mediated MDMX silencing was associated with p53 signaling pathway activation, growth inhibition, and apoptotic cell death. Genomic aberration of MDMX has also been observed in salivary gland cancer (SGC) and high expression of MDMX was positively correlated with poor outcomes in patients.68 All these studies emphasize the therapeutic importance of MDMX in tumor therapy. It suggests that targeting MDMX may provide a new treatment method for cancer therapy.
Amplification and Overexpression of MDMX in Pan-Cancer
The abnormal amplification and expression of genes in cancer often suggest their involvement in the process of tumor initiation and development. We downloaded copy number variation (CNV) and gene expression data for 33 cancer types from The Cancer Genome Atlas (TCGA) pan-cancer database and conducted respective analyses using the R programming package. As shown in Figure 3, we analyzed the amplification levels of MDMX in different cancer types based on the TCGA database. The results showed that MDMX was upregulated in most cancers, including breast cancer(BRCA), cholangiocellularcarcinoma (CHOL), Colon Cancer (COAD), Glioblastoma (GBM), Head and Neck Cancer (HNSC), Kidney Clear Cell Carcinoma (KIRC), Kidney Papillary Cell Carcinoma (KIRP), Liver Cancer (LIHC), Lung Adenocarcinoma (LUAD), Lung Squamous Cell Carcinoma (LUSC), Prostate Cancer (PRAD), Rectal Cancer (READ), Stomach Cancer (STAD), Thyroid Cancer (THCA), and conversely, down-regulated in Kidney Chromophobe (KICH). The results of CNV in Figure 3B are generally consistent with the expression levels, except for copy number deletion in KICH, where MDMX exhibited abnormal amplification in a substantial number of cancer samples. We subsequently conducted a correlation analysis of the amplification and expression of MDMX. The results revealed a positive correlation trend across the majority of cancer types, with statistical significance set at p < 0.01 and R > 0.25. Notably, within THYM alone, CNV and mRNA expression manifest a discernible negative correlation trend (Figure 3C), this may be attributed to the insufficient availability of samples with mRNA expression level information in THYM. Furthermore, Table 1 summarizes the amplification and expression status of MDMX among various reported cancer types. The purpose of this endeavor is to furnish valuable clinical relevance, particularly in light of the development of MDMX inhibitors.
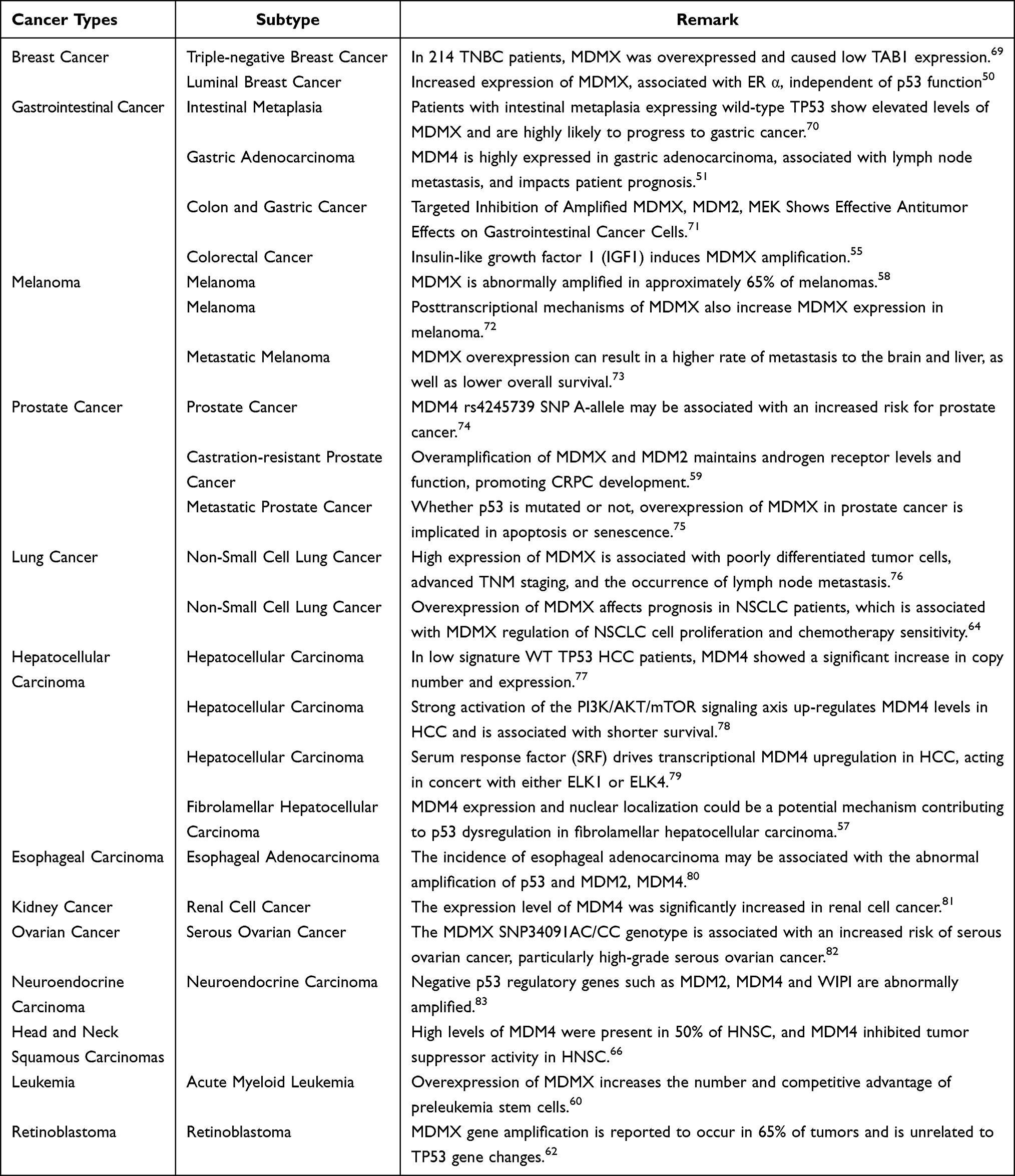 |
Table 1 Amplification and Overexpression of MDMX in Pancancer |
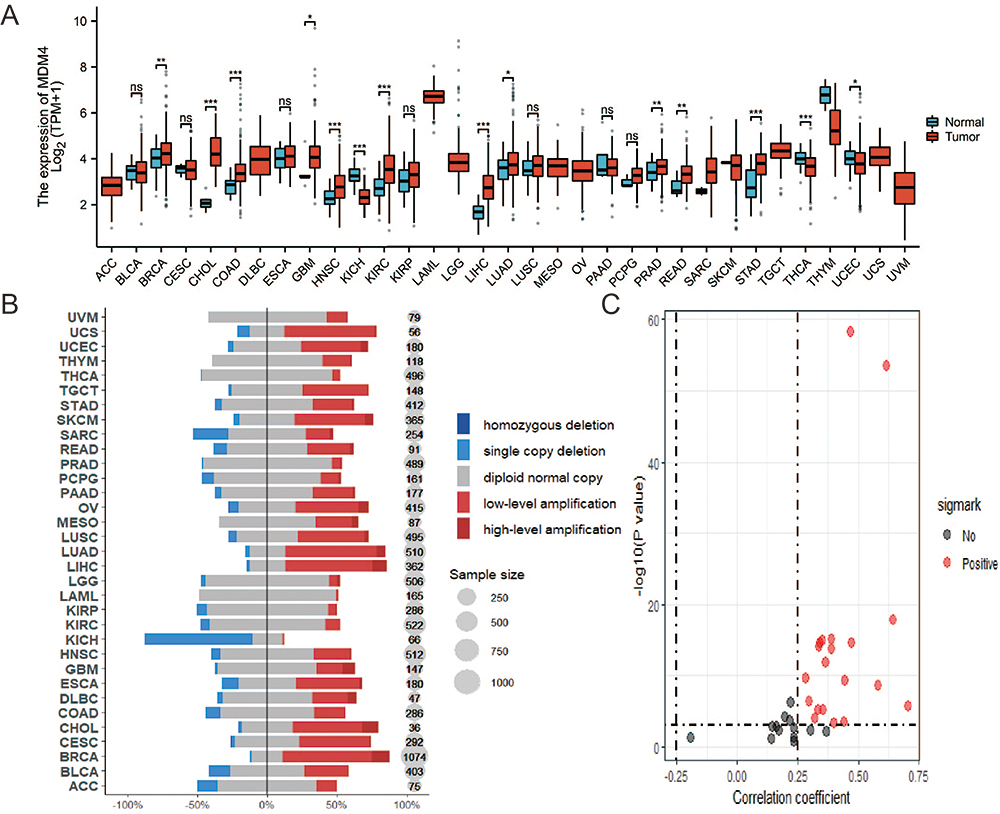 |
Figure 3 Differential amplification and expression of MDMX in various cancers. (A). The expression levels of MDMX across pan-cancer. (B). CNV of MDMX across pan-cancer. (C). Correlation Analysis of MDMX CNV and Expression Levels (*P<0.05; **P<0.01; ***P<0.001; ns, P>0.05, not significant). |
4.2 p53-Independent MDMX Functions
Controversy of p53-Independent Functions of MDMX in Tumorigenesis
The expression levels of MDM2 and MDMX are altered in many type tumor cells, and their role in tumorigenesis independent of p53 has been a significant research subject. Knockout of MDM2 in p53-negative or mutant p53-expressing tumor cell lines has been shown to inhibit cell growth.84 MDM2 knockout also reduces primary tumor volume in some tumors, such as breast cancer.52 MDMX in cancer cells can also affect the tumorigenesis and development of cancer independently through p53. In thymomas with p53 deletion, MDMX can inhibit tumor proliferation,85 and the specific inhibition of mitosis by MDMX’s zinc-finger domain prevents chromosome loss in p53-deficient tumors and inhibits the growth of p53-mutated breast tumors. MDMX’s RING domain also inhibits the proliferation of p53-deficient cancer cells.86 Overexpression of the MDMX gene in p53-deficient mice also reduced survival and increased tumor number.87 In addition, MDMX binds to retinoblastoma protein (RB) in an Mdm2-dependent manner and promotes RB degradation. The RING domain of MDMX would bind to the C-terminal domain of RB to promote the interaction of MDM2 with RB. MDMX would interact with RB to regulate the level of RB protein in vivo and would inhibit RB-mediated E2F1 suppression and cellular senescence. Knockdown of MDMX results in the accumulation of RB protein, arrest of cell cycle G1, inhibition of non-anchored growth, and retardation of tumor growth in vivo in an RB-dependent manner.88 Alternatively, MDMX may be involved in cell cycle regulation in a p53-independent manner by promoting the activity of E2F family members and p73,89 suggesting that MDMX can be used as a potential chemotherapeutic target in cancers lacking wild-type p53.
The Effects of MDMX on DNA Damage and Replication
MDMX can regulate the cellular response to DNA damage. In this regard, MDMX seems to cause greater genomic instability through its association with Nbs1, which MDMX binds to inhibit DNA break repair when ectopically expressed.90 Overexpression of MDMX inhibitsthe interaction between MDMX and ATM and slows the DNA damage response. Interestingly, this function of MDMX is independent of p53 and MDM2.91 However, the exact molecular mechanism needs extra exploration. MDMX may also play a key role in DNA replication. Studies have shown that the processing and replication of DNA in tumor-derived cells and primary cells lacking wild-type p53 are dependent on MDMX.91 In the absence of MDMX, DNA replication is severely damaged, but the overexpression of MDMX and RNF2 (RING finger protein 2, also known as RING1B) can restore DNA replication. Therefore, MDMX can act as a promoter of DNA replication independent of p53. In addition, the absence of MDMX can delay the progression of replication forks and make tumor cells sensitive to gemcitabine, which suggests that MDMX may play a role in malignant tumors.91 MDMX can also interact with members of the Polycomb Repressor Complexes and promote the ubiquitination of H2A, thereby prevent the accumulation of DNA/RNA-hybrids, and support DNA replication.91
Tumor Inhibitors Related to MDMX
There is increasing evidence that p53 inactivation in these cancers is usually the result of upregulation of MDM2 and MDMX protein levels. Therefore, the development of related inhibitors has become an effective method of anticancer treatment. MDM2 inhibitors have been used in clinical trials alone or in combination, but clinical experience with these drugs remains limited. So far, most MDM2 inhibitors treat p53 wild-type cancer by inhibiting the interaction between MDM2 and p53, and a lot of such inhibitors have entered the clinical trial stage. The targeted toxicity and effects of these agents in lymphatic organs and the gastrointestinal tract have been reported in preclinical studies.92 Previous studies have shown us the positive application prospects of MDM2 inhibitors. However, more in vitro experiments and clinical trials are needed to prove the role of MDM2 inhibitors in cancer treatment, especially for the toxicity of inhibitors to normal cells.93 Similar to MDM2, the MDMX inhibitors are explored and some of them have been used in clinical trials, Figure 4 illustrates the mechanisms of three types MDMX inhibitors. The information of MDMX inhibitors is summarized in Table 2.
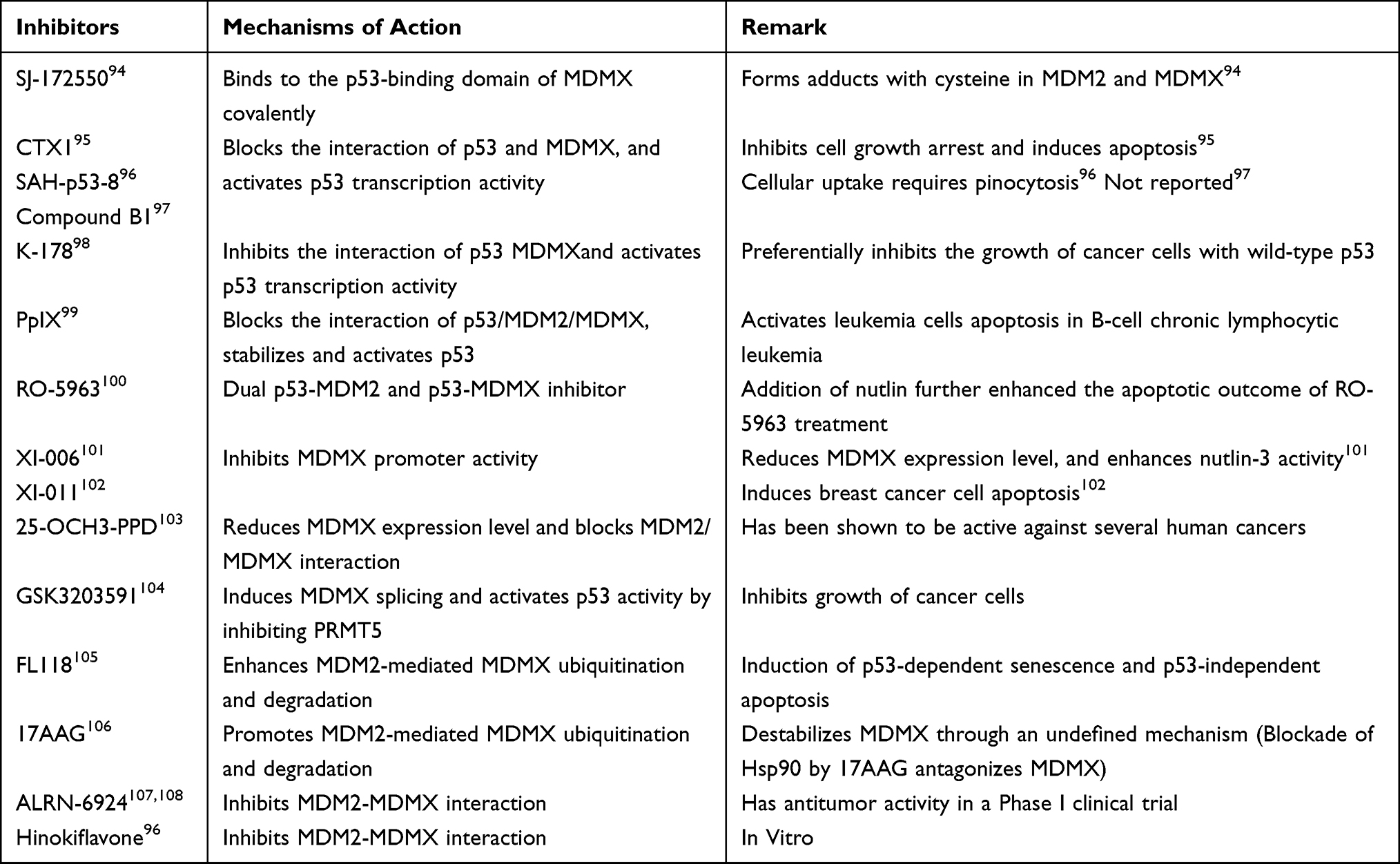 |
Table 2 A List of Current Available MDMX Inhibitors |
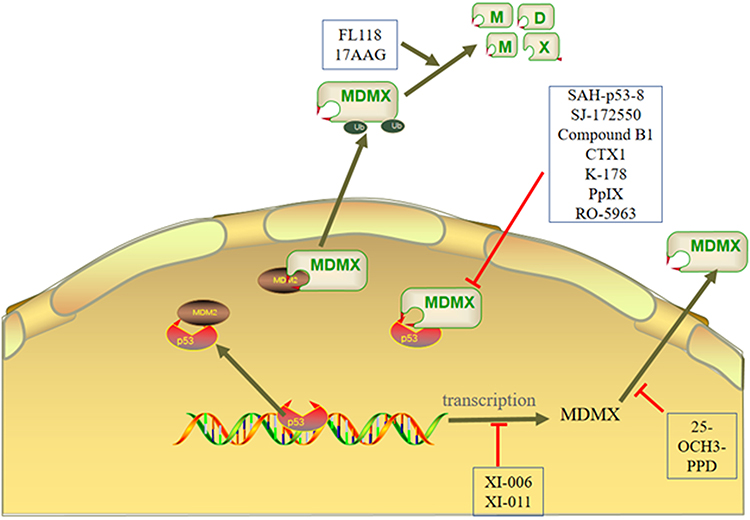 |
Figure 4 The mechanisms of MDMX inhibitors are mainly divided into three types. First, the inhibitor inhibits the expression of MDMX. Second, the inhibitor affects the formation of p53-MDM2/MDMX complex. Third, the inhibitor activates the E3 ligase activity of MDM2 to degrade MDMX. |
Inhibiting p53-MDMX Binding
The binding mechanism of p53 MDM2/MDMX complex has been clarified. Therefore, many inhibitors that inhibit the interaction between p53 and MDMX have been developed to inhibit tumor growth.109,110 So far, at least a dozen inhibitors that can directly inhibit the binding of MDMX and p53 have been found such as SAH-p53-8, SJ-172550, mSF-SAH, WK298, RO-5963, pyrrolopyrimidine 3a, Compound B1, CTX1, and K-178, which have been shown to have anticancer effects in vivo and in vitro. Among them, RO-5963 and pyrrolopyrimidine 3a are dual inhibitors of p53-MDM2 and p53-MDMX. Some inhibitors can activate p53 in vitro, while some inhibitors have been proved to have anti-cancer effects in vivo. SAH-p53-8 can block the combination of p53-MDMX and activate the p53 signaling pathway, thereby inhibiting the growth of cancer cells in vitro and in vivo, inducing apoptosis and overcoming MDMX-mediated cancer chemoresistance. Hinokiflavone has been proved to inhibit the interaction between MDMX and MDM2 by combining with the RING domain of MDMX. In addition, in different cancer cells, Hinokiflavone can inhibit the expression of MDMX and MDM2, thus activating p53 to induce apoptosis.96 ALRN-6924 is the first cell-penetrating stapled α-helical peptide, consistent with Hinokiflavone, the mechanism of action of ALRN-6924 is interfering with the interaction between p53 and MDMX/MDM2. It is worth noting that ALRN-6924 has shown antitumor activity in a phase I clinical trial in patients with solid tumors.107,108 SJ-172550 can bind to the p53 binding domain of MDMX, thereby blocking p53-MDMX binding, activating wild-type p53, and inducing apoptosis of retinoblastoma cells overexpressing MDMX.94 Compound B1 can also inhibit the binding of MDMX-p53 detected by the FP-based method, but the anticancer efficacy of Compound B1 has not yet been determined.95 Karan et al screened an MDMX inhibitor CTX1 through cell experiment. In breast cancer cell line MCF7, CTX1 can promote cell apoptosis by restoring p53 activity.95 Further studies showed that CTX1 will competitively inhibit the combination of p53 and MDMX through interaction with MDMX, thus restoring the p53 activity destroyed by MDMX.95 Uesato et al screened a class of oaminothiophenal derivatives, proving that s-2-isobutylamide phenyl 2-methyl- propylthioate (K-178) can specifically activate p53 and inhibit some cancer cell lines by inhibiting the interaction between p53 and MDMX, including human breast cancer (MCF-7), lung cancer (A427), and colon cancer (HCT116) cell lines.98 PpIX can inhibit the interaction of p53 and MDM2/MDMX at the same time and promote apoptosis in B-cell chronic lymphocytic leukemia cells in the absence of light and without affecting normal cells. However, the exact mechanism of PpIX inhibiting the interaction between proteins is still unclear, which may be related to the N-terminal domain of p53 and p73.99 RO-5963 inhibits the interaction of p53 with MDM2 and MDMX by inducing the formation of dimeric protein complex maintained by dimeric small molecule core. However, RO-5963 can lead to an increase of MDM2 content. Therefore, it may be a feasible method to use RO-5963 and nutlin in the synergistic treatment of cancer. This function effectively stabilizes p53 and activates p53 signaling pathway in cancer cells, leading to cell cycle arrest and cell apoptosis.100
Inhibiting the Expression of MDMX
In addition to inhibiting the interaction between p53 and MDMX, directly inhibiting the expression of MDMX to enhance the expression of p53 target genes (such as p21, PUMA) is also an effective way to activate the anticancer activity of p53. Wang et al screened the NCI diversity chemical library for promoters and discovered a compound XI-006, which reduces MDMX mRNA and protein levels by inhibiting the activity of the MDMX promoter.111 In addition, XI-006 can promote the expression of related transcription factors downstream of p53 in breast cancer cell MCF7, resulting in MCF-7 apoptosis. XI-006 can also enhance p53 activation induced by nutlin-3 (an MDM2 antagonist) and its inhibitory effect on cancer cell viability in vitro.111 Pishas et al further demonstrated the anticancer activity of XI-006 in Ewing’s sarcoma and osteosarcoma cell lines in vitro.101 However, the apoptosis of MCF7 cells has little relationship with the change of MDMX expression level, which indicates that XI-006 may not only participate in the regulation of MDMX but also affect the regulation of MDMX on p53 activity.101 In addition, studies have discovered another MDMX inhibitor called XI-011, which can induce breast cancer cell line MCF7 apoptosis by activating p53.102 XI-011 reduces the mRNA level of MDMX by inhibiting the activity of the MDMX promoter, and the expression level of MDMX plays an critical role in the drug activity of XI-011. The exact mechanism of XI-006 and XI-011 needs to be further studied. Luteolin also inhibits the occurrence and development of cancer by inhibiting the expression of MDMX. In A549 and H460, luteolin promotes the expression of miR-34a-5p, and overexpression of miR-34a-5p inhibits the expression of MDMX, which increases the expression level of p53 and induces apoptosis of cancer cells. Luteolin also inhibited tumor growth in H460 xenotransplantation mice.112 In addition, tanshinone IIA has been identified as a new MDMX inhibitor. Studies have found that tanshinone IIA inhibits the expression of MDMX by inhibiting MDMX mRNA synthesis, which leads to a decrease of inhibitor of apoptosis 3(IAP3), and sensitizes MDMX-overexpressing cells to apoptosis.113 Further studies have shown that tanshinone IIA can both induce apoptosis of H1299 cells and enhance apoptosis induced by doxorubicin.113 Ginsenosides are a kind of natural product with multiple biological activities. 25-OCH3-PPD, a new ginsenoside, has strong anticancer activity in cancer research models in vitro and in vivo, and the host toxicity is minimal.103 The RING domain of MDM2 is the target of 25-OCH3-PPD. In prostate cells, 25-OCH3-PPD can affect the stability of MDM2 by destroying the formation of MDMX-MDM2 complex, thereby strongly inhibiting the growth and metastasis of prostate cancer cells.114 Protein arginine methyltransferase 5(PRMT5) regulates MDMX abundance by affecting its alternative splicing.115 Studies have found that GSK3203591, a specific inhibitor of PRMT5, can induce the alternative splicing of MDMX and activate the p53 activity. Further studies have shown that GSK3203591 inhibits the growth of cancer cells in vitro and inhibits tumor growth in mouse model of lymphoma transplantation in vivo.104
Promotion of Ubiquitination and Degradation of MDMX
Inhibiting the combination of MDMX and MDM2 can also affect the regulation of MDM2 on p53. Ling et al reported that FL118 can inhibit p53 ubiquitination and promote the degradation of the MDMX protein.116 FL118 can degrade MDMX by enhancing the E3 ubiquitin ligase activity of MDM2. In addition, FL118 can inhibit the combination of p53 and MDM2 and increase the interaction between MDM2 and MDMX, thus restoring the anti-tumor activity of p53. Interestingly, FL118 can also promote p53 independent apoptosis in cancers with p53 deletion. In HCT116 cells, FL118 can induce p53 independent apoptosis.116 How FL118 changes the biochemical properties of the MDM2-MDMX E3 complex is still an open question. It remains to be determined whether FL118 binds directly to MDM2 or MDMX, E2 enzymes that alter ubiquitination, or whether FL118 binds to other MDM2/MDMX complex-related proteins. Sesquiterpenoids are a kind of natural product with diverse structures and pharmacological activities, and they have strong anticancer activity. Among them, chrysanthemum lactone A will also destroy the interaction between MDMX and MDM2 by acting on the RING domain of MDM2 disrupting the interaction between MDM2 and MDMX and promoting the ubiquitination and degradation of these two proteins.105 Vaseva et al found that the Hsp90 inhibitor 17-allylamino-17-desmeth oxygelnamycin (17AAG) induces apoptosis in cancer cells.106 Studies have found that 17AAG can induce the degradation of the MDMX protein, however, the molecular mechanism of 17AAG-induced degradation of MDMX remains to be further studied.
Tumor Immunotherapy Related to MDMX
Cancer often accompanies uncontrolled cell proliferation and immune evasion, which are key hallmarks of tumorigenesis. The importance of immunotherapy in cancer treatment has become increasingly evident, and in recent years, there has been a growing body of literature focusing on the role of MDMX in immunotherapy. Immune checkpoint inhibition (ICI) is a form of immunotherapy that involves the inhibition of immune checkpoint proteins, such as PD-1, CTLA-4, and others, to restore a patient’s intrinsic immune response, enable more effective recognition and attack of cancer cells. However, not all cancer patients respond to ICI, and some may experience hyperprogressive disease (HPD) following ICI treatment, resulting in a rapid increase in tumor burden.
Degradation of MDM2/MDMX via ubiquitin-dependent pathways has been shown to effectively restore the function of p53 and p73 both in vitro and in vivo, leading to the activation of CD8+ T cells and rendering them more responsive to anti-PD-1 immunotherapy.117 Patients with melanoma liver metastases exhibit limited responses to anti-PD-1 monotherapy, but can benefit from MDM2/X inhibitors when used in combination with anti-PD-1 therapy.73 MDMX-Ser314 phosphorylation induces the downregulation of p53 in tumor-associated cells, creating an immunosuppressive tumor microenvironment. However, blocking MDMX-Ser314 phosphorylation reduces immunosuppression and significantly delays tumor growth.49 It is worth noting that not only can ICI therapy potentially benefit from MDMX inhibition, but MDMX amplification may also be associated with HPD. MDM4 amplification has been linked to reduced survival rates in NSCLC patients treated with ICI.118 In the first-line combined immunotherapy for HCC, MDMX amplification was found to be an effective predictive marker of HPD in HCC patients.119 To date, MDMX amplification has been demonstrated to serve as a negative biomarker for ICI treatment in various types of cancer, potentially leading to rapid disease progression.120
In summary, the role of MDMX in the future of cancer immunotherapy is a topic of utmost significance. Further research will aid in a better understanding of MDMX’s function in various cancer types and its potential as a biomarker for predicting responses to ICI treatment. This holds the promise of helping physicians tailor immunotherapy regimens more effectively, thereby improving the chances of survival and quality of life for cancer patients. By delving into the mechanisms of MDMX and its association with HPD, we may gain a deeper insight into overcoming the limitations of ICI therapy, enabling the broader application of this vital immunotherapeutic approach.
The Splicing Body of MDMX
Alternative splicing is one of the fundamental mechanisms that regulates gene expression and plays an important role in cell biology, including cancer cell biology, where alternative splicing contributes to the diversity of proteins that directly determine the cell state.121 In humans, approximately 95% of multiexon genes have alternative splicing; therefore, it is important to study alternative splicing and to understand the function of different splice isoforms of individual genes under normal physiological or pathological conditions.122
Although missense mutations of MDMX are rarely observed in human tumors, alterations in the MDMX sequence due to alternative splicing have been reported in tumors and normal cell lines. Using optional promoters and optional splicing, MDMX generates multiple protein isoforms, further increasing complexity. MDMX splice variants are mainly found in tumors and cancer cell lines. For example, MDMX-B splicing variants have been found in human gliomas,123 and their expression levels are related to cancer stages. The WWW inhibitory sequence in MDMX is unique to MDMX and does not exist in MDM2, and it competes with p53 TAD 1 for binding to the N-terminal domain of MDMX.22 MDMX splicing variants lacking WWW inhibitory sequences have potential carcinogenic effects and exist in some aggressive tumors. Many studies have confirmed the MDMX splice variant (MDMX-S) as a possible target for tumorigenesis.72,123–126 The ectopic expression of the MDMX-S protein spliceosome has a higher binding affinity to p53 and has a stronger inhibitory effect on p53 than full-length MDMX. The overexpression of MDMX and MDMX-S has also been confirmed in mantle cell lymphoma, which inhibits p21, thereby promoting cell cycle progression.127 Although the expression of MDMX-S mRNA is often observed, the endogenous MDMX-S protein is not detectable in any normal or cancer cell lines, which means that endogenous MDMX-S may be very unstable There may be a mutual conversion between the protein and MDMX-FL. MDMX-S may play a key role in controlling the level of MDMX-FL protein and activating p53. However, although MDMX-S is also overexpressed in B-cell chronic lymphocytic leukemia (B-CLL), it does not cause tumor formation, nor does it cause tumor invasiveness; in contrast, overexpression of MDMX-S in B-CLL is a consequence of tumorigenesis.128 In addition, another abnormal splicing form of the MDMX spliceosome, MDMX-211, was also found to bind to the MDM2 protein.129 Although MDMX-211 cannot directly bind to p53, MDMX-211 can stabilize p53 by inhibiting MDM2-mediated degradation of p53. MDMX-A was detected in the cervical cancer cell line C33A.130 This variant lacks an in-frame encoded exon-9, and most of the acidic region is missing. Furthermore, this substitutional deletion of exon-9 is associated with MDM2 degradative activity and may control the stability of MDMX-FL. At the same time, the deletion of the acidic domain of MDMX can release the acidic domain of MDM2, further promoting its degradation function, especially the degradation of p53.130 However, it has not yet been reported whether MDMX-A is associated with cancer. To date, the detection of protein isoforms remains a challenge. There are still many unsolved mysteries about the existence and expression levels of MDM2 and MDMX splicing variants in tumors.
Summary
When MDMX was first discovered, it was only considered as a cofactor of MDM2, but now MDMX is considered as a key negative regulator of p53. Since MDMX is often amplified and/or overexpressed in various types of cancer and plays an important role in controlling the p53-MDM2/MDMX cycle, MDMX has been proved to be a promising molecular target for cancer treatment. Therefore, targeting MDMX to treat cancer is very meaningful research. p53-MDMX binding inhibitors play an anti-cancer role by inhibiting the binding of p53 and MDMX to activate p53 and downstream transcription factors. However, p53 is missing or mutated in many cancers, so the research on p53-MDMX binding inhibitors has limited significance.131 Inhibitors such as ALRN-6924 and XI-011 that inhibit the combination of MDMX and MDM2 or the transcription of MDMX have entered the clinical trial stage. However, we should not ignore the side effects of these inhibitors. It is worth noting that the regulatory mechanism of MDMX expression has not been fully elucidated, which is crucial for the development of such MDMX inhibitors. Promoting MDM2 mediated ubiquitination and degradation of MDMX is the main regulation mechanism of MDMX. In addition, it is also a feasible method to degrade MDMX through protein targeted chimera. Recently discovered splicing dependent mechanism of MDMX overexpression in tumors provides another option for inhibiting MDMX.132 MDMX will destroy the response of p53 dependent cells to external stimulation and MDMX inhibitor treatment.27 Because these MDMX spliceosomes play a key role in regulating the protein stability and activity of MDMX, MDM2 and p53, they may also become targets for the development of anti-cancer drugs. Paradoxically, more and more evidence show that MDMX may also have tumor inhibition effect under stress. As a response to DNA damage, MDMX is expressed in mitochondria, and MDMX can enhance the phosphorylation of p53 and its mitochondrial localization under stress, promote the combination of p53 and Bcl2, and promote p53 mediated apoptosis.133 In addition, under stress such as DNA damage, virus infection, c-Abl tyrosine kinase can be activated, then phosphorylate MDMX at tyrosine 99 (Tyr99), thereby inhibiting MDMX-p53 binding and activating p53-dependent cell apoptosis.134
In summary, MDMX is not only a carcinogen dependent on p53, but also has a carcinogenic function independent of p53. Exploring the function of MDMX may provide a new research direction for developing effective MDMX inhibitors. New drugs designed to effectively inhibit MDMX represent a promising future direction in the targeted treatment and surveillance of those tumors with MDMX overexpression.
Funding
This study was supported by grants from the National Natural Science Foundation of China (82172911, 82173180), Fujian Provincial Health Technology Project (2020CXA049), the Scientific Research Foundation for Advanced Talents of Fujian Medical University (XRCZX2020028), and the Natural Science Foundation of Fujian province(2021J01668).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Wade M, Li Y-C, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13(2):83–96. doi:10.1038/nrc3430
2. Feki A, Irminger-Finger I. Mutational spectrum of p53 mutations in primary breast and ovarian tumors. Crit Rev Oncol Hematol. 2004;52(2):103–116. doi:10.1016/j.critrevonc.2004.07.002
3. Oliner JD, Saiki AY, Caenepeel S. The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb Perspect Med. 2016;6(6):a026336. doi:10.1101/cshperspect.a026336
4. Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7(7):1126–1132. doi:10.1101/gad.7.7a.1126
5. Freedman DA, Wu L, Levine AJ. Functions of the MDM2 oncoprotein. Cell Mol Life Sci. 1999;55(1):96–107. doi:10.1007/s000180050273
6. Shvarts A, Steegenga WT, Riteco N, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996;15(19):5349–5357. doi:10.1002/j.1460-2075.1996.tb00919.x
7. Wang W, Qin -J-J, Rajaei M, et al. Targeting MDM2 for novel molecular therapy: beyond oncology. Med Res Rev. 2020;40(3):856–880. doi:10.1002/med.21637
8. Finch RA, Donoviel DB, Potter D, et al. mdmx is a negative regulator of p53 activity in vivo. Cancer Res. 2002;62(11):3221–3225.
9. Tackmann NR, Zhang Y. Mouse modelling of the MDM2/MDMX-p53 signalling axis. J Mol Cell Biol. 2017;9(1):34–44. doi:10.1093/jmcb/mjx006
10. Wang X, Wang J, Jiang X. MdmX protein is essential for Mdm2 protein-mediated p53 polyubiquitination. J Biol Chem. 2011;286(27):23725–23734. doi:10.1074/jbc.M110.213868
11. Marine J-C, Jochemsen AG. MDMX (MDM4), a promising target for p53 reactivation therapy and beyond. Cold Spring Harb Perspect Med. 2016;6(7):a026237. doi:10.1101/cshperspect.a026237
12. Haupt S, Mejía-Hernández JO, Vijayakumaran R, Keam SP, Haupt Y. The long and the short of it: the MDM4 tail so far. J Mol Cell Biol. 2019;11(3):231–244. doi:10.1093/jmcb/mjz007
13. Wang S, Zhao Y, Aguilar A, Bernard D, Yang C-Y. Targeting the MDM2-p53 protein-protein interaction for new cancer therapy: progress and challenges. Cold Spring Harb Perspect Med. 2017;7(5):a026245. doi:10.1101/cshperspect.a026245
14. Qin -J-J, Li X, Hunt C, Wang W, Wang H, Zhang R. Natural products targeting the p53-MDM2 pathway and mutant p53: recent advances and implications in cancer medicine. Genes Dis. 2018;5(3):204–219. doi:10.1016/j.gendis.2018.07.002
15. Gupta A, Shah K, Oza MJ, Behl T. Reactivation of p53 gene by MDM2 inhibitors: a novel therapy for cancer treatment. Biomed Pharmacother. 2019;109:484–492. doi:10.1016/j.biopha.2018.10.155
16. Popowicz GM, Czarna A, Holak TA. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle. 2008;7(15):2441–2443. doi:10.4161/cc.6365
17. Okamoto K, Kashima K, Pereg Y, et al. DNA damage-induced phosphorylation of MdmX at serine 367 activates p53 by targeting MdmX for Mdm2-dependent degradation. Mol Cell Biol. 2005;25(21):9608–9620. doi:10.1128/MCB.25.21.9608-9620.2005
18. Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A. 2008;105(10):3933–3938. doi:10.1073/pnas.0708917105
19. Huang Q, Chen L, Yang L, et al. MDMX acidic domain inhibits p53 DNA binding in vivo and regulates tumorigenesis. Proc Natl Acad Sci U S A. 2018;115(15):E3368–77. doi:10.1073/pnas.1719090115
20. Chen L, Borcherds W, Wu S, et al. Autoinhibition of MDMX by intramolecular p53 mimicry. Proc Natl Acad Sci U S A. 2015;112(15):4624–4629. doi:10.1073/pnas.1420833112
21. Uchida C, Miwa S, Isobe T, et al. Effects of MdmX on Mdm2-mediated downregulation of pRB. FEBS Lett. 2006;580(7):1753–1758. doi:10.1016/j.febslet.2006.02.029
22. Bista M, Petrovich M, Fersht AR. MDMX contains an autoinhibitory sequence element. Proc Natl Acad Sci U S A. 2013;110(44):17814–17819. doi:10.1073/pnas.1317398110
23. Leslie PL, Ke H, Zhang Y. The MDM2 RING domain and central acidic domain play distinct roles in MDM2 protein homodimerization and MDM2-MDMX protein heterodimerization. J Biol Chem. 2015;290(20):12941–12950. doi:10.1074/jbc.M115.644435
24. Uldrijan S, Pannekoek W-J, Vousden KH. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J. 2007;26(1):102–112. doi:10.1038/sj.emboj.7601469
25. Kawai H, Lopez-Pajares V, Kim MM, Wiederschain D, Yuan Z-M. RING domain-mediated interaction is a requirement for MDM2’s E3 ligase activity. Cancer Res. 2007;67(13):6026–6030. doi:10.1158/0008-5472.CAN-07-1313
26. Koo N, Sharma AK, Narayan S. Therapeutics targeting p53-MDM2 interaction to induce cancer cell death. Int J Mol Sci. 2022;23(9):5005. doi:10.3390/ijms23095005
27. Toledo F, Wahl GM. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int J Biochem Cell Biol. 2007;39(7–8):1476–1482. doi:10.1016/j.biocel.2007.03.022
28. Fu T, Min H, Xu Y, Chen J, Li G. Molecular dynamic simulation insights into the normal state and restoration of p53 function. Int J Mol Sci. 2012;13(8):9709–9740. doi:10.3390/ijms13089709
29. Fang Y, Liao G, Yu B. Small-molecule MDM2/X inhibitors and PROTAC degraders for cancer therapy: advances and perspectives. Acta Pharm Sin B. 2020;10(7):1253–1278. doi:10.1016/j.apsb.2020.01.003
30. Song Q, Liu X-Q, Rainey JK. 1H, 15N and 13C backbone resonance assignments of the acidic domain of the human MDMX protein. Biomol NMR Assign. 2022;16(1):171–178. doi:10.1007/s12104-022-10081-8
31. Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R. The MDM2-p53 pathway revisited. J Biomed Res. 2013;27(4):254–271. doi:10.7555/JBR.27.20130030
32. Levav-Cohen Y, Goldberg Z, Tan KH, et al. The p53-Mdm2 loop: a critical juncture of stress response. Subcell Biochem. 2014;85:161–186.
33. Yu D-H, Xu Z-Y, Mo S, Yuan L, Cheng X-D, Qin -J-J. Targeting MDMX for cancer therapy: rationale, strategies, and challenges. Front Oncol. 2020;10:1389. doi:10.3389/fonc.2020.01389
34. Zauberman A, Flusberg D, Haupt Y, Barak Y, Oren M. A functional p53-responsive intronic promoter is contained within the human mdm2 gene. Nucleic Acids Res. 1995;23(14):2584–2592. doi:10.1093/nar/23.14.2584
35. Huang L, Yan Z, Liao X, et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc Natl Acad Sci U S A. 2011;108(29):12001–12006. doi:10.1073/pnas.1102309108
36. Chen L, Li C, Pan Y, Chen J. Regulation of p53-MDMX interaction by casein kinase 1 alpha. Mol Cell Biol. 2005;25(15):6509–6520. doi:10.1128/MCB.25.15.6509-6520.2005
37. Di Conza G, Mancini F, Buttarelli M, Pontecorvi A, Trimarchi F, Moretti F. MDM4 enhances p53 stability by promoting an active conformation of the protein upon DNA damage. Cell Cycle. 2012;11(4):749–760. doi:10.4161/cc.11.4.19208
38. Yang J, Jin A, Han J, Chen X, Zheng J, Zhang Y. MDMX recruits UbcH5c to facilitate MDM2 E3 ligase activity and subsequent p53 degradation in vivo. Cancer Res. 2021;81(4):898–909. doi:10.1158/0008-5472.CAN-20-0790
39. Dueber EC, Schoeffler AJ, Lingel A, et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science. 2011;334(6054):376–380. doi:10.1126/science.1207862
40. Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008;15(5):841–848. doi:10.1038/sj.cdd.4402309
41. Li D, Tavana O, Sun S-C, Gu W. Peli1 modulates the subcellular localization and activity of mdmx. Cancer Res. 2018;78(11):2897–2910. doi:10.1158/0008-5472.CAN-17-3531
42. Pan Y, Chen J. Modification of MDMX by sumoylation. Biochem Biophys Res Commun. 2005;332(3):702–709. doi:10.1016/j.bbrc.2005.05.012
43. Zuckerman V, Lenos K, Popowicz GM, et al. c-Abl phosphorylates Hdmx and regulates its interaction with p53. J Biol Chem. 2009;284(6):4031–4039. doi:10.1074/jbc.M809211200
44. Chen X, Gohain N, Zhan C, Lu W-Y, Pazgier M, Lu W. Structural basis of how stress-induced MDMX phosphorylation activates p53. Oncogene. 2016;35(15):1919–1925. doi:10.1038/onc.2015.255
45. Elias B, Laine A, Ronai Z. Phosphorylation of MdmX by CDK2/Cdc2(p34) is required for nuclear export of Mdm2. Oncogene. 2005;24(15):2574–2579. doi:10.1038/sj.onc.1208488
46. Wu S, Chen L, Becker A, Schonbrunn E, Chen J. Casein kinase 1α regulates an MDMX intramolecular interaction to stimulate p53 binding. Mol Cell Biol. 2012;32(23):4821–4832. doi:10.1128/MCB.00851-12
47. Wei X, Wu S, Song T, et al. Secondary interaction between MDMX and p53 core domain inhibits p53 DNA binding. Proc Natl Acad Sci U S A. 2016;113(19):E2558–2563. doi:10.1073/pnas.1603838113
48. de Polo A, Luo Z, Gerarduzzi C, Chen X, Little JB, Yuan Z-M. AXL receptor signalling suppresses p53 in melanoma through stabilization of the MDMX-MDM2 complex. J Mol Cell Biol. 2017;9(2):154–165. doi:10.1093/jmcb/mjw045
49. Wang B, Lim C-B, Yan J, et al. MDMX phosphorylation-dependent p53 downregulation contributes to an immunosuppressive tumor microenvironment. J Mol Cell Biol. 2020;12(9):713–722. doi:10.1093/jmcb/mjaa038
50. Swetzig WM, Wang J, Das GM. Estrogen receptor alpha (ERα/ESR1) mediates the p53-independent overexpression of MDM4/MDMX and MDM2 in human breast cancer. Oncotarget. 2016;7(13):16049–16069. doi:10.18632/oncotarget.7533
51. Bao J, Nanding A, Song H, Xu R, Qu G, Xue Y. The overexpression of MDM4: an effective and novel predictor of gastric adenocarcinoma lymph node metastasis. Oncotarget. 2016;7(41):67212–67222. doi:10.18632/oncotarget.11971
52. Gao C, Xiao G, Piersigilli A, Gou J, Ogunwobi O, Bargonetti J. Context-dependent roles of MDMX (MDM4) and MDM2 in breast cancer proliferation and circulating tumor cells. Breast Cancer Res. 2019;21(1):5. doi:10.1186/s13058-018-1094-8
53. Gao C, Xiao G, Bargonetti J. Contemplations on MDMX (MDM4) driving triple negative breast cancer circulating tumor cells and metastasis. Oncotarget. 2019;10(49):5007–5010. doi:10.18632/oncotarget.27134
54. Bauer M, Kantelhardt EJ, Stiewe T, et al. Specific allelic variants of SNPs in the MDM2 and MDMX genes are associated with earlier tumor onset and progression in Caucasian breast cancer patients. Oncotarget. 2019;10(20):1975–1992. doi:10.18632/oncotarget.26768
55. Gilkes DM, Pan Y, Coppola D, Yeatman T, Reuther GW, Chen J. Regulation of MDMX expression by mitogenic signaling. Mol Cell Biol. 2008;28(6):1999–2010. doi:10.1128/MCB.01633-07
56. Suda T, Yoshihara M, Nakamura Y, et al. Rare MDM4 gene amplification in colorectal cancer: the principle of a mutually exclusive relationship between MDM alteration and TP53 inactivation is not applicable. Oncol Rep. 2011;26(1):49–54. doi:10.3892/or.2011.1270
57. Karki A, Putra J, Kim SS, et al. MDM4 expression in fibrolamellar hepatocellular carcinoma. Oncol Rep. 2019;42(4):1487–1496. doi:10.3892/or.2019.7241
58. Gembarska A, Luciani F, Fedele C, et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat Med. 2012;18(8):1239–1247. doi:10.1038/nm.2863
59. Chopra H, Khan Z, Contreras J, Wang H, Sedrak A, Zhu Y. Activation of p53 and destabilization of androgen receptor by combinatorial inhibition of MDM2 and MDMX in prostate cancer cells. Oncotarget. 2018;9(5):6270–6281. doi:10.18632/oncotarget.23569
60. Ueda K, Kumari R, Schwenger E, et al. MDMX acts as a pervasive preleukemic-to-acute myeloid leukemia transition mechanism. Cancer Cell. 2021;39(4):529–547.e7. doi:10.1016/j.ccell.2021.02.006
61. Leventaki V, Rodic V, Tripp SR, et al. TP 53 pathway analysis in paediatric Burkitt lymphoma reveals increased MDM4 expression as the only TP53 pathway abnormality detected in a subset of cases. Br J Haematol. 2012;158(6):763–771. doi:10.1111/j.1365-2141.2012.09243.x
62. Zhang J, Schweers B, Dyer MA. The first knockout mouse model of retinoblastoma. Cell Cycle. 2004;3(7):952–959. doi:10.4161/cc.3.7.1002
63. Laurie NA, Donovan SL, Shih C-S, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi:10.1038/nature05194
64. Zhao H, Xie Y-Z, Xing R, Sun M, Chi F, Zeng Y-C. MDMX is a prognostic factor for non-small cell lung cancer and regulates its sensitivity to cisplatin. Cell Oncol. 2017;40(4):357–365. doi:10.1007/s13402-017-0325-9
65. Han X, Garcia-Manero G, McDonnell TJ, et al. HDM4 (HDMX) is widely expressed in adult pre-B acute lymphoblastic leukemia and is a potential therapeutic target. Mod Pathol. 2007;20(1):54–62. doi:10.1038/modpathol.3800727
66. Valentin-Vega YA, Barboza JA, Chau GP, El-Naggar AK, Lozano G. High levels of the p53 inhibitor MDM4 in head and neck squamous carcinomas. Hum Pathol. 2007;38(10):1553–1562. doi:10.1016/j.humpath.2007.03.005
67. Sinatkas V, Stathopoulou K, Xagoraris I, et al. MDMX/MDM4 is highly expressed and contributes to cell growth and survival in anaplastic large cell lymphoma. Leuk Lymphoma. 2021;62(7):1563–1573. doi:10.1080/10428194.2021.1876871
68. Ach T, Schwarz-Furlan S, Ach S, et al. Genomic aberrations of MDM2, MDM4, FGFR1 and FGFR3 are associated with poor outcome in patients with salivary gland cancer. J Oral Pathol Med. 2016;45(7):500–509. doi:10.1111/jop.12394
69. Fan Y, Li M, Ma K, et al. Dual-target MDM2/MDMX inhibitor increases the sensitization of doxorubicin and inhibits migration and invasion abilities of triple-negative breast cancer cells through activation of TAB1/TAK1/p38 MAPK pathway. Cancer Biol Ther. 2019;20(5):617–632. doi:10.1080/15384047.2018.1539290
70. Busuttil RA, Zapparoli GV, Haupt S, et al. Role of p53 in the progression of gastric cancer. Oncotarget. 2014;5(23):12016–12026. doi:10.18632/oncotarget.2434
71. Wang X, Yamamoto Y, Imanishi M, et al. Enhanced G1 arrest and apoptosis via MDM4/MDM2 double knockdown and MEK inhibition in wild-type TP53 colon and gastric cancer cells with aberrant KRAS signaling. Oncol Lett. 2021;22(1):558. doi:10.3892/ol.2021.12819
72. Dewaele M, Tabaglio T, Willekens K, et al. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J Clin Invest. 2016;126(1):68–84. doi:10.1172/JCI82534
73. Arnoff TE, El-Deiry WS. MDM2/MDM4 amplification and CDKN2A deletion in metastatic melanoma and glioblastoma multiforme may have implications for targeted therapeutics and immunotherapy. Am J Cancer Res. 2022;12(5):2102–2117.
74. Stegeman S, Moya L, Selth LA, Spurdle AB, Clements JA, Batra J. A genetic variant of MDM4 influences regulation by multiple microRNAs in prostate cancer. Endocr Relat Cancer. 2015;22(2):265–276. doi:10.1530/ERC-15-0013
75. Mejía-Hernández JO, Raghu D, Caramia F, et al. Targeting MDM4 as a novel therapeutic approach in prostate cancer independent of p53 status. Cancers. 2022;14(16):3947. doi:10.3390/cancers14163947
76. Wang D, Zhang S, Zhao M, Chen F. LncRNA MALAT1 accelerates non-small cell lung cancer progression via regulating miR-185-5p/MDM4 axis. Cancer Med. 2020;9(23):9138–9149. doi:10.1002/cam4.3570
77. Ally A, Balasundaram M, Carlsen R; The Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169(7):1327–1341.e23. doi:10.1016/j.cell.2017.05.046
78. Pellegrino R, Calvisi DF, Neumann O, et al. EEF1A2 inactivates p53 by way of PI3K/AKT/mTOR-dependent stabilization of MDM4 in hepatocellular carcinoma. Hepatology. 2014;59(5):1886–1899. doi:10.1002/hep.26954
79. Pellegrino R, Thavamani A, Calvisi DF, et al. Serum Response Factor (SRF) drives the transcriptional upregulation of the MDM4 oncogene in HCC. Cancers. 2021;13(2):199. doi:10.3390/cancers13020199
80. Ten Kate FJC, Suzuki L, Dorssers LCJ, et al. Pattern of p53 protein expression is predictive for survival in chemoradiotherapy-naive esophageal adenocarcinoma. Oncotarget. 2017;8(61):104123–104135. doi:10.18632/oncotarget.22021
81. Jiang K, Sun F, Zhu J, Luo G, Ban Y, Zhang P. miR-33a inhibits cell growth in renal cancer by downregulation of MDM4 expression. Mol Genet Genomic Med. 2019;7(8):e833. doi:10.1002/mgg3.833
82. Gansmo LB, Bjørnslett M, Halle MK, et al. The MDM4 SNP34091 (rs4245739) C-allele is associated with increased risk of ovarian-but not endometrial cancer. Tumour Biol. 2016;37(8):10697–10702. doi:10.1007/s13277-016-4940-2
83. Hu W, Feng Z, Modica I, et al. Gene amplifications in well-differentiated pancreatic neuroendocrine tumors inactivate the p53 pathway. Genes Cancer. 2010;1(4):360–368. doi:10.1177/1947601910371979
84. Feeley KP, Adams CM, Mitra R, Eischen CM. Mdm2 is required for survival and growth of p53-deficient cancer cells. Cancer Res. 2017;77(14):3823–3833. doi:10.1158/0008-5472.CAN-17-0809
85. Matijasevic Z, Steinman HA, Hoover K, Jones SN. MdmX promotes bipolar mitosis to suppress transformation and tumorigenesis in p53-deficient cells and mice. Mol Cell Biol. 2008;28(4):1265–1273. doi:10.1128/MCB.01108-07
86. Matijasevic Z, Krzywicka-Racka A, Sluder G, Gallant J, Jones SN. The Zn-finger domain of MdmX suppresses cancer progression by promoting genome stability in p53-mutant cells. Oncogenesis. 2016;5(10):e262. doi:10.1038/oncsis.2016.62
87. Xiong S, Pant V, Zhang Y, et al. The p53 inhibitor Mdm4 cooperates with multiple genetic lesions in tumourigenesis. J Pathol. 2017;241(4):501–510. doi:10.1002/path.4854
88. Zhang H, Hu L, Qiu W, et al. MDMX exerts its oncogenic activity via suppression of retinoblastoma protein. Oncogene. 2015;34(44):5560–5569. doi:10.1038/onc.2015.11
89. Klein AM, Biderman L, Tong D, et al. MDM2, MDMX, and p73 regulate cell-cycle progression in the absence of wild-type p53. Proc Natl Acad Sci U S A. 2021;118(44):e2102420118. doi:10.1073/pnas.2102420118
90. Carrillo AM, Bouska A, Arrate MP, Eischen CM. Mdmx promotes genomic instability independent of p53 and Mdm2. Oncogene. 2015;34(7):846–856. doi:10.1038/onc.2014.27
91. Wohlberedt K, Klusmann I, Derevyanko PK, et al. Mdm4 supports DNA replication in a p53-independent fashion. Oncogene. 2020;39(25):4828–4843. doi:10.1038/s41388-020-1325-1
92. Holzer P, Masuya K, Furet P, et al. Discovery of a dihydroisoquinolinone derivative (NVP-CGM097): a highly potent and selective MDM2 inhibitor undergoing Phase 1 clinical trials in p53wt tumors. J Med Chem. 2015;58(16):6348–6358. doi:10.1021/acs.jmedchem.5b00810
93. Konopleva M, Martinelli G, Daver N, et al. MDM2 inhibition: an important step forward in cancer therapy. Leukemia. 2020;34(11):2858–2874. doi:10.1038/s41375-020-0949-z
94. Bista M, Smithson D, Pecak A, et al. On the mechanism of action of SJ-172550 in inhibiting the interaction of MDM4 and p53. PLoS One. 2012;7(6):e37518. doi:10.1371/journal.pone.0037518
95. Karan G, Wang H, Chakrabarti A, et al. Identification of a small molecule that overcomes HdmX-mediated suppression of p53. Mol Cancer Ther. 2016;15(4):574–582. doi:10.1158/1535-7163.MCT-15-0467
96. Ilic VK, Egorova O, Tsang E, et al. Hinokiflavone inhibits MDM2 activity by targeting the MDM2-MDMX RING domain. Biomolecules. 2022;12(5):643. doi:10.3390/biom12050643
97. Boltjes A, Huang Y, van de Velde R, et al. Fragment-based library generation for the discovery of a peptidomimetic p53-Mdm4 inhibitor. ACS Comb Sci. 2014;16(8):393–396. doi:10.1021/co500026b
98. Uesato S, Matsuura Y, Matsue S, et al. Discovery of new low-molecular-weight p53-Mdmx disruptors and their anti-cancer activities. Bioorg Med Chem. 2016;24(8):1919–1926. doi:10.1016/j.bmc.2016.03.021
99. Jiang L, Malik N, Acedo P, Zawacka-Pankau J. Protoporphyrin IX is a dual inhibitor of p53/MDM2 and p53/MDM4 interactions and induces apoptosis in B-cell chronic lymphocytic leukemia cells. Cell Death Discov. 2019;5:77. doi:10.1038/s41420-019-0157-7
100. Graves B, Thompson T, Xia M, et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc Natl Acad Sci U S A. 2012;109(29):11788–11793. doi:10.1073/pnas.1203789109
101. Pishas KI, Adwal A, Neuhaus SJ, et al. XI-006 induces potent p53-independent apoptosis in Ewing sarcoma. Sci Rep. 2015;5(1):11465. doi:10.1038/srep11465
102. Wang H, Yan C. A small-molecule p53 activator induces apoptosis through inhibiting MDMX expression in breast cancer cells. Neoplasia. 2011;13(7):611–619. doi:10.1593/neo.11438
103. Voruganti S, Qin -J-J, Sarkar S, et al. Oral nano-delivery of anticancer ginsenoside 25-OCH3-PPD, a natural inhibitor of the MDM2 oncogene: nanoparticle preparation, characterization, in vitro and in vivo anti-prostate cancer activity, and mechanisms of action. Oncotarget. 2015;6(25):21379–21394. doi:10.18632/oncotarget.4091
104. Gerhart SV, Kellner WA, Thompson C, et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci Rep. 2018;8(1):9711. doi:10.1038/s41598-018-28002-y
105. Qin -J-J, Li X, Wang W, Zi X, Zhang R. Targeting the NFAT1-MDM2-MDMX network inhibits the proliferation and invasion of prostate cancer cells, independent of p53 and androgen. Front Pharmacol. 2017;8:917. doi:10.3389/fphar.2017.00917
106. Vaseva AV, Yallowitz AR, Marchenko ND, Xu S, Moll UM. Blockade of Hsp90 by 17AAG antagonizes MDMX and synergizes with Nutlin to induce p53-mediated apoptosis in solid tumors. Cell Death Dis. 2011;2(5):e156. doi:10.1038/cddis.2011.39
107. Pairawan S, Zhao M, Yuca E, et al. First in class dual MDM2/MDMX inhibitor ALRN-6924 enhances antitumor efficacy of chemotherapy in TP53 wild-type hormone receptor-positive breast cancer models. Breast Cancer Res. 2021;23(1):29. doi:10.1186/s13058-021-01406-x
108. Saleh MN, Patel MR, Bauer TM, et al. Phase 1 trial of ALRN-6924, a dual inhibitor of MDMX and MDM2, in patients with solid tumors and lymphomas bearing wild-type TP53. Clin Cancer Res. 2021;27(19):5236–5247. doi:10.1158/1078-0432.CCR-21-0715
109. Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol. 2017;10(1):133. doi:10.1186/s13045-017-0500-5
110. Espadinha M, Barcherini V, Lopes EA, Santos MMM. An Update on MDMX and Dual MDM2/X Inhibitors. Curr Top Med Chem. 2018;18(8):647–660. doi:10.2174/1568026618666180604080119
111. Wang H, Ma X, Ren S, Buolamwini JK, Yan C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol Cancer Ther. 2011;10(1):69–79. doi:10.1158/1535-7163.MCT-10-0581
112. Jiang Z-Q, M-H L, Qin Y-M, Jiang H-Y, Zhang X, Wu M-H. Luteolin inhibits tumorigenesis and induces apoptosis of non-small cell lung cancer cells via regulation of microRNA-34a-5p. Int J Mol Sci. 2018;19(2):447. doi:10.3390/ijms19020447
113. Zu Y, Wang J, Ping W, Sun W. Tan IIA inhibits H1299 cell viability through the MDM4‑IAP3 signaling pathway. Mol Med Rep. 2018;17(2):2384–2392. doi:10.3892/mmr.2017.8152
114. Wang W, Qin -J-J, Li X, et al. Prevention of prostate cancer by natural product MDM2 inhibitor GS25: in vitro and in vivo activities and molecular mechanisms. Carcinogenesis. 2018;39(8):1026–1036. doi:10.1093/carcin/bgy063
115. Bezzi M, Teo SX, Muller J, et al. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013;27(17):1903–1916. doi:10.1101/gad.219899.113
116. Ling X, Xu C, Fan C, Zhong K, Li F, Wang X. FL118 induces p53-dependent senescence in colorectal cancer cells by promoting degradation of MdmX. Cancer Res. 2014;74(24):7487–7497. doi:10.1158/0008-5472.CAN-14-0683
117. Zheng X, Yan J, You W, et al. De novo nano-erythrocyte structurally braced by biomimetic Au(I)-peptide Skeleton for MDM2/MDMX predation toward augmented pulmonary adenocarcinoma immunotherapy. Small. 2021;17(20):e2100394. doi:10.1002/smll.202100394
118. El-Deiry WS, Arnoff T, Sahin I, et al. A pancancer analysis of impact of MDM2/MDM4 on immune checkpoint blockade (ICB). JCO. 2022;40(16_suppl):2630. doi:10.1200/JCO.2022.40.16_suppl.2630
119. Cheng J, Li Y, Wang X, et al. Response stratification in the first-line combined immunotherapy of hepatocellular carcinoma at genomic, transcriptional and immune repertoire levels. J Hepatocell Carcinoma. 2021;8:1281–1295. doi:10.2147/JHC.S326356
120. Fang W, Zhou H, Shen J, et al. MDM2/4 amplification predicts poor response to immune checkpoint inhibitors: a pan-cancer analysis. ESMO Open. 2020;5(1):4. doi:10.1136/esmoopen-2019-000614
121. Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17(2):100–107. doi:10.1016/S0168-9525(00)02176-4
122. Bardot B, Toledo F. Targeting MDM4 Splicing in Cancers. Genes (Basel). 2017;8(2):82. doi:10.3390/genes8020082
123. Grawenda AM, Møller EK, Lam S, et al. Interaction between p53 mutation and a somatic HDMX biomarker better defines metastatic potential in breast cancer. Cancer Res. 2015;75(4):698–708. doi:10.1158/0008-5472.CAN-14-2637
124. Bartel F, Schulz J, Böhnke A, et al. Significance of HDMX-S (or MDM4) mRNA splice variant overexpression and HDMX gene amplification on primary soft tissue sarcoma prognosis. Int J Cancer. 2005;117(3):469–475. doi:10.1002/ijc.21206
125. Lenos K, Grawenda AM, Lodder K, et al. Alternate splicing of the p53 inhibitor HDMX offers a superior prognostic biomarker than p53 mutation in human cancer. Cancer Res. 2012;72(16):4074–4084. doi:10.1158/0008-5472.CAN-12-0215
126. Liu L, Fan L, Fang C, et al. S-MDM4 mRNA overexpression indicates a poor prognosis and marks a potential therapeutic target in chronic lymphocytic leukemia. Cancer Sci. 2012;103(12):2056–2063. doi:10.1111/cas.12008
127. Liang M, Han X, Vadhan-Raj S, et al. HDM4 is overexpressed in mantle cell lymphoma and its inhibition induces p21 expression and apoptosis. Mod Pathol. 2010;23(3):381–391. doi:10.1038/modpathol.2009.170
128. Pant V, Larsson CA, Aryal N, et al. Tumorigenesis promotes Mdm4-S overexpression. Oncotarget. 2017;8(16):25837–25847. doi:10.18632/oncotarget.15552
129. Giglio S, Mancini F, Gentiletti F, et al. Identification of an aberrantly spliced form of HDMX in human tumors: a new mechanism for HDM2 stabilization. Cancer Res. 2005;65(21):9687–9694. doi:10.1158/0008-5472.CAN-05-0450
130. de Graaf P, Little NA, Ramos YFM, Meulmeester E, Letteboer SJF, Jochemsen AG. Hdmx protein stability is regulated by the ubiquitin ligase activity of Mdm2. J Biol Chem. 2003;278(40):38315–38324. doi:10.1074/jbc.M213034200
131. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18(2):89–102. doi:10.1038/nrc.2017.109
132. Rallapalli R, Strachan G, Tuan RS, Hall DJ. Identification of a domain within MDMX-S that is responsible for its high affinity interaction with p53 and high-level expression in mammalian cells. J Cell Biochem. 2003;89(3):563–575. doi:10.1002/jcb.10535
133. Mancini F, Pieroni L, Monteleone V, et al. MDM4/HIPK2/p53 cytoplasmic assembly uncovers coordinated repression of molecules with anti-apoptotic activity during early DNA damage response. Oncogene. 2016;35(2):228–240. doi:10.1038/onc.2015.76
134. Pazgier M, Liu M, Zou G, et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc Natl Acad Sci U S A. 2009;106(12):4665–4670. doi:10.1073/pnas.0900947106
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.