")
Back to Journals » Journal of Inflammation Research » Volume 17
Maresin-1 Attenuates Sepsis-Associated Acute Kidney Injury via Suppressing Inflammation, Endoplasmic Reticulum Stress and Pyroptosis by Activating the AMPK/SIRT3 Pathway
Authors Sun M, Wang F, Li H, Li M, Wang Y, Wang C, Zhang Y, Zhang D, Li J, Yao S
Received 9 November 2023
Accepted for publication 16 February 2024
Published 27 February 2024 Volume 2024:17 Pages 1349—1364
DOI https://doi.org/10.2147/JIR.S442729
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Adam D Bachstetter
Miaomiao Sun,1– 3,* Fuquan Wang,1– 3,* Haopeng Li,1– 3 Mengyu Li,1– 3 Yu Wang,1– 3 Chenchen Wang,1– 3 Yan Zhang,1– 3 Dingyu Zhang,1– 4 Jianhua Li,5 Shanglong Yao1– 3
1Department of Anesthesiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430022, People’s Republic of China; 2Institute of Anesthesia and Critical Care Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430022, People’s Republic of China; 3Key Laboratory of Anesthesiology and Resuscitation (Huazhong University of Science and Technology), Ministry of Education, Wuhan, 430022, People’s Republic of China; 4Wuhan Jinyintan Hospital, Wuhan, 430023, People’s Republic of China; 5Department of Critical Care Medicine, Chongqing University Jiangjin Hospital, Chongqing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shanglong Yao; Jianhua Li, Email [email protected]; [email protected]
Background: Sepsis-associated acute kidney injury (SA-AKI) is a common complication in patients with sepsis, triggering high morbidity and mortality. Maresin-1 (MaR1) is a pro-resolution lipid mediator that promotes the resolution of acute inflammation and protects organs from inflammation.
Methods: In this study, we established an SA-AKI model using cecal ligation and puncture (CLP) and investigated the effect and mechanism of MaR1. The blood and kidneys were harvested 24 hours after surgery. The blood biochemical/routine indicators, renal function, SA-AKI-related pathophysiological processes, and AMPK/SIRT3 signaling in septic mice were observed by histological staining, immunohistochemical staining, Western blot, qPCR, ELISA and TUNEL Assay.
Results: MaR1 treatment alleviated kidney injury in septic mice, reflected in improved pathological changes in renal structure and renal function. MaR1 treatment decreased the levels of serum creatinine (sCr) and blood urea nitrogen (BUN) and the expressions of KIM-1, NGAL and TIMP-2, which were related to kidney injury, while inhibited the expressions of inflammatory factors TNF-α, IL-1β and IL-6. The expression of endoplasmic reticulum stress-related indicators p-PERK/PERK, GRP78, p-EIF2α/EIF2α, ATF4, CHOP, and pyroptosis-related indicators Caspase-1, NLRP3, GSDMD, IL-18, and IL-1β also decreased after MaR1 treatment. The mechanism may be related to the activation of the AMPK/SIRT3 signaling pathway, and an AMPK inhibitor (compound C) partially reverses MaR1’s protective effects in septic mice.
Conclusion: Taken together, these findings suggest that MaR1 may partially ameliorate SA-AKI by activating the AMPK/SIRT3 signaling pathway, providing a potential new perspective for research on SA-AKI.
Keywords: Maresin-1, pyroptosis, AMPK/SIRT3, endoplasmic reticulum stress, sepsis-associated acute kidney injury, inflammation
Introduction
The kidney is one of the earliest organs to suffer damage in sepsis.1,2 A worse prognosis was predicted by SA-AKI than by the two symptoms separately.3,4 SA-AKI might have a direct mechanism caused by an infection or the host’s response to an infection, or it can have an indirect mechanism brought on by sepsis or the unfavorable effects of sepsis treatment.5,6 In addition, recent studies have shown that the pathological process of SA-AKI may be related to excessive inflammatory response, ER stress, and pyroptosis, especially pyroptosis of tubule cells, which is highly distinct from other types of acute kidney injury (AKI).7–9 Given the significant mortality and morbidity caused by SA-AKI, it is becoming more and more important to fully comprehend its cellular pathophysiological mechanisms and persistently investigate new therapeutic approaches for SA-AKI.3
Maresin-1 (MaR1), a pro-resolution lipid mediator produced from docosahexaenoic acid, was initially identified in human macrophages. Its anti-inflammatory and pro-resolution properties help to promote the resolution of acute inflammation and protect organs from inflammation.10,11 By reducing the inflammatory response, oxidative stress, ER stress, pyroptosis, and apoptosis, these protective effects are primarily accomplished.11–15 MaR1 has been thoroughly studied, but its regulatory mechanism, notably in SA-AKI, is yet unknown.
Adenosine monophosphate activated protein kinase (AMPK) plays an important role in regulating energy homeostasis and a variety of signaling pathways, and studies have shown that AMPK can be involved in the regulation of inflammation and oxidative stress.16,17 Similarly, the protein Sirtuin 3 (SIRT3) also regulates inflammation levels and the activity of antioxidant enzymes.18,19 Previous studies have reported a potential link between AMPK and SIRT3.20,21 We speculate that AMPK/SIRT3 signaling pathway plays a crucial role in SA-AKI.
Therefore, this study aimed to investigate the effect of MaR1 and clarify its underlying mechanism on SA-AKI, aiming to provide sufficient experimental basis and theoretical support for seeking new targets for SA-AKI.
Materials and Methods
Experimental Animals
Specific pathogen-free (SPF) wild-type male BALB/C mice (aged 8–10 weeks; weighing 22 ± 4 g) were obtained from Vital River Laboratory Animal Technology Co., Ltd., Beijing, China. All animals were housed in a constant-temperature room (22–24°C) with a 12-hour dark/12-hour light cycle and fed sufficient food and water. Before starting, all experimental operations were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the Laboratory Animal Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China) (IACUC Number: 3036).
Reagents and Drugs
MaR1 was purchased from Cayman Chemical (10878, Ann Arbor, MI, USA). Dorsomorphin dihydrochloride (Compound C, CC) (Cat: HY-13418) was purchased from MedChemExpress (Monmouth Junction, NJ, USA). Phosphate buffered saline (PBS) was obtained from Gibco. The Creatinine (Cr) Assay Kit (sarcosine oxidase) and the Urea Assay Kit were obtained from Jiancheng Bioengineering Institute (Nanjing, China). Trizol reagent was supplied by Thermo Fisher Scientific. HiScript® RT SuperMix and AceQ qPCR SYBR® Green Master Mix were obtained from Vazyme (Nanjing, China). The BCA Protein Assay Kit was purchased from Beijing ComWin Biotech Co., Ltd. The enhanced chemiluminescent substrate (ECL) (BL520A) was obtained from Biosharp (Beijing, China). The following antibodies were used in this study: Anti-PERK (#3192S), Anti-p-EIF2α (3398S), Anti-EIF2α (5324S), Anti-ATF4 (11815S), Anti-CHOP (5554S), Anti-NLRP3 (#15101S), and Anti-p-AMPK (Thr172, #2535T) were purchased from Cell Signaling Technology (dilution 1:1000, Danvers, MA, USA). Anti-GRP78 (11587-1-AP), Anti-Caspase-1 (22915-1-AP), Anti-IL-1β (26048-1-AP), Anti-IL-18 (10663-1-AP), Anti-AMPK (10929-2-AP), and Anti-Sirt3 (10099-1-AP) were purchased from Proteintech (dilution 1:1000, IL, USA). Anti-GADMD (ab209845) was purchased from Abcam (dilution 1:1000, Cambridge, MA, USA). Anti-HMGB1 (A2553) was purchased from ABclonal (dilution 1:1000, Wuhan, China). Anti-p-PERK (YP1055) was purchased from ImmunoWay Biotechnology (dilution 1:1000, Plano, TX, USA). Anti-α-Tubulin (ANT329s) and HRP Goat Anti-Rabbit IgG (H+L) (ANT020) were purchased from Antgene Biotechnology (dilution 1:5000, Wuhan, China). Anti-β-actin (66009-1-Ig) and HRP-conjugated Affinipure Goat Anti-Mouse IgG (H+L) (SA00001-1) were purchased from Proteintech (dilution 1:5000, IL, USA).
Experimental Design
The model of SA-AKI was established in vivo. In brief, the mice were anesthetized with 2% sodium pentobarbital intraperitoneally at a dose of 80 mg/kg. The incision (0.75–1.0 cm) was made in the midline of the abdomen, and the cecum was exposed. The cecum was tightly ligated with sterile 4–0 silk thread at 2/3 of the distal cecum. A sterile 18G needle was used to pierce through the middle of the distal cecum ligated (through-and-through) and extrude a small amount of feces, trying to avoid damage to blood vessels. Then, the cecum was pushed back into the abdominal cavity to close the cavity. Sham surgery animals underwent the same laparotomy and externalization of the cecum, but no ligation or perforation. Animals were resuscitated after the operation with 1 mL of normal saline (NS) administered subcutaneously, recovered on warming blankets, and fed sufficient food and water.
BALB/C male mice were randomly divided into the sham group, the CLP group, the CLP+MaR1 (0.04 ug/kg) group, the CLP+MaR1 (0.4 ug/kg) group, and the CLP+MaR1 (4 ug/kg) group. After CLP, the mice were injected intraperitoneally with different doses of MaR1 (0.04 ug/kg, 0.4 ug/kg, and 4 ug/kg) dissolved in normal saline. Meanwhile, the sham group and the CLP group were injected intraperitoneally with an equal volume of normal saline. The blood and kidneys were harvested 24 hours after surgery, with the mice anesthetized by pentobarbital sodium injection and then sacrificed humanely.
BALB/C male mice were randomly divided into the sham group, the CLP group, the CLP+MaR1 group, and the CLP+MaR1+CC (AMPK antagonist, dissolved in normal saline) group. CC (20 mg/kg) was administered 2 hours before surgery by intraperitoneal injection, with the dosage based on previous studies.22 After CLP, the mice were injected intraperitoneally with MaR1 (4 ug/kg) dissolved in normal saline. Meanwhile, the sham group and the CLP group were injected intraperitoneally with an equal volume of normal saline. The blood and kidneys were harvested 24 hours after surgery, with the mice anesthetized by pentobarbital sodium injection and then sacrificed humanely.
Renal Function and Histologic Examination
Blood samples were taken and centrifuged at 3500 rpm for 15 minutes at ambient temperature in order to get serum. The levels of serum creatinine (sCr) and blood urea nitrogen (BUN) were determined with the Creatinine (Cr) Assay Kit (sarcosine oxidase) and the Urea Assay Kit. These procedures were strictly in accordance with the instructions. One kidney was fixed with paraformaldehyde for 24 hours and then paraffin embedded, and kidney sections of 4 μm were used for hematoxylin–eosin (HE) and periodic acid-Schiff (PAS) reagent staining. HE-stained tissue sections and PAS-stained tissue sections were viewed by light microscopy (Olympus Optical, Tokyo, Japan) at magnifications of ×400. Tissue damage was scored in a blinded manner. The percentage of injured renal tubules and histological injury were indicated by brush border loss, tubular degeneration, tubular dilation/flattening, tubular cast formation, and vacuolization. Renal tubular injury was assessed on a semiquantitative scale of 0–4, as described below: 0, normal; 1, <25% injury; 2, 25–50% injury; 3, 51–75% injury; and 4, >75% injury.21
Immunohistochemistry
After paraffin sections were dewaxed, antigen repair was carried out in a microwave oven. Tissue sections were blocked with 2.5% normal goat serum and inoculated with the primary antibodies anti-GRP78, anti-p-PERK, anti-IL-1β, and anti-IL-18 diluted 200:1 in IHC antibody diluent at a temperature of 4 °C overnight. After washing three times with PBS, sections were incubated at room temperature with biotinylated secondary antibody for 30 min, which were then visualized under 400x magnification through light microscopy (Olympus Optical, Tokyo, Japan).
TUNEL Assay
Kidney sections were embedded in paraffin, cut into 4-μm-thick sections, and incubated with the reagents of the TUNEL kit according to the instructions of the manufacturer (Roche, Basel, Switzerland). The nuclei were counterstained with DAPI. The positive staining was detected by fluorescence microscopy (Olympus Optical, Tokyo, Japan). The TUNEL-positive cells were counted in six randomly selected fields from each slide at a magnification of 400× to determine the number of dead cells.
Enzyme-Linked Immunosorbent Assay
The levels of TNF-α, IL-1β, IL-6, and IL-18 in serum were evaluated using enzyme-linked immunosorbent assay (ELISA) kits (Neobioscience, Shanghai, China). These procedures were strictly in accordance with the manufacturer’s instructions.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
The total RNA was harvested from kidney tissues using Trizol, and the total RNA was reverse transcribed into cDNA with HiScript® RT SuperMix, and then RT-PCR was performed with the AceQ qPCR SYBR® Green Master Mix according to the manufacturer’s instructions in Bio-Rad CFX Connect (Hercules, CA, USA). The cycle threshold (Ct) values were recorded, and the relative expression of the target genes was normalized to GAPDH as the internal control using the 2-∆∆Cq method. PCR primers were synthesized by TsingKe Biological Technology (Wuhan, China), as listed in Table 1.
 |
Table 1 Primer Sequences of RT-PCR Test |
Western Blotting
The total protein was extracted from kidney tissue using RIPA lysis buffer, and the protein concentration was determined using a BCA protein assay kit. Total kidney lysates (40 μg proteins in each well) were separated electrophoretically by 10% acrylamide SDS-PAGE and transferred to a PVDF membrane after the proteins of different molecular weights were separated. The PVDF membrane was blocked with 5% non-fat milk at room temperature for 1 hour and then washed three times with TBST for 10 minutes each. Subsequently, the membranes were incubated with primary antibodies according to the manufacturer’s instructions at 4°C overnight. On the second day, membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. The specific immunoreactive protein bands were visualized using enhanced chemiluminescence (ECL) with a UVP imaging system (Upland, CA, USA). Quantification was performed using ImageJ software.
Molecular Docking
Chemdraw software was used to draw the MaR1 structure and MOE 2019.0102 software was run to minimize its energy and finally save it in mdb format in preparation for molecular docking. Download the AMPK (PDB ID: 5UFU) protein structure from the PDB database (RCSB PDB: Homepage). Then the protein was imported into MOE 2019.0102 software, the water molecule and the original ligand in the ligand were removed, and then “QuickPrep” was run to quickly process the protein, and finally the processed protein was saved in mdb format. The “Dock” module in MOE 2019.0102 software is used for molecular docking. The docking mode is all-atom docking and small molecules are imported into it. The rest Settings are default.
Statistical Analysis
GraphPad Prism 7.0 (GraphPad Inc., USA) was used for the visualization of graphs and data analysis. All data were presented as the means ± standard deviations. Statistical analysis among multiple groups was done by one-way analysis of variance (ANOVA). P < 0.05 was considered statistically significant.
Results
MaR1 Reduced Sepsis-Associated Acute Kidney Injury and Improved Renal Function
We investigated the concentration gradient to examine the effects of MaR1 on renal function in order to find a suitable dose that may be delivered to mice. The pretest HE staining results revealed that the renal sections of mice in the sham group had normal morphology, clear histological structure, no degeneration, atrophy, swelling, or necrosis of renal tubular cells, and no dilatation of the renal tubular lumen. Renal tubular epithelial cell edema and shedding, vacuolar degeneration, a disappearance of the brush border, protein tubule type, and dilatation of the renal tubular lumen were observed in SA-AKI mice, whereas MaR1 at doses of 0.4 ug/kg and 4 ug/kg could significantly improve the renal histopathological damage in SA-AKI mice (Figure 1A and B). The serum indicators of kidney injury, sCr and BUN, were clearly elevated in the SA-AKI mice, whereas MaR1 at a dose of 4 ug/kg could significantly reduce their levels after treatment (Figure 1C and D). However, at doses of 0.04 ug/kg and 0.4 ug/kg, MaR1 had no effect on renal function, as seen by sCr and BUN levels in the CLP group (Figure 1C and D). Consistent with these results, mRNA levels of kidney injury molecule-1 (KIM-1), neutrophil gelatinase-associated lipocalin (NGAL), and tissue inhibitor of metalloproteinases 2 (TIMP2) were also significantly decreased after MaR1 administration (Figure 1E–G). The above results confirmed that MaR1 treatment can effectively reduce pathological injury and renal dysfunction in mice after the establishment of the SA-AKI model.
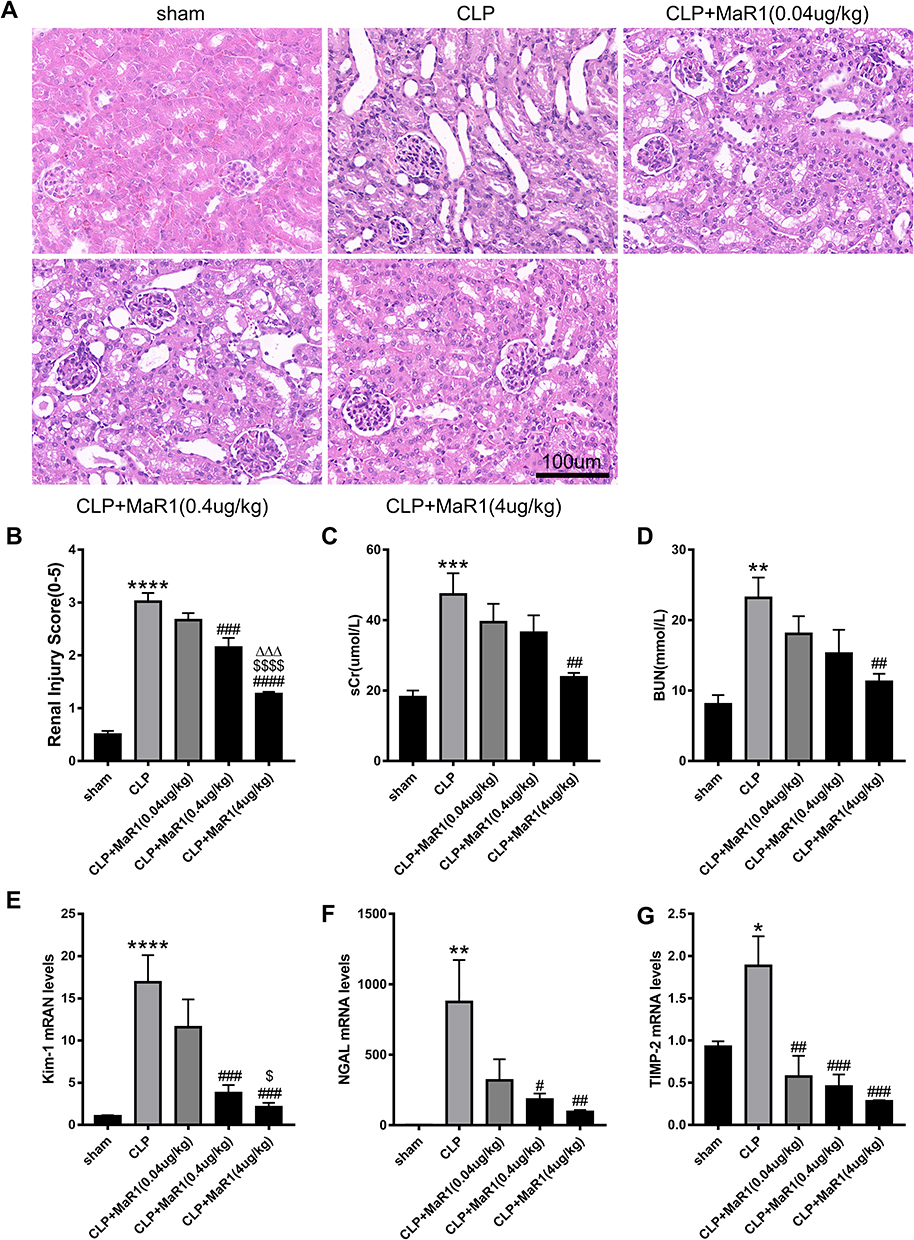 |
Figure 1 Effects of different doses of Maresin1 on renal histopathology and renal function in SA-AKI mice. Representative pictures of H&E staining of renal tissues (magnification 400 ×) in (A). (B) Pathological renal tissue damage score. Serum creatinine (C) and blood urea nitrogen (D) levels at 24 h after CLP in mice. mRNA levels of Kim-1 (E), NGAL (F) and TIMP-2 (G) in renal tissues were examined by real-time PCR. The data are presented as the means ± SD (n = 4–6 per group). *P<0.05, **P<0.01, ***P<0.001, ****P < 0.0001 vs sham group; #P<0.05, ##P<0.01, ###P<0.001, ####P<0.0001 vs CLP group; $P<0.05, $$$$P<0.0001 vs CLP+MaR1 (0.04 ug/kg) group. ∆∆∆P<0.001 vs CLP+MaR1 (0.4 ug/kg) group. |
MaR1 Ameliorated the Inflammatory Response in SA-AKI Mice
Our results suggested that the levels of the pro-inflammatory factors TNF-α, IL-1β, and IL-6 in serum in the SA-AKI mice were significantly higher than those in the sham group, while serum levels of pro-inflammatory factors in the CLP+MaR1 (0.4 ug/kg) and CLP+MaR1 (4 ug/kg) groups were significantly lower than the CLP group (Figure 2A–C). Consistent with these results, mRNA levels of TNF-α, IL-1β and IL-6 in SA-AKI mice were also significantly decreased after MaR1 (4 ug/kg) treatment compared with those in the CLP group (Figure 2D–F). Therefore, a dose of 4 ug/kg MaR1 was selected for the following experiment to study the mechanism of MaR1.
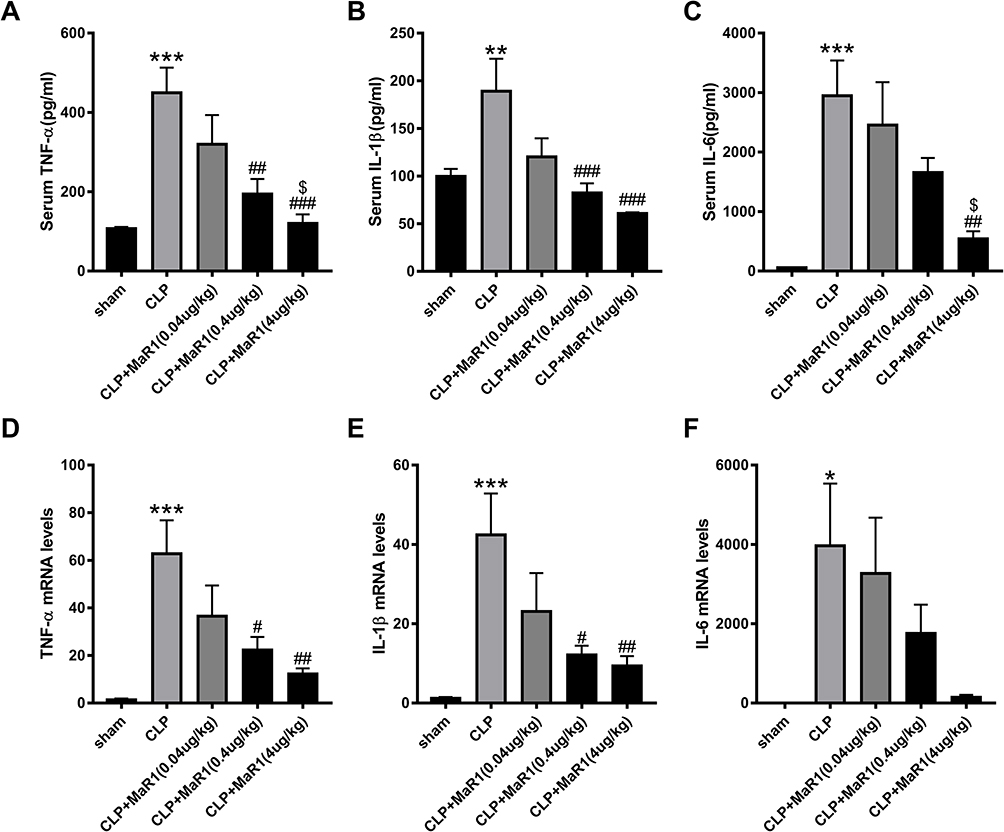 |
Figure 2 Effects of different doses of Maresin1 on inflammatory response in SA-AKI mice. The levels of cytokines in serum (A–C), including TNF-α, IL-1β and IL-6, were examined as markers of inflammation. mRNA levels of TNF-α (D), IL-1β (E) and IL-6 (F) in renal tissues were examined by real-time PCR. The data are presented as the means ± SD (n = 4–6 per group). *P<0.05, **P<0.01, ***P<0.001 vs sham group; #P<0.05, ##P<0.01, ###P<0.001 vs CLP group; $P<0.05 vs CLP+MaR1 (0.04 ug/kg) group. |
MaR1 Can Activate AMPK/SIRT3 Pathway in SA-AKI Mice
Previous studies have shown that AMPK/SIRT3 pathway is involved in the regulation of excessive inflammation, oxidative stress and cell apoptosis.21,23 MaR1 has also been reported to activate AMPK to regulate ER stress.15 In order to better study the correlation between MaR1 and AMPK domains, we used PyMOL2.3.0 and Ligplot v2.2.5 software to predict the molecular docking between MaR1 and AMPK (Figure 3A). The results showed that the binding energy of MaR1 and AMPK was −6.1634 kcal/mol, indicating that MaR1 and AMPK had a good binding effect. In Figure 3B, MaR1 interacts with AMPK mainly through hydrogen bonding and hydrophobic force, forming hydrogen bonds with Asp157 and Gly175 residues of AMPK protein, with the length of hydrogen bonds being 2.91Å and 3.33Å, respectively. In addition, MaR1 also interacts with Val24, Glu100, Glu143, Lys141, Asn144, Asp139, Leu160, Cys174, Pro177, Ser173, Gly25 and Thr26 via hydrophobic interactions, which are shown as arcs (red) in Figure 3B. We then examined the expression levels of AMPK and Sirt3. We found that p-AMPK/AMPK and Sirt3 expression levels were reduced in SA-AKI mice, while MaR1 activated the AMPK/SIRT3 pathway (Figure 3C–E). To further confirm the results mentioned above, Compound C, an AMPK inhibitor that can result in AMPK inhibition, was injected intraperitoneally two hours prior to the CLP procedure to further support the results mentioned above. We discovered that CC counteracted MaR1’s activation of the AMPK and inhibited SIRT3 protein expression (Figure 3F–H). Meanwhile, the mRNA level of SIRT3 matched its protein levels (Figure 3I). Together, these data indicated that MaR1 can activate AMPK/SIRT3 pathway in SA-AKI mice.
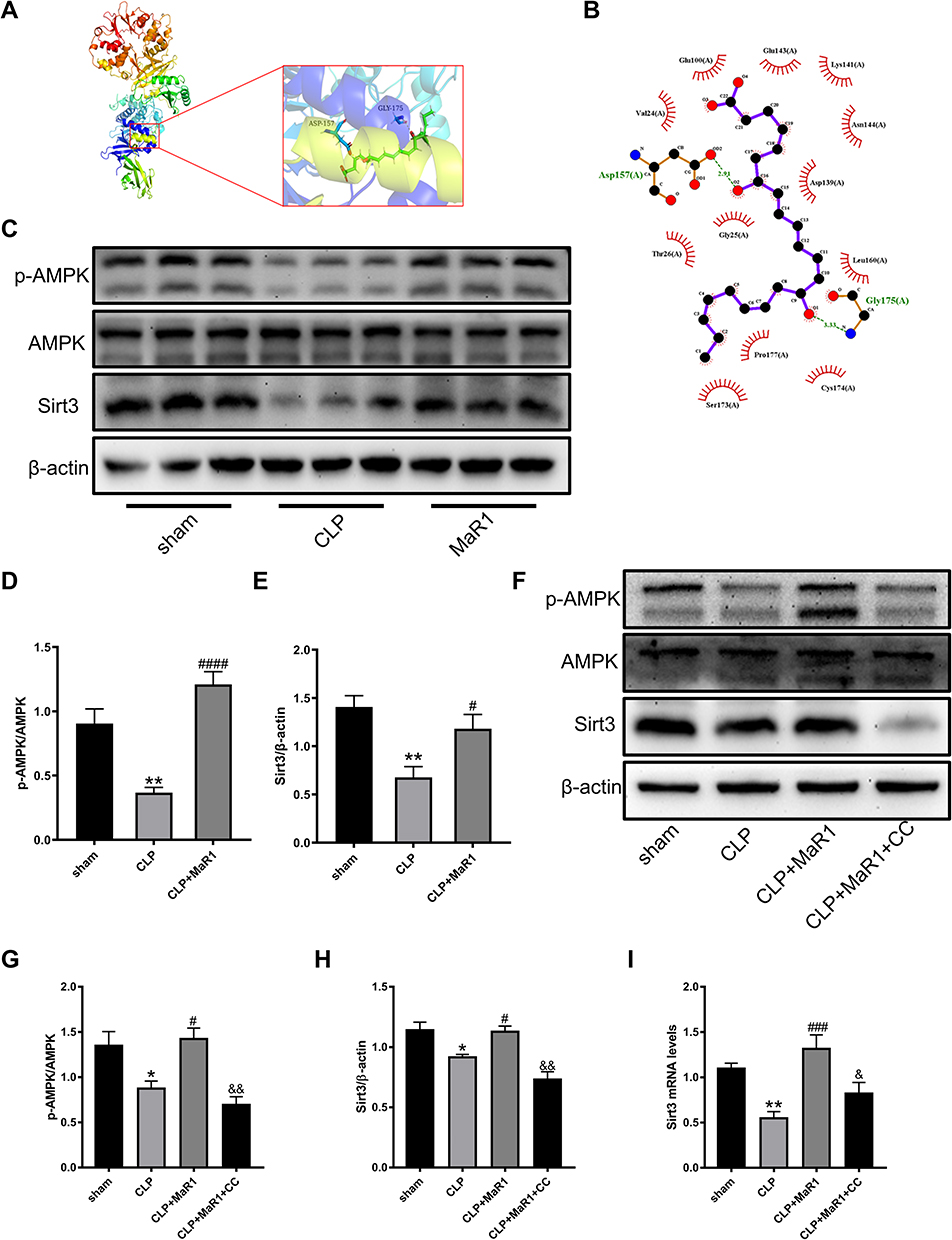 |
Figure 3 MaR1 upregulated the protein expression of AMPK/Sirt3 pathways in SA-AKI mice. (A and B) Predicted docking pose of MaR1 in complex with AMPK (PDB ID: 5UFU). (A) 3D rendering of MaR1 (green structure) docking with AMPK. (B) Interface plane rendering of MaR1 (purple structure) and AMPK. Green dashed lines indicate hydrogen bonds. (C–E) Levels of p-AMPK, AMPK and Sirt3 in renal tissues were measured using Western blotting. (F–H) AMPK inhibitor was intraperitoneally injected 2h before surgery and levels of p-AMPK, AMPK and Sirt3 in renal tissues were measured using Western blotting again. β-actin was used as the internal loading control. (I) mRNA level of Sirt3 in renal tissues was examined by real-time PCR. The data are presented as the means ± SD (n = 4–6 per group). *P<0.05, **P<0.01 vs sham group; #P<0.05, ###P<0.001, ####P < 0.0001 vs CLP group; &P<0.05, &&P<0.01 vs CLP+MaR1 group. |
Compound C Reversed the Effect of MaR1 on Improving Renal Tissue Pathological Injury and Renal Function
SA-AKI mice receiving 4 μg/kg MaR1 displayed improvements in renal pathological lesion by both H&E staining and PAS staining (Figure 4A). Renal tissue damage scores were also confirmed (Figure 4B). Compared with MaR1 alone, CC reversed the effect of MaR1 on improving renal tissue pathological injury and renal function (Figure 4A–D). mRNA levels of KIM-1, NGAL, and TIMP2 in the renal tissues were significantly increased in the CLP+MaR1+CC group compared with those in the CLP+MaR1 group (Figure 4E–G). CC reverses the effect of MaR1 in SA-AKI.
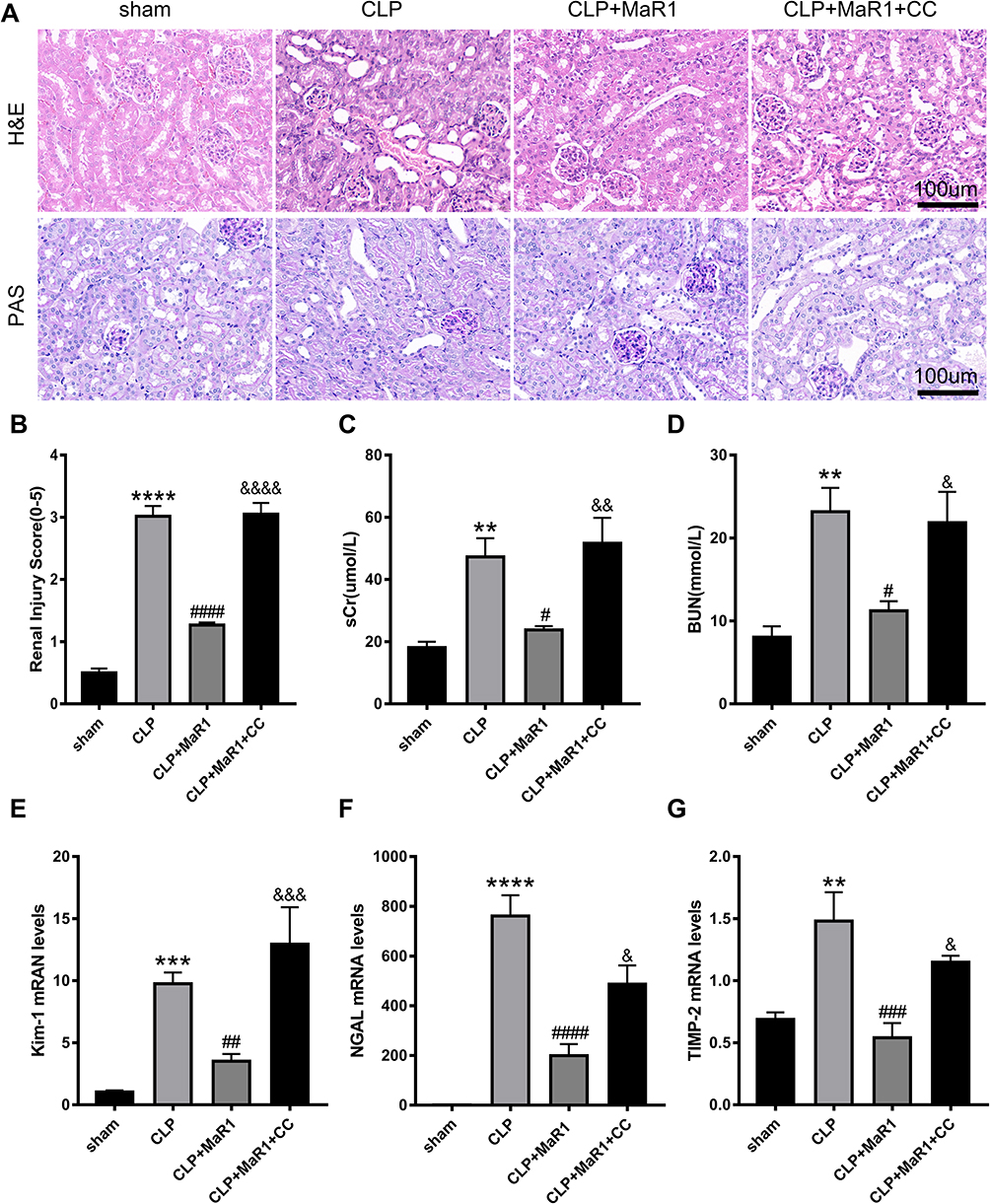 |
Figure 4 Compound C reversed the effect of MaR1 on improving renal tissue pathological injury and renal function. Representative pictures of H&E and PAS staining of renal tissues (magnification 400 ×) in (A). (B) Pathological renal tissue damage score. Serum creatinine (C) and blood urea nitrogen (D) levels at 24 h after CLP in mice. mRNA levels of Kim-1 (E), NGAL (F) and TIMP-2 (G) in renal tissues were examined by real-time PCR. The data are presented as the means ± SD (n = 4–6 per group). **P<0.01, ***P<0.001, ****P < 0.0001 vs sham group; #P<0.05, ##P<0.01, ###P<0.001, ####P < 0.0001 vs CLP group; &P<0.05, &&P<0.01, &&&P<0.001, &&&&P < 0.0001 vs CLP+MaR1 group. |
Compound C Reversed the Effect of MaR1 on Improving the Inflammatory Response
According to our findings, the levels of the pro-inflammatory factors TNF-α, IL-1β, and IL-6 in serum in the SA-AKI mice were significantly higher than those in the sham group. Meanwhile, the treatment of MaR1 can reduce these inflammatory factors in the serum. However, CC reversed the effect of MaR1 on improving the inflammatory response (Figure 5A–C). These changes of the pro-inflammatory factors TNF-α, IL-1β, and IL-6 in the renal tissues were further confirmed at the mRNA level by qRT-PCR (Figure 5D–F). Results from Western blotting (Figure 5G and H) demonstrated the degree of high mobility group box protein 1 (HMGB1) expression in the renal tissues of the four groups of mice. Among them, the CLP group had the highest expression of the HMGB1, the CLP+MaR1 group was lower than that of the CLP group, and the CLP+MaR1 +CC group was higher than that of the CLP group.
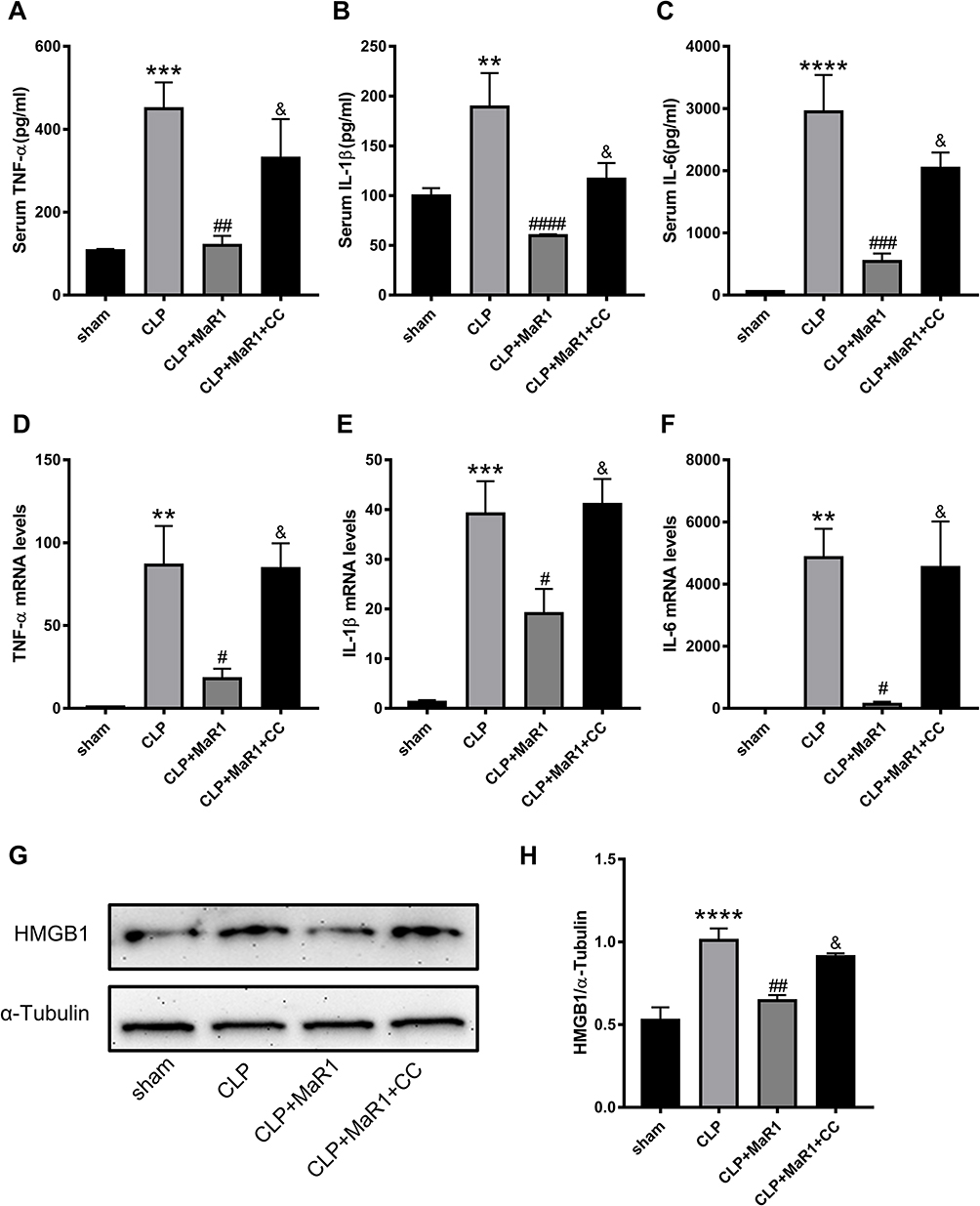 |
Figure 5 Compound C reversed the effect of MaR1 on improving the inflammatory response. The levels of cytokines in serum (A–C), including TNF-α, IL-1β and IL-6, were examined as markers of inflammation. mRNA levels of TNF-α (D), IL-1β (E) and IL-6 (F) in renal tissues were examined by real-time PCR. Protein level of HMGB1 (G and H) in renal tissues were measured using Western blotting. α-Tubulin was used as the internal loading control. The data are presented as the means ± SD (n = 4–6 per group). **P<0.01, ***P<0.001, ****P < 0.0001 vs sham group; #P<0.05, ##P<0.01, ###P<0.001, ####P < 0.0001 vs CLP group; &P<0.05 vs CLP+MaR1 group. |
Compound C Suppress MaR1’s Effect on ER Stress in SA-AKI Mice
We performed Western blot and qRT-PCR analyses to detect specific markers related to ER stress in SA-AKI mice. By detecting protein levels, we could clearly see that ER stress-specific markers, including GRP78, p-PERK, p-EIF2α, ATF4, and CHOP, were significantly activated following CLP. The results, on the other hand, confirmed that MaR1 employment significantly reduced the ER stress caused by sepsis development, as indicated by the significantly reduced protein levels of GRP78, p-PERK, p-EIF2α, ATF4, and CHOP. In contrast to the CLP+MaR1 group, CC reversed MaR1’s inhibitory effect on ER stress, resulting in higher expression of ER stress-specific proteins (Figure 6A–F). qRT-PCR analysis further confirmed the effect of MaR1 on inhibiting ER stress induced by sepsis (Figure 6G–I). Consistent with the results of Western blot, we also found increased expression of GRP78 and p-PERK in the kidney tissue of sepsis mice by immunohistochemical detection (Figure 6J).
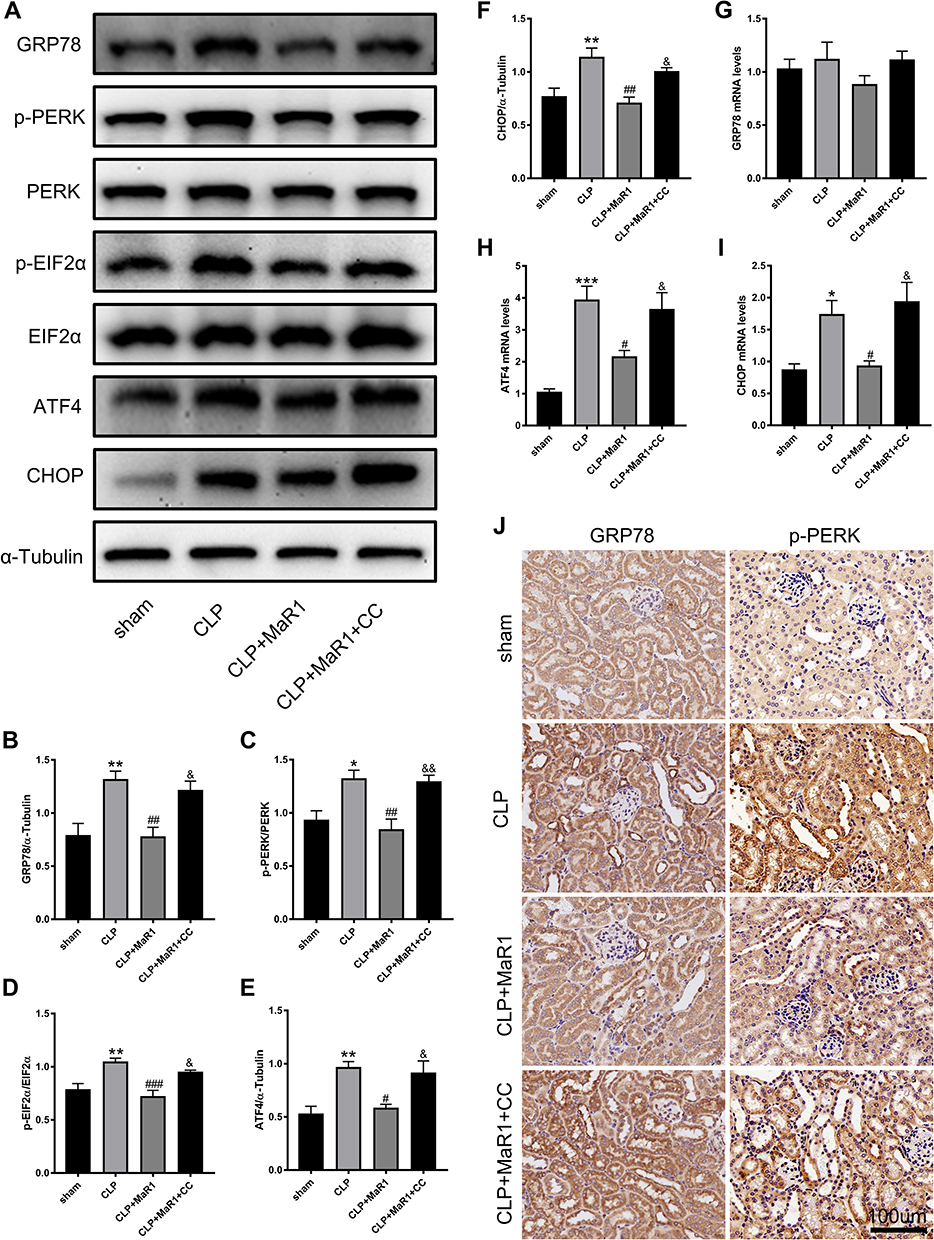 |
Figure 6 Compound C suppress MaR1’s effect on ER stress in SA-AKI mice. (A–F) Levels of GRP78, p-PERK, PERK, p- EIF2α, EIF2α, ATF4, and CHOP in renal tissues were measured using Western blotting. α-Tubulin was used as the internal loading control. mRNA levels of GRP78 (G), ATF4 (H), and CHOP (I) in renal tissues were examined by real-time PCR. (J) Immunostaining for GRP78 and p-PERK in mouse renal tissues (magnification 400 ×). The data are presented as the means ± SD (n = 4–6 per group). *P<0.05, **P<0.01, ***P<0.001 vs sham group; #P<0.05, ##P<0.01, ###P<0.001 vs CLP group; &P<0.05, &&P<0.01 vs CLP+MaR1 group. |
Compound C Suppress MaR1’s Effect on Pyroptosis in SA-AKI Mice
Next, pyroptosis-associated markers were further detected. According to the findings, CLP dramatically activated the levels of the pyroptosis-associated proteins Caspase-1, NLRP3, GSDMD, and IL-18. The data also showed that Caspase-1, NLRP3, GSDMD, and IL-18 protein levels were dramatically reduced when MaR1 was employed, which reduced pyroptosis in septic mice. MaR1 had an inhibitory effect on pyroptosis in the CLP+MaR1 group, whereas CC reversed this effect, increasing the expression of proteins related to pyroptosis (Figure 7A and C–F). The mRNA levels of Caspase-1, NLRP3, and GSDMD were consistent with their Western blot results. The mRNA level of IL-18 was not statistically significant (Figure 7G–J). By using immunohistochemistry analysis, we also discovered elevated expression of IL-1β and IL-18 in the kidney tissue of septic mice. MaR1 therapy can decrease the expression of IL-1β and IL-18, while CC counteracts this effect (Figure 7M). The levels of the pro-inflammatory factors IL-1β and IL-18 in serum further confirmed the effect of MaR1 on inhibiting pyroptosis induced by sepsis (Figures 5B and 7K). We next used the TUNEL assay to detect kidney injury. TUNEL staining results indicated that the number of TUNEL-positive renal tubule epithelial cells in SA-AKI mice was obviously higher than that in sham mice, whereas TUNEL-positive cells were markedly decreased in the MaR1 treatment group compared to the CLP group. However, the CC treatment reversed it (Figure 7B and L).
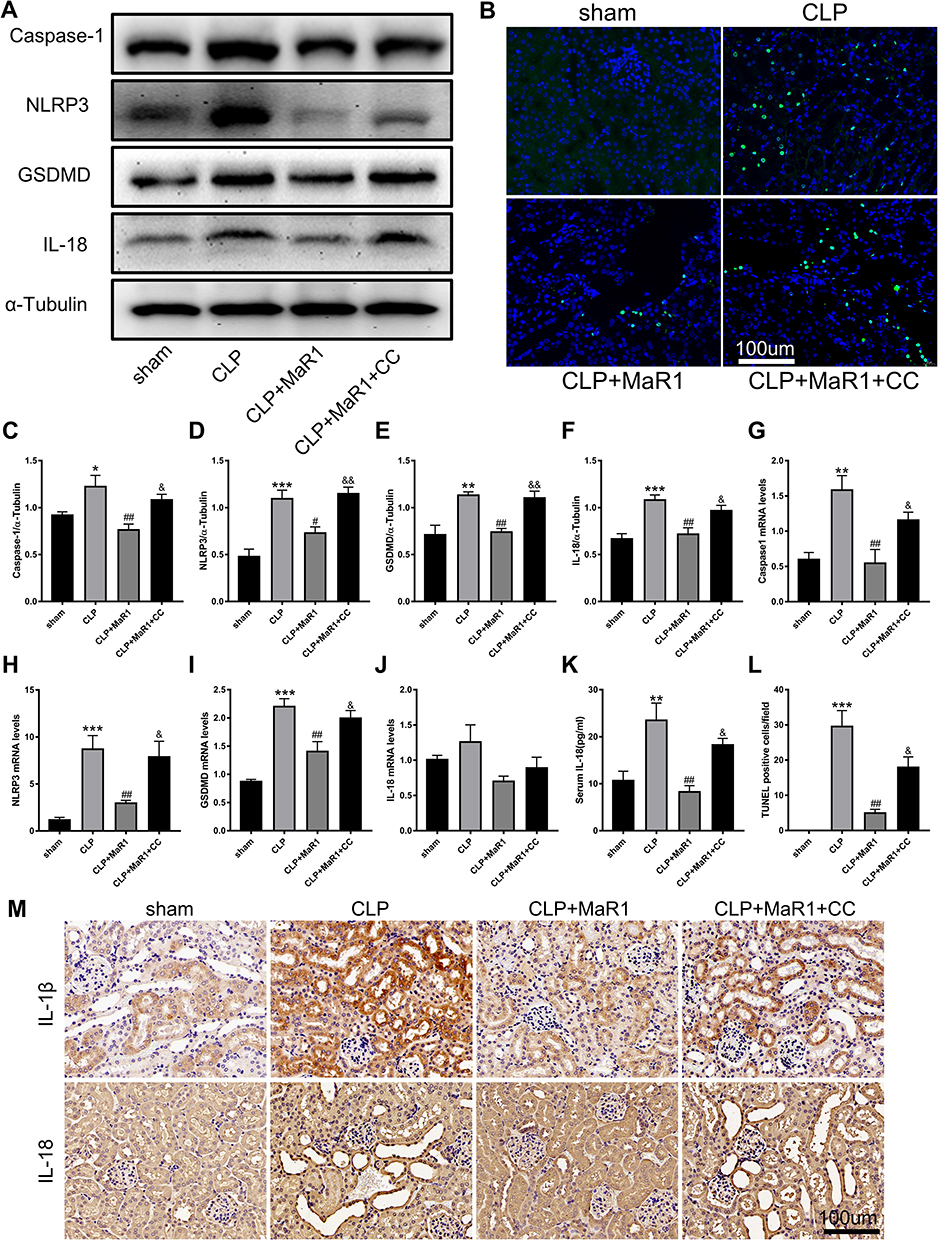 |
Figure 7 Compound C suppress MaR1’s effect on pyroptosis in SA-AKI mice. (A and C–F) Levels of Caspase-1, NLRP3, GSDMD, and IL-18 in renal tissues were measured using Western blotting. α-Tubulin was used as the internal loading control. mRNA levels of Caspase-1 (G), NLRP3 (H), GSDMD (I), and IL-18 (J) in renal tissues were examined by real-time PCR. (K) The level of cytokine IL-18 in serum was examined as marker of pyroptosis. (M) Immunostaining for IL-1β and IL-18 in mouse renal tissues (magnification 400 ×). (B and L) TUNEL staining of renal tissue and the number of TUNEL positive cells (n=3, magnification 400 ×). The data are presented as the means ± SD (n = 4–6 per group). *P<0.05, **P<0.01, ***P<0.001 vs sham group; #P<0.05, ##P<0.01 vs CLP group; &P<0.05, &&P<0.01 vs CLP+MaR1 group. |
Discussion
Our research demonstrated that MaR1 therapy can attenuate SA-AKI by activating the AMPK/SIRT3 signaling pathway to reduce inflammatory responses, endoplasmic reticulum stress, and pyroptosis. In addition, CC, an inhibitor of AMPK, inhibited the protective effect of MaR1 against SA-AKI. These results may provide a new perspective for the investigation of SA-AKI (Figure 8).
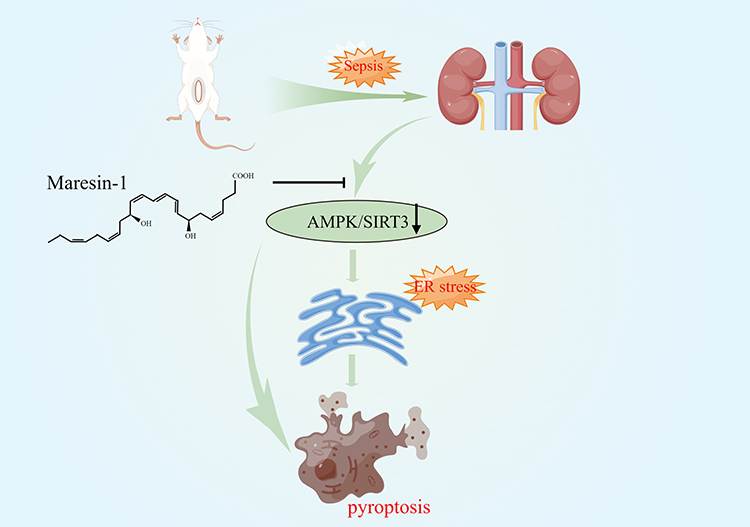 |
Figure 8 Maresin-1 therapy can attenuate SA-AKI by activating the AMPK/SIRT3 signaling pathway to reduce inflammatory responses, endoplasmic reticulum stress, and pyroptosis. |
Sepsis is a well-known global health issue24 and it’s the cause of 45–70% of AKI cases in severely ill individuals.3,25 However, there are no novel therapeutic medications available.26 It is reassuring to know that studies have shown that some therapeutic approaches may be able to reduce the severity of SA-AKI in mice by preventing pathological processes such as inflammation, ER stress, pyroptosis, and apoptosis.7–9,27–29 Maresin-1, a derivative of a typical omega-3 fatty acid, has demonstrated positive effects in models of inflammatory diseases.12,14,15,30–32 However, the protective effects and mechanisms of MaR1 protection in SA-AKI have not been elucidated. In our study, MaR1 was discovered to dose-dependently ameliorate the pathological alterations and malfunction of kidney tissue and reduce the levels of damage markers Kim-1, NGAL, and TIMP-2 in SA-AKI mice. In the early stages of sepsis, the innate immune system was overactivated and up-regulated the expression of genes associated with inflammation, including TNF-α, IL-1β, and IL-6. Our results showed that MaR1 can significantly reduce the expression of inflammatory cytokines TNF-α, IL-1β, and IL-6 to minimize excessive inflammatory response.
Omega-3 fatty acids, including eicosapentaenoic acid and DHA, have been reported to activate AMPK and consequently improve lipid metabolism in the liver and skeletal muscle. However, it remains elusive whether MaR1 activates AMPK directly or indirectly in hepatocytes.15,33 We simulated the molecular docking between MaR1 and AMPK using PyMOL2.3.0 and Ligplot v2.2.5 software, and showed that MaR1 and AMPK had a good binding effect. But how exactly MaR1 activates AMPK needs further research in the future. AMPK is a heterotrimeric complex made up of two regulatory subunits, β and γ, as well as a catalytic subunit.34,35 SIRT3 is localized in the mitochondrial matrix.34,36 The mutual regulation between AMPK and SIRT3 has been reported. SIRT3 is the downstream target of AMPK under inflammatory conditions.21,22 Previous studies have shown that AMPK/SIRT3 pathway is involved in the regulation of excessive inflammation, oxidative stress and cell apoptosis.21,23 We found that the AMPK/SIRT3 pathway was inhibited in SA-AKI mice, while MaR1 activated it. Interestingly, an AMPK inhibitor, Compound C, not only inhibited AMPK, but also inhibited downstream SIRT3 expression. Also, Compound C reversed the effect of MaR1 on improving renal tissue pathological injury and renal function and reducing inflammatory response. Thus, our results confirm that AMPK/SIRT3 pathway may play an important role in MaR1’s defense against SA-AKI.
Excessive inflammatory response, ER stress, and pyroptosis are considered important and interrelated molecular events in the pathogenesis of SA-AKI.7–9,37 HMGB1, a ubiquitous nuclear protein, is closely associated with excessive inflammation, endoplasmic reticulum stress, and pyroptosis, as well as disease progression.38,39 Previous studies have shown increased expression of ER stress-related markers in lung, heart, liver, and kidney tissues in septic mice, and elevated expression of these markers is directly related to the degree of organ dysfunction.40–42 Our study found that MaR1 can significantly inhibit the expression levels of ER stress-related protein markers GRP78, p-PERK, p-EIF2α, ATF4 and CHOP in kidney tissue of sepsis mice. ER plays an important role in the occurrence of pyroptosis. The resting NLRP3 is located in the ER. When the inflammasome is activated, pyroptosis is initiated.37,43 Previous experiments also showed that pyroptosis played an important role in both the CLP-constructed sepsis model and the LPS-induced kidney injury, and inhibition of pyroptosis could reduce SA-AKI.9,44 In our study, elevated expression of the pyroptosis-related markers, including caspase-1, NLRP3, GSDMD, IL-1β, and IL-18 in animal models, proved that pyroptosis clearly plays a role in SA-AKI. MaR1 treatment in SA-AKI mice significantly reduced excessive inflammatory and ER stress while also effectively containing the pyroptosis outbreak. Interestingly, CC can inhibit the effect of MaR1, indicating that the protective mechanism of MaR1 may be closely related to AMPK/SIRT3 pathway.
There are some limitations to our research. The severity of our CLP model may be higher than cases typically reported in clinical trials. At the same time, the model was not supported with mechanical ventilation or vasoactive drugs, which may increase the mortality of the model compared to the clinical setting. Despite the high severity, the phenotype of organ dysfunction in our model does generalize to human SA-AKI, so the biological findings may still apply to the human disease.
Conclusion
In conclusion, our results suggest that MaR1 has a protective effect on the SA-AKI mouse model. MaR1 may partially ameliorate kidney injury by activating the AMPK/SIRT3 signaling pathway, inhibiting excessive inflammation, inhibiting ER stress, and reducing pyroptosis. In addition, our study reveals a new mechanism by which MaR1 protects the kidney by blocking excessive inflammation, ER stress and pyroptosis, providing a potential new target for research on SA-AKI. More and more studies have shown that ER stress is closely related to pyroptosis, and we need to further explore the mechanism of action between them in SA-AKI in the future.
Data Sharing Statement
The data used to support the results of this study are available from the corresponding author upon reasonable request.
Acknowledgment
Picture 8 was prepared by Figraw (www.figraw.com).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by the Hubei Technological Innovation Special Fund (2019ACA167).
Disclosure
The authors declare that they have no known competing financial interests.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
2. Poston JT, Koyner JL. Sepsis associated acute kidney injury. BMJ. 2019;364:k4891. doi:10.1136/bmj.k4891
3. Hoste EA, Bagshaw SM, Bellomo R, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. 2015;41(8):1411–1423. doi:10.1007/s00134-015-3934-7
4. Peerapornratana S, Manrique-Caballero CL, Gómez H, Kellum JA. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019;96(5):1083–1099. doi:10.1016/j.kint.2019.05.026
5. Mehta RL, Bouchard J, Soroko SB, et al. Sepsis as a cause and consequence of acute kidney injury: program to improve care in acute renal disease. Intensive Care Med. 2011;37(2):241–248. doi:10.1007/s00134-010-2089-9
6. Zarbock A, Nadim MK, Pickkers P, et al. Sepsis-associated acute kidney injury: consensus report of the 28th Acute Disease Quality Initiative Workgroup. Nat Rev Nephrol. 2023;19(6):401–417. doi:10.1038/s41581-023-00683-3
7. Jiang N, Huang R, Zhang J, et al. TIMP2 mediates endoplasmic reticulum stress contributing to sepsis-induced acute kidney injury. FASEB J. 2022;36(4):e22228. doi:10.1096/fj.202101555RR
8. Ferrè S, Deng Y, Huen SC, et al. Renal tubular cell spliced X-box binding protein 1 (Xbp1s) has a unique role in sepsis-induced acute kidney injury and inflammation. Kidney Int. 2019;96(6):1359–1373. doi:10.1016/j.kint.2019.06.023
9. Sun J, Ge X, Wang Y, Niu L, Tang L, Pan S. USF2 knockdown downregulates THBS1 to inhibit the TGF-β signaling pathway and reduce pyroptosis in sepsis-induced acute kidney injury. Pharmacol Res. 2022;176:105962. doi:10.1016/j.phrs.2021.105962
10. Im DS. Maresin-1 resolution with RORα and LGR6. Prog Lipid Res. 2020;78:101034. doi:10.1016/j.plipres.2020.101034
11. Zhao M, Li C, Zhang J, et al. Maresin-1 and its receptors RORα/LGR6 as potential therapeutic target for respiratory diseases. Pharmacol Res. 2022;182:106337. doi:10.1016/j.phrs.2022.106337
12. Saito-Sasaki N, Sawada Y, Nakamura M. Maresin-1 and inflammatory disease. Int J Mol Sci. 2022;23(3):1367. doi:10.3390/ijms23031367
13. Xiao J, Yang Q, Zhang Y, et al. Maresin conjugates in tissue regeneration-1 suppresses ferroptosis in septic acute kidney injury. Cell Biosci. 2021;11(1):221. doi:10.1186/s13578-021-00734-x
14. Yang W, Tao K, Zhang P, Chen X, Sun X, Li R. Maresin 1 protects against lipopolysaccharide/d-galactosamine-induced acute liver injury by inhibiting macrophage pyroptosis and inflammatory response. Biochem Pharmacol. 2022;195:114863. doi:10.1016/j.bcp.2021.114863
15. Jung TW, Kim HC, Abd El-Aty AM, Jeong JH. Maresin 1 attenuates NAFLD by suppression of endoplasmic reticulum stress via AMPK-SERCA2b pathway. J Biol Chem. 2018;293(11):3981–3988. doi:10.1074/jbc.RA117.000885
16. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19(2):121–135. doi:10.1038/nrm.2017.95
17. Zhang H, Zhang C, Pan L, et al. Low-level Nd:YAG laser inhibiting inflammation and oxidative stress in human gingival fibroblasts via AMPK/SIRT3 axis. J Photochem Photobiol B. 2024;251:112845. doi:10.1016/j.jphotobiol.2024.112845
18. Miller FJ. Hypertension and mitochondrial oxidative stress revisited: sirtuin 3, the improved “Antioxidant”. Circ Res. 2020;126(4):453–455. doi:10.1161/CIRCRESAHA.120.316567
19. Wu QJ, Zhang TN, Chen HH, et al. The sirtuin family in health and disease. Signal Transduct Target Ther. 2022;7(1):402.
20. Zhu M, He J, Xu Y, et al. AMPK activation coupling SENP1-Sirt3 axis protects against acute kidney injury. Mol Ther. 2023;31(10):3052–3066. doi:10.1016/j.ymthe.2023.08.014
21. Yang L, Wang B, Guo F, et al. FFAR4 improves the senescence of tubular epithelial cells by AMPK/SirT3 signaling in acute kidney injury. Signal Transduct Target Ther. 2022;7(1):384. doi:10.1038/s41392-022-01254-x
22. Pan JS, Huang L, Belousova T, et al. Stanniocalcin-1 inhibits renal ischemia/reperfusion injury via an AMP-activated protein kinase-dependent pathway. J Am Soc Nephrol. 2015;26(2):364–378. doi:10.1681/ASN.2013070703
23. Liu Z, Liu H, Xiao L, Liu G, Sun L, He L. STC-1 ameliorates renal injury in diabetic nephropathy by inhibiting the expression of BNIP3 through the AMPK/SIRT3 pathway. Lab Invest. 2019;99(5):684–697. doi:10.1038/s41374-018-0176-7
24. Fleischmann C, Scherag A, Adhikari NK, et al. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med. 2016;193(3):259–272. doi:10.1164/rccm.201504-0781OC
25. Uchino S, Kellum JA, Bellomo R, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294(7):813–818. doi:10.1001/jama.294.7.813
26. Cohen J, Vincent JL, Adhikari NK, et al. Sepsis: a roadmap for future research. Lancet Infect Dis. 2015;15(5):581–614.
27. Porter AW, Nguyen DN, Clayton DR, et al. The molecular chaperone GRP170 protects against ER stress and acute kidney injury in mice. JCI Insight. 2022;7(5). doi:10.1172/jci.insight.151869.
28. Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol. 2014;25(12):2689–2701. doi:10.1681/ASN.2014030262
29. Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney Int. 2016;89(4):779–791. doi:10.1016/j.kint.2015.11.021
30. Ohuchi K, Ono Y, Joho M, et al. A docosahexaenoic acid-derived pro-resolving agent, maresin 1, protects motor neuron cells death. Neurochem Res. 2018;43(7):1413–1423. doi:10.1007/s11064-018-2556-1
31. Jung TW, Park HS, Choi GH, et al. Maresin 1 attenuates pro-inflammatory reactions and ER stress in HUVECs via PPARα-mediated pathway. Mol Cell Biochem. 2018;448(1–2):335–347. doi:10.1007/s11010-018-3392-y
32. Wang YH, Li Y, Wang JN, et al. Maresin 1 attenuates radicular pain through the inhibition of NLRP3 inflammasome-induced pyroptosis via NF-κB signaling. Front Neurosci. 2020;14:831. doi:10.3389/fnins.2020.00831
33. Figueras M, Olivan M, Busquets S, López-Soriano FJ, Argilés JM. Effects of eicosapentaenoic acid (EPA) treatment on insulin sensitivity in an animal model of diabetes: improvement of the inflammatory status. Obesity. 2011;19(2):362–369. doi:10.1038/oby.2010.194
34. Clark AJ, Parikh SM. Targeting energy pathways in kidney disease: the roles of sirtuins, AMPK, and PGC1α. Kidney Int. 2021;99(4):828–840. doi:10.1016/j.kint.2020.09.037
35. Li A, Yi B, Han H, et al. Vitamin D-VDR (vitamin D receptor) regulates defective autophagy in renal tubular epithelial cell in streptozotocin-induced diabetic mice via the AMPK pathway. Autophagy. 2022;18(4):877–890. doi:10.1080/15548627.2021.1962681
36. Huang C, Jiang S, Gao S, et al. Sirtuins: research advances on the therapeutic role in acute kidney injury. Phytomedicine. 2022;101:154122. doi:10.1016/j.phymed.2022.154122
37. Li N, Wang Y, Wang X, Sun N, Gong YH. Pathway network of pyroptosis and its potential inhibitors in acute kidney injury. Pharmacol Res. 2022;175:106033. doi:10.1016/j.phrs.2021.106033
38. Zhang YY, Ning BT. Signaling pathways and intervention therapies in sepsis. Signal Transduct Target Ther. 2021;6(1):407.
39. Deng C, Zhao L, Yang Z, et al. Targeting HMGB1 for the treatment of sepsis and sepsis-induced organ injury. Acta Pharmacol Sin. 2022;43(3):520–528. doi:10.1038/s41401-021-00676-7
40. Huang M, Cai S, Su J. The pathogenesis of sepsis and potential therapeutic targets. Int J Mol Sci. 2019;20(21):5376. doi:10.3390/ijms20215376
41. Luo X, Lin B, Gao Y, et al. Genipin attenuates mitochondrial-dependent apoptosis, endoplasmic reticulum stress, and inflammation via the PI3K/AKT pathway in acute lung injury. Int Immunopharmacol. 2019;76:105842. doi:10.1016/j.intimp.2019.105842
42. Ma Y, Liu J, Liu H, Han X, Sun L, Xu H. Podocyte protection by Angptl3 knockout via inhibiting ROS/GRP78 pathway in LPS-induced acute kidney injury. Int Immunopharmacol. 2022;105:108549. doi:10.1016/j.intimp.2022.108549
43. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi:10.1038/nature09663
44. Li Y, Zhang J, Zhai P, et al. The potential biomarker TIFA regulates pyroptosis in sepsis-induced acute kidney injury. Int Immunopharmacol. 2023;115:109580. doi:10.1016/j.intimp.2022.109580
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.