")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Maraviroc: a review of its use in HIV infection and beyond
Authors Woollard S, Kanmogne G
Received 16 June 2015
Accepted for publication 28 July 2015
Published 1 October 2015 Volume 2015:9 Pages 5447—5468
DOI https://doi.org/10.2147/DDDT.S90580
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Shawna M Woollard, Georgette D Kanmogne
Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE, USA
Abstract: The human immunodeficiency virus-1 (HIV-1) enters target cells by binding its envelope glycoprotein gp120 to the CD4 receptor and/or coreceptors such as C-C chemokine receptor type 5 (CCR5; R5) and C-X-C chemokine receptor type 4 (CXCR4; X4), and R5-tropic viruses predominate during the early stages of infection. CCR5 antagonists bind to CCR5 to prevent viral entry. Maraviroc (MVC) is the only CCR5 antagonist currently approved by the United States Food and Drug Administration, the European Commission, Health Canada, and several other countries for the treatment of patients infected with R5-tropic HIV-1. MVC has been shown to be effective at inhibiting HIV-1 entry into cells and is well tolerated. With expanding MVC use by HIV-1-infected humans, different clinical outcomes post-approval have been observed with MVC monotherapy or combination therapy with other antiretroviral drugs, with MVC use in humans infected with dual-R5- and X4-tropic HIV-1, infected with different HIV-1 genotype or infected with HIV-2. This review discuss the role of CCR5 in HIV-1 infection, the development of the CCR5 antagonist MVC, its pharmacokinetics, pharmacodynamics, drug–drug interactions, and the implications of these interactions on treatment outcomes, including viral mutations and drug resistance, and the mechanisms associated with the development of resistance to MVC. This review also discusses available studies investigating the use of MVC in the treatment of other diseases such as cancer, graft-versus-host disease, and inflammatory diseases.
Keywords: chemokine receptors, human immunodeficiency virus, CCR5 antagonists, pharmacokinetics, pharmacodynamics, drug interactions, mutations, resistance, AIDS
A Letter to the Editor has been received and published for this article
Introduction
The human immunodeficiency virus-1 (HIV-1) enters target cells by binding its envelope glycoprotein gp120 to the CD4 receptor and/or coreceptors such as the C-C chemokine receptor type 5 (CCR5; R5) and C-X-C chemokine receptor type 4 (CXCR4; X4).1 R5-tropic and X4-tropic viral strains use CCR5 and CXCR4, respectively, as coreceptor to enter and infect target cells. Some HIV-1 strains are dual tropic and can use CCR5 and/or CXCR4.2 CCR5 is expressed on several cell types, including T-cells, dendritic cells, and leukocytes.2,3 In HIV-infected humans, R5-tropic viruses predominate during the early stages of infection, whereas X4-tropic viruses usually emerge during the later stages.1,2 The importance of CCR5 in HIV/AIDS was demonstrated by studies showing that a 32-base-pair deletion in the CCR5 gene results in resistance to HIV-1 infection or slower progression to AIDS.4,5
Given CCR5’s importance in HIV-1 transmission, infection, and AIDS progression, drugs targeting CCR5 have been an important area of research. In a short-term trial with HIV-infected patients, aplaviroc, the first CCR5 antagonist to enter clinical trials, demonstrated significant antiretroviral activity.6 However, in subsequent Phase II trials, testing was discontinued due to increased cases of idiosyncratic hepatotoxicity.7 Vicriviroc (VCV) significantly reduced viral loads (VL).8 Two subsequent Phase II trials confirmed VCV antiretroviral activity and safety.9,10 However, in a third Phase II and two Phase III trials, VCV showed higher rates of virological failure than other antiretroviral drugs,11,12 and its further development was terminated. Cenicriviroc, a CCR5 and CCR2 inhibitor, has completed Phase IIb trials and showed potent antiretroviral activity in vitro and in vivo.13,14 Maraviroc (MVC, Pfizer) is a small molecule, reversible CCR5 antagonist,15 currently approved for treatment of patients infected with R5-tropic HIV-1.15 This paper will review MVC discovery and development, its efficacy against HIV-1/AIDS, pharmacokinetics, pharmacodynamics and drug resistance, and its use in other diseases.
MVC discovery
MVC, originally called UK-427,857 (empirical formula: C29H41F2N5O), was developed by Pfizer during CCR5 ligand studies.16 High-throughput screening to identify small molecules that could inhibit the binding of macrophage inflammatory protein-1-beta to CCR5 stably expressed in HEK-293 cells lead to the discovery of imidazopyridine, UK-107,543.17 UK-107,543 displayed efficient and potent inhibition of macrophage inflammatory protein-1-beta binding to CCR5, with a half-maximal inhibitory concentration of 650 nM. However, UK-107,543 had no antiretroviral activity. MVC was the result of UK-107,543 optimization for binding potency against CCR5, antiretroviral activity, absorption, pharmacokinetics, and selectivity for the human ERG channel.16 This optimization is summarized in Figure 1. Modifications of UK-107,543 to UK-372,673 resulted in increased binding to CCR5 and antiretroviral activity, with 90% inhibitory concentration (IC90) of 75 nM.18 Further modifications to make UK-382,055 increased its antiretroviral activity (IC90: 3 nM), but blocked potassium channels.18 Modifications to make UK-396,794 further increased antiretroviral activity (IC90: 0.6 nM) and increased absorption, but UK-396,794 was rapidly metabolized.18 In total, 956 analogues were screened before finally getting MVC, which displayed good antiretroviral activity (IC90 below 2 nM), did not block potassium channels, was not rapidly metabolized, and had good absorption.18
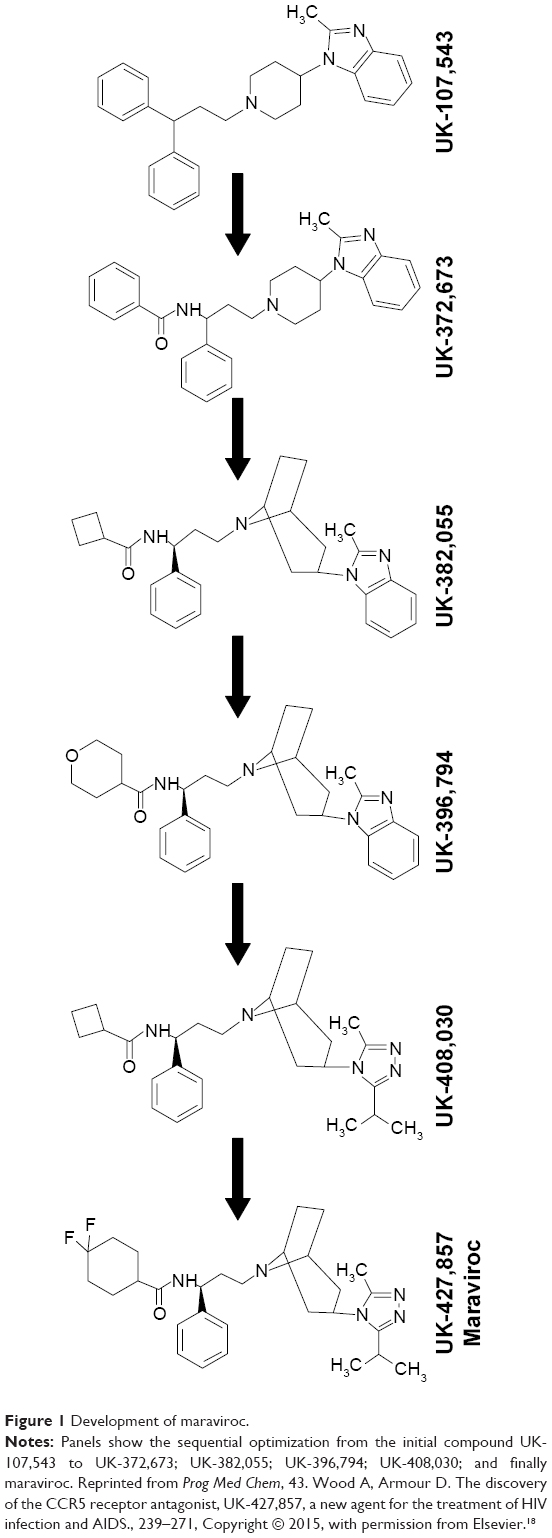 | Figure 1 Development of maraviroc. |
MVC clinical trials
In Phase I trial, MVC reached steady-state plasma concentrations after 7 days’ treatment and was well tolerated at clinically relevant doses (<900 mg/kg).19 A phase II trial in asymptomatic HIV-1-infected humans receiving MVC, 300 mg/kg twice daily showed that VL decreased by 1.6 log10 copies/mL compared to 0.02 log10 copies/mL in infected humans receiving placebo.20 These results validated MVC as a non-viral target antiretroviral therapy (ART). The MOTIVATE trials were two Phase III trials to determine the efficacy and safety of MVC in patients infected with R5-tropic HIV-1.21 Patients received oral MVC or placebo once or twice daily, with doses adjusted based on other drugs in the patient’s ART regimen. MVC decreased VL by -1.84 log10 copies/mL compared to -0.79 log10 copies/mL in placebo-treated patients.21 MVC showed efficacy throughout the 96 weeks of treatment; 86.7% and 81.4% of patients receiving MVC twice daily and once daily, respectively, who had VL below 50 copies/mL at week 48, maintained virological suppression up to week 96.22 From week 48 to week 96, the median CD4 count increased by 89 and 113 cells/mm3 in patients receiving MVC once daily and twice daily, respectively.22 The incidences of adverse events were similar for patients on ART regimens containing MVC twice daily, MVC once daily, or placebo, at week 96 and year 5.23 Subgroup analyses of pooled data from week 48 of the MOTIVATE trials were performed based on parameters such as ethnicity, baseline VL, CD4 count, tropism, genetic polymorphisms, and ART regimens. Results showed that MVC was effective in multiple patients infected with R5-tropic HIV-1 who had previously been treated with other ART drugs, including patients with low baseline CD4 count or high VL.24 Furthermore, more patients receiving MVC who failed treatment had X4-tropic HIV-1 at treatment failure compared to those receiving placebo, but MVC-treated patients showed no decrease in CD4 count.24
The MERIT study was a Phase IIb/III trial to determine the efficacy of MVC versus efavirenz (EFV) in treatment-naïve patients infected with R5-tropic HIV-1.25 MVC treatment did not initially demonstrate noninferiority for patients with VL below 50 copies/mL, with 65.3% and 69.3% of patients receiving EFV and MVC, respectively, having VL below 50 copies/mL. However, in post hoc reanalysis after removal of patients with previously undetectable X4-tropic viruses, MVC was noninferior to EFV, with 68.3% and 68.5% of patients in the EFV and MVC groups, respectively, having VL below 50 copies/mL.25 Analysis at 5 years showed that 50.8% and 45.9% of patients receiving MVC and EFV, respectively, had maintained VL below 50 copies/mL.26 The mean increased in CD4 count was 293 cells/μL in MVC-treated patients compared to 271 cells/μL in EFV-treated patients;26 compared to the EFV group, fewer MVC-treated patients discontinued treatment due to adverse events.26 Following these studies, MVC was approved by United States Food and Drug Administration and the European Commission for treatment-experienced HIV-1-infected humans, and was subsequently also approved for treatment-naïve HIV-1-infected humans.15
MVC pharmacokinetics
Dosage, absorption, and bioavailability
Standard MVC dosage is 300 mg/kg twice daily;15 for patients receiving a cytochrome P450-3A4 (CYP3A4) inhibitor or inducer, dosage is adjusted to 150 or 600 mg/kg twice daily, respectively.15 In an effort to reduce possible toxicity associated with high drug concentrations, simplify drug regimens, and reduce costs, the clinical outcomes of 150 or 300 mg/kg MVC once daily have been examined. In the MOTIVATE trials, once daily MVC showed virological suppression (mean change in plasma HIV-1 RNA levels of −1.68 log10 copies/mL), although this was not as robust as the levels of virological suppression in subjects receiving MVC twice daily.21 The Maraviroc Once Daily with Darunavir Enhanced by Ritonavir in a New Regimen study, comparing the efficacy of 150 mg/kg MVC plus darunavir/ritonavir (DRV/RTV) once daily versus tenofovir (TDF) plus DRV/RTV once daily, was terminated due to inferiority of the MVC-containing regimen.27 However, other studies have shown that 150 mg/kg MVC plus DRV/RTV once daily can effectively suppress HIV-1 infection.28 Virological suppression was also demonstrated with nucleoside reverse-transcriptase inhibitor (NRTI)-sparing regimens of 150 mg/kg MVC once daily plus lopinavir (LPV)/RTV and atazanavir (ATV)/RTV.29,30 Another study examined the effect of MVC-containing regimens with TDF-containing regimens on bone loss; the authors used a dose of 150 mg/kg MVC once daily in combination with DRV/RTV and emtricitabine (FTC), and showed that, although bone mineral density decreased in both groups, the magnitude of decrease was less in patients receiving MVC.31 However, this latter study did not analyze the effects of treatment regimens on viremia.
MVC (molecular weight: 513.67 g/mol) is moderately lipophilic, with a distribution constant at pH 7.4 (log D7.4) of 2.1.32 Drug lipophilicity can increase its ability to penetrate lipid membranes such as the blood–brain barrier;33 and the optimal log D7.4 for diffusion from plasma into cerebrospinal fluid (CSF) is 1–10.34 MVC is a weak base with a pKa of 7.3,35 is highly soluble at pH 1–7.5,15 and 76% of MVC binds to plasma proteins.35 At a single MVC dose of 300 mg/kg, time to maximum concentration (Tmax) occurred by 2 hours post-treatment in humans;32 with higher MVC levels in plasma (median maximum concentration [Cmax: 800 ng/g]) compared to whole blood (Cmax: 489 ng/g).32 MVC exposure (area under the curve; AUC) was 4,497 ng h/L for plasma and 2,251 ng h/L for blood.32 MVC AUC decreased by 33%–37% after a high fat meal,15,36 but this did not affect MVC antiviral activity.15 At 100 and 300 mg/kg, the absolute MVC oral bioavailability is 23.1% and 33%, respectively.32
Distribution
MVC is distributed throughout the body and can be detected in the seminal plasma (SP), vaginal tissues (VT), cervicovaginal fluids (CF), rectum, and CSF.15,37–47 Pharmacokinetic data are summarized in Table 1. MVC concentrations and AUC in the SP are lower than in the blood plasma (plasma).37 However, the median protein binding in the SP is 9% compared to 76% in the plasma; more pharmacologically available MVC are found in the SP,37 and most patients on MVC had undetectable VL in SP,38 suggesting that MVC might reduce sexual HIV-1 transmission. MVC has higher concentrations in vaginal tissue and CF than in plasma, and also has lower protein binding (7.6%) in CF than in plasma.40
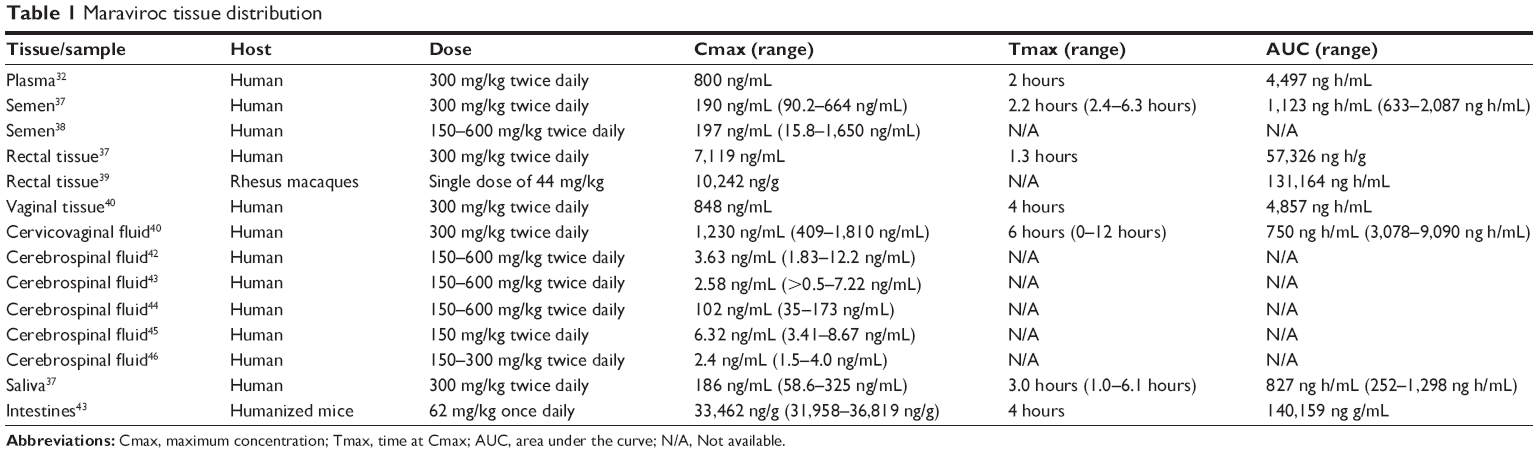 | Table 1 Maraviroc tissue distribution |
Compared to plasma, MVC concentrations in rectal tissues are 7.5-fold higher after a single dose and 26-fold higher after multiple doses.37 These results were corroborated by animal studies showing that MVC concentrations in macaques’ rectal tissues were 23% higher than in plasma;39 however, despite high concentrations in rectal tissues, MVC could not prevent simian-HIV (SHIV) rectal transmission.39 MVC CSF concentrations are often low but vary; studies of asymptomatic HIV-1-infected humans42,43,45,46 showed lower concentrations than a study of HIV-1-infected patients with central nervous system (CNS) impairment.44 These discrepancies could be due to differences in drug regimens or increased drug entry into the brain due to blood–brain barrier inflammation in patients with CNS impairment. Despite low CSF concentrations, MVC suppressed CSF VL.42,43 MVC is detectable in humans’ saliva and correlates with its plasma concentrations;37 thus, measuring MVC saliva concentrations could be a less invasive option of monitoring adherence. Quantification of MVC in humans’ intestines has not been done, however, in MVC-treated humanized mice, the highest drug concentrations were in the intestine.47 This is likely due to MVC excretion through feces.35
Metabolism
Unmetabolized MVC constitutes 42% of circulating drug.32 The most abundant metabolite in the plasma is a secondary amine product of N-dealkylation (UK-408,027).32 MVC is metabolized by CYP3A4 and CYP3A5;35,48 CYP3A4 is its major enzyme,35 while CYP3A5 is involved in the formation of mono-oxygenated metabolites.48,49 In fact, ketoconazole, a potent CYP3A4 inhibitor, reduced MVC metabolism and UK-408,207 formation by over 80%,50 while specific inhibitors of CYP1A2, CYP2C9, CYP2C19, and CYP2D6 had no significant effect.50 MVC is a P-glycoprotein (Pgp) substrate; in MVC-treated Pgp-knockout-friend-leukemia-virus-B mice Cmax and AUC increased by 108% and 183%, respectively, compared to MVC-treated wild-type mice.35
Genetic polymorphisms
CYP3A5 is involved in the oxidative metabolism of MVC, and it has been suggested that CYP3A5 genetic polymorphisms may influence MVC pharmacokinetics.49 The wild-type homozygous allele, CYP3A5*1, is associated with the highest CYP3A5 protein expression, whereas the CYP3A5*3, CYP3A5*6, and CYP3A5*7 alleles are associated with nonfunctional or dysfunctional CYP3A5 expression.49,51–54 CYP3A5 expression varies by ethnicity, 80%–90% of Caucasians are non-expressers due to a high prevalence of CYP3A5*3, while the majority of African–Americans are expressers, with 45% being homozygous for CYP3A5*1.51,55 Individuals with no CYP3A5*1 had similar MVC plasma concentrations as those with heterozygous CYP3A5*1.49 However, compared to humans with no CYP3A5*1 and heterozygous subjects, individuals with homozygous CYP3A5*1 allele had lower MVC exposures and lower MVC plasma concentrations, with half of the individuals in the homozygous group having average MVC concentrations below 100 ng/mL, the threshold value below which there is increased risk of virological failure.49 Compared to individuals with no CYP3A5*1 and heterozygous individuals, MVC Cmax in homozygous individuals decreased by 33% and 22%, respectively; and MVC exposure also decreased in homozygous individuals by 41% and 30%.49 Individuals with no CYP3A5*1 showed decreased exposure to the predominant MVC oxidative metabolite compared to the homozygous group.49
MVC is a substrate for the organic anion transporter polypeptide-1B1, an influx transporter, also known as solute carrier organic anion transporter family member-1B1.56 The solute carrier organic anion transporter family member-1B1 gene has two functional single nucleotide polymorphisms (SNPs), c.521 T>C and c.388 A>G.56–58 The c.521 T>C SNP contains two haplotypes, *5 (388A-521 C) and *15 (388G-521 C), that have been associated with reduced activity and increased plasma concentrations of several drugs.58–60 HIV-1-infected patients who were heterozygous for the c.521 T>C SNP had a 55% increase in MVC concentrations compared to patients who were homozygous for the wild-type allele.56
Excretion
MVC is primarily excreted through feces, with 76% excreted through this route, and 19%–20% excreted through urine at 36–96 hours post-dose.32,35 Unmetabolized MVC accounts for 33% of excreted drug,35 major excreted metabolites include a product of hydroxylation of the methyl group of the triazole moiety, four products of mono-oxidation in the difluorocyclohexyl ring, and N-dealkylation adjacent to the tropane ring, resulting in a secondary amine.32,35
MVC pharmacodynamics
In radioligand-binding assays, MVC inhibited the binding of chemokine (C-C motif) ligand (CCL)-3, CCL4, and CCL5 to CCR5 with a half-maximal inhibitory concentration of 3.3, 7.2, and 5.2 nM, respectively.35 Furthermore, MVC potently inhibited downstream CCR5 signaling and did not induce CCR5 internalization, suggesting that MVC is a functional CCR5 antagonist.35 MVC showed no significant affinity for other receptors, including CCR2, which has sequence similarity to CCR5 and is susceptible to other CCR5 antagonists.61,62 MVC inhibits HIV-1 binding to peripheral blood mononuclear cells (PBMC) (IC90: 3.1 nM), and up to 10 μM MVC had no effect on cell proliferation.35 MVC effectively inhibited infection of several R5-tropic HIV-1 strains, but not X4-tropic HIV-1.35
MVC concentrations increase by 11%–32% in humans with mild-to-moderate hepatic impairment, but with proper monitoring, no dose adjustment is necessary.63 In individuals with severe renal impairment, MVC renal clearance was fourfold lower than in patients with normal renal function (27 versus 110 mg/min, respectively).64 In a patient with end-stage liver disease and renal impairment receiving 300 mg/kg MVC, raltegravir (RAL), and enfuvirtide (ENF) twice daily, MVC serum concentrations were 25-fold higher than expected, and remained high after doses were adjusted to every 48 hours.65 No dose adjustment is necessary in patients with mild-to-moderate renal impairment, but in patients with severe renal impairment or end-stage renal disease, MVC should not be given if the patient is also taking CYP3A4 inducers or inhibitors.15 Compared to untreated patients, MVC treatment of humans infected with R5-tropic HIV-1 resulted in improved lipid profiles, increased CD4 count, and decreased levels of soluble endothelial protein-C receptor.66,67
MVC and immune cell restoration
MVC improves T-cell counts, and higher baseline CD4 counts correlate with better response to MVC treatment.68 In the MOTIVATE trials, patients receiving MVC had larger CD4+ T-cell increases than those receiving placebo, even after adjusting for the greater virological potency of MVC-containing regimens.69 Short-term MVC treatment of patients with persistent virological failure resulted in increased CD4+ and CD8+ T-cells and positively correlated with improved antiviral activity.70 Following virological failure, infected patients treated for 96 weeks with RAL, etravirine (ETR), and MVC showed a greater increase in CD4+ T-cells than patients receiving RAL, ETR, and DRV/RTV.71 Patients on MVC intensification showed increased CD4 count slope,72 maintained higher CD4 counts after MVC discontinuation,73 and showed decreased VL in memory T-cells.74 In vitro studies also showed that MVC blocks CCR5 and CCR2 internalization and inhibits T-cell chemotaxis,74 increases CCR5 expression on activated T-cells, decreases the expression of CD25, CD38, and HLA-DR, and increases the expression of CD69 in CD4+ and CD8+ T-lymphocytes.75
In patients with suppressed viremia, MVC treatment modestly increased CD4 and CD8 counts and reduced the percentages of regulatory T-cells (Tregs).76 ART regimens containing MVC or MVC monotherapy decreased the frequency of Tregs in treatment-naïve HIV-1-infected patients from day 8 post-treatment initiation, and dose-dependently reduced Tregs frequency in PBMC.77 Treatment of virologically suppressed patients with ART regimens containing MVC decreased the expression of natural killer cell activation markers, decreased the frequency of inflammatory monocytes, including CD16+ monocytes, and this was associated with improved neuropsychological performance.78,79 However, in some HIV-1-infected patients with low baseline T-cells counts, MVC intensification had limited-to-no effect on CD4 counts, compared to patients on ART regimens without MVC;80–82 although patients on MVC-containing ART regimens showed increased CD8 counts.80
MVC and HIV-1 mother-to-child transmission
Mother-to-child transmission (MTCT) is the most common route of pediatric HIV-1 transmission and occurs in utero, intrapartum, or during breastfeeding.83 The mother’s plasma VL can predict the likelihood of HIV-1 transmission to the child; in developed countries where ART is readily available, MTCT is below 2%.84 A study of MVC placental transfer in macaques showed that after a single MVC dose 2 hours pre-delivery, maximal plasma MVC concentrations in infants occurred immediately after birth, Cmax: 22 ng/mL.85 Infants’ Cmax were 44-fold lower than mothers’ Cmax, and MVC was not detectable in infants 3.5 hours after birth.85 This suggests that a single MVC dose may not protect babies during vaginal delivery. Previous World Health Organization (WHO) recommendation for reducing MTCT, when access to ART is insufficient for a complete antepartum, intrapartum, and postpartum regimen, was a single-dose nevirapine (NVP) during labor.85 This single NVP dose reduced MTCT by approximately 50%,86 but increased drug resistance.87 The macaque study85 sought to determine whether MVC could replace NVP as a single intrapartum dose option. However, while WHO guidelines have since been revised, many low-income countries continue to use single-dose NVP.88 There have been no other major studies on MVC placental transfer after multiple doses. The low MVC placental transfer was confirmed using an ex vivo human perfused cotyledon model, which reproduces the third-trimester pregnancy conditions, and is considered the gold standard method for measuring fetal exposure risk during maternal treatment.83 The MVC fetal transfer rate was 8% and its clearance index was 0.26;83 low MVC transfer was associated with transporters-mediated drug efflux across placental membranes.83 However, an ART regimen consisting of 600/100 mg DRV/RTV, 150 mg MVC, and 200 mg ETR twice daily, with TDF/FTC prevented HIV-1 MTCT.89 In this case, MVC concentration in the umbilical cord blood was 69 ng/mL and the umbilical cord-to-plasma ratio was 0.37.89 Studies are needed to determine MVC levels in breast milk, quantify MVC transfer after multiple/prolonged dosing, and determine the long-term efficacy of MVC in preventing HIV-1 MTCT.
MVC drug–drug interactions
MVC interactions with protease inhibitors
Protease inhibitors (PIs) bind to HIV-1 protease, preventing its cleavage of Gag and GagPol precursors into their mature/infective forms.90 PIs, including saquinavir (SQV), RTV, LPV, ATV, tipranavir (TPV), amprenavir (APV), and DRV, are common CYP3A4 inhibitors91 and would increase MVC concentrations. Co-administration of MVC with SQV, SQV/RTV, LPV/RTV, RTV, ATV, ATV/RTV, or DRV/RTV increased MVC plasma Cmax by 332%, 423%, 161%, 128%, 209%, 267%, and 229%, respectively.92,93 However, TPV/RTV had no effect on MVC plasma concentrations.92 Fosamprenavir (FPV), an APV pro-drug, is used as a slow-release version of APV. In a treatment regimen including FPV, RTV, and MVC, MVC AUC and Cmax increased by 249% and 152%, respectively;94 APV AUC and Cmax decreased by 35% and 34%, respectively;94 RTV AUC and Cmax decreased by 34% and 39%, respectively.94 While FPV/RTV increasing MVC concentrations is not surprising, since both are CYP3A4 inhibitors, the mechanisms of MVC reducing RTV and FPV concentrations are not known. This may involve protein-binding displacement, which is associated with increase in unbound FPV and RTV ready to be metabolized. Overall, adjustments of MVC doses to 150 mg/kg twice daily are recommended when co-administered with PIs, except for TPV/RTV15 (Table 2).
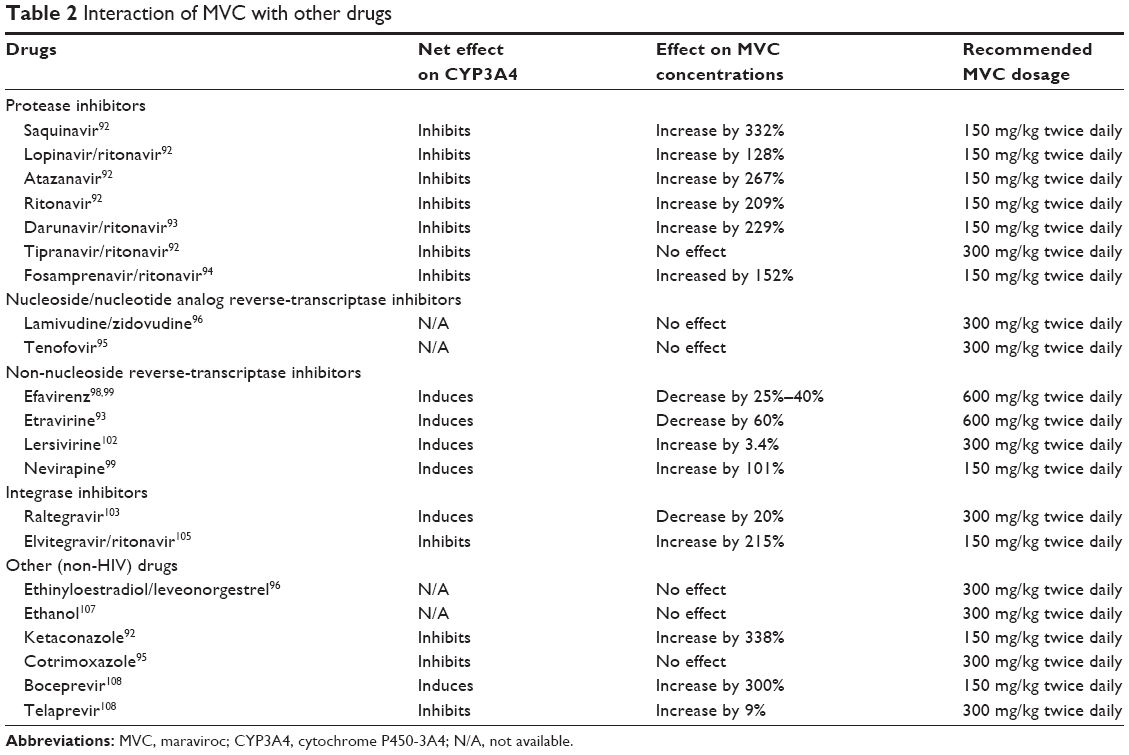 | Table 2 Interaction of MVC with other drugs |
MVC interactions with nucleoside/nucleotide analog reverse-transcriptase inhibitors
NRTIs and nucleotide analog reverse-transcriptase inhibitors (NtRTIs) are nucleoside or nucleotide analogs that block the reverse transcriptase activity by incorporating themselves into viral RNA. TDF, a NtRTI, does not affect MVC concentration;95 MVC treatment had no effect on lamivudine/zidovudine (3TC/ZDV) concentrations, both NRTIs.96 NRTIs are mostly cleared renally and are not metabolized by CYPs enzymes;97 therefore, no dose adjustments are necessary when NRTIs are co-administered with MVC15 (Table 2).
MVC interactions with non-nucleoside reverse-transcriptase inhibitors
Non-nucleoside reverse-transcriptase inhibitors (NNRTIs) bind to the reverse transcriptase to prevent RNA conversion to cDNA. EFV, a CYP3A4 inducer, reduced MVC concentrations by over 50% in healthy subjects98 and by 25%–40% in HIV-1-infected patients.99 ETR, another CYP3A4 inducer100,101 decreased MVC Cmax by 60%, but co-administration of MVC with ETR/DRV/RTV increased MVC Cmax by 176%.93 MVC dosage should be increased to 600 mg/kg twice daily when co-administered with EFV or ETR.15 Lersivirine is a weak CYP3A4 inducer, although its further development was stopped in 2013.102 In healthy subjects given 300 mg MVC and 500 mg Lersivirine twice daily, MVC Cmax and AUC increased by 3.4% and 6.2%, respectively, compared to subjects given MVC plus placebo.102 NVP increased MVC Cmax and AUC by 101% and 154%, respectively99 (Table 2).
MVC interactions with integrase inhibitors
Integrase inhibitors are a newer class of ART drugs that inhibit integrase to prevent insertion of the HIV-1 genome into cellular DNA. Co-administration of RAL with MVC decreased RAL and MVC plasma concentrations, respectively, by 33% and 20%.103 However, another study showed no MVC effects on RAL concentrations.104 Elvitegravir (EVG) is often given in combination with RTV, and is metabolized by CYP3A4.105,106 Co-administration of EVG/RTV with MVC resulted in a 215% increase in MVC plasma concentration, while EVG and RTV concentrations remained unchanged.105 No dose adjustments are necessary when integrase inhibitors are co-administered with MVC.15
MVC interactions with non-HIV-1 drugs
MVC does not affect the pharmacokinetics of midazolam, a benzodiazepine metabolized by CYP3A4, and had no effect on oral contraceptives ethinyloestradiol and levonorgestrel.96 When used with alcohol, MVC increased alcohol concentrations by 12%, but alcohol use did not affect MVC plasma concentrations.107 Ketoconazole, an antifungal commonly used for the treatment of opportunistic infections in HIV-1/AIDS patients, and a CYP3A4 inhibitor, increased MVC plasma Cmax by 338%; thus, MVC dosage should be decreased to 150 mg/kg twice daily when given with ketoconazole.92 Cotrimoxazole, an antibiotic commonly used to treat HIV-1-associated pneumocystis jiroveci infections, had no effect on MVC plasma concentrations or its clearance.95 Boceprevir, an antiviral used for hepatitis-C treatment, increased MVC plasma concentrations by 300%, thus, MVC dosage should be decreased to 150 mg/kg twice daily when given with boceprevir.108 Telaprevir is a hepatitis-C PI and inhibitor of CYP3A and Pgp.108 Co-administration of telaprevir and MVC to healthy subjects increased MVC AUC and Cmax, respectively, by ninefold and tenfold, compared to subjects treated only with MVC.108 Data on MVC interactions with other drugs are summarized in Table 2.
MVC drug resistance
Resistance due to the outgrowth of X4-tropic HIV-1 strains
Resistance to MVC often occurs when previously undetectable X4-tropic HIV-1 are selected under pressure from drug treatment.25 This occurs when R5-tropic viruses, which previously constitute the majority of viral species, are sufficiently repressed. Viruses capable of using X4, which previously constituted a small minority, then multiply and become the dominant viral species.109 In the MOTIVATE trials, patients who were unresponsive to MVC treatment often had detectable X4-tropic viruses at treatment failure;24 76 (57%) of MVC-treated subjects that failed treatment had X4-tropic or dual-R5X4-tropic HIV-1 at the time of treatment failure, whereas only 6 (6%) of subjects receiving optimized background therapy plus placebo had detectable X4-tropic or dual-R5X4-tropic viral strains.24 In deep-sequencing analysis of plasma samples from subjects who experienced treatment failure, subjects who had a tropism shift had multiple amino acid mutations in the gp120 third variable (V3) region; the most common mutations included substitutions of glycine (G) to arginine (R) at position 11 (G11R), proline (P) to R at position 13 (P13R), and alanine to lysine (K) at position 25 (A25K).110 Deep sequencing further showed that most of these patients had some non-R5-tropic viruses at the time of initial screening.110 In a study of 62 individuals infected with R5-tropic HIV-1 who had been receiving MVC for 10 days, only two showed emergence of dual-X4R5-tropic HIV-1 strains.20,109 Phenotype and genotype screening showed that one patient had dual-tropic strains at baseline, and HIV-1 variants using X4 were genetically distinct from variants using R5.109 In one patient on MVC-containing regimen who had failed treatment, X4-tropic HIV-1 strains were rescued from previously stored PBMC samples, and became the main circulating strain under selective pressure from MVC.20
Mutations associated with resistance to MVC
Mutations found in vitro
Variable mutations in the V3 loop of gp120 can occur but may not confer complete resistance to MVC,111 and there has been no signature pattern of mutations described that can help predict resistance. Substitutions from alanine to threonine at position 316 (A316T), isoleucine to valine at position 323 (I323V), and alanine to serine at position 319 (A319S) were shown by genetic sequencing of clades-B and -G primary HIV-1 isolates following sequential passage.112 Deletions of isoleucine and serine at position 315 and position 317, respectively, also occurred in some viral isolates.112 Viral site-directed mutagenesis of the substitutions at position 316 and position 323 back to their original sequence, alanine to threonine and isoleucine to valine for position 316 and position 323, respectively, had full MVC response.112 Site-directed mutagenesis of either substitution alone only partially restored MVC response.112 That study also showed mutations outside the V3 loop: in the V1; V2; V4; and constant domains (C)-3, C4, and C5 regions of gp120, and in gp41.112 The authors suggested that mutations outside the V3 loop could help accommodate the resistance mutations occurring inside the V3 loop. Mutations due to serial passage of CC1/85 and Bal HIV-1 strains in vitro include valine to methionine (M) at position 169 (V169M) and asparagine (N) to K at position 192 (N192K) in the V2 region, leucine (L) to tryptophan (W) at position 317 (L317W) in the V3 region, I408A in the V4 region, aspartate (D) to N at position 462 (D462N), N463T, S464T, and N465aD in the V5 region, and L820I, I829V, and tyrosine (Y) to cysteine (C) at position 837 (Y837C) in gp41.113 Other mutations include T199K and T275M in the C2 region and I304V/F312W/T314A/ glutamic acid (E) to D at position 317 (E317D)/I318V in the V3 loop.114 The T199K mutation has been shown to increase viral fitness;114 combination of F312W/T314A/E317D mutations with either I304V or I318V mutation was necessary for V3 loop binding to CCR5 in the presence of MVC.114 These mutations resulted in changes in the V3 loop configuration and secondary structure that enabled gp120 binding to drug-bound CCR5.114 In vitro selected mutations of a subtype-A HIV-1 showed mutations in the C4 but not the V3 region.115 Common mutations associated with resistance to MVC are summarized in Table 3.
 | Table 3 Mutations associated with resistance to MVC |
Mutations found in clinical specimens
Mutations seen in patients enrolled in the MOTIVATE trials include G11S + I26V, S18G + A22T, A19S + I26V, I20F + A25D + I26V, and I20F + Y21I in the V3 region.110 In a patient resistant to MVC, the mutations present included P/T to histidine (H) at position308 (P/T308H), T320H, and I322aV in the V3 loop; D407G and loss of a glycosylation site at residue-386 in the V4 region; and V489I in the C5 region.116 The P/T308H mutation seemed to be the most important, resulting in complete resistance to MVC treatment, while the H320T and V322aI mutations resulted in lesser resistance.116 Insertion of the resistant V3 loop into a MVC-sensitive ENV clone resulted in partial resistance to MVC, while addition of mutations from the V4 region showed complete resistance.116 However, when V4 mutations were added without V3 mutations, there was no resistance to MVC.116 Studies of MVC-naïve patients harboring commonly seen resistance mutations showed that 9.8% (93/951 subjects) had V3 mutations associated with MVC resistance.117 Common mutations included G11S/I26V (6.4%) and I20F/A25D/I26V (2.2%); 14 isolates with these mutation patterns were selected for MVC sensitivity tests, 13 were fully sensitive to MVC; the MVC-resistant isolate had I20F/Y21I mutations and reversing this mutation restored the sensitivity to MVC.117 However, when V3 MVC-resistant mutations from one isolate were cloned into other isolates, there was lesser or no resistance to MVC, suggesting that resistance due to V3 mutations may be dependent on which env sequence they occur in.117 Common mutations associated with resistance to MVC are summarized in Table 3.
Resistance-associated mutations and viral subtype
It has been suggested that naturally occurring mutations to MVC might be more common in subtype-C than subtype-B HIV-1. Analysis of 65 samples showed that 52.3% (75% of subtype-C and 18.2% of subtype-B) had at least one mutation associated with MVC resistance.118 A commonly seen mutation was A316T in the gp120 region, occurring in 67.8% of subtype-C samples and 18.2% of subtype-B samples.118 The I323V mutation in the gp120 region occurred in 7.14% of subtype-C and was not seen in subtype-B samples.118 This observation was further confirmed in treatment-naïve HIV-1 infected patients in Zambia, all with subtype-C HIV-1.119 In an analysis of patients’ PBMC from three different study populations, the A316T mutation was found in 68%, 80.7%, and 64.5% of the 28, 32, and 90 samples analyzed, respectively.119 The I323V mutation in the gp120 was rare but was only present with the A316T mutation.119 A high prevalence of the A316T mutation, which results in partial resistance to MVC, was found in Zambian mother–infant pairs infected with HIV-1 subtype-C.120 A study of 80 HIV-infected patients in Brazil who had failed treatment showed that 27.5% harbored the the A316T, I323V, and/or S405A mutations in the gp120.121 However, in another study of 498 individuals infected with R5-tropic, subtype-B HIV-1, mutation patterns associated with MVC resistance were less than 5%.122 Single mutations were more commonly observed, but their significance on MVC resistance was not examined.122
Mechanisms involved in resistance to MVC
Resistance to MVC is typically non-competitive, characterized by a decrease in maximal percentage inhibition (MPI), with most resistant viruses having 80%–95% MPI.111,123 The mechanisms through which resistance to MVC evolves have not been fully elucidated, but involve mutations that result in increased affinity of gp120 to MVC-bound CCR5, enabling gp120 binding to CCR5 despite conformational changes from MVC binding.124 Resistant viruses could interact with CCR5 in the presence of MVC through increased binding to the CCR5 N-terminal domain.111 In fact, CCR5 antagonists work by binding to a hydrophobic pocket in the CCR5 transmembrane region;111 this results in conformational change in the extracellular loop (ECL) region, but little-to-no conformational change in the N-terminal domain.111 Therefore, mutations resulting in increased binding to the N-terminal domain would enable binding to MVC-bound CCR5. This could cause broad cross-resistance among several CCR5 antagonists, since most do not affect N-terminal conformation.111 In fact, three HIV-1 isolates resistant to VCV also displayed resistance to MVC, as well as resistance to TAK-779, another small-molecule CCR5 inhibitor.125 Another model for resistance to MVC is the requirement of both the N-terminal and ECL domains for viral binding; mutations in the V3 loop would thus allow its binding to the ECL in the presence of MVC, due to increased affinity for the binding site.124 Conformational change to the ECL region differs among CCR5 antagonists, thus, mutations resulting in the V3 loop binding to MVC-bound CCR5 may not result in binding in the presence of other CCR5 antagonists.116 The I322a residue is in contact with D11, Y10, and Y14, three key amino acid residues of the CCR5 N-terminal domain that are predicted to interact with the V3 loop.126 The hydrophobic side chain of I322a intercalates between all three residues, making hydrophobic contacts with the aromatic rings of both Y residues and the backbone of D11.126 Replacement of I with V likely alters the hydrophobic packing of these interactions and thereby modulates how V3 interacts with the CCR5 N-terminal.126
Use of MVC in HIV-1 infection
Routine treatment
Several studies have examined MVC use for treatment of HIV-1 infected humans. In a study of 32 patients, the most common reasons for MVC initiation were treatment failure, intolerance to previous ART regimens, and treatment intensification.127 After 3 and 6 months MVC treatment, 75% and 78% of patients, respectively, had fully suppressed viremia;127 the median increase in CD4+ count was 141 and 124 cells/μL at 3 and 6 months, respectively.127 In a study of 27 patients (20 with R5-tropic HIV-1, 1 with dual-R5X4-tropic HIV-1, and 6 with undetermined viral tropism) receiving MVC-containing ART regimens, 59% (10/17) of patients with detectable viremia before MVC initiation achieved viral suppression; HIV-1 remained undetectable in 60% (6/10) of patients who had undetectable virus before MVC initiation.128 CD4 count increased in 78%, remained unchanged in 11%, and decreased in 11% of patients.128 Another study of 25 patients infected with R5-tropic HIV-1 who had failed treatment showed that 12 months’ MVC intensification resulted in increased CD4 count and undetectable VL in 21 patients, and two patients without suppressed viremia showed a switch to X4-using HIV-1 by 24 months.129 Another study of 27 patients receiving MVC who had experienced treatment failure showed that at the time of treatment failure, 12 patients had X4-using viruses and 15 had R5-using viruses.130 Of the 12 patients with X4-tropic HIV-1 at treatment failure, four were infected with X4-tropic and dual-R5X4-tropic HIV-1 before MVC initiation.130 Resistance profiles of four patients with R5-using viruses at treatment failure showed that two patients had MVC-resistant HIV-1.130
Drug monitoring
The United States Department of Health and Human Services guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents suggest a minimum MVC plasma concentrations of 50 ng/mL.131 Intent-to-treat analysis of the MERIT study found that the probability of virological suppression decreased when average plasma concentrations were below 75 ng/mL.132 Additional studies are needed to determine which concentrations are optimal for accurate drug monitoring.
Since several drugs, including other ART drugs, can affect MVC concentrations, monitoring is important to ensure that patients maintain sufficient plasma drug concentrations.92,95,96,98 Several groups have developed high-performance liquid chromatography (HPLC) with ultra violet (UV) detection or with tandem mass spectrometry (MS/MS) for drug quantification.133–139 There are several drawbacks with many of these methods, including not being able to quantify low amounts of drug, the high cost of tests and equipment, and complicated procedures that may not be suitable for routine clinical practice, especially clinical practices in resource-limited settings. In the past few years, newer methods have been developed to address some of these concerns.140 Simiele et al developed a method that used less sample volume (could quantify MVC in 100 μL, compared to 500 μL of sample required with similar HPLC methods), could be completed in a shorter time (20 minutes), and used a less expensive HPLC-MS instrument that had a UV detector instead of a more costly HPLC-MS/MS instrument.140 This method had a similar lower limit of MVC quantification as other methods (4.9 ng/mL) and did not show any significant interference with other drugs likely to be used concomitantly with MVC.140 Emory et al developed a LC-MS/MS method that could quantify MVC levels from 0.5 ng/mL.138 This method can be performed using a 96-well plate, making it suitable for high-throughput screening.138 Overall, progress is being made in developing methods that will make drug monitoring easier in the future.
Pre-exposure and post-exposure prophylaxis
Pre-exposure prophylaxis (PrEP) can reduce the risk of HIV-1 infection by over 90% when taken consistently.141 The only drug currently approved for use as HIV-1 PrEP is tenofovir/emtricitabine (Truvada).142 Investigation of MVC for PrEP has been mostly limited to animal studies. MVC treatment, 62 mg/kg by oral gavage, protected humanized Rag22/2cc2/2(RAG-hu) mice against vaginal HIV-1 challenge;143 MVC-treated mice had undetectable HIV-1 RNA and DNA, while all placebo-treated mice became infected.143 MVC protective effects were further confirmed by 10 weeks monitoring data showing stable CD4+ T-cells in MVC-treated mice while placebo-treated mice had decreased CD4+ T-cells.39,143 However, MVC treatment, 44 mg/kg by oral gavage, did not protect macaques against rectal SHIV transmission, despite high MVC concentrations in rectal tissues.39 This could be due to the high density of CCR5+ cells and activated memory CD4+ T-cells in macaques’ gastrointestinal mucosa, which would suggest that higher MVC concentrations are needed to prevent rectal SHIV transmission.39 These macaques received one dose 24 hours pre- and post-SHIV rectal challenge,39 and accumulation of MVC in tissues from daily or twice daily use may be more effective at preventing rectal viral transmission.
ART drugs can also be used as microbicides in PrEP, often as gels or time-released rings applied vaginally or rectally. A MVC gel protected against SHIV and HIV-1 infection in macaques and RAG-hu mice, respectively.144,145 In macaques, vaginal application of MVC gel prevented SHIV infection in a time-dependent and dose-dependent manner, with half-maximal protection at 4 hours and 0.5 mM.144 MVC gels were also able to prevent HIV-1 via rectal transmission in macaques.146 Vaginal application of MVC gel to RAG-hu mice (5 mM 1 hours before HIV-1 vaginal challenge) protected all mice against infection.145
An ex vivo model sought to determine whether MVC oral administration could block HIV-1 infection in Langerhans cells (LCs) and viral transmission to CD4+ T-cells.147 Twenty healthy volunteers were given 300 mg/kg MVC twice daily for 1, 2, 3, and 14 days and epithelial tissue explants infected with HIV-1.147 One or 2 days MVC pretreatment partially inhibited infection of LCs within epithelial tissues and 3 or 14 days pretreatment completely inhibited LCs infection.147 MVC treatment also prevented HIV-1 transmission from LCs to co-cultured CD4+ T-cells.147
A Phase I clinical trial (MTN-013/IPM 026) evaluated the pharmacokinetics and pharmacodynamics of vaginal rings containing MVC.148 The trial enrolled 48 HIV-1 negative individuals using vaginal rings containing 100 mg MVC and 25 mg dapivirine (DPV) or 100 mg MVC for 28 days. MVC concentrations in the CF peaked at day 2 of ring use (Cmax: 22×106 pg/mL and AUC: 3.7×109 pg h/mL with the MVC-only ring).148 With the MVC/DPV ring, MVC concentrations in the CF peaked at day 1 of ring use and were higher than in subjects with MVC ring alone (Cmax: 97×106 pg/mL and AUC: 6.2×109 pg h/mL).148 MVC was only detectable in the cervical tissues (CT) in 4 out of 12 individuals with the MVC-only ring and undetectable in all individuals with the MVC/DPV ring.148 MVC plasma concentrations were below the limit of detection in both MVC and MVC/DPV rings subject groups.148 Ex vivo HIV-1 challenge of CT from MVC ring users did not show any drug-associated viral inhibition.148 Overall, although data are limited to animal and ex vivo models, MVC (oral or microbicides) could be useful in HIV-1 PrEP.
The only reported case of MVC use in post-exposure prophylaxis was a student exposed to multidrug-resistant HIV-1 using a needlestick.149 The student received LPV/RTV, FPV, TDF, and 3TC 15 minutes after injury, and 3 days later LPV/RTV was replaced with MVC.149 The student remained HIV-1-negative after a 6-month follow-up and the drug regimen was well tolerated.149
Dual therapy
The MVC + DRV/RTV (MIDAS) study assessed the efficacy of MVC (150 mg) + DRV/RTV (800/100 mg) once daily, in 24 ART-naïve R5-tropic HIV-1-infected subjects.150 At week 48, 92% had VL below 50 copies/mL; their median CD4 count increased by 216 cells/mm3 at week 96.150 A retrospective cohort study of 60 treatment-experienced R5-tropic HIV-1-infected patients28 corroborated these findings; in this second study patients received MVC (150 mg) + DRV/RTV (800/100 mg) once daily. At week 48, 78% of patients had VL below 50 copies/mL and median CD4 count increased by 70 cells/μL.28 This suggests that MVC + DRV/RTV may be a viable NRTI-sparing regimen,28 however, additional studies with larger sample sizes and appropriate controls are needed to confirm this hypothesis.
Combination of MVC (600 mg), RAL (400 mg), and ETR (200 mg) twice daily is effective in treatment-experienced R5-tropic HIV-1-infected patients.151 At week 48, all 28 patients had VL below 400 copies/mL; 93% had VL below 50 copies/mL and the median CD4 count increased by 267 cells/μL.151 The long-term efficacy of this regimen was confirmed at week 204, where 96% of patients had VL below 50 copies/mL and median CD4 count increased by 267 cells/mm3.152 In both studies, the treatment regimen was well tolerated.151,152 A 24-week study of 26 treatment-experienced patients given RAL (400 mg) + MVC (300 mg) twice daily showed higher than expected levels of viral rebound.153 In the ROCnRAL study, RAL + MVC treatment of HIV-1-infected patients with lipoatrophy did not suppress viremia, despite improvements in lipid profile and bone density.154 Another study assessed the efficacy of LPV/RTV + MVC (150 mg, once daily) or TDF/FTC, in HIV-1-infected patients with VL>1,000 copies/mL.155 At week 48, VL was below 50 copies/mL in all patients receiving MVC + LPV/RTV, and in 96% of patients receiving TDF/FTC + LPV/RTV.155 Furthermore, patients receiving MVC + LPV/RTV showed a larger median increase in CD4 counts (MVC group: 286 cells/μL versus TDF/FTC group: 199 cells/μL).155 MVC (150 mg) + ATV/RTV (300/100 mg) once daily had beneficial antiviral activity comparable to TDF/FTC (300/200 mg) + ATV/RTV; at week 48 74.6% (44/60) of patients had VL below 50 copies/mL, median CD4 count increased by 173 cells/mm3 in the MVC-treated group and 187 cells/mm3 in the TDC/FTC-treated group.156
NRTIs are associated with endothelial dysfunction and increased inflammation.157 Previously treated R5-HIV-1-infected patients with undetectable VL receiving a NTRI-sparing dual regimen (MVC (150 mg once daily) + RTV-boosted PI) showed improved endothelial function.157 Treatment of human PBMC and polymorphonuclear neutrophils with MVC and DRV decreased cell apoptosis and migration.158 The OPTIPRIM study159 compared the standard three-drug regimen versus a five-drug regimen in reducing viral DNA load in HIV-1-infected patients. Patients in the five drugs group were given RAL (400 mg) + MVC (150 mg) twice daily with a fixed-dose combination of TDF (300 g) + FTC (200 g) + DRV (800 g) + RTV (100 g) once daily; patients in the three drugs group were given TDF (300 g) + FTC (200 g) + DRV (800 g) + RTV (100 g) once daily.159 After 24 weeks treatment, there was no significant difference in HIV-1 DNA loads of patients in the three or five drugs regimen (2.25 log10 per 106 PBMC versus 2.35 log10 per 106 PBMC), suggesting that there was no virological benefit to the intensive five-drug regimen.159
MVC and dual-tropic HIV-1 infection
MVC efficacy in dual/mixed-R5X4-tropic HIV-1 infection was examined in a randomized, placebo-controlled, double-blind, Phase II study of treatment-experienced patients (A4001029 study).160 Patients received MVC (150 or 300 mg) or placebo plus optimized background therapy once or twice daily.160 By week 24, 61% (38/62), 60% (38/63), and 52% (32/61) in the placebo, MVC once daily, and MVC twice daily groups, respectively, had discontinued treatment because of inefficacy.160 The mean decrease in VL was 0.97 log10, 0.91 log10, and 1.2 log10 copies/mL in patients receiving placebo, once daily MVC, and twice daily MVC, respectively;160 the mean CD4 count increased by 36, 60, and 62 cells/μL in patients receiving placebo, once daily MVC, and twice daily MVC, respectively.160 At treatment failure more patients receiving MVC had X4-tropic HIV-1 compared to patients receiving placebo.160 These results showed that MVC treatment of humans infected with dual/mixed-R5X4-tropic HIV-1 have little-to-no virological benefit. Deep sequencing showed that patients receiving MVC twice daily had greater virological response when X4-using virus constituted less than 10% of the total viral population compared to patients with higher percentages of X4-using virus.161
In vitro studies showed that dual-R5X4-tropic HIV-1 strains that preferably use R5 are genetically and phenotypically similar to R5-tropic strains and can be inhibited by MVC.162,163 Human studies correlating HIV-1 tropism to virological response to short-term MVC exposure (8 days treatment, 300 mg/kg twice daily) showed that patients in the control group and patients with dual/mixed-tropic HIV-1 had no significant change in VL, but in patients with R5-tropic HIV-1, VL decreased by 1.41 log10 copies/mL.164 There was 93.5% concordance between virological response to MVC and viral tropism, with positive virological response in 95% (19/20) of patients with R5-tropic HIV-1 and negative virological response in 90.9% (10/11) of patients with dual/mixed virus.164 These results suggested that short-term MVC exposure could help determine/confirm the genotypic or phenotypic HIV-1 tropism, particularly in patients with non-reportable results by a Trofile assay.164 However, subsequent studies found no concordance between standard V3-based genotypic tropism assays and virological response to MVC monotherapy,165 and determined that short-term MVC treatment of HIV-1-infected, treatment-naïve patients could not predict viral tropism.166 In this later study, following 10 days MVC treatment (300 mg/kg twice daily) of 30 patients infected with R5-tropic and 10 patients infected with dual/mixed-tropic HIV-1, VL decreased by 1.52 log10 and 1.62 log10 copies/mL in patients with R5-tropic and dual/mixed-tropic virus, respectively;166 thus, short-term MVC exposure cannot predict viral tropism in treatment-naïve patients. The discrepancies between findings in these different studies could be due to differences in the study population.166,164 It is also possible that short-term MVC treatment could help predict viral tropism in treatment-experienced patients, but not in treatment-naïve patients.
MVC and HIV-1/hepatitis-C co-infection
After 6-months MVC treatment of HIV-1/hepatitis-C virus (HCV) co-infected patients, there was no significant increase in serum mediators of fibrogenesis and fibrosis, such as transforming growth factor beta-1, tissue inhibitors of metalloproteinases-1, and matrix metalloproteinase-2.167 The GUSTA study examined the effect of MVC/DRV/RTV dual therapy on hepatic injury in HIV-1/HCV co-infected patients;168 patients on dual therapy did not show any increases in the incidence of adverse events or severe liver abnormalities.168 MVC also reduced the progression of hepatic fibrosis in HIV-1/HCV co-infected patients.169
MVC and immune reconstitution inflammatory syndrome
Immune reconstitution inflammatory syndrome (IRIS) occurs when a suppressed immune system begins to recover, and produces a massive inflammatory response to previously acquired pathogens. ART initiation in a treatment-naïve HIV-1 and polyomavirus-JC (JCV) co-infected patient with high VL and low CD4 count resulted in IRIS and decreased cognitive impairment, and this was markedly improved after ENF/MVC treatment.170 MVC treatment of HIV-1/JCV co-infected patients with IRIS, cognitive impairments, and progressive multifocal leukoencephalopathy (PML) resulted in decreased levels of CCR5+ immune cells in the CSF and improved patients’ conditions;171 this was reversed by treatment interruption, and restarting MVC treatment again improved the patients’ conditions.171 However, another HIV-1/JCV + patient with virological failure, low CD4 count and PML-associated IRIS, treated with MVC displayed rapid clinical deterioration and died 21 days after MVC initiation.172 This difference in MVC treatment outcomes could be due to advanced AIDS and PML in this latter patient.
Synergistic antiviral effects of MVC and antibodies or peptides
Antibodies directed against the second CCR5 ECL such as HGS004 and HGS101 had greater antiviral activity against MVC-bound than MVC-free CCR5, and inhibited infection by MVC-resistant HIV-1 more potently with MVC-bound than with free CCR5.173,174 HGS004 and MVC have potent antiviral synergy against R5-tropic HIV-1,173 and the IgG-CD4-gp120(Bal) fusion protein synergizes with MVC.175 The CCL5-derived R4.0 peptide, CCL5, and MVC exhibited concomitant interactions with CCR5 and promoted synergic inhibition of HIV-1 in acute-infection assays.176 Another CCR5 antibody, PRO-140, also showed a synergistic antiviral effect with MVC.177
MVC and HIV-2 infection
Of the 34 million HIV-infected individuals worldwide, one-to-two million (mostly in West Africa) are infected with HIV-2.178 Although HIV-2 can progress to AIDS, VL are often lower and disease progression is slower compared to HIV-1 infection.179 Treatment is complex due to limited clinical trials with HIV-2-infected patients; in fact, studies on current ART drugs were done using mostly HIV-1 isolates and HIV-1-infected patients. HIV-2 is resistant to NNRTIs and has reduced sensitivity to PIs.180 Like HIV-1, the major HIV-2 coreceptors are CCR5 and CXCR4.181,182 In vitro studies showed that similar concentrations of MVC and other CCR5 antagonists that inhibit HIV-1 could also inhibit infection by primary CCR5-tropic HIV-2, with comparable MPI.183,184
Treatment of an HIV-2-infected AIDS patient with cognitive impairment and resistant to NRTIs, NNRTIs, and PIs, with a salvage therapy consisting of TDF + FTC + TPV/RTV + RAL + MVC, decreased blood VL to undetectable levels, but CSF VL remained high, neurological impairments continued, and blood VL later increased.185 Genotyping analyses suggested that treatment failure was not due to viral escape, but to poor CNS penetration of ART drugs.185 However, salvage therapy containing MVC successfully repressed blood VL in other HIV-2-infected patients resistant to other ART drugs.186,187 The presence of neurocognitive impairment and high CSF VL before initiation of MVC salvage therapy may have contributed to treatment failure in the first study,185 likely because of a large CNS/CSF viral reservoir, but it is not known whether this factor alone could explain the failure of MVC salvage therapy in that patient. The latter two studies186,187 did not indicate the patients’ CSF VL or their neurocognitive status. Furthermore, none of these studies tested the tropism of HIV-2 strains circulating in the patients, therefore, it is possible that the patient who failed MVC salvage therapy185 had mixed-/dual-R5X4-tropic HIV-2 strains, or HIV-2 strains using coreceptors other than CCR5. In fact, in addition to CCR5 and CXCR4, HIV-2 can use CCR3, G-protein-coupled receptor-15, or CXCR6 to enter and infect target cells.181,182 Overall, in vitro and in vivo evidence suggests that MVC can be effective against R5-tropic HIV-2.
Summary of MVC use in HIV-1 therapy
MVC is a small-molecule CCR5 antagonist used for the treatment of R5-tropic HIV-1 infection in both treatment-naïve and treatment-experienced patients.15 MVC has favorable safety, pharmacokinetic and pharmacodynamic profiles. MVC is a substrate for CYP3A4, CYP3A5, Pgp, and organic anion transporter polypeptide-1B1, but is primarily metabolized by CYP3A4.35,48,56 As a substrate for CYP3A4, MVC’s pharmacokinetics is affected by the concurrent use of CYP3A4 inhibitors and inducers.49 Typical dosage is 300 mg/kg twice daily.15 However, when combined with a CYP3A4 inducer the dosage is increased to 600 mg/kg twice daily; and when combined with a CYP3A4 inhibitor the dosage in decreased to 150 mg/kg twice daily.15 MVC has been used successfully for routine treatment of HIV-1-infected patients, with both decreases in VL and increases in CD4+ cell levels observed in treated individuals.127,128 MVC has shown promise in pre- and post-exposure prophylaxis, although current data are conflicting. Microbicide gels containing MVC reduced vaginal and rectal SHIV transmission in macaques;144,146 but women using vaginal rings containing MVC had low drug concentrations in their CF, and their CT was not resistant to ex vivo HIV-1 infection.148 NRTIs are often associated with harsh side effects and because MVC has a favorable safety profile, MVC has been used in NRTI- and PI-sparing dual regimens.28,150,156 MVC could reduce hepatic fibrosis in HIV-1/HCV co-infected patients, reduce the severity of IRIS, and suppress VL in HIV-2-infected humans.169–171,186,187 In addition to its virological benefits, MVC has been shown to increase the levels of immune cells, including CD4+ and CD8+ cells, suggesting an additional immunological benefit.70,76 Resistance to MVC is often associated with the outgrowth of previously undetectable X4-tropic viral strains.109 Overall, current studies showed that MVC is safe and efficacious against infection with R5-tropic virus in vivo, ex vivo, and in vitro.
Use of MVC in other diseases
CCR5 has been implicated in other diseases besides HIV-1/AIDS, including cancer, inflammatory and graft-versus-host diseases (GVHDs).188–206 Therefore, it was proposed that blocking CCR5 can attenuate the severity or progression of these diseases.
Cancer
CCL5 and CCR5 expression are increased in breast cancer cells, correlate with poor prognosis,188 and breast cancer patients with CCR5-delta32 have longer metastasis-free survival.207 MVC blocked CCR5 on breast cancer cells and this was associated with decreased CCL5-induced calcium signaling and cell invasion.188 In vivo studies also showed that MVC significantly reduces the number and size of breast cancer metastasis in mice,188 increases survival, reduces weight loss, attenuates liver damage, and reduces the number and size of hepatic tumors in a mouse model of hepatocellular carcinoma.189 CCR5 expression negatively correlates with gastric cancer progression, and MVC reduced the number of peritoneal and mesenteric nodules, and decreased tumor burden in mice.190 CCR5 is also involved in prostate cancer metastasis; the proto-oncogene tyrosine-protein kinase induces CCR5 signaling in prostate epithelial cells and there is increased CCR5 expression in humans with prostate cancer, with higher CCR5 expression in metastases, compared to primary tumors.191 MVC treatment of mice injected with a prostate cancer cell line reduced the bone and brain metastasis burden by 80% and 60%, respectively.191 Perineural invasion in salivary adenoid cystic carcinoma (SACC) is also associated with increased CCR5 and CCL5 expression,192 and MVC blocked CCL5-induced increase in migration and perineural invasion of salivary adenoid cystic carcinoma.192
Graft-versus-host disease
GVHD often occurs following an allogenic transplant, and its incidence is 30%–70% among transplant patients.193 During GVHD activated donor leukocytes recognize the recipient’s antigens as foreign, resulting in tissue damage. CCR5 is involved in GVHD pathology as it binds CCL5 and recruits leukocytes to tissue sites;193,194 humans without genetically functional CCR5 have longer survival rates after renal transplantation.193 MVC blocks T-lymphocyte chemotaxis in vitro, and adding MVC to the standard therapy of patients undergoing hematopoietic stem-cell transplantation resulted in lower GVHD incidence.194 The use of MVC for GVHD prevention is currently in Phase II trials.193
Heart and lung diseases
Pulmonary arterial hypertension (PH) is partly caused by chronic lung inflammation, and chemokines, including CCL5, play a major role in pulmonary vascular remodeling, a PH hallmark pathological feature.195,196 CCR5 expression in the lungs of humans with PH is increased compared to healthy donors.197 MVC treatment decreased PH development, or reversed PH in CCR5-knockout mice that had murine CCR5 replaced by human CCR5, and in animals subjected to PH-inducing conditions.197 Cardiac dysfunction is prevalent among HIV-1-infected patients,198 resulting from damage to cardiomyocytes by proinflammatory mediators and viral proteins.199 In MVC-treated Simian Immunodeficiency Virus (SIV)-infected macaques, diastolic function was similar to uninfected animals, whereas untreated and infected animals displayed diastolic dysfunction.199 Diastolic dysfunction was associated with myocardial macrophages activation and MVC decreased the expression of the macrophage marker CD163 compared to untreated animals, but CD68 expression remained unchanged.199 CCR5 antagonists also reduced the atherosclerotic burden and secretion of proinflammatory Th1-cytokines in dyslipidemia mouse models.208,209 RTV induces inflammation in adipose tissues, and this is associated with increased expression of proinflammatory cytokines and accelerated formation of atherosclerotic aortic plaques.210 C57BL6/J ApoE-/- mice treated with RTV + MVC were protected against aortic plaque progression; and showed less macrophage infiltration into the aortic wall, decreased levels of intercellular adhesion molecule-1, vascular cell-adhesion protein-1, CCL2, interleukin-17A, CCL5, and tumor necrosis factor-alpha, compared to mice treated with RTV alone.211 These data suggest that MVC has protective and anti-inflammatory properties on the vasculature.
Hemorrhage
Trauma-induced hemorrhage often results in increased inflammation and liver damage.202 This likely involves CCR5, as MVC treatment attenuates liver injury in rats subjected to trauma-induced hemorrhage by increasing peroxisome proliferator-activated receptor-gamma activity and decreasing proinflammatory factors.202 Peroxisome proliferator-activated receptor-gamma activation also improved liver function during trauma-induced hemorrhage;201 and CCR5-deficient mice have lower inflammatory pain under chemical or inflammatory stimuli.212
Rheumatoid arthritis
CCR5 ligands and other chemokines are increased in synovial fluids during rheumatoid arthritis (RA), resulting in tissue and joint damage.204,205 Therefore, blocking CCR5 could reduce inflammation at synovial joints and reduce RA symptoms. In support of this hypothesis, previous work demonstrated that CCR5-delta32 mutation had protective effects in patients with RA.206 However, in a Phase IIa study, MVC showed no efficacy in the treatment of RA.203
Liver disease
CCL5 has been implicated in the pathogenesis of different hepatic disorders: HCV-infected patients with advanced stage liver injury have high levels of CCL5 mRNA,213 and hepatic CCL5 expression is increased in a mouse model of hepatic steatosis.214 Furthermore, CCL5 promotes hepatic inflammation and fibrosis in experimental fibrogenesis models.215 Mice on high fat diet treated with MVC showed less weight gain or liver damage compared to untreated mice on a similar diet.215 Furthermore, MVC-treated mice had lower hepatic triglycerides, lower degrees of steatosis, and decreased CCL5 expression compared to untreated mice on high fat diet.216
Summary
MVC, a CCR5 antagonist, is effective against infection with R5-tropic HIV-1 in vivo and in vitro;15,20,25 it is well tolerated and safe for most individuals. MVC has a good pharmacokinetic profile, with relatively low protein binding and high bioavailability.32 It also has a wide distribution throughout the body, with high concentrations found in the vaginal and rectal tissues.15,37–41,43–47,143,145 MVC appears to be safe in patients with mild-to-moderate hepatic and renal disorders.39,63 Limited studies have examined MVC effects on HIV-1 MTCT; however, MVC placental MVC is low.83,85 MVC is metabolized by CYP3A4, and dosing must be adjusted when given with CYP3A4 inducers or inhibitors.15 True resistance to MVC is rare, as most cases of virological failure seen are associated with outgrowth of previously undetectable X4-tropic HIV-1 strains.25,109 Current evidence suggests that MVC is not effective against dual-/mixed-R5X4-tropic HIV-1 infections. Although there have been mutations associated with resistance to MVC, mutation patterns observed vary with HIV-1 strains and no specific mutation patterns identified can predict resistance to MVC. In addition to its role against HIV-1 infection, clinical trial data and animal studies suggest that MVC may help in the treatment of other diseases, including cancer, GVHD, and inflammatory diseases.188–206 Additional studies are needed to further our knowledge about the safety of long-term MVC use, its effectiveness against HIV-1 MTCT, its antiviral efficacy in HIV-1 reservoirs such as the CNS and lymphoid tissues, and its potential role in post-exposure prophylaxis.
Acknowledgments
This work was partly supported by grants from the National Institute of Health, National Institute of Mental Health, to GDK (MH081780 and MH094160).
Disclosure
The authors report no conflicts of interest in this work.
References
Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harb Perspect Med. 2012;2(8). | ||
Naif HM. Pathogenesis of HIV infection. Infect Dis Rep. 2013;5(Suppl 1):e6. | ||
Lee B, Ratajczak J, Doms RW, Gewirtz AM, Ratajczak MZ. Coreceptor/chemokine receptor expression on human hematopoietic cells: biological implications for human immunodeficiency virus-type 1 infection. Blood. 1999;93(4):1145–1156. | ||
Huang Y, Paxton WA, Wolinsky SM, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nature Med. 1996;2(11):1240–1243. | ||
Samson M, Libert F, Doranz BJ, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382(6593):722–725. | ||
Lalezari J, Thompson M, Kumar P, et al. Antiviral activity and safety of 873140, a novel CCR5 antagonist, during short-term monotherapy in HIV-infected adults. AIDS. 2005;19(14):1443–1448. | ||
Nichols WG, Steel HM, Bonny T, et al. Hepatotoxicity observed in clinical trials of aplaviroc (GW873140). Antimicrob Agents Chemother. 2008;52(3):858–865. | ||
Schurmann D, Fatkenheuer G, Reynes J, et al. Antiviral activity, pharmacokinetics and safety of vicriviroc, an oral CCR5 antagonist, during 14-day monotherapy in HIV-infected adults. AIDS. 2007;21(10):1293–1299. | ||
Gulick RM, Su Z, Flexner C, et al. Phase 2 study of the safety and efficacy of vicriviroc, a CCR5 inhibitor, in HIV-1-Infected, treatment-experienced patients: AIDS clinical trials group 5211. J Infect Dis. 2007;196(2):304–312. | ||
Suleiman J, Zingman BS, Diaz RS, et al. Vicriviroc in combination therapy with an optimized regimen for treatment-experienced subjects: 48-week results of the VICTOR-E1 phase 2 trial. J Infect Dis. 2010;201(4):590–599. | ||
Landovitz RJ, Angel JB, Hoffmann C, et al. Phase II study of vicriviroc versus efavirenz (both with zidovudine/lamivudine) in treatment-naive subjects with HIV-1 infection. J Infect Dis. 2008;198(8):1113–1122. | ||
Caseiro MM, Nelson M, Diaz RS, et al. Vicriviroc plus optimized background therapy for treatment-experienced subjects with CCR5 HIV-1 infection: final results of two randomized phase III trials. J Infect. 2012;65(4):326–335. | ||
Baba M, Takashima K, Miyake H, et al. TAK-652 inhibits CCR5-mediated human immunodeficiency virus type 1 infection in vitro and has favorable pharmacokinetics in humans. Antimicrob Agents Chemother. 2005;49(11):4584–4591. | ||
Lalezari J, Gathe J, Brinson C, et al. Safety, efficacy, and pharmacokinetics of TBR-652, a CCR5/CCR2 antagonist, in HIV-1-infected, treatment-experienced, CCR5 antagonist-naive subjects. J Acquir Immune Defic Syndr. 2011;57(2):118–125. | ||
Pfizer. Selzentry. Available from: http://www.viivhealthcare.com/media/70429/us_selzentry.pdf. Accessed February 25, 2015. | ||
Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2004;26(8):639–663. | ||
Dorr P, Westby M, Dobbs S, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49(11):4721–4732. | ||
Wood A, Armour D. The discovery of the CCR5 receptor antagonist, UK-427,857, a new agent for the treatment of HIV infection and AIDS. Prog Med Chem. 2005;43:239–271. | ||
Abel S, van der Ryst E, Rosario MC, et al. Assessment of the pharmacokinetics, safety and tolerability of maraviroc, a novel CCR5 antagonist, in healthy volunteers. Br J Clin Pharmacol. 2008;65(Suppl 1):5–18. | ||
Fatkenheuer G, Pozniak AL, Johnson MA, et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nature Med. 2005;11(11):1170–1172. | ||
Gulick RM, Lalezari J, Goodrich J, et al. Maraviroc for previously treated patients with R5 HIV-1 infection. New Engl J Med. 2008;359(14):1429–1441. | ||
Hardy WD, Gulick RM, Mayer H, et al. Two-year safety and virologic efficacy of maraviroc in treatment-experienced patients with CCR5-tropic HIV-1 infection: 96-week combined analysis of MOTIVATE 1 and 2. J Acquir Immune Defic Syndr. 2010;55(5):558–564. | ||
Gulick RM, Fatkenheuer G, Burnside R, et al. Five-year safety evaluation of maraviroc in HIV-1-infected treatment-experienced patients. J Acquir Immune Defic Syndr. 2014;65(1):78–81. | ||
Fatkenheuer G, Nelson M, Lazzarin A, et al. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. New Engl J Med New Engl J Med. 2008;359(14):1442–1455. | ||
Cooper DA, Heera J, Goodrich J, et al. Maraviroc versus efavirenz, both in combination with zidovudine–lamivudine, for the treatment of antiretroviral-naive subjects with CCR5-tropic HIV-1 infection. J Infect Dis. 2010;201(6):803–813. | ||
Cooper DA, Heera J, Ive P, et al. Efficacy and safety of maraviroc vs. efavirenz in treatment-naive patients with HIV-1: 5-year findings. AIDS. 2014;28(5):717–725. | ||
Stellbrink HP, Szlavik J, Murphy D, et al. Maraviroc (MVC) dosed once daily with darunavir/ritonavir (DRV/r) in a 2 drug-regimen compared to emtricitabine/tenofovir (TDF/FTC) with DRV/r; 48-week results from MODERN (Study A4001095). In: 20th International AIDS Conference; July 20–25, 2014; 2014; Melbourne, Australia. | ||
Macias J, Recio E, Marquez M, et al. Efficacy and safety of once-daily maraviroc plus ritonavir-boosted darunavir in pretreated HIV-infected patients in a real-life setting. HIV Med. 2014;15(7):417–424. | ||
Nozza S, Galli L, Chiappetta S, et al. Maraviroc 150 mg QD plus lopinavir/ritonavir, a NRTI-sparing regimen for HIV-infected naive patients: 48-weeks final results. In: Eleventh International Congress on Drug Therapy in HIV Infection; November 2–6, 2014; 2012; Glasgow, Scotland. | ||
Mills A, Mildvan D, Podzamzer G, et al. Once-daily maraviroc in combination with ritonavir-boosted atazanavir in treatment-naïve patients infected with CCR5 tropic HIV-1 (study A4001078): 96-week results. In: 19th International AIDS Conference; Washington DC, July 22–27, 2012. | ||
Taiwo BO, Chan ES, Fichtenbaum CJ, et al. Less bone loss with maraviroc- versus tenofovir-containing antiretroviral therapy in the AIDS Clinical Trials Group A5303 Study. Clin Infect Dis. Epub 2015 Jun 9. | ||
Abel S, Russell D, Whitlock LA, Ridgway CE, Nedderman AN, Walker DK. Assessment of the absorption, metabolism and absolute bioavailability of maraviroc in healthy male subjects. Br J Clin Pharmacol. 2008;65(Suppl 1):60–67. | ||
Nau R, Sorgel F, Eiffert H. Penetration of drugs through the blood–cerebrospinal fluid/blood–brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23(4):858–883. | ||
Nau R, Kinzig M, Dreyhaupt T, Kolenda H, Sorgel F, Prange HW. Kinetics of ofloxacin and its metabolites in cerebrospinal fluid after a single intravenous infusion of 400 milligrams of ofloxacin. Antimicrob Agents Chemother. 1994;38(8):1849–1853. | ||
Walker DK, Abel S, Comby P, Muirhead GJ, Nedderman AN, Smith DA. Species differences in the disposition of the CCR5 antagonist, UK-427,857, a new potential treatment for HIV. Drug Metab Disposit. 2005;33(4):587–595. | ||
Chan PL, Weatherley B, McFadyen L. A population pharmacokinetic meta-analysis of maraviroc in healthy volunteers and asymptomatic HIV-infected subjects. Br J Clin Pharmacol. 2008;65(Suppl 1):76–85. | ||
Brown KC, Patterson KB, Malone SA, et al. Single and multiple dose pharmacokinetics of maraviroc in saliva, semen, and rectal tissue of healthy HIV-negative men. J Infect Dis. 2011;203(10):1484–1490. | ||
Tiraboschi JM, Niubo J, Curto J, Podzamczer D. Maraviroc concentrations in seminal plasma in HIV-infected patients. J Acquir Immune Defic Syndr. 2010;55(5):e35–e36. | ||
Massud I, Aung W, Martin A, et al. Lack of prophylactic efficacy of oral maraviroc in macaques despite high drug concentrations in rectal tissues. J Virol. 2013;87(16):8952–8961. | ||
Dumond JB, Patterson KB, Pecha AL, et al. Maraviroc concentrates in the cervicovaginal fluid and vaginal tissue of HIV-negative women. J Acquir Immune Defic Syndr. 2009;51(5):546–553. | ||
Malcolm RK, Forbes CJ, Geer L, et al. Pharmacokinetics and efficacy of a vaginally administered maraviroc gel in rhesus macaques. J Antimicrob Chemother. 2013;68(3):678–683. | ||
Yilmaz A, Watson V, Else L, Gisslen M. Cerebrospinal fluid maraviroc concentrations in HIV-1 infected patients. AIDS. 2009;23(18):2537–2540. | ||
Tiraboschi JM, Niubo J, Curto J, Podzamczer D. Maraviroc concentrations in cerebrospinal fluid in HIV-infected patients. J Acquir Immune Defic Syndr. 2010;55(5):606–609. | ||
Melica G, Canestri A, Peytavin G, et al. Maraviroc-containing regimen suppresses HIV replication in the cerebrospinal fluid of patients with neurological symptoms. AIDS. 2010;24(13):2130–2133. | ||
Garvey L, Nelson M, Latch N, et al. CNS effects of a CCR5 inhibitor in HIV-infected subjects: a pharmacokinetic and cerebral metabolite study. J Antimicrob Chemother. 2012;67(1):206–212. | ||
Croteau D, Best BM, Letendre S, et al. Lower than expected maraviroc concentrations in cerebrospinal fluid exceed the wild-type CC chemokine receptor 5-tropic HIV-1 50% inhibitory concentration. AIDS. 2012;26(7):890–893. | ||
Veselinovic M, Yang KH, LeCureux J, et al. HIV pre-exposure prophylaxis: mucosal tissue drug distribution of RT inhibitor tenofovir and entry inhibitor maraviroc in a humanized mouse model. Virology. 2014;464–465:253–263. | ||
Lu Y, Hendrix CW, Bumpus NN. Cytochrome P450 3A5 plays a prominent role in the oxidative metabolism of the anti-human immunodeficiency virus drug maraviroc. Drug Metab Disposit. 2012;40(12):2221–2230. | ||
Lu Y, Fuchs EJ, Hendrix CW, Bumpus NN. CYP3A5 genotype impacts maraviroc concentrations in healthy volunteers. Drug Metab Disposit. 2014;42(11):1796–1802. | ||
Hyland R, Dickins M, Collins C, Jones H, Jones B. Maraviroc: in vitro assessment of drug-drug interaction potential. Br J Clin Pharmacol. 2008;66(4):498–507. | ||
Hustert E, Haberl M, Burk O, et al. The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics. 2001;11(9):773–779. | ||
Kuehl P, Zhang J, Lin Y, et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001;27(4):383–391. | ||
Xie HG, Wood AJ, Kim RB, Stein CM, Wilkinson GR. Genetic variability in CYP3A5 and its possible consequences. Pharmacogenomics. 2004;5(3):243–272. | ||
Lamba JK, Lin YS, Schuetz EG, Thummel KE. Genetic contribution to variable human CYP3A-mediated metabolism. Adv Drug Deliv Rev. 2002;54(10):1271–1294. | ||
van Schaik RH, van der Heiden IP, van den Anker JN, Lindemans J. CYP3A5 variant allele frequencies in Dutch Caucasians. Clin Chem. 2002;48(10):1668–1671. | ||
Siccardi M, D’Avolio A, Nozza S, et al. Maraviroc is a substrate for OATP1B1 in vitro and maraviroc plasma concentrations are influenced by SLCO1B1 521 T>C polymorphism. Pharmacogenet Genom. 2010;20(12):759–765. | ||
Kameyama Y, Yamashita K, Kobayashi K, Hosokawa M, Chiba K. Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genom. 2005;15(7):513–522. | ||
Katz DA, Carr R, Grimm DR, et al. Organic anion transporting polypeptide 1B1 activity classified by SLCO1B1 genotype influences atrasentan pharmacokinetics. Clin Pharmacol Ther. 2006;79(3):186–196. | ||
Kohlrausch FB, de Cassia Estrela R, Barroso PF, Suarez-Kurtz G. The impact of SLCO1B1 polymorphisms on the plasma concentration of lopinavir and ritonavir in HIV-infected men. Br J Clin Pharmacol. 2010;69(1):95–98. | ||
Niemi M, Schaeffeler E, Lang T, et al. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1). Pharmacogenetics. 2004;14(7):429–440. | ||
Paterlini MG. Structure modeling of the chemokine receptor CCR5: implications for ligand binding and selectivity. Biophys J. 2002;83(6):3012–3031. | ||
Zhang Y, Lou B, Lal RB, Gettie A, Marx PA, Moore JP. Use of inhibitors to evaluate coreceptor usage by simian and simian/human immunodeficiency viruses and human immunodeficiency virus type 2 in primary cells. J Virol. 2000;74(15):6893–6910. | ||
Abel S, Davis JD, Ridgway CE, Hamlin JC, Vourvahis M. Pharmacokinetics, safety and tolerability of a single oral dose of maraviroc in HIV-negative subjects with mild and moderate hepatic impairment. Antivir Ther. 2009;14(6):831–837. | ||
Vourvahis M, Fang J, Checchio T, et al. Pharmacokinetics, safety, and tolerability of maraviroc in HIV-negative subjects with impaired renal function. HIV Clin Trials. 2013;14(3):99–109. | ||
Pau AK, Penzak SR, Boyd SD, McLaughlin M, Morse CG. Impaired maraviroc and raltegravir clearance in a human immunodeficiency virus-infected patient with end-stage liver disease and renal impairment: a management dilemma. Pharmacotherapy. 2012;32(1):e1–e6. | ||
Nozza S, Pogliaghi M, Chiappetta S, et al. Levels of soluble endothelial protein C receptor are associated with CD4+ changes in Maraviroc-treated HIV-infected patients. PloS One. 2012;7(6):e37032. | ||
MacInnes A, Lazzarin A, Di Perri G, et al. Maraviroc can improve lipid profiles in dyslipidemic patients with HIV: results from the MERIT trial. HIV Clin Trials. 2011;12(1):24–36. | ||
Schapiro JM, Boucher CA, Kuritzkes DR, et al. Baseline CD4(+) T-cell counts and weighted background susceptibility scores strongly predict response to maraviroc regimens in treatment-experienced patients. Antivir Ther. 2011;16(3):395–404. | ||
Asmuth DM, Goodrich J, Cooper DA, et al. CD4+ T-cell restoration after 48 weeks in the maraviroc treatment-experienced trials MOTIVATE 1 and 2. J Acquir Immune Defic Syndr. 2010;54(4):394–397. | ||
Pulido I, Machmach K, Romero-Sanchez MC, et al. T-cell changes after a short-term exposure to maraviroc in HIV-infected patients are related to antiviral activity. J Infect. 2012;64(4):417–423. | ||
Cossarini F, Galli A, Galli L, et al. Immune recovery and T cell subset analysis during effective treatment with maraviroc. J Antimicrob Chemother. 2012;67(10):2474–2478. | ||
Cuzin L, Trabelsi S, Delobel P, et al. Maraviroc intensification of stable antiviral therapy in HIV-1-infected patients with poor immune restoration: MARIMUNO-ANRS 145 study. J Acquir Immune Defic Syndr. 2012;61(5):557–564. | ||
Kawana-Tachikawa A, Llibre JM, Bravo I, et al. Effect of maraviroc intensification on HIV-1-specific T cell immunity in recently HIV-1-infected individuals. PloS One. 2014;9(1):e87334. | ||
Yuan J, Ren HY, Shi YJ, Liu W. In vitro immunological effects of blocking CCR5 on T cells. Inflammation. 2015;38(2):902–910. | ||
Arberas H, Guardo AC, Bargallo ME, et al. In vitro effects of the CCR5 inhibitor maraviroc on human T cell function. J Antimicrob Chemother. 2013;68(3):577–586. | ||
Beliakova-Bethell N, Jain S, Woelk CH, et al. Maraviroc intensification in patients with suppressed HIV viremia has limited effects on CD4+ T cell recovery and gene expression. Antiviral Res. 2014;107:42–49. | ||
Pozo-Balado MM, Martinez-Bonet M, Rosado I, et al. Maraviroc reduces the regulatory T-cell frequency in antiretroviral-naive HIV-infected subjects. J Infect Dis. 2014;210(6):890–898. | ||
Dentone C, Di Biagio A, Parodi A, et al. Innate immunity cell activation in virologically suppressed HIV-infected maraviroc-treated patients. AIDS. 2014;28(7):1071–1074. | ||
Ndhlovu LC, Umaki T, Chew GM, et al. Treatment intensification with maraviroc (CCR5 antagonist) leads to declines in CD16-expressing monocytes in cART-suppressed chronic HIV-infected subjects and is associated with improvements in neurocognitive test performance: implications for HIV-associated neurocognitive disease (HAND). J Neurovirol. 2014;20(6):571–582. | ||
Hunt PW, Shulman NS, Hayes TL, et al. The immunologic effects of maraviroc intensification in treated HIV-infected individuals with incomplete CD4+ T-cell recovery: a randomized trial. Blood. 2013;121(23):4635–4646. | ||
Stepanyuk O, Chiang TS, Dever LL, et al. Impact of adding maraviroc to antiretroviral regimens in patients with full viral suppression but impaired CD4 recovery. AIDS. 2009;23(14):1911–1913. | ||
Wilkin TJ, Lalama CM, McKinnon J, et al. A pilot trial of adding maraviroc to suppressive antiretroviral therapy for suboptimal CD4(+) T-cell recovery despite sustained virologic suppression: ACTG A5256. J Infect Dis. 2012;206(4):534–542. | ||
Vinot C, Gavard L, Treluyer JM, et al. Placental transfer of maraviroc in an ex vivo human cotyledon perfusion model and influence of ABC transporter expression. Antimicrob Agents Chemother. 2013;57(3):1415–1420. | ||
Mofenson LM, Brady MT, Danner SP, et al. Guidelines for the prevention and treatment of opportunistic infections among HIV-exposed and HIV-infected children: recommendations from CDC, the National Institutes of Health, the HIV Medicine Association of the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the American Academy of Pediatrics. Morbid Mortal Weekly Rep. 2009;58(RR-11):1–166. | ||
Winters MA, Van Rompay KK, Kashuba AD, Shulman NS, Holodniy M. Maternal–fetal pharmacokinetics and dynamics of a single intrapartum dose of maraviroc in rhesus macaques. Antimicrob Agents Chemother. 2010;54(10):4059–4063. | ||
Guay LA, Musoke P, Fleming T, et al. Intrapartum and neonatal single-dose nevirapine compared with zidovudine for prevention of mother-to-child transmission of HIV-1 in Kampala, Uganda: HIVNET 012 randomised trial. Lancet. 1999;354(9181):795–802. | ||
Eshleman SH, Jackson JB. Nevirapine resistance after single dose prophylaxis. AIDS Rev. 2002;4(2):59–63. | ||
Ciaranello AL, Perez F, Keatinge J, et al. What will it take to eliminate pediatric HIV? Reaching WHO target rates of mother-to-child HIV transmission in Zimbabwe: a model-based analysis. PLoS Med. 2012;9(1):e1001156. | ||
Calcagno A, Trentini L, Marinaro L, et al. Transplacental passage of etravirine and maraviroc in a multidrug-experienced HIV-infected woman failing on darunavir-based HAART in late pregnancy. J Antimicrob Chemother. 2013;68(8):1938–1939. | ||
Pang X, Liu Z, Zhai G. Advances in non-peptidomimetic HIV protease inhibitors. Curr Med Chem. 2014;21(17):1997–2011. | ||
Cupp MJ, Tracy TS. Cytochrome P450: new nomenclature and clinical implications. Am Family Phys. 1998;57(1):107–116. | ||
Abel S, Russell D, Taylor-Worth RJ, Ridgway CE, Muirhead GJ. Effects of CYP3A4 inhibitors on the pharmacokinetics of maraviroc in healthy volunteers. Br J Clin Pharmacol. 2008;65(Suppl 1):27–37. | ||
Kakuda TN, Abel S, Davis J, et al. Pharmacokinetic interactions of maraviroc with darunavir–ritonavir, etravirine, and etravirine–darunavir–ritonavir in healthy volunteers: results of two drug interaction trials. Antimicrob Agents Chemother. 2011;55(5):2290–2296. | ||
Vourvahis M, Plotka A, Mendes da Costa L, Fang A, Heera J. Pharmacokinetic interaction between maraviroc and fosamprenavir–ritonavir: an open-label, fixed-sequence study in healthy subjects. Antimicrob Agents Chemother. 2013;57(12):6158–6164. | ||
Abel S, Russell D, Whitlock LA, Ridgway CE, Muirhead GJ. The effects of cotrimoxazole or tenofovir co-administration on the pharmacokinetics of maraviroc in healthy volunteers. Br J Clin Pharmacol. 2008;65(Suppl 1):47–53. | ||
Abel S, Russell D, Whitlock LA, Ridgway CE, Muirhead GJ. Effect of maraviroc on the pharmacokinetics of midazolam, lamivudine/zidovudine, and ethinyloestradiol/levonorgestrel in healthy volunteers. Br J Clin Pharmacol. 2008;65(Suppl 1):19–26. | ||
Barry M, Mulcahy F, Merry C, Gibbons S, Back D. Pharmacokinetics and potential interactions amongst antiretroviral agents used to treat patients with HIV infection. Clin Pharmacokinet. 1999;36(4):289–304. | ||
Abel S, Jenkins TM, Whitlock LA, Ridgway CE, Muirhead GJ. Effects of CYP3A4 inducers with and without CYP3A4 inhibitors on the pharmacokinetics of maraviroc in healthy volunteers. Br J Clin Pharmacol. 2008;65(Suppl 1):38–46. | ||
Pozniak AL, Boffito M, Russell D, Ridgway CE, Muirhead GJ. A novel probe drug interaction study to investigate the effect of selected antiretroviral combinations on the pharmacokinetics of a single oral dose of maraviroc in HIV-positive subjects. Br J Clin Pharmacol. 2008;65(Suppl 1):54–59. | ||
Katlama C, Haubrich R, Lalezari J, et al. Efficacy and safety of etravirine in treatment-experienced, HIV-1 patients: pooled 48 week analysis of two randomized, controlled trials. AIDS. 2009;23(17):2289–2300. | ||
Scholler-Gyure M, Kakuda TN, Sekar V, et al. Pharmacokinetics of darunavir/ritonavir and TMC125 alone and coadministered in HIV-negative volunteers. Antivir Ther. 2007;12(5):789–796. | ||
Vourvahis M, Langdon G, Labadie RR, et al. Pharmacokinetic effects of coadministration of lersivirine with raltegravir or maraviroc in healthy subjects. Antimicrob Agents Chemother. 2012;56(2):887–892. | ||
Andrews E, Glue P, Fang J, Crownover P, Tressler R, Damle B. Assessment of the pharmacokinetics of co-administered maraviroc and raltegravir. Br J Clin Pharmacol. 2010;69(1):51–57. | ||
Baroncelli S, Villani P, Weimer LE, et al. Raltegravir plasma concentrations in treatment-experienced patients receiving salvage regimens based on raltegravir with and without maraviroc coadministration. Ann Pharmacother. 2010;44(5):838–843. | ||
Ramanathan S, Abel S, Tweedy S, West S, Hui J, Kearney BP. Pharmacokinetic interaction of ritonavir-boosted elvitegravir and maraviroc. J Acquir Immune Defic Syndr. 2010;53(2):209–214. | ||
Zolopa AR, Berger DS, Lampiris H, et al. Activity of elvitegravir, a once-daily integrase inhibitor, against resistant HIV Type 1: results of a phase 2, randomized, controlled, dose-ranging clinical trial. J Infect Dis. 2010;201(6):814–822. | ||
Gruber VA, Rainey PM, Lum PJ, Beatty GW, Aweeka FT, McCance-Katz EF. Interactions between alcohol and the HIV entry inhibitor Maraviroc. J Int Assoc Providers AIDS Care. 2013;12(6):375–377. | ||
Vourvahis M, Plotka A, Kantaridis C, Fang A, Heera J. The effects of boceprevir and telaprevir on the pharmacokinetics of maraviroc: an open-label, fixed-sequence study in healthy volunteers. J Acquir Immune Defic Syndr. 2014;65(5):564–570. | ||
Westby M, Lewis M, Whitcomb J, et al. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J Virol. 2006;80(10):4909–4920. | ||
Swenson LC, Chui CK, Brumme CJ, et al. Genotypic analysis of the V3 region of HIV from virologic nonresponders to maraviroc-containing regimens reveals distinct patterns of failure. Antimicrob Agents Chemother. 2013;57(12):6122–6130. | ||
Roche M, Salimi H, Duncan R, et al. A common mechanism of clinical HIV-1 resistance to the CCR5 antagonist maraviroc despite divergent resistance levels and lack of common gp120 resistance mutations. Retrovirology. 2013;10:43. | ||
Westby M, Smith-Burchnell C, Mori J, et al. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J Virol. 2007;81(5):2359–2371. | ||
Asin-Milan O, Chamberland A, Wei Y, Haidara A, Sylla M, Tremblay CL. Mutations in variable domains of the HIV-1 envelope gene can have a significant impact on maraviroc and vicriviroc resistance. AIDS Res Ther. 2013;10(1):15. | ||
Yuan Y, Yokoyama M, Maeda Y, et al. Structure and dynamics of the gp120 V3 loop that confers noncompetitive resistance in R5 HIV-1(JR-FL) to maraviroc. PloS One. 2013;8(6):e65115. | ||
Ratcliff AN, Shi W, Arts EJ. HIV-1 resistance to maraviroc conferred by a CD4 binding site mutation in the envelope glycoprotein gp120. J Virol. 2013;87(2):923–934. | ||
Tilton JC, Wilen CB, Didigu CA, et al. A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J Virol. 2010;84(20):10863–10876. | ||
Delobel P, Cazabat M, Saliou A, et al. Primary resistance of CCR5-tropic HIV-1 to maraviroc cannot be predicted by the V3 sequence. J Antimicrob Chemother. 2013;68(11):2506–2514. | ||
Araujo LA, Junqueira DM, de Medeiros RM, Matte MC, Almeida SE. Naturally occurring resistance mutations to HIV-1 entry inhibitors in subtypes B, C, and CRF31_BC. J Clin Virol. 2012;54(1):6–10. | ||
Gonzalez S, Gondwe C, Tully DC, et al. Short communication: antiretroviral therapy resistance mutations present in the HIV type 1 subtype C pol and env regions from therapy-naive patients in Zambia. AIDS Res Hum Retroviruses. 2010;26(7):795–803. | ||
Guo H, Liu C, Liu B, Wood C, Kong X. Analysis of primary resistance mutations to HIV-1 entry inhibitors in therapy naive subtype C HIV-1 infected mother–infant pairs from Zambia. J Clin Virol. 2013;58(1):233–239. | ||
Alencar CS, Nishiya AS, Ferreira S, Giret MT, Diaz RS, Sabino EC. Evaluation of primary resistance to HIV entry inhibitors among brazilian patients failing reverse transcriptase/protease inhibitors treatment reveal high prevalence of maraviroc resistance-related mutations. AIDS Res Human Retrovir. 2010;26(12):1267–1271. | ||
Seclen E, Gonzalez Mdel M, Lapaz M, et al. Primary resistance to maraviroc in a large set of R5-V3 viral sequences from HIV-1-infected patients. J Antimicrob Chemother. 2010;65(12):2502–2504. | ||
Pugach P, Marozsan AJ, Ketas TJ, Landes EL, Moore JP, Kuhmann SE. HIV-1 clones resistant to a small molecule CCR5 inhibitor use the inhibitor-bound form of CCR5 for entry. Virology. 2007;361(1):212–228. | ||
Trkola A, Kuhmann SE, Strizki JM, et al. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc Natl Acad Sci U S A. 2002;99(1):395–400. | ||
Putcharoen O, Lee SH, Henrich TJ, et al. HIV-1 clinical isolates resistant to CCR5 antagonists exhibit delayed entry kinetics that are corrected in the presence of drug. J Virol. 2012;86(2):1119–1128. | ||
Berro R, Klasse PJ, Jakobsen MR, Gorry PR, Moore JP, Sanders RW. V3 determinants of HIV-1 escape from the CCR5 inhibitors Maraviroc and Vicriviroc. Virology. 2012;427(2):158–165. | ||
Reuter S, Braken P, Jensen B, et al. Maraviroc in treatment-experienced patients with HIV-1 infection – experience from routine clinical practice. Eur J Med Res. 2010;15(6):231–237. | ||
Babiker ZO, Douthwaite ST, Collier LE, Pennell A, Uriel AJ, Wilkins E. Real-life outcomes of maraviroc-based regimens in HIV-1-infected individuals. J Int Assoc Provid AIDS Care. 2013;12(1):12–14. | ||
Bon I, Clo A, Borderi M, et al. Prevalence of R5 strains in multi-treated HIV subjects and impact of new regimens including maraviroc in a selected group of patients with CCR5-tropic HIV-1 infection. Int J Infect Dis. 2013;17(10):e875–e882. | ||
Raymond S, Maillard A, Amiel C, et al. Virological failure of patients on maraviroc-based antiretroviral therapy. J Antimicrob Chemother. 2015;70(6):1858–1864. | ||
National Institutes of Health. Federally approved HIV/AIDS medical practice guidelines; Available from: https://aidsinfo.nih.gov/contentfiles/adultandadolescentgl003093.pdf. 2015: H18. | ||
Levin J. Maraviroc exposure-efficacy (<50 copies/mL) analysis in HIV-1-infected treatment-naive subjects. In: ITT Population (MERIT Study) XVII International AIDS Conference; August 3–8, 2008; Mexico City, Mexico. | ||
Fayet A, Beguin A, Zanolari B, et al. A LC-tandem MS assay for the simultaneous measurement of new antiretroviral agents: raltegravir, maraviroc, darunavir, and etravirine. J Chromatogr B, Anal Technol Biomed Life Sci. 2009;877(11–12):1057–1069. | ||
Martin J, Deslandes G, Dailly E, et al. A liquid chromatography-tandem mass spectrometry assay for quantification of nevirapine, indinavir, atazanavir, amprenavir, saquinavir, ritonavir, lopinavir, efavirenz, tipranavir, darunavir and maraviroc in the plasma of patients infected with HIV. J Chromatogr B, Anal Technol Biomed Life Sci. 2009;877(27):3072–3082. | ||
D’Avolio A, Simiele M, Baietto L, et al. A validated high-performance liquid chromatography-ultraviolet method for quantification of the CCR5 inhibitor maraviroc in plasma of HIV-infected patients. Therap Drug Monitor. 2010;32(1):86–92. | ||
Else L, Watson V, Tjia J, et al. Validation of a rapid and sensitive high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) assay for the simultaneous determination of existing and new antiretroviral compounds. J Chromatogr B, Anal Technol Biomed Life Sci. 2010;878(19):1455–1465. | ||
Brewer E, Felix T, Clarke P, Edgington A, Muirhead D. An LC-MS-MS method for quantitative determination of maraviroc (UK-427,857) in human plasma, urine and cerebrospinal fluid. Biomed Chromatogr. 2010;24(12):1316–1323. | ||
Emory JF, Seserko LA, Marzinke MA. Development and bioanalytical validation of a liquid chromatographic-tandem mass spectrometric (LC-MS/MS) method for the quantification of the CCR5 antagonist maraviroc in human plasma. Clin Chim Acta. 2014;431:198–205. | ||
Takahashi M, Hirano A, Okubo N, Kinoshita E, Nomura T, Kaneda T. Development and application of a simple LC-MS method for the determination of plasma maraviroc concentrations. J Med Invest. 2010;57(3–4):245–250. | ||
Simiele M, Baietto L, Audino A, et al. A validated HPLC-MS method for quantification of the CCR5 inhibitor maraviroc in HIV+ human plasma. J Pharmaceut Biomed Anal. 2014;94:65–70. | ||
Centers for Disease Control and Prevention. Pre-Exposure Prophylaxis (PrEP). Atlanta: Centers for Disease Control and Prevention. Available from: http://www.cdc.gov/hiv/prevention/research/prep/. Accessed March 18, 2015. | ||
Sciences G. Truvada prescribing information. Available from: http://www.gilead.com/~/media/Files/pdfs/medicines/hiv/truvada/truvada_pi.PDF. Accessed March 18, 2015. | ||
Neff CP, Ndolo T, Tandon A, Habu Y, Akkina R. Oral pre-exposure prophylaxis by anti-retrovirals raltegravir and maraviroc protects against HIV-1 vaginal transmission in a humanized mouse model. PloS One. 2010;5(12):e15257. | ||
Veazey RS, Ketas TJ, Dufour J, et al. Protection of rhesus macaques from vaginal infection by vaginally delivered maraviroc, an inhibitor of HIV-1 entry via the CCR5 co-receptor. J Infect Dis. 2010;202(5):739–744. | ||
Neff CP, Kurisu T, Ndolo T, Fox K, Akkina R. A topical microbicide gel formulation of CCR5 antagonist maraviroc prevents HIV-1 vaginal transmission in humanized RAG-hu mice. PloS One. 2011;6(6):e20209. | ||
Dobard CW, Taylor A, Sharma S, et al. Protection against rectal chimeric simian/human immunodeficiency virus transmission in Macaques by rectal-specific gel formulations of maraviroc and tenofovir. J Infect Dis. Epub 2015 Jun 12. | ||
Matsuzawa T, Kawamura T, Ogawa Y, et al. Oral administration of the CCR5 inhibitor, maraviroc, blocks HIV ex vivo infection of Langerhans cells within the epithelium. J Invest Dermatol. 2013;133(12):2803–2805. | ||
Chen BA, Panther L, Marzinke MA, et al. Phase 1 safety, pharmacokinetics, and pharmacodynamics of dapivirine and maraviroc vaginal rings: a double-blind randomized trial. J Acquir Immune Defic Syndr. Epub 2015 May 28. | ||
Mechai F, Quertainmont Y, Sahali S, Delfraissy JF, Ghosn J. Post-exposure prophylaxis with a maraviroc-containing regimen after occupational exposure to a multi-resistant HIV-infected source person. J Med Virol. 2008;80(1):9–10. | ||
Taiwo B, Acosta EP, Ryscavage P, et al. Virologic response, early HIV-1 decay, and maraviroc pharmacokinetics with the nucleos(t)ide-free regimen of maraviroc plus darunavir/ritonavir in a pilot study. J Acquir Immune Defic Syndr. 2013;64(2):167–173. | ||
Nozza S, Galli L, Visco F, et al. Raltegravir, maraviroc, etravirine: an effective protease inhibitor and nucleoside reverse transcriptase inhibitor-sparing regimen for salvage therapy in HIV-infected patients with triple-class experience. AIDS. 2010;24(6):924–928. | ||
Nozza S, Galli L, Bigoloni A, et al. Four-year outcome of a PI and NRTI-sparing salvage regimen: maraviroc, raltegravir, etravirine. New Microbiol. 2014;37(2):145–151. | ||
Nozza S, Bigoloni A, Calcagno A, et al. Viral rebound after switch to maraviroc/raltegravir dual therapy in highly experienced and virologically suppressed patients with HIV-1 infection. J Antimicrob Chemother. 2014;69(5):1436–1439. | ||
Katlama C, Assoumou L, Valantin MA, et al. Maraviroc plus raltegravir failed to maintain virological suppression in HIV-infected patients with lipohypertrophy: results from the ROCnRAL ANRS 157 study. J Antimicrob Chemother. 2014;69(6):1648–1652. | ||
Nozza S, Galli L, Antinori A, et al. Maraviroc 150 mg daily plus lopinavir/ritonavir, a nucleoside/nucleotide reverse transcriptase inhibitor-sparing regimen for HIV-infected naive patients: 48-week final results of VEMAN study. Clin Microbiol Infect. 2014;21(5):510.e1–e9. | ||
Mills A, Mildvan D, Podzamczer D, et al. Maraviroc once-daily nucleoside analog-sparing regimen in treatment-naive patients: randomized, open-label pilot study. J Acquir Immune Defic Syndr. 2013;62(2):164–170. | ||
Bernal E, Verdu JM, Vera F, et al. Improvement of endothelial function after switching previously treated HIV-infected patients to an NRTI-sparing bitherapy with maraviroc. J Int AIDS Soc. 2014;17(4 Suppl 3):19726. | ||
Ermocida A, Mengoni F, Sauzullo I, et al. Extravirologic modulation of immune response by an NRTI-sparing antiretroviral regimen including darunavir and maraviroc. New Microbiol. 2014;37(2):225–229. | ||
Cheret A, Nembot G, Melard A, et al. Intensive five-drug antiretroviral therapy regimen versus standard triple-drug therapy during primary HIV-1 infection (OPTIPRIM-ANRS 147): a randomised, open-label, phase 3 trial. Lancet Infect Dis. 2015;15(4):387–396. | ||
Saag M, Goodrich J, Fatkenheuer G, et al. A double-blind, placebo-controlled trial of maraviroc in treatment-experienced patients infected with non-R5 HIV-1. J Infect Dis. 2009;199(11):1638–1647. | ||
Surdo M, Alteri C, Puertas MC, et al. Effect of maraviroc on non-R5 tropic HIV-1: refined analysis of subjects from the phase IIb study A4001029. Clin Microbiol Infect. 2015;21(1):e101–e106. | ||
Symons J, van Lelyveld SF, Hoepelman AI, et al. Maraviroc is able to inhibit dual-R5 viruses in a dual/mixed HIV-1-infected patient. J Antimicrob Chemother. 2011;66(4):890–895. | ||
Svicher V, Balestra E, Cento V, et al. HIV-1 dual/mixed tropic isolates show different genetic and phenotypic characteristics and response to maraviroc in vitro. Antiviral Res. 2011;90(1):42–53. | ||
Genebat M, Ruiz-Mateos E, Leon JA, et al. Correlation between the Trofile test and virological response to a short-term maraviroc exposure in HIV-infected patients. J Antimicrob Chemother. 2009;64(4):845–849. | ||
Gonzalez-Serna A, McGovern RA, Harrigan PR, et al. Correlation of the virological response to short-term maraviroc monotherapy with standard and deep-sequencing-based genotypic tropism prediction methods. Antimicrob Agents Chemother. 2012;56(3):1202–1207. | ||
Hernandez-Novoa B, Madrid-Elena N, Dronda F, et al. Virological response to short-course maraviroc monotherapy does not predict viral tropism in HIV-1-infected treatment-naive patients. J Antimicrob Chemother. 2014;69(7):1916–1919. | ||
Macias J, Viloria MM, Rivero A, et al. Lack of short-term increase in serum mediators of fibrogenesis and in non-invasive markers of liver fibrosis in HIV/hepatitis C virus-coinfected patients starting maraviroc-based antiretroviral therapy. Eur J Clin Microbiol Infect Dis. 2012;31(8):2083–2088. | ||
Gagliardini R, Rossetti B, Bianco C, et al. Safety and therapeutic efficacy of the switch to maraviroc + darunavir/ritonavir in HIV/HCV coinfected patients: initial results from GUSTA study. J Int AIDS Soc. 2014;17(4 Suppl 3):19818. | ||
Gonzalez EO, Boix V, Deltoro MG, et al. The effects of Maraviroc on liver fibrosis in HIV/HCV co-infected patients. J Int AIDS Soc. 2014;17(4 Suppl 3):19643. | ||
Martin-Blondel G, Cuzin L, Delobel P, et al. Is maraviroc beneficial in paradoxical progressive multifocal leukoencephalopathy-immune reconstitution inflammatory syndrome management? AIDS. 2009;23(18):2545–2546. | ||
Giacomini PS, Rozenberg A, Metz I, et al. Maraviroc and JC virus-associated immune reconstitution inflammatory syndrome. New Engl J Med. 2014;370(5):486–488. | ||
Rodriguez M, Silva-Sanchez FA, Luna-Rivero C, Vega-Barrientos R, Alvarado-de la Barrera C, Reyes-Teran G. Maraviroc failed to control progressive multifocal leukoencephalopathy-associated IRIS in a patient with advanced HIV infection. Case Rep Med. 2014;2014:381480. | ||
Latinovic O, Le N, Reitz M, et al. Synergistic inhibition of R5 HIV-1 by maraviroc and CCR5 antibody HGS004 in primary cells: implications for treatment and prevention. AIDS. 2011;25(9):1232–1235. | ||
Latinovic O, Reitz M, Le NM, et al. CCR5 antibodies HGS004 and HGS101 preferentially inhibit drug-bound CCR5 infection and restore drug sensitivity of Maraviroc-resistant HIV-1 in primary cells. Virology. 2011;411(1):32–40. | ||
Latinovic O, Schneider K, Szmacinski H, Lakowicz JR, Heredia A, Redfield RR. Binding of fusion protein FLSC IgG1 to CCR5 is enhanced by CCR5 antagonist Maraviroc. Antiviral Res. 2014;112:80–90. | ||
Secchi M, Vassena L, Morin S, Schols D, Vangelista L. Combination of the CCL5-derived peptide R4.0 with different HIV-1 blockers reveals wide target compatibility and synergic cobinding to CCR5. Antimicrob Agents Chemother. 2014;58(10):6215–6223. | ||
Murga JD, Franti M, Pevear DC, Maddon PJ, Olson WC. Potent antiviral synergy between monoclonal antibody and small-molecule CCR5 inhibitors of human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2006;50(10):3289–3296. | ||
Gottlieb GS, Eholie SP, Nkengasong JN, et al. A call for randomized controlled trials of antiretroviral therapy for HIV-2 infection in West Africa. AIDS. 2008;22(16):2069–2072. | ||
Benard A, van Sighem A, Taieb A, et al; The ACHIEV2E Collaboration Study Group. Immunovirological response to triple nucleotide reverse-transcriptase inhibitors and ritonavir-boosted protease inhibitors in treatment-naive HIV-2-infected patients. Clin Infect Dis. 2011;52(10):1257–1266. | ||
Ntemgwa ML, d’Aquin Toni T, Brenner BG, Camacho RJ, Wainberg MA. Antiretroviral drug resistance in human immunodeficiency virus type 2. Antimicrob Agents Chemother. 2009;53(9):3611–3619. | ||
Morner A, Bjorndal A, Albert J, et al. Primary human immunodeficiency virus type 2 (HIV-2) isolates, like HIV-1 isolates, frequently use CCR5 but show promiscuity in coreceptor usage. J Virol. 1999;73(3):2343–2349. | ||
Shi Y, Brandin E, Vincic E, et al. Evolution of human immunodeficiency virus type 2 coreceptor usage, autologous neutralization, envelope sequence and glycosylation. J Gen Virol. 2005;86(Pt 12):3385–3396. | ||
Espirito-Santo M, Santos-Costa Q, Calado M, Dorr P, Azevedo-Pereira JM. Susceptibility of HIV type 2 primary isolates to CCR5 and CXCR4 monoclonal antibodies, ligands, and small molecule inhibitors. AIDS Res Human Retrovir. 2012;28(5):478–485. | ||
Visseaux B, Charpentier C, Hurtado-Nedelec M, et al. In vitro phenotypic susceptibility of HIV-2 clinical isolates to CCR5 inhibitors. Antimicrob Agents Chemother. 2012;56(1):137–139. | ||
Armstrong-James D, Stebbing J, Scourfield A, et al. Clinical outcome in resistant HIV-2 infection treated with raltegravir and maraviroc. Antiviral Res. 2010;86(2):224–226. | ||
Caixas U, Ferreira J, Marinho AT, et al. Long-term maraviroc use as salvage therapy in HIV-2 infection. J Antimicrob Chemother. 2012;67(10):2538–2539. | ||
Stegmann S, Manea ME, Charpentier C, et al. Foscarnet as salvage therapy in HIV-2-infected patient with antiretroviral treatment failure. J Clin Virol. 2010;47(1):79–81. | ||
Velasco-Velazquez M, Jiao X, De La Fuente M, et al. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012;72(15):3839–3850. | ||
Ochoa-Callejero L, Perez-Martinez L, Rubio-Mediavilla S, Oteo JA, Martinez A, Blanco JR. Maraviroc, a CCR5 antagonist, prevents development of hepatocellular carcinoma in a mouse model. PloS One. 2013;8(1):e53992. | ||
Mencarelli A, Graziosi L, Renga B, et al. CCR5 antagonism by maraviroc reduces the potential for gastric cancer cell dissemination. Trans Oncol. 2013;6(6):784–793. | ||
Sicoli D, Jiao X, Ju X, et al. CCR5 receptor antagonists block metastasis to bone of v-Src oncogene-transformed metastatic prostate cancer cell lines. Cancer Res. 2014;74(23):7103–7114. | ||
Shen Z, Li T, Chen D, et al. The CCL5/CCR5 axis contributes to the perineural invasion of human salivary adenoid cystic carcinoma. Oncol Rep. 2014;31(2):800–806. | ||
Choi SW, Hildebrandt GC, Olkiewicz KM, et al. CCR1/CCL5 (RANTES) receptor–ligand interactions modulate allogeneic T-cell responses and graft-versus-host disease following stem-cell transplantation. Blood. 2007;110(9):3447–3455. | ||
Reshef R, Luger SM, Hexner EO, et al. Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. New Engl J Med. 2012;367(2):135–145. | ||
Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med. 2007;28(1):23–42, vii. | ||
Dorfmuller P, Zarka V, Durand-Gasselin I, et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Critl Care Med. 2002;165(4):534–539. | ||
Amsellem V, Lipskaia L, Abid S, et al. CCR5 as a treatment target in pulmonary arterial hypertension. Circulation. 2014;130(11):880–891. | ||
Barbaro G, Barbarini G, Di Lorenzo G; The Gruppo Italiano Per lo Studio Cardiologico dei Pazienti Affetti da AIDS. Early impairment of systolic and diastolic function in asymptomatic HIV-positive patients: a multicenter echocardiographic and echo-Doppler study. AIDS Res Human Retrovir. 1996;12(16):1559–1563. | ||
Kelly KM, Tocchetti CG, Lyashkov A, et al. CCR5 inhibition prevents cardiac dysfunction in the SIV/macaque model of HIV. J Am Heart Assoc. 2014;3(2):e000874. | ||
Liu FC, Yu HP, Hwang TL, Tsai YF. Protective effect of tropisetron on rodent hepatic injury after trauma-hemorrhagic shock through P38 MAPK-dependent hemeoxygenase-1 expression. PloS One. 2012;7(12):e53203. | ||
Suzuki T, Kawasaki T, Choudhry MA, Chaudry IH. Role of PPARgamma in the salutary effects of 17beta-estradiol on Kupffer cell cytokine production following trauma-hemorrhage. J Cell Physiol. 2011;226(1):205–211. | ||
Liu FC, Tsai YF, Yu HP. Maraviroc attenuates trauma-hemorrhage-induced hepatic injury through PPAR gamma-dependent pathway in rats. PloS One. 2013;8(10):e78861. | ||
Fleishaker DL, Garcia Meijide JA, Petrov A, et al. Maraviroc, a chemokine receptor-5 antagonist, fails to demonstrate efficacy in the treatment of patients with rheumatoid arthritis in a randomized, double-blind placebo-controlled trial. Arthritis Res Ther. 2012;14(1):R11. | ||
Haringman JJ, Smeets TJ, Reinders-Blankert P, Tak PP. Chemokine and chemokine receptor expression in paired peripheral blood mononuclear cells and synovial tissue of patients with rheumatoid arthritis, osteoarthritis, and reactive arthritis. Ann Rheumat Dis. 2006;65(3):294–300. | ||
Patel A, Kesler B, Wise RA. Persistent pneumomediastinum in interstitial fibrosis associated with rheumatoid arthritis: treatment with high-concentration oxygen. Chest. 2000;117(6):1809–1813. | ||
Pokorny V, McQueen F, Yeoman S, et al. Evidence for negative association of the chemokine receptor CCR5 d32 polymorphism with rheumatoid arthritis. Ann Rheumat Dis. 2005;64(3):487–490. | ||
Span PN, Pollakis G, Paxton WA, et al. Improved metastasis-free survival in nonadjuvantly treated postmenopausal breast cancer patients with chemokine receptor 5 del32 frameshift mutations. Int J Cancer. 2015;136(1):91–97. | ||
Braunersreuther V, Steffens S, Arnaud C, et al. A novel RANTES antagonist prevents progression of established atherosclerotic lesions in mice. Arterioscler Thromb Vasc Biol. 2008;28(6):1090–1096. | ||
Veillard NR, Kwak B, Pelli G, et al. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res. 2004;94(2):253–261. | ||
Mencarelli A, Cipriani S, Renga B, et al. The bile acid sensor FXR protects against dyslipidemia and aortic plaques development induced by the HIV protease inhibitor ritonavir in mice. PloS One. 2010;5(10):e13238. | ||
Cipriani S, Francisci D, Mencarelli A, et al. Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation. 2013;127(21):2114–2124. | ||
Lee YK, Choi DY, Jung YY, et al. Decreased pain responses of C-C chemokine receptor 5 knockout mice to chemical or inflammatory stimuli. Neuropharmacology. 2013;67:57–65. | ||
Nischalke HD, Nattermann J, Fischer HP, Sauerbruch T, Spengler U, Dumoulin FL. Semiquantitative analysis of intrahepatic CC-chemokine mRNas in chronic hepatitis C. Mediators Inflamm. 2004;13(5–6):357–359. | ||
Desai MS, Mariscalco MM, Tawil A, Vallejo JG, Smith CW. Atherogenic diet-induced hepatitis is partially dependent on murine TLR4. J Leukoc Biol. 2008;83(6):1336–1344. | ||
Seki E, De Minicis S, Gwak GY, et al. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest. 2009;119(7):1858–1870. | ||
Perez-Martinez L, Perez-Matute P, Aguilera-Lizarraga J, et al. Maraviroc, a CCR5 antagonist, ameliorates the development of hepatic steatosis in a mouse model of non-alcoholic fatty liver disease (NAFLD). J Antimicrob Chemother. 2014;69(7):1903–1910. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.