")
Back to Journals » OncoTargets and Therapy » Volume 9
Low concentrations of 5-aza-2'-deoxycytidine induce breast cancer stem cell differentiation by triggering tumor suppressor gene expression
Authors Phan NL, Trinh NV , Pham PV
Received 13 September 2015
Accepted for publication 16 November 2015
Published 23 December 2015 Volume 2016:9 Pages 49—59
DOI https://doi.org/10.2147/OTT.S96291
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Nhan Lu-Chinh Phan, Ngu Van Trinh, Phuc Van Pham
Laboratory of Stem Cell Research and Application, University of Science, Vietnam National University, Ho Chi Minh City, Vietnam
Background: Breast cancer stem cells (BCSCs) are considered the cause of tumor growth, multidrug resistance, metastasis, and recurrence. Therefore, differentiation therapy to reduce self-renewal of BCSCs is a promising approach. We have examined the effects of 5-aza-2'-deoxycytidine (DAC) on BCSC differentiation.
Materials and methods: BCSCs were treated with a range of DAC concentrations from 0.625 to 100 µM. The differentiation status of DAC-treated BCSCs was graded by changes in cell proliferation, CD44+CD24- phenotype, expression of tumor suppressor genes, including BRCA1, BRCA2, p15, p16, p53, and PTEN, and antitumor drug resistance.
Results: DAC treatment caused significant BCSC differentiation. BCSCs showed a 15%–23% reduction in proliferation capacity, 3.0%–21.3% decrease in the expression of BCSC marker CD44+/CD24-, activation of p53 expression, and increased p15, p16, BRCA1, and BRCA2 expression. Concentrations of DAC ranging from 0.625 to 40 µM efficiently induce cell cycle arrest in S-phase. ABCG2, highly expressed in BCSCs, also decreased with DAC exposure. Of particular note, drug-sensitivity of BCSCs to doxorubicin, verapamil, and tamoxifen also increased 1.5-, 2.0-, and 3.7-fold, respectively, after pretreatment with DAC.
Conclusion: DAC reduced breast cancer cell survival and induced differentiation through reexpression of tumor suppressor genes. These results indicate the potential of DAC in targeting specific chemotherapy-resistant cells within a tumor.
Keywords: breast cancer, breast cancer stem cells, differentiation, epigenetics, 5-aza-2'-deoxycytidine
Introduction
Breast cancer stem cells (BCSCs) were discovered in 2003 by Al-Hajj et al.1 BCSCs are recognized as a subpopulation expressing CD44+CD24−/low ESA+ and Lin− markers. Another candidate marker that fits the CSC concept is aldehyde dehydrogenase 1 (ALDH1).2 A number of cancer cell lines also express CD44+, such as colon cancer,3 liver cancer,4 renal cancer,5 bladder cancer,6 cervical cancer,7 gallbladder cancer,8 hepatocellular carcinoma,9 and human nasopharyngeal carcinoma.10 However a combination of CD44+ and CD24− was found in a BCSC subpopulation within a breast tumor, and is responsible for initiation, progression, chemotherapy resistance, and metastasis.11–15 Therefore, targeting BCSCs is a promising therapeutic approach, and the best strategy is differentiation therapy to reduce the stemness of BCSCs.
Differentiation therapy could be used to differentiate CSCs terminally and make them lose their self-renewal property, a hallmark of the CSC phenotype. Inducing differentiation also reduces their drug resistance. To date, there are different strategies to induce differentiation of BCSCs using antitumor drugs, signaling pathway inhibitors, or gene knockdowns.
Some drugs such as acetaminophen, cisplatin, and retinoic acid induce differentiation of BCSCs; for example, Takehara et al16 reduced the tumorigenic ability of MDA-MB-231 cells using acetaminophen treatment in nude mice. Similarly, cisplatin treatment at 10 and 20 μM also reduced BCSC viability by 36%–51%, proliferation capacity by 36%–67%, and stem cell markers (CD49f, SSEA4) by 12%–67%, while upregulating the differentiation markers, CK18, SMA, and β-tubulin, by 10%–130%.17 Exposure to retinoic acid (2 μM) or vorinostat combined with 6 Gy irradiation also reduced by 30% and 70%, respectively, mammosphere survival compared to the irradiated control. In combination with paclitaxel (0.5 μM), retinoic acid and vorinostat decreased by 70% and 60%, respectively, mammosphere survival compared to paclitaxel alone.18
IMD-0354, the NF-κB inhibitor, targets to BCSCs in a combination therapy of doxorubicin encapsulated in targeted nanoparticles. IMD-0354 induced differentiation of BCSCs, a decrease in the side-population of cells, inhibiting dye/drug efflux, reducing ABC transporters, reducing colony formation on soft agar, causing low attachment to plates, and decreasing gene expression of stem cell markers, including Oct4, Nanog, and Sox2, and apoptosis resistance.19
Using a different strategy, Pham et al20 induced differentiation of BCSCs by knocking down CD44 gene expression with siRNA. CD44 is an important factor contributing to properties of CSC; in association with Wnt, it maintains the immortality of CSC.21 Hedgehog and Notch signaling pathway also have a close relationship with CD44 in regulating the self-renewal of CSC.22–26 In vitro, CD44 knockdown of BCSCs abolished stemness and increased susceptibility to chemotherapy.20,27 In vivo, a combination of CD44 downregulation and doxorubicin strongly suppressed tumor growth, significantly reducing tumor size and weight.28
5-aza-2′-deoxycytidine (DAC) can be used as an epigenetic drug that utilizes a demethylation mechanism; it has been approved for use in malignant disease and cancer treatment by the US Food and Drug Administration.29–31 DAC is incorporated into DNA where it inhibits activation of DNA methyltransferase. DAC induces differentiation, apoptosis, and senescence in leukemic cells in vitro32–34 and also other cancer cell types.35–37 These results show the potential of DAC in treating malignant disease, and thus we have examined the effects of DAC on the differentiation of BCSCs in vitro.
Materials and methods
Cell culture
BCSCs with phenotype CD44+CD24− were isolated as previously reported.20 Cells were cultured in T25 culture flasks (Sigma-Aldrich, St Louis, MO, USA) for RNA extraction, flow cytometry, and an E-plate 96 (ACEA Biosciences, Inc., San Diego, CA, USA) for cell proliferation and drug sensitivity assays. The cells were cultured at 37°C in air with 5% CO2 in Dulbecco’s Modified Eagle’s Medium/F12 (Sigma-Aldrich) supplemented with 10% fetal bovine serum and 1% antibiotic–antimycotic (GeneWorld, Ho Chi Minh City, Vietnam). The medium was replaced every 3 days. When 70%–80% confluence was reached, cells were detached with 0.5% trypsin/0.2% EDTA in Dulbecco’s phosphate-buffered saline (PBS; Sigma-Aldrich). The MCF-7 cell line is used as a control breast cancer cell line. This study was approved by the ethics committee of the Institutional Review Board, Vietnam National University, Vietnam and the ethics committee of Oncology Hospital, Vietnam.
Determination of cell proliferation and drug sensitive by xCELLigence
Cells were seeded on an E-plate 96 (1,000 cells/well) and cultured for 24 hours before adding DAC. Cells were treated with DAC alone or in combination with verapamil, doxorubicin, and tamoxifen (all purchased from Sigma-Aldrich). The drugs were added to the medium every 24 hours. Initially, cells were treated with ten different concentrations of DAC (0.1, 0.625, 1.25, 2.5, 5, 10, 20, 40, 60, 80, and 100 μM) for 114 hours to determinate, the most effectively inhibited DAC concentration. Then, the concentration of DAC that most effectively inhibited proliferation was chosen to be combined with verapamil, doxorubicin, and tamoxifen to treat cells for 48 hours. Proliferation in each sample was calculated by comparison with the untreated control, and this was monitored every 15 minutes using the Real-Time Cell Analyzer xCELLigence System (Roche-Applied Science, Indianapolis, IN, USA).
Gene expression analysis
To determine if DAC is effective in DNA demethylation and reactivating silenced genes, real-time polymerase chain reaction (RT-PCR) was used to detect changes in the expression of p15, p16, p53, PTEN, BRCA1, and BRCA2 genes silenced in BCSCs by hypermethylation in their promoters. RNA was extracted using an easy-BLUE TM Total RNA Extraction Kit (Intron Biotechnology, Seongnam, South Korea) after cells were exposed to DAC at inhibitory concentration in 72 hours. A Brilliant III Ultra Fast SYBR Green QRT-PCR master mix kit (Agilent Technology, Santa Clara, CA, USA) was used for reverse transcription and quantitative RT-PCR. The experiment was monitored using an Eppendorf Mastercycler® RealPlex2 (Eppendorf, Hamburg, Germany) and then gene expression was calculated by the 2−DDCT method. The PCR primer sequences used in this study are shown in Table 1.
 | Table 1 Primer sequences used for reverse transcription polymerase chain reaction |
Flow cytometry assays
Differentiation of CSCs was identified by the CD44+/CD24− level. The cells were treated with DAC, trypsinized, and washed in PBS before being stained with monoclonal antibodies anti-CD44 and anti-CD24 (BD Biosciences, Franklin Lakes, NJ, USA). Tubes were incubated in the dark at room temperature for 30 minutes before FACSflow solution (BD Biosciences) was added. Using CellQuest Pro software (BD Biosciences), the CD44+CD24− level was identified by quadrant analysis.
To analyze ABCG2 expression, BCSCs were fixed with FCM Fixation Buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 30 minutes and washed in PBS. Cold FCM Permeabilization Buffer (Santa Cruz Biotechnology) was added for 5 minutes at room temperature. Approximately 105 cells were labeled with 2 μL ABCG2-FITC antibody (Santa Cruz Biotechnology) at 37°C for 30 minutes. Labeled cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences).
To analyze cell cycle, BCSCs were harvested by trypsinization, washed with PBS, and resuspended in 0.5 mL PBS. The tubes were gently vortexed, and 4.5 mL ice cold 70% ethanol was added dropwise over 30–60 seconds before incubation for 2 hours at 4°C. Cells were washed and stained with PI staining solution (Sigma-Aldrich) and analyzed using a FACSCalibur flow cytometer (BD Biosciences).
Statistical analysis
All graphs and statistical procedures were done using GraphPad 6 (GraphPad Software, San Diego, CA, USA). One-way analysis of variance and t-tests were used for data analysis, and results are expressed as mean ± standard deviation. Statistical significance was set at P<0.05.
Results
DAC inhibits BCSC proliferation at low concentrations
DAC inhibited BCSC proliferation (Figure 1A). At the highest concentration (100 μM) of DAC for 114 hours; no cell death was observed, indicating lack of cytotoxicity across the whole range, but there was a decrease in proliferation at all concentrations. The doubling times were significantly greater than the control group, which had a doubling time of 24.9±0.5 hours (Figure 2A). DAC clearly did not inhibit MCF-7 cells (Figure 1B); indeed, proliferation increased in MCF-7 cells treated with 1.25, 2.5, 5, 10, 40, 60, 80, and 100 μM DAC. At 0.625 and 20 μM, DAC increased the doubling times to 26.1±0.5 and 26.5±0.6 hours, respectively, with doubling times in the control group being 25.9±0.4 hours (Figure 2B). The data in Figure 1A indicate that low doses of DAC effectively inhibited BCSC proliferation. Based on these results, we used a low concentration of DAC (<10 μM) for all the future experiments.
 | Figure 1 Cell proliferation curve after exposure to DAC for 114 hours. |
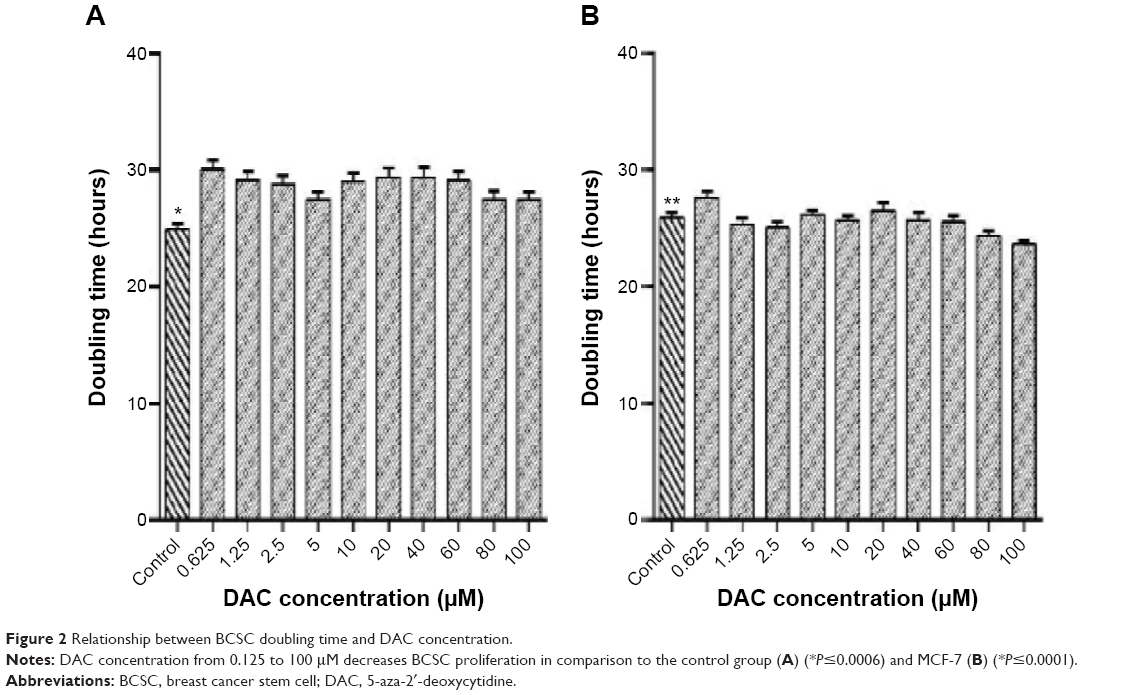 | Figure 2 Relationship between BCSC doubling time and DAC concentration. |
DAC altered tumor suppressor gene expression
The effect of DAC exposure at 5 and 10 μM on the expression of p15, p16, p53, BRCA1, and BRCA2 genes was determined and compared to the control group. At both concentrations, DAC reactivated p53 in comparison with control group, which did not express this gene. Otherwise, p15, p16, BRCA1, and BRCA2 genes increased their expression on treatment with DAC. When treated with 5 μM DAC, p15, p16, BRCA1, and BRCA2 gene expression increased 2.95-, 5.00-, 106.30-, and 1.40-fold, respectively, in comparison with the control group. At 10 μM DAC concentration, the results were similar, with corresponding 7.46-, 1.03-, 3.00-, and 1.50–fold increase in gene expression (Figure 3). However, PTEN expression was unchanged when treated with 5 and 10 μM DAC.
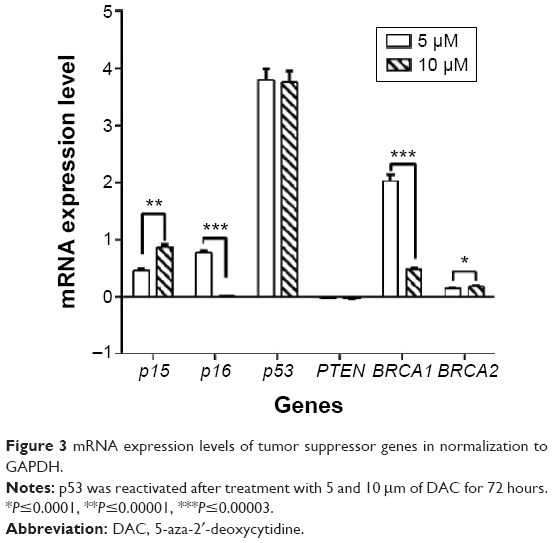 | Figure 3 mRNA expression levels of tumor suppressor genes in normalization to GAPDH. |
DAC arrests BCSCs in S phase
The cell cycle of BCSCs changed after DAC exposure (Figure 4); in the control group the distribution of cells was 63.5%±3.2% in the G1 phase, 9.3%±0.47% in the S phase, and 21.0%±1.1% in the G2/M phase (Figure 4A). This ratio changed after exposure to DAC because of arrest mainly in the S phase in a dose-dependent manner. The distribution of cells in the S phase in the control group was 9.3%±0.47% (Figure 4H). This increased after DAC exposure at 0.625, 1.25, 5, 10, 20, and 40 μM DAC, with cells in the S phase accounting for 11.2%±0.6%, 14.2%±0.8%, 13.2%±0.7%, 19.1%±0.95%, 19.2%±0.96%, and 18.2%±0.91%.
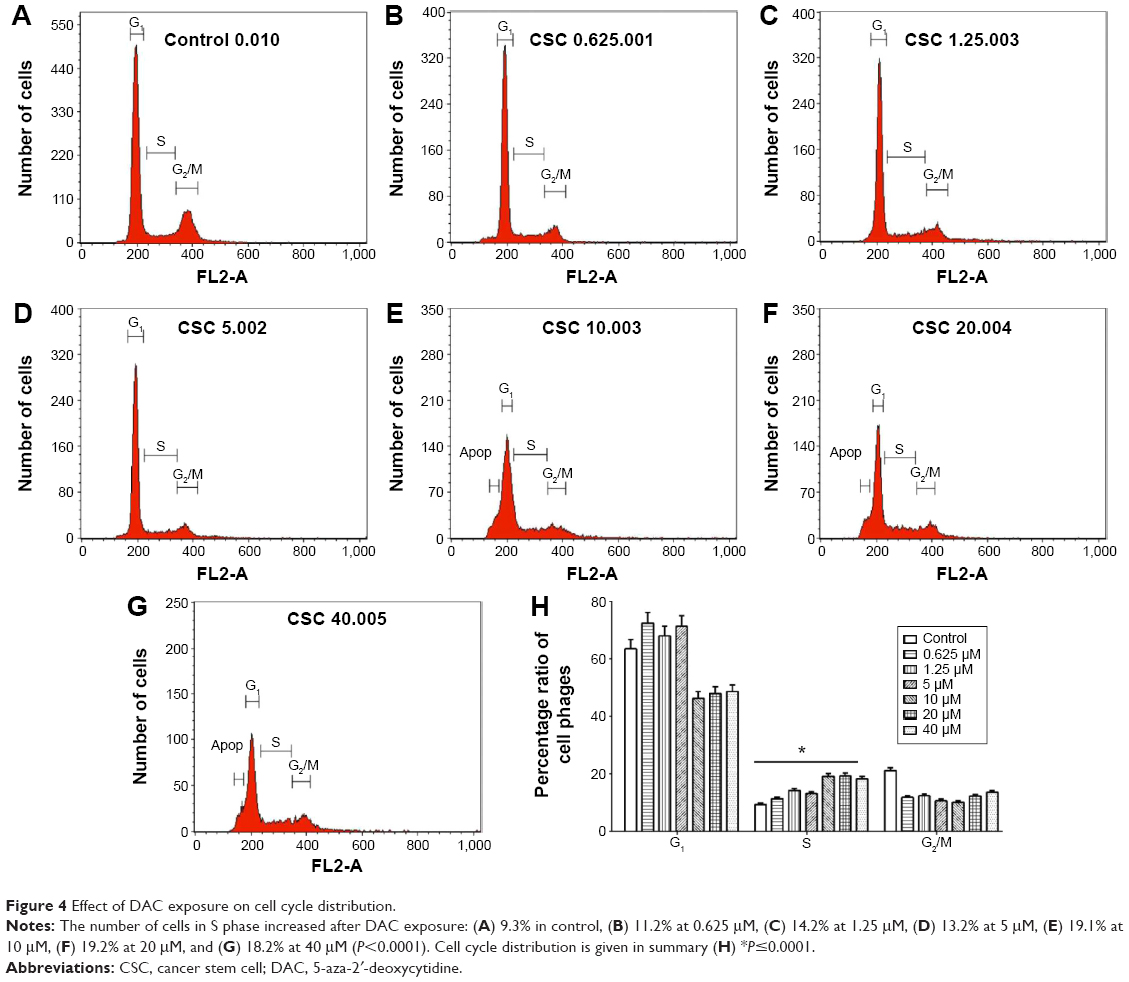 | Figure 4 Effect of DAC exposure on cell cycle distribution. |
DAC decreases the BCSC population
To determine the effect of DAC exposure on the differentiation of BCSCs, we treated BCSCs for 72 hours and assessed the CD44+/CD24− population; the number of cells in the CD44+/CD24− population significantly decreased compared to the control group (Figure 5). The percentage of CD44+/CD24− cells was 52.7% in the untreated group, and 17.8% in the 5 μM DAC group and 25.1% in the 10 μM DAC group.
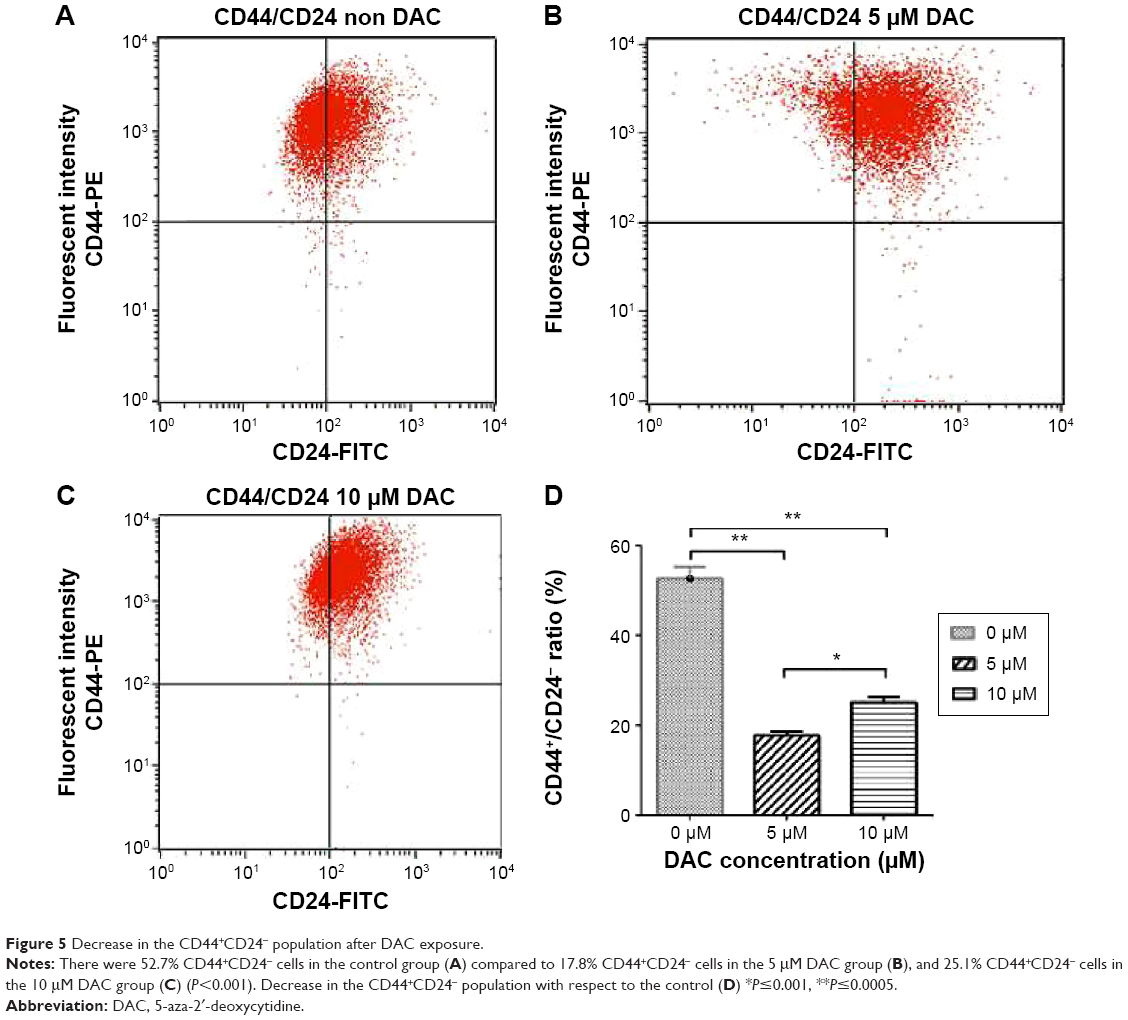 | Figure 5 Decrease in the CD44+CD24− population after DAC exposure. |
DAC reduces ABCG2 expression of BCSCs
To verify the effect of DAC on BCSC drug resistance, the level of ABCG2, a key protein expressed in CSCs, was measured. In the control population (Figure 6), ABCG2 expression was 99.9%±5.0%, which changed after exposure to DAC. A concentration of 2.5, 5, 10, and 20 μM DAC strongly reduced the expression by 15.8%±0.6%, 63%±3.0%, 19.8%±1.0%, and 19.5%±1.0%, respectively. The results provide a supplementary explanation for the antitumor drug sensitivity of BCSCs treated with DAC.
 | Figure 6 ABCG2 expression analyzed by flow cytometry. |
BCSCs with DAC are sensitized to antitumor drugs
Half-maximal inhibitory concentration (IC50) values in the DAC–doxorubicin, DAC–verapamil, and DAC–tamoxifen groups were reduced in comparison to the control group (Figure 7). The IC50 of doxorubicin was 0.89 and 0.24 μg/mL in groups with and without 10 μM DAC, respectively, resulting in a 3.7-fold reduction. The effectiveness of verapamil was also increased by 10 μM DAC, the IC50 of verapamil with and without 10 μM DAC was reduced 1.97-fold from 35.3 to 17.9 μM, respectively. On the other hand, the IC50 of tamoxifen changed from 150 to 101 μM, a 1.48-fold reduction in the presence of DAC. The results, which were statistically significant, show that DAC had the strongest effect in combination with doxorubicin followed by verapamil and then tamoxifen. The doubling times of BCSCs in samples pretreated with DAC were longer than those treated with antitumor drugs alone. Doxorubicin at 0.05, 0.1, 0.25, 0.5, and 1 μg/mL resulted in BCSC doubling times of 20.1±0.18, 20.1±0.09, 26.4±0.16, 29.3±0.28, and 29.4±0.31 hours, respectively, and in combination with 10 μM DAC, the BCSC doubling times were 45.36±0.14, 48.2±0.19, 44.6±0.17, 49.4±0.18, and 54.9±0.01 hours, respectively (Figure 8). Similarly, verapamil at 10, 20, 40, 60, and 80 μM resulted in doubling times of 32.1±0.14, 32.6±0.12, 36.4±0.13, 61.7±0.35, and 54.8±0.44 hours, respectively. Addition of 10 μM DAC resulted in an increase of doubling times to 49.5±0.3, 50.9±0.3, 48.8±0.29, 48.5±0.31, and 66.7±0.95 hours, respectively. Tamoxifen effectively inhibited BCSCs; at 25, 50, 100, 125, and 150 μM DAC, doubling times were 23.4±0.14, 22.8±0.22, 39.9±1, 56.7±1.8, and 75.3±8.2 hours, respectively. At 10 μM DAC, the doubling times changed to 52.4±0.33, 60.0±0.32, and 113.2±11.5 at 25, 50, and 100 μM tamoxifen, respectively. Interestingly, we found that 125 and 150 μM tamoxifen combined with DAC caused BCSC death. These results demonstrate that the combination of antitumor drugs with DAC increased the doubling time and as well as the drug sensitivity of BCSCs.
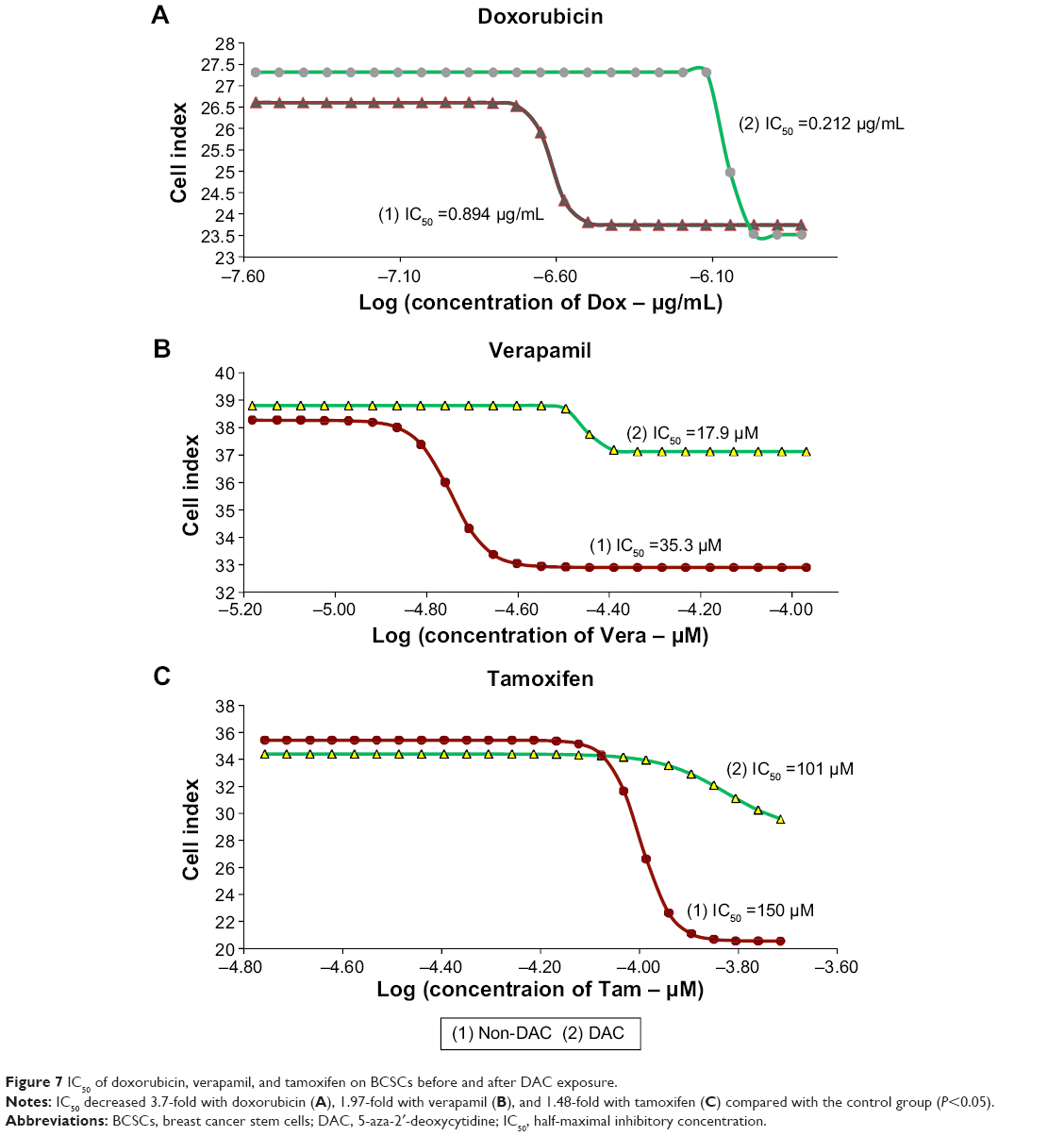 | Figure 7 IC50 of doxorubicin, verapamil, and tamoxifen on BCSCs before and after DAC exposure. |
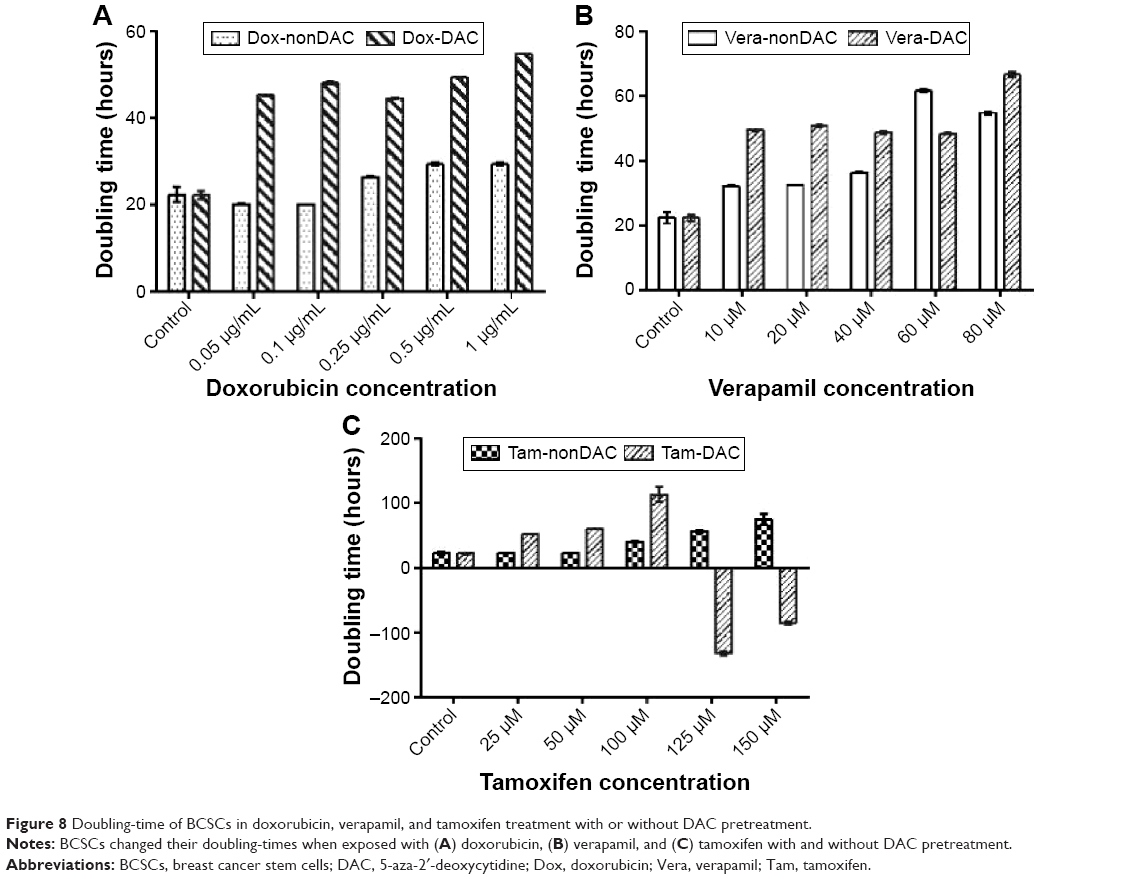 | Figure 8 Doubling-time of BCSCs in doxorubicin, verapamil, and tamoxifen treatment with or without DAC pretreatment. |
Discussion
Previous studies have investigated the ability of DAC to induce expression of tumor suppressor genes in a variety of cell lines. Tumor suppressor genes that are reported to increase their expression in response to DAC exposure include Rb,38 p53,39 p21,40 p27, p15,41,42 p16,29,43,44 Apaf-1,45 PTEN, BRCA1, and BRCA2.46,47 In this study, the effect of DAC exposure on p15, p16, p53, PTEN, BRCA1, and BRCA2 expression was similar to previous reports. Interestingly, we found that p53 expression in BCSC was restored by DAC treatment, which raises the hypothesis that the demethylation of p53 caused reexpression and vice versa in BCSC. Moreover, it has been determined that hypermethylation in the promoter region leads to tumorigenesis where there are no p53 gene mutations.48 Collectively, demethylation of p53 is important in regulating this expression. Reexpression of p53 inhibited cell proliferation and induced proapoptotic gene expression in human cancer cells.49 In addition to p53, the results also showed that p15, p16, PTEN, BRCA1, and BRCA2 increased their expression in response to DAC exposure. The p15 gene is inactivated in response to promoter region hypermethylation, and treatment with DAC activates p15 mRNA.50 Hypermethylation of p16 promoters, which are strongly associated with the risk of breast cancer,51 may be involved in the pathogenesis of breast cancer.52 Demethylation of BRCA1 and BRCA2 by DAC resulted in reexpression of these genes.47 PTEN promoter methylation may have decreased the expression of PTEN,53 since expression of the PTEN gene was associated with low methylation levels.54
p15, p16,55,56 p53,57 PTEN, BRCA1, and BRCA258 are involved in the cell cycle regulation. Increased expression of these genes could induce cell cycle arrest, thereby inhibiting cell proliferation.39 Our results were similar to that of Hurtubise and Momparler,59 who showed that DAC affects the S phase of breast cancer cells. Using DAC as a demethylating agent could either reactivate silenced genes or increase the expression of hypermethylated genes. Expression of tumor suppressor genes, such as p15, p16, p53, and PTEN, was increased. It is known that DAC acts as a proliferation inhibitor.60–62 Consequently, BCSCs treated with DAC are affected both in proliferation and cell cycle regulation.
Low doses of DAC have inhibitory effects on some cancer cell lines. Singh et al63 reported that cell proliferation of the MCF-7 cell line was decreased 65% on treatment with 5 μM DAC. Treating MCF-7 cells with 0.5, 1, 2, and 5 μM DAC resulted in a 7%, 15%, 37%, and 45% decrease in proliferation, respectively.45 Dai et al64 used 102.4 μM DAC to decrease cell growth by 58.6%.
An important characteristic of BCSCs is their resistance to chemotherapeutic drugs; resistance is related to high expression of ATP-binding cassette transporters.65–67 ABC transporters are membrane transporters that can pump many structurally unrelated small molecules (such as cytotoxic drugs and dyes) out of cells.68,69 Therefore, increase in ABC transporters enables CSCs to resist many cancer therapies.65,70 ABCG2 is the most notable breast cancer resistance protein of this superfamily;70 its expression is decreased during the differentiation of BCSCs to non-CSCs.71 It also helps protect cells from cytotoxic agents.72,73 As a result of epigenetic changes, BCSCs treated with DAC have significantly lower ABCG2 levels that increase the antitumor drug sensitivity to drugs such as doxorubicin, verapamil, and tamoxifen. Our results showed that drug sensitivity of BCSCs was increased when antitumor drugs were combined with DAC, giving similar results to those obtained by Mirza et al,74 who showed that the combination of DAC with doxorubicin, paclitaxel, and 5-fluorouracil resulted in a 15%, 16%, and 13% decrease in cell proliferation, respectively, in MCF-7 and MDA cell lines when compared to the drugs being used alone.
Conclusion
Low-dose DAC acts as a demethylating agent causing aberrant gene expression. Its exposure induces epigenetic changes in BCSCs, such as p53 reactivation and increased expression of tumor suppressor genes p15, p16, PTEN, BRCA1, and BRCA2. DAC-treated BCSCs have significantly decreased expression of ABCG2, are arrested in S phase of the cell cycle, and are more sensitive to doxorubicin, tamoxifen, and verapamil. Most importantly, DAC exposure inhibits the BCSC population. These findings suggest that DAC could potentially be used in epigenetic therapies targeting BCSC differentiation.
Acknowledgment
This study was funded by Ministry of Science and Technology under grant No. DTDL.2011-T/30, Vietnam.
Disclosure
The authors report no conflicts of interest in this work.
References
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. | ||
Sophos NA, Vasiliou V. Aldehyde dehydrogenase gene superfamily: the 2002 update. Chem Biol Interact. 2003;143–144:5–22. | ||
Herrlich P, Pals S, Ponta H. CD44 in colon cancer. Eur J Cancer. 1995;31A(7–8):1110–1112. | ||
Jonas S, Allenmersh T. Expression of messenger-RNA cd44 splice variants in primary colon-cancer and liver metastases. Oncol Rep. 1995;2(6):1103–1106. | ||
Kan M, Aki M, Akiyama K, Naruo S, Kanayama H, Kagawa S. High-level expression of the CD44 variant sharing exon v10 in renal cancer. Jpn J Cancer Res. 1995;86(9):847–853. | ||
Matsumura Y, Sugiyama M, Matsumura S, et al. Unusual retention of introns in CD44 gene transcripts in bladder cancer provides new diagnostic and clinical oncological opportunities. J Pathol. 1995;177(1):11–20. | ||
Kainz C, Tempfer C, Kohlberger P, et al. Immunohistochemical detection of adhesion molecule CD44 splice variants in lymph node metastases of cervical cancer. Int J Cancer. 1996;69(3):170–173. | ||
Shi C, Tian R, Wang M, et al. CD44+ CD133+ population exhibits cancer stem cell-like characteristics in human gallbladder carcinoma. Cancer Biol Ther. 2010;10(11):1182–1190. | ||
Zhu Z, Hao X, Yan M, et al. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer. 2010;126(9):2067–2078. | ||
Su J, Xu XH, Huang Q, et al. Identification of cancer stem-like CD44+ cells in human nasopharyngeal carcinoma cell line. Arch Med Res. 2011;42(1):15–21. | ||
Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66(19):9339–9344. | ||
Croker AK, Allan AL. Cancer stem cells: implications for the progression and treatment of metastatic disease. J Cell Mol Med. 2008;12(2):374–390. | ||
Monteiro J, Fodde R. Cancer stemness and metastasis: therapeutic consequences and perspectives. Eur J Cancer. 2010;46(7):1198–1203. | ||
Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist. 2010;15(Suppl 5):39–48. | ||
Sampieri K, Fodde R. Cancer stem cells and metastasis. Semin Cancer Biol. 2012;22(3):187–193. | ||
Takehara M, Hoshino T, Namba T, Yamakawa N, Mizushima T. Acetaminophen-induced differentiation of human breast cancer stem cells and inhibition of tumor xenograft growth in mice. Biochem Pharmacol. 2011;81(9):1124–1135. | ||
Prabhakaran P, Hassiotou F, Blancafort P, Filgueira L. Cisplatin induces differentiation of breast cancer cells. Front Oncol. 2013;3:134. | ||
Roy R, Willan PM, Clarke R, Farnie G. Differentiation therapy: targeting breast cancer stem cells to reduce resistance to radiotherapy and chemotherapy. Breast Cancer Res. 2010;12(Suppl 1):05. | ||
Gomez-Cabrero A, Wrasidlo W, Reisfeld RA. IMD-0354 targets breast cancer stem cells: a novel approach for an adjuvant to chemotherapy to prevent multidrug resistance in a murine model. PLoS One. 2013;8(8):e73607. | ||
Pham PV, Phan NL, Nguyen NT, et al. Differentiation of breast cancer stem cells by knockdown of CD44: promising differentiation therapy. J Transl Med. 2011;9:209. | ||
Zeilstra J, Joosten SP, Dokter M, Verwiel E, Spaargaren M, Pals ST. Deletion of the WNT target and cancer stem cell marker CD44 in Apc(Min/+) mice attenuates intestinal tumorigenesis. Cancer Res. 2008;68(10):3655–3661. | ||
Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063–6071. | ||
Chen BY, Liu JY, Chang HH, et al. Hedgehog is involved in prostate basal cell hyperplasia formation and its progressing towards tumorigenesis. Biochem Biophys Res Commun. 2007;357(4):1084–1089. | ||
Farnie G, Clarke RB. Mammary stem cells and breast cancer – role of Notch signalling. Stem Cell Rev. 2007;3(2):169–175. | ||
Tanaka H, Nakamura M, Kameda C, et al. The Hedgehog signaling pathway plays an essential role in maintaining the CD44+CD24−/low subpopulation and the side population of breast cancer cells. Anticancer Res. 2009;29(6):2147–2157. | ||
Sharma A, Paranjape AN, Rangarajan A, Dighe RR. A monoclonal antibody against human Notch1 ligand-binding domain depletes subpopulation of putative breast cancer stem-like cells. Mol Cancer Ther. 2012;11(1):77–86. | ||
Van Phuc P, Nhan PL, Nhung TH, et al. Downregulation of CD44 reduces doxorubicin resistance of CD44CD24 breast cancer cells. Onco Targets Ther. 2011;4:71–78. | ||
Van Pham P, Vu NB, Duong TT, et al. Suppression of human breast tumors in NOD/SCID mice by CD44 shRNA gene therapy combined with doxorubicin treatment. Onco Targets Ther. 2012;5:77–84. | ||
Lemaire M, Chabot GG, Raynal NJ, et al. Importance of dose-schedule of 5-aza-2′-deoxycytidine for epigenetic therapy of cancer. BMC Cancer. 2008;8:128. | ||
Momparler RL. Epigenetic therapy of non-small cell lung cancer using decitabine (5-aza-2′-deoxycytidine). Front Oncol. 2013;3:188. | ||
Naldi I, Taranta M, Gherardini L, et al. Novel epigenetic target therapy for prostate cancer: a preclinical study. PLoS One. 2014;9(5):e98101. | ||
Pinto A, Attadia V, Fusco A, Ferrara F, Spada OA, Di Fiore PP. 5-Aza-2′-deoxycytidine induces terminal differentiation of leukemic blasts from patients with acute myeloid leukemias. Blood. 1984;64(4):922–929. | ||
Fulda S, Kufer MU, Meyer E, van Valen F, Dockhorn-Dworniczak B, Debatin KM. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene. 2001;20(41):5865–5877. | ||
Schnekenburger M, Grandjenette C, Ghelfi J, et al. Sustained exposure to the DNA demethylating agent, 2′-deoxy-5-azacytidine, leads to apoptotic cell death in chronic myeloid leukemia by promoting differentiation, senescence, and autophagy. Biochem Pharmacol. 2011;81(3):364–378. | ||
Patra A, Deb M, Dahiya R, Patra SK. 5-Aza-2′-deoxycytidine stress response and apoptosis in prostate cancer. Clin Epigenetics. 2011;2(2):339–348. | ||
Yao TT, Mo SM, Liu LY, Lu HW, Huang ML, Lin ZQ. 5-Aza-2′-deoxycytidine may influence the proliferation and apoptosis of cervical cancer cells via demethylation in a dose- and time-dependent manner. Genet Mol Res. 2013;12(1):312–318. | ||
Wongtrakoongate P, Li J, Andrews PW. Aza-deoxycytidine induces apoptosis or differentiation via DNMT3B and targets embryonal carcinoma cells but not their differentiated derivatives. Br J Cancer. 2014;110(8):2131–2138. | ||
Zheng Z, Li L, Liu X, et al. 5-Aza-2′-deoxycytidine reactivates gene expression via degradation of pRb pocket proteins. FASEB J. 2012;26(1):449–459. | ||
Zhu WG, Hileman T, Ke Y, et al. 5-aza-2′-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. J Biol Chem. 2004;279(15):15161–15166. | ||
Amatori S, Papalini F, Lazzarini R, et al. Decitabine, differently from DNMT1 silencing, exerts its antiproliferative activity through p21 upregulation in malignant pleural mesothelioma (MPM) cells. Lung Cancer. 2009;66(2):184–190. | ||
Farinha NJ, Shaker S, Lemaire M, Momparler L, Bernstein M, Momparler RL. Activation of expression of p15, p73 and E-cadherin in leukemic cells by different concentrations of 5-aza-2′-deoxycytidine (Decitabine). Anticancer Res. 2004;24(1):75–78. | ||
Guo Y, Engelhardt M, Wider D, Abdelkarim M, Lubbert M. Effects of 5-aza-2′-deoxycytidine on proliferation, differentiation and p15/INK4b regulation of human hematopoietic progenitor cells. Leukemia. 2006;20(1):115–121. | ||
Lu Y, Zhang X, Zhang J. Inhibition of breast tumor cell growth by ectopic expression of p16/INK4A via combined effects of cell cycle arrest, senescence and apoptotic induction, and angiogenesis inhibition. J Cancer. 2012;3:333–344. | ||
Liu SV, Fabbri M, Gitlitz BJ, Laird-Offringa IA. Epigenetic therapy in lung cancer. Front Oncol. 2013;3:135. | ||
Xiong H, Qiu H, Zhuang L, Xiong H, Jiang R, Chen Y. Effects of 5-Aza-CdR on the proliferation of human breast cancer cell line MCF-7 and on the expression of Apaf-1 gene. J Huazhong Univ Sci Technolog Med Sci. 2009;29(4):498–502. | ||
Wei M, Grushko TA, Dignam J, et al. BRCA1 promoter methylation in sporadic breast cancer is associated with reduced BRCA1 copy number and chromosome 17 aneusomy. Cancer Res. 2005;65(23):10692–10699. | ||
Lee MN, Tseng RC, Hsu HS, et al. Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non-small cell lung cancer. Clin Cancer Res. 2007;13(3):832–838. | ||
Kang JH, Kim SJ, Noh DY, et al. Methylation in the p53 promoter is a supplementary route to breast carcinogenesis: correlation between CpG methylation in the p53 promoter and the mutation of the p53 gene in the progression from ductal carcinoma in situ to invasive ductal carcinoma. Lab Invest. 2001;81(4):573–579. | ||
Pulukuri SM, Rao JS. Activation of p53/p21Waf1/Cip1 pathway by 5-aza-2′-deoxycytidine inhibits cell proliferation, induces pro-apoptotic genes and mitogen-activated protein kinases in human prostate cancer cells. Int J Oncol. 2005;26(4):863–871. | ||
Herman JG, Jen J, Merlo A, Baylin SB. Hypermethylation-associated inactivation indicates a tumor suppressor role for p15INK4B. Cancer Res. 1996;56(4):722–727. | ||
Askari M, Sobti RC, Nikbakht M, Sharma SC. Promoter hypermethylation of tumour suppressor genes (p14/ARF and p16/INK4a): case-control study in North Indian population. Mol Biol Rep. 2013;40(8):4921–4928. | ||
Lee JJ, Ko E, Cho J, et al. Methylation and immunoexpression of p16(INK4a) tumor suppressor gene in primary breast cancer tissue and their quantitative p16(INK4a) hypermethylation in plasma by real-time PCR. Korean J Pathol. 2012;46(6):554–561. | ||
Zhang HY, Liang F, Jia ZL, Song ST, Jiang ZF. PTEN mutation, methylation and expression in breast cancer patients. Oncol Lett. 2013;6(1):161–168. | ||
Song D, Ni J, Xie H, Ding M, Wang J. DNA demethylation in the PTEN gene promoter induced by 5-azacytidine activates PTEN expression in the MG-63 human osteosarcoma cell line. Exp Ther Med. 2014;7(5):1071–1076. | ||
Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371(6494):257–261. | ||
Yin D, Xie D, Hofmann WK, Miller CW, Black KL, Koeffler HP. Methylation, expression, and mutation analysis of the cell cycle control genes in human brain tumors. Oncogene. 2002;21(54):8372–8378. | ||
Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci U S A. 1992;89(16):7491–7495. | ||
Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95(11):866–871. | ||
Hurtubise A, Momparler RL. Effect of histone deacetylase inhibitor LAQ824 on antineoplastic action of 5-Aza-2′-deoxycytidine (decitabine) on human breast carcinoma cells. Cancer Chemother Pharmacol. 2006;58(5):618–625. | ||
Han X, Tan ZJ, Guo RD, Li ZJ, Wang YL. Effect of 5-aza-2′-deoxycytidine on growth and methylation of RUNX3 gene in human pancreatic cancer cell line MiaPaca2. Zhonghua Zhong Liu Za Zhi. 2013;35(1):17–21. Chinese. | ||
Han XA, Zeng HL, Han YP, Sun EW. Effects of decitabine on proliferation and apoptosis of NB4 and K562 cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2013;21(2):356–360. Chinese. | ||
Uchida N, Hsieh MM, Platner C, Saunthararajah Y, Tisdale JF. Decitabine suspends human CD34+ cell differentiation and proliferation during lentiviral transduction. PLoS One. 2014;9(8):e104022. | ||
Singh KP, Treas J, Tyagi T, Gao W. DNA demethylation by 5-aza-2-deoxycytidine treatment abrogates 17 beta-estradiol-induced cell growth and restores expression of DNA repair genes in human breast cancer cells. Cancer Lett. 2012;316(1):62–69. | ||
Dai Z-J, Kang HF, Lu WF, et al. RUNX3 gene promoter demethylation by 5-Aza-CdR induces apoptosis in breast cancer MCF-7 cell line. Onco Targets Ther. 2013;6:411–417. | ||
Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007;67(10):4827–4833. | ||
Matsui W, Wang Q, Barber JP, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008;68(1):190–197. | ||
Debeb B, Zhang X, Krishnamurthy S, et al. Characterizing cancer cells with cancer stem cell-like features in 293T human embryonic kidney cells. Mol Cancer. 2010;9(1):180. | ||
Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2− cancer cells are similarly tumorigenic. Cancer Res. 2005;65(14):6207–6219. | ||
Christgen M, Ballmaier M, Bruchhardt H, et al. Identification of a distinct side population of cancer cells in the Cal-51 human breast carcinoma cell line. Mol Cell Biochem. 2007;306(1–2):201–212. | ||
Deeley RG, Westlake C, Cole SP. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol Rev. 2006;86(3):849–899. | ||
Yu F, Yao H, Zhu P, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131(6):1109–1123. | ||
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. | ||
Durmus S, Sparidans RW, van Esch A, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the PARP inhibitor rucaparib (AG-014699). Pharm Res. 2014;32(1):37–46. | ||
Mirza S, Sharma G, Pandya P, Ralhan R. Demethylating agent 5-aza-2-deoxycytidine enhances susceptibility of breast cancer cells to anticancer agents. Mol Cell Biochem. 2010;342(1–2):101–109. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.