")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Lipopolysaccharide-induced caveolin-1 phosphorylation-dependent increase in transcellular permeability precedes the increase in paracellular permeability
Authors Wang N, Zhang D, Sun G, Zhang H, You Q, Shao M, Yue Y
Received 17 November 2014
Accepted for publication 7 January 2015
Published 28 August 2015 Volume 2015:9 Pages 4965—4977
DOI https://doi.org/10.2147/DDDT.S77646
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Wei Duan
Nan Wang,1,2 Dan Zhang,1,2 Gengyun Sun,1 Hong Zhang,1,2 Qinghai You,1 Min Shao,1 Yang Yue1
1Department of Respiration, 2Department of Emergency, The First Affiliated Hospital of Anhui Medical University, Hefei, People’s Republic of China
Background: Lipopolysaccharide (LPS) was shown to induce an increase in caveolin-1 (Cav-1) expression in endothelial cells; however, the mechanisms regarding this response and the consequences on caveolae-mediated transcellular transport have not been completely investigated. This study aims to investigate the role of LPS-induced Cav-1 phosphorylation in pulmonary microvascular permeability in pulmonary microvascular endothelial cells (PMVECs).
Methods: Rat PMVECs were isolated, cultured, and identified. Endocytosis experiments were employed to stain the nuclei by DAPI, and images were obtained with a fluorescence microscope. Permeability of endothelial cultures was measured to analyze the barrier function of endothelial monolayer. Western blot assay was used to examine the expression of Cav-1, pCav-1, triton-insoluble Cav-1, and triton-soluble Cav-1 protein.
Results: The LPS treatment induced phosphorylation of Cav-1, but did not alter the total Cav-1 level till 60 min in both rat and human PMVECs. LPS treatment also increased the triton-insoluble Cav-1 level, which peaked 15 min after LPS treatment in both rat and human PMVECs. LPS treatment increases the intercellular cell adhesion molecule-1 expression. Src inhibitors, including PP2, PP1, Saracatinib, and Quercetin, partially inhibited LPS-induced phosphorylation of Cav-1. In addition, both PP2 and caveolae disruptor MβCD inhibited LPS-induced increase of triton-insoluble Cav-1. LPS induces permeability by activating interleukin-8 and vascular endothelial growth factor and targeting other adhesion markers, such as ZO-1 and occludin. LPS treatment also significantly increased the endocytosis of albumin, which could be blocked by PP2 or MβCD. Furthermore, LPS treatment for 15 min significantly elevated Evans Blue-labeled BSA transport in advance of a decrease in transendothelial electrical resistance of PMVEC monolayer at this time point. After LPS treatment for 30 min, transendothelial electrical resistance decreased significantly. Moreover, PP2 and MβCD blocked LPS-induced increase in Evans Blue-labeled BSA level.
Conclusion: Our study demonstrates that LPS-induced Cav-1 phosphorylation may lead to the increase of transcellular permeability prior to the increase of paracellular permeability in a Src-dependent manner. Thus, LPS-induced Cav-1 phosphorylation may be a therapeutic target for the treatment of inflammatory lung disease associated with elevated microvascular permeability.
Keywords: caveolin-1, paracellular permeability, phosphorylation, pulmonary microvascular permeability, transcellular permeability
Introduction
Pulmonary microvascular endothelial cells (PMVECs), which form the intimal surface of the pulmonary microvascular as monolayer, provide a dynamic barrier that is critical for lung gas exchange and regulation of fluid and solute passage between the blood and interstitial compartments in the lung.1 An increase of pulmonary microvascular permeability due to the impairment of this barrier and the following pulmonary interstitial and alveolar edema are key hallmarks of inflammation and have been implicated in the pathogenesis of many diseases, such as acute respiratory distress syndrome.2 Since acute respiratory distress syndrome is a severe form of diffuse lung disease that imposes a substantial health burden throughout the world,3 the regulation of pulmonary microvascular permeability continuous to be a heavily studied research area worldwide.
Vascular permeability is regulated via paracellular and transcellular transport pathways. The paracellular transport is only applicable for small molecules, such as glucose, while the transfer of larger solutes, such as albumin, is mediated by transcellular transport via caveolae-mediated vesicular transport, which plays a crucial role in the maintenance of normal colloid osmotic pressure.4,5 Caveolae are 50-nm- to 100-nm-diameter plasma membrane invaginations with a characteristic “flask-shaped” morphology. Caveolin-1 (Cav-1), a structural protein of caveolae, regulates the vesicle carriers involved in the transcytosis of albumin across the endothelial barrier.6 It has been shown that overexpression of Cav-1 in endothelial cells is associated with increased transcytosis of albumin.7 Furthermore, an increase in Cav-1 phosphorylation is associated with both increased albumin transcytosis and decreased transendothelial electric resistance of pulmonary endothelial cells.4 Bacterial lipopolysaccharide (LPS), a glycoprotein in the outer membrane of gram-negative bacilli, is associated with increased lung microvascular endothelial permeability and pulmonary edema formation.8 Although LPS was shown to induce the increase of Cav-1 expression in endothelial cells9,10 and murine macrophages,11,12 the mechanism of the response and its consequences in regulating caveolae-mediated transcellular transport have not been completely investigated. Therefore, in the current study, we investigated the effect of LPS on the transcytosis of albumin across PMVECs and the underlying mechanisms.
Materials and methods
Isolation, culture, and identification of rat PMVECs
Adult Sprague-Dawley rats (250–300 g) were purchased from the Experimental Animal Center of Anhui Medical University. All animal experiments were performed after approval from the Animal Care and Use Committee of Anhui Medical University. Rat PMVECs were isolated from rat lungs according to previously reported method.13 Unless otherwise specified, all chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA). Rat PMVECs were incubated at 37°C in a humidified air containing 5% CO2 with Dulbecco’s Modified Eagle’s Medium (DMEM) medium supplemented with 10% fetal bovine serum. For experiments, the passage 4–6 cells were used at 80%–90% confluence. After cells reached the desired confluency, they were harvested by treating with a 0.25% solution of trypsin–EDTA (37°C, 2 min).
Human lung epithelial cells were cultured with complete DMEM medium at 37°C in a humidified air containing 5% CO2. CD34 antigen in the cultured cells was detected by immunocytochemical staining. Cells were fixed with paraformaldehyde for 10 min at room temperature, washed in PBS, incubated with 3% H2O2 to neutralize endogenous peroxidase for 10 min, washed in PBS, and blocked with 2% BSA in PBS for 15 min. After three washes with PBS, cells were incubated with 1:200 polyclonal antibody of CD34 at 37°C for 1 h and then incubated with the secondary antibody at 37°C for 15 min. After washing with PBS three times, the staining was visualized by adding diaminobenzidine (ZSGB-BIO, Beijing, People’s Republic of China) according to the manufacturer’s instructions. Hematoxylin staining was performed to show the nucleoli.
In addition, fluorescein isothiocyanate-conjugated bandeiraea simplicifolia I isolectin B4 binding to the cultured cells was observed with fluorescence microscope.
Endocytosis of Alexa 488-albumin
Endocytosis experiments were performed as described previously.14 Rat PMVECs were seeded in six-well plates. When the cells grew to 80%–90% confluence, the culture medium was removed and the cells were cultured in DMEM medium without fetal bovine serum for 5 h. After removal of the medium, each well was washed and incubated with PBS buffer containing Alexa 488 albumin (50 μg/mL), and the cells were incubated at 37°C for 30 min. The wells were then washed with PBS three times, and the cells were fixed with 4% paraformaldehyde for 20 min at room temperature and washed in PBS for another three times. The nuclei were stained by DAPI, and images were obtained with a fluorescence microscope.
Translocation assay
Twenty microliters of PMVEC suspension was dissolved in buffer solution containing 0.5 μL of full-length Tat-AF633. The solution was again added to an avidin-coated silicon substrate to bind the Cav-1 (in PMVECs) onto the substrate. The fluorescence images of the Cav-1 were obtained in a time-resolved manner. The same experiments were performed in the presence of the water-soluble dyes Alexa Fluor 488. The images were captured with a confocal laser scanning microscope (Olympus Corporation, Tokyo, Japan).
Assay for barrier function of endothelial monolayer
Permeability of endothelial cultures was measured as described previously.15 In brief, 100-μL cells were seeded (2×105 cells/mL) onto transwell polycarbonate inserts. The filters were pretreated for 24 h with DMEM medium before seeding endothelial cells. The 600-μL medium was added into the wells and changed every day. Experiments were performed with cells at 80%–90% confluency. A tracer solution containing Evans Blue-labeled BSA (EBA) (0.67 mg/mL) was prepared in complete medium and was used to assess monolayer permeability. In the upper compartment of the Transwell insert, plasma mix was replaced with 100 μL of tracer solution, and culture medium in the lower compartment was renewed with 600 μL of fresh medium containing 4% BSA. After incubation for 30 min at 37°C, 200-μL samples were withdrawn from the lower compartment, and the absorption was measured on a Microplate Reader at 620 nm.
Western blot
Membrane and cytosolic proteins were isolated with a Membrane and Cytosol Extraction Kit (Beyotime, Haimen, Jiangsu, People’s Republic of China). Triton-soluble and -insoluble proteins were isolated with a lysis buffer containing Triton-100 (Beyotime), and protein concentrations were determined using a BCA protein assay kit (Biosynthesis Biotechnology Co., Ltd, Beijing, People’s Republic of China). Equal amounts of protein were separated by 10% SDS gel electrophoresis under denaturing and nonreducing conditions and then transferred to a nitrocellulose membrane. The membrane was blocked with 5% nonfat milk in Tris-buffered saline and Tween 20 at room temperature for 1 h and then incubated with primary antibody at 4°C overnight. After washing in Tris-buffered saline and Tween 20, the blots were incubated with horseradish-coupled secondary antibody. The signal was detected using enhanced chemiluminescence and recorded on X-ray films. The relative density of the target bands was quantified using Image J acquisition and analysis software.
Statistical analysis
Data were analyzed using the SPSS 17.0 software (SPSS, Chicago, IL, USA). The results are presented as means ± standard deviations. Data were compared by one-way analysis of variance followed by LSD post hoc test. A P-value of less than 0.05 was considered significant.
Results
Identification of rat PMVECs
The cultured rat PMVECs were polygon or short and fusiform in shape and showed a typical cobblestone-like appearance under inverted optical microscope (Figure 1A). Immunocytochemical staining (brown staining color) showed that CD34 antigen was expressed in the cytoplasm of cultured rat PMVECs (Figure 1B and C), whereas the pulmonary epithelial cells were negative (Figure 1D). PMVECs were also positively stained by fluorescein isothiocyanate-conjugated bandeiraea simplicifolia I isolectin (Figure 1E), suggesting the isolated cells have the typical characteristics of PMVEC. The identification of human PMVECs was also performed equal to the rat PMVECs (data not shown).
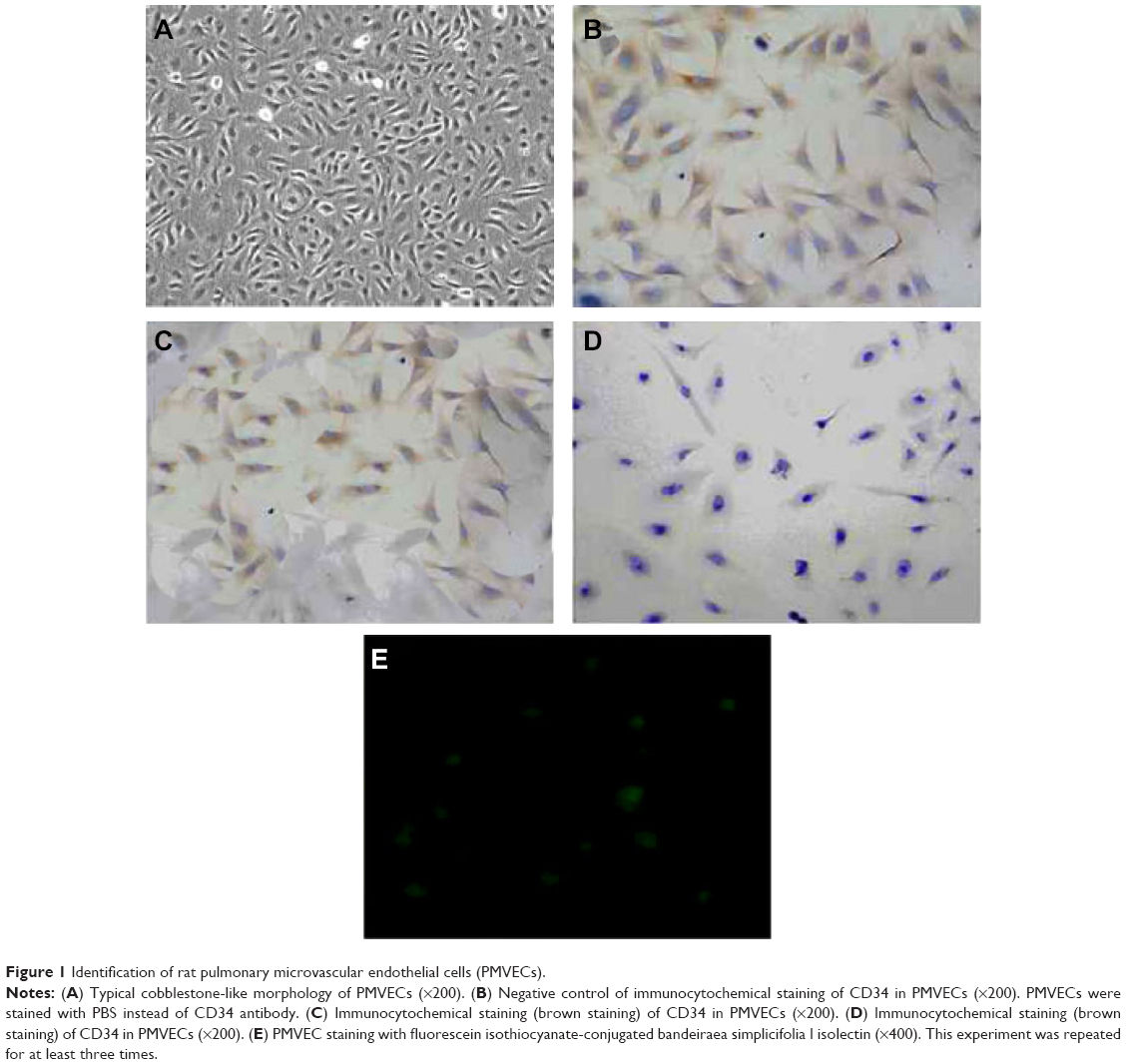 | Figure 1 Identification of rat pulmonary microvascular endothelial cells (PMVECs). |
LPS treatment induced Cav-1 activation and translocation
To investigate the effect of LPS on Cav-1 expression and activation, we performed Western blot analysis. The results showed that the phosphorylation of Cav-1 on Tyr-14 was significantly increased upon treatment with LPS for up to 30 min. Although the phosphorylation of Cav-1 decreased at the 60-min time point compared with that at 30 min, it was still higher than control (Figure 2A and B). Total Cav-1 levels were not altered after 60-min LPS treatment. Cav-1 is considered as a marker protein for small invaginations of the plasma membrane referred to as caveolae. Hallmarks of these membrane microdomains are their distinct protein and lipid composition as well as detergent insolubility.16 To investigate the distribution of Cav-1 in cells after stimulation with LPS, the triton-soluble and triton-insoluble proteins were extracted after LPS stimulation for indicated times. Western blot showed that the triton-insoluble Cav-1 level increased after LPS treatment and reached a peak at 15 min (Figure 2C and D). Confocal laser scanning microscope showed that LPS treatment for 15 min induced the translocation of Cav-1 from cell membrane to the cytoplasm (Figure 2E and F).
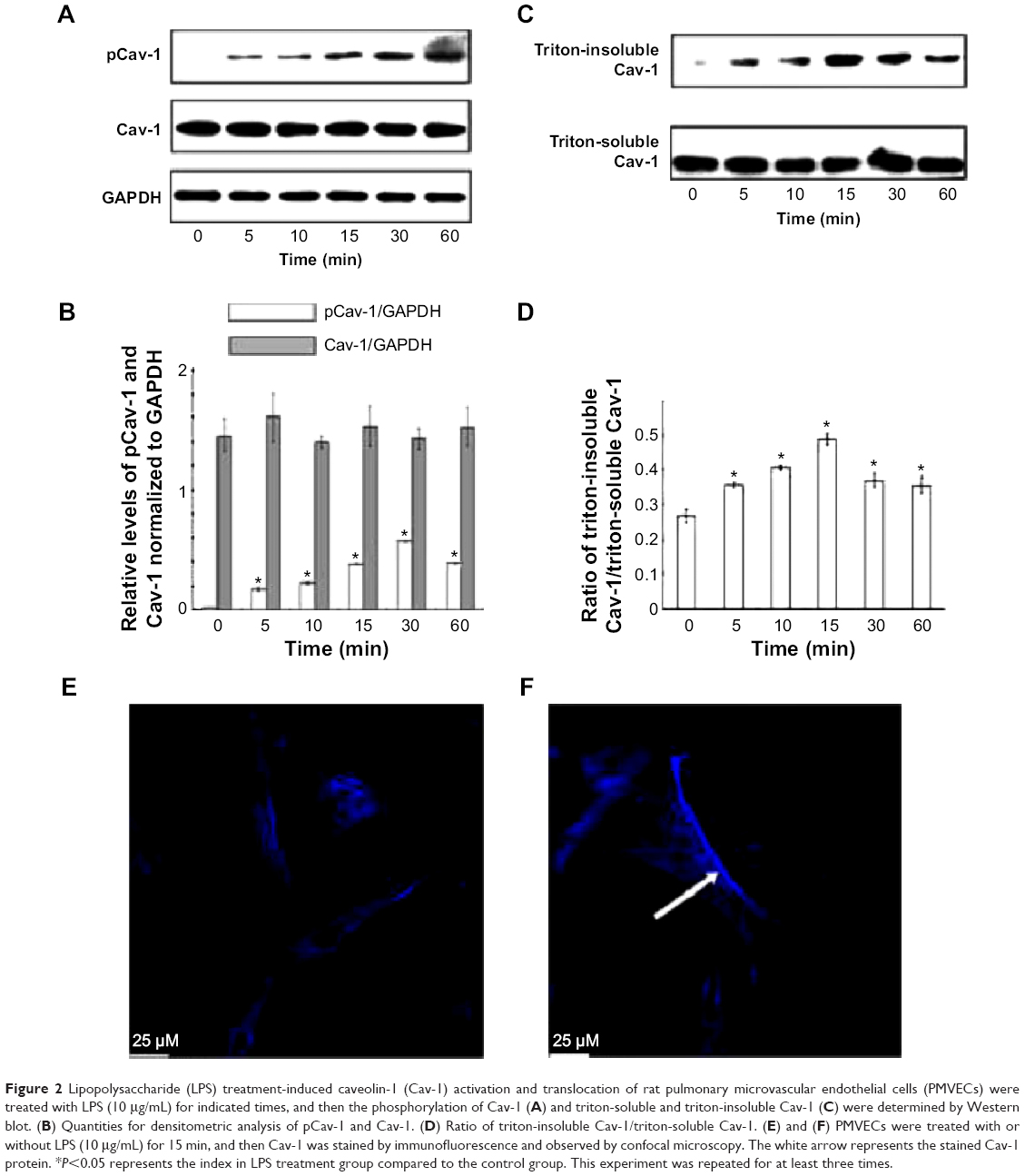 | Figure 2 Lipopolysaccharide (LPS) treatment-induced caveolin-1 (Cav-1) activation and translocation of rat pulmonary microvascular endothelial cells (PMVECs) were treated with LPS (10 μg/mL) for indicated times, and then the phosphorylation of Cav-1 (A) and triton-soluble and triton-insoluble Cav-1 (C) were determined by Western blot. (B) Quantities for densitometric analysis of pCav-1 and Cav-1. (D) Ratio of triton-insoluble Cav-1/triton-soluble Cav-1. (E) and (F) PMVECs were treated with or without LPS (10 μg/mL) for 15 min, and then Cav-1 was stained by immunofluorescence and observed by confocal microscopy. The white arrow represents the stained Cav-1 protein. *P<0.05 represents the index in LPS treatment group compared to the control group. This experiment was repeated for at least three times. |
Furthermore, the LPS-induced Cav-1 phosphorylation was also examined in human PMVECs. The phosphorylation of Cav-1 decreased at the 60-min time point compared with that at 30 min; however, it was still higher compared to the control (Figure 3A and B). The Western blot results also showed that the triton-insoluble Cav-1 level increased after LPS treatment and reached a peak at 15 min (Figure 3C and D). Also, the LPS treatment for 15 min induced the translocation of Cav-1 from cell membrane to the cytoplasm (data not shown). Therefore, the LPS treatment induced Cav-1 phosphorylation and translocation both in rat and human PMVECs.
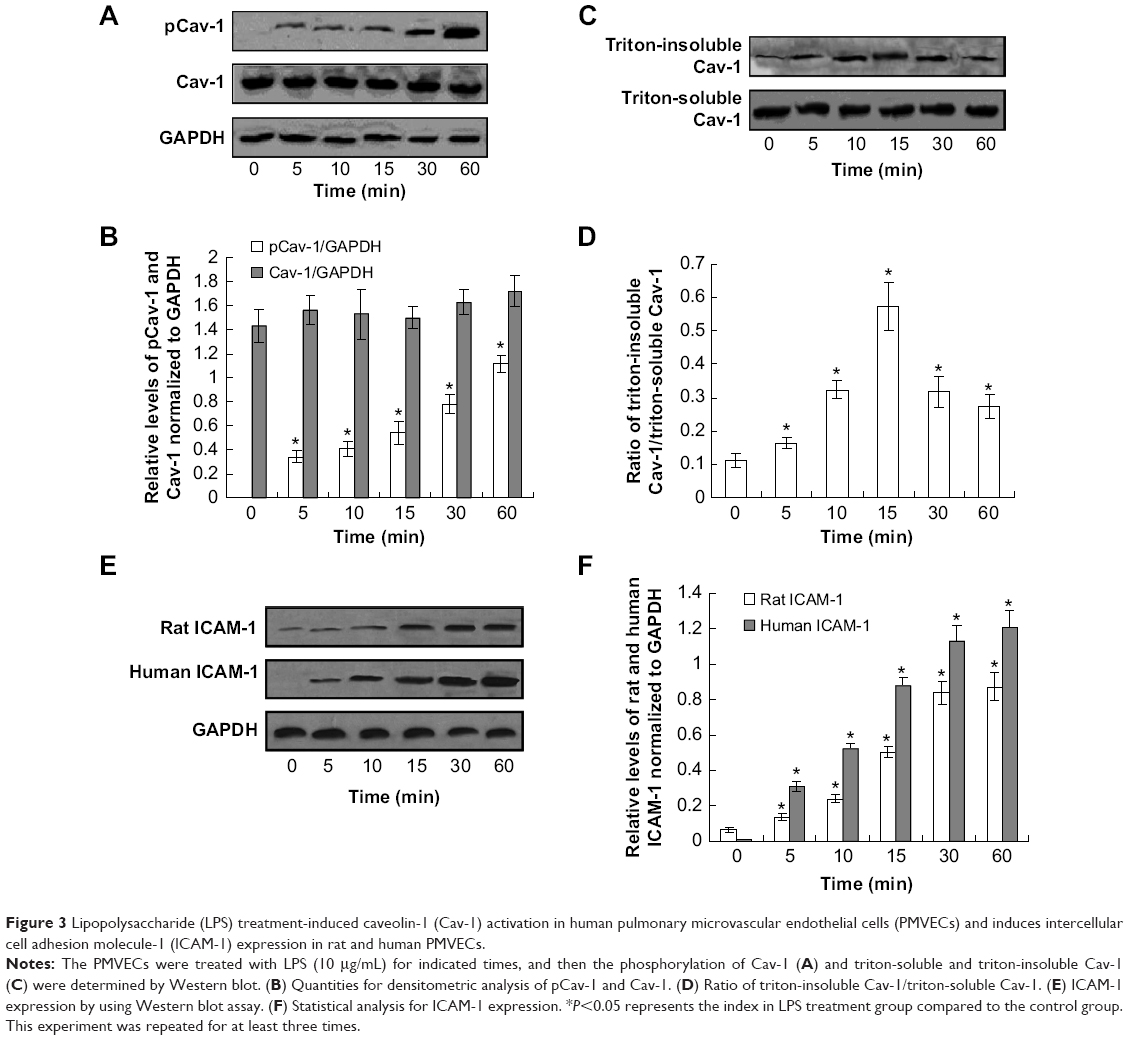 | Figure 3 Lipopolysaccharide (LPS) treatment-induced caveolin-1 (Cav-1) activation in human pulmonary microvascular endothelial cells (PMVECs) and induces intercellular cell adhesion molecule-1 (ICAM-1) expression in rat and human PMVECs. |
As already known, clathrin-mediated and caveolae-mediated endocytosis of vascular endothelial cadherin contribute to LPS-induced vascular hyperpermeability. Clathrin phosphorylation was also examined. The results indicated that there were no changes for the levels of p-clathrin and clathrin protein in LPS treatment compared to control (Figure S1).
LPS treatment increases the intercellular cell adhesion molecule-1 expression
In order to explore the mechanism of the translocation after LPS treatment, the intercellular cell adhesion molecule-1 protein was detected in both rat and human PMVECs. The results indicated that the intercellular cell adhesion molecule-2 protein was increased significantly after LPS treatment compared to the control both in rat and human PMVECs (Figure 3E and F) (P<0.05) and also increased following an increase in the concentration of LPS.
The effect of LPS on Cav-1 was Src-dependent
Src family protein tyrosine kinases have been implicated in the upstream signaling pathways of vascular hyperpermeability and activated Src is known to phosphorylate Cav-1 to initiate plasmalemmal vesicle fission and transendothelial vesicular transport of albumin.17 Therefore, we next examined whether the effect of LPS on Cav-1 activation and translocation was Src-dependent. Pretreatment with a Src inhibitor PP2 partly inhibited LPS-induced phosphorylation of Cav-1 (Figure 4A). In addition, pretreatment with PP2 or MβCD also inhibited LPS-induced increase in triton-insoluble Cav-1 level (Figure 4B).
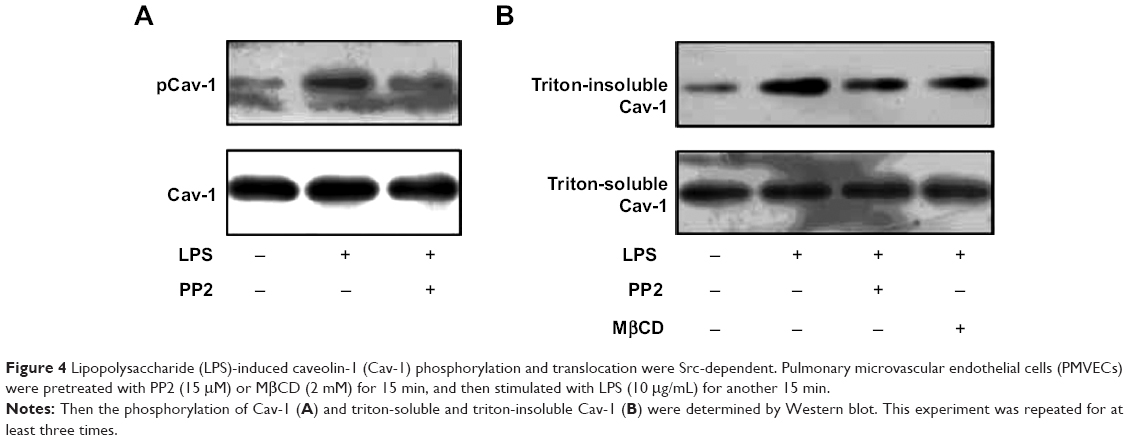 | Figure 4 Lipopolysaccharide (LPS)-induced caveolin-1 (Cav-1) phosphorylation and translocation were Src-dependent. Pulmonary microvascular endothelial cells (PMVECs) were pretreated with PP2 (15 μM) or MβCD (2 mM) for 15 min, and then stimulated with LPS (10 μg/mL) for another 15 min. |
Furthermore, in order to confirm the effect of LPS on Cav-1 was Src-dependent, the other Src inhibitors, such as PP1, Saracatinib, and Quercetin, were detected by Western blot assay. The results showed that pretreatment with the Src inhibitor PP1, Saracatinib and Quercetin, could also inhibit the LPS-induced phosphorylation of Cav-1 (Figure S2).
LPS induces permeability by activating interleukin-8 and vascular endothelial growth factor and targeting other adhesion markers
In order to investigate the mechanism of the LPS-induced permeability, the endothelial injury or permeability-related cytokines, interleukin-8 and vascular endothelial growth factor (VEGF), were examined. The Western blot assay results indicated that interleukin-8 and VEGF were increased significantly when treated with LPS compared to the control (Figure 5A, P<0.05). Meanwhile, the adhesion markers, ZO-1 and Occludin, were also detected. The results showed the levels of ZO-1 and occludin were also increased after LPS treatment compared to control (Figure 5B, P<0.05). However, the increase of the ZO-1 and occludin was initiated at 15 min after LPS treatment.
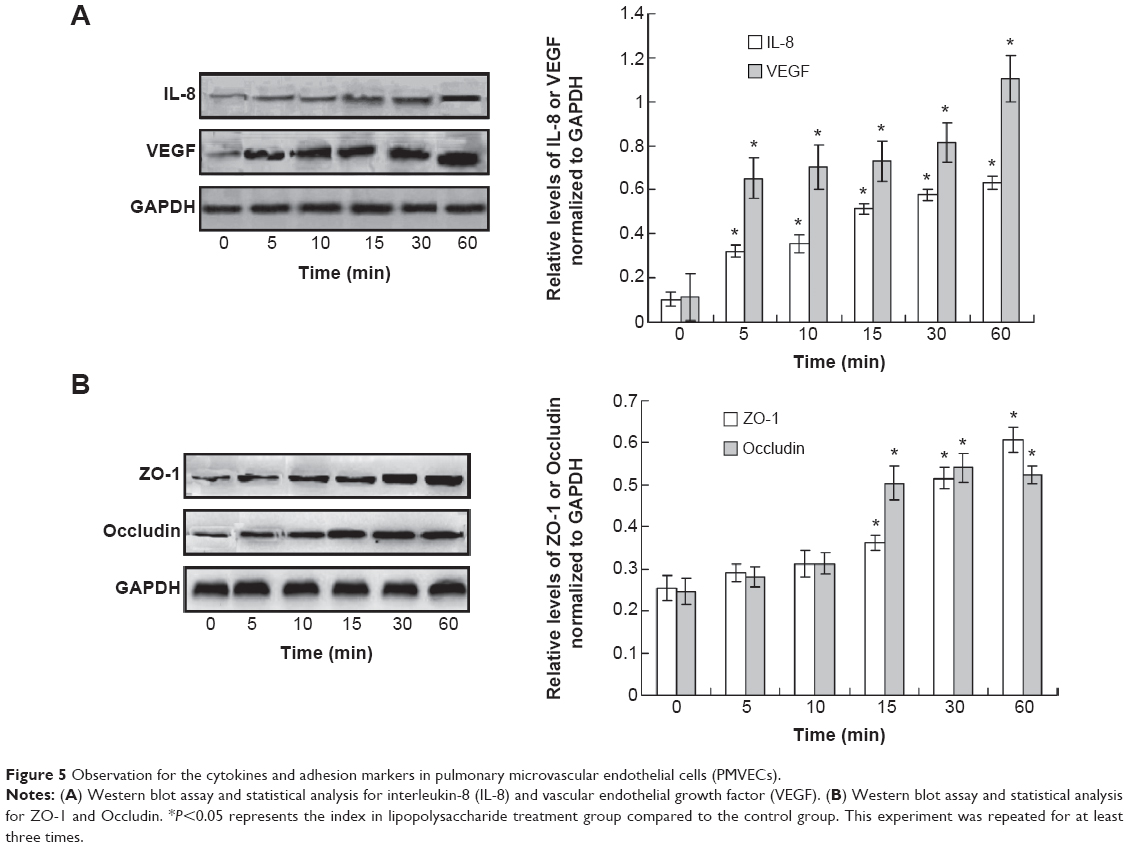 | Figure 5 Observation for the cytokines and adhesion markers in pulmonary microvascular endothelial cells (PMVECs). |
LPS increased the transcellular permeability through the phosphorylation of Cav-1
Endocytosis is the initial step of the transcellular transport of macromolecules including albumin. The control group (Figure 6A) illustrated the normal endocytosis of Alexa 488-labeled albumin. LPS treatment (Figure 6B) significantly increased the endocytosis of Alexa 488-labeled albumin (with obvious light fluorescence, which was marked with white arrows). Pretreatment with MβCD (Figure 6C) or PP2 (Figure 6D) suppressed LPS-induced endocytosis of albumin. We next tested the effect of LPS on the microvascular permeability. The results showed that LPS treatment for 15 min significantly elevated the EBA level in the bottom of the Transwell chamber (Figure 7A). However, the transendothelial electrical resistance of the monolayer PMVECs was not altered at this time point (Figure 7B). After LPS treatment for 30 min, the transendothelial electrical resistance of PMVECs was decreased significantly, indicating that the increase of transcellular permeability occurred prior to the increase in paracellular permeability. Pretreatment of PMVECs with PP2 or MβCD for 15 min significantly blocked LPS-induced increase in EBA transport (Figure 7C).
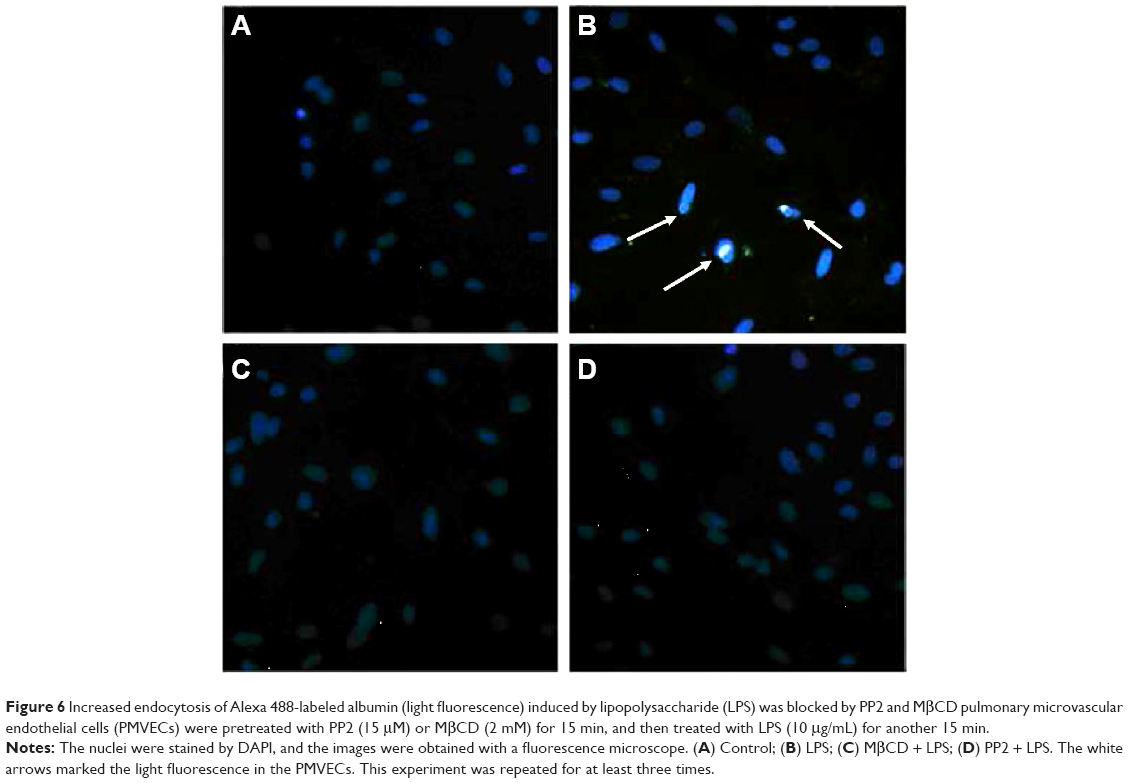 | Figure 6 Increased endocytosis of Alexa 488-labeled albumin (light fluorescence) induced by lipopolysaccharide (LPS) was blocked by PP2 and MβCD pulmonary microvascular endothelial cells (PMVECs) were pretreated with PP2 (15 μM) or MβCD (2 mM) for 15 min, and then treated with LPS (10 μg/mL) for another 15 min. |
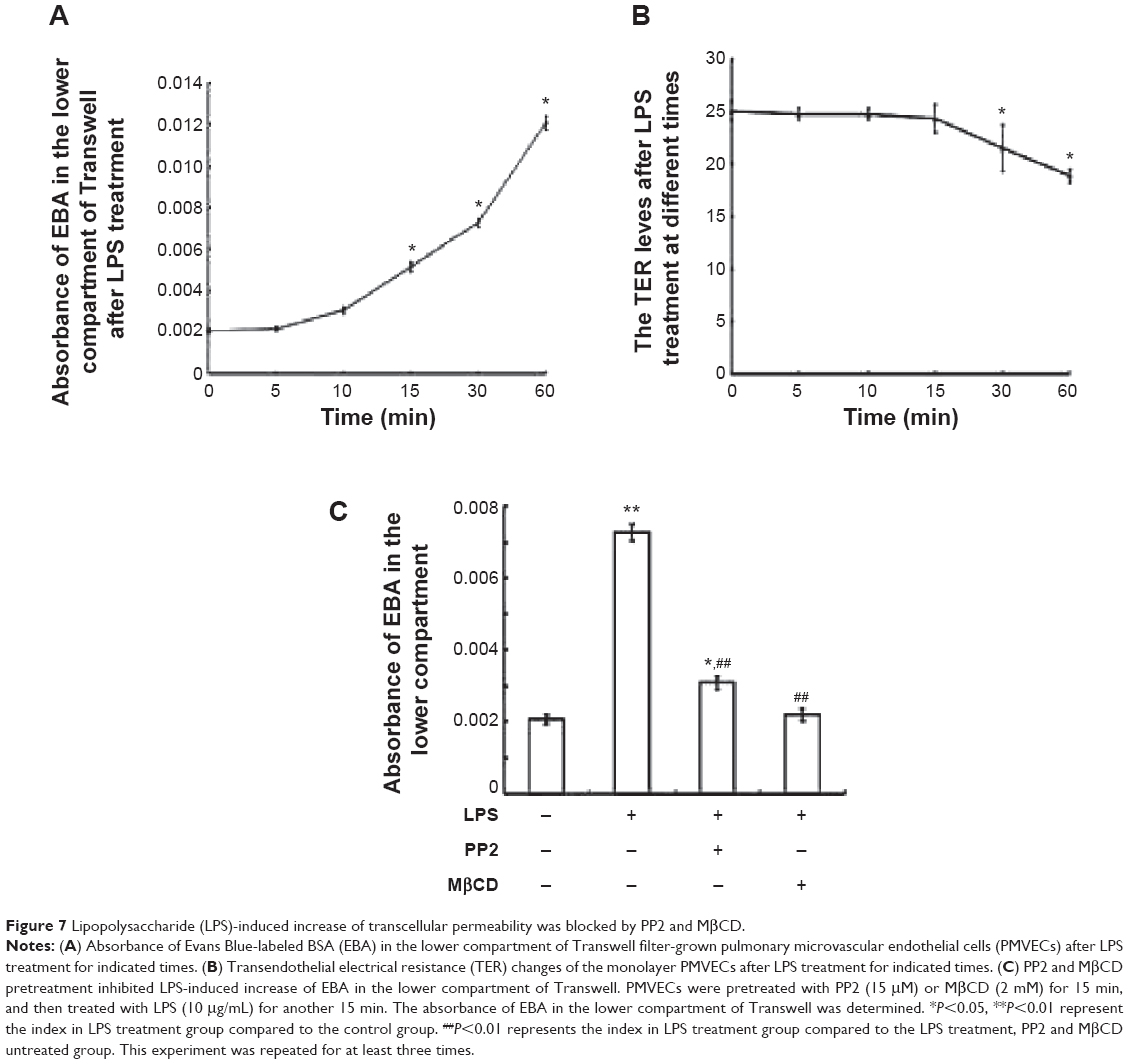 | Figure 7 Lipopolysaccharide (LPS)-induced increase of transcellular permeability was blocked by PP2 and MβCD. |
Discussion
Cav-1, a major structural and signaling protein of caveolae, has been reported to be upregulated in human PMVECs treated with LPS.10 In the present study, we demonstrated that LPS treatment also induced the phosphorylation and translocation of Cav-1 from cell membrane to the cytoplasm in rat PMVECs. This effect of LPS was blocked by the Src inhibitor, PP2, indicating that LPS-induced Cav-1 phosphorylation is Src-dependent. Furthermore, LPS treatment also increased the endocytosis of albumin and transcellular transport of EBA across PMVEC monolayers. Pretreatment with PP2 or MβCD, which is known to disrupt caveolae, inhibited LPS-induced increase of albumin transport. Moreover, the increase in EBA transport through the PMVEC monolayers occurred prior to the decrease in monolayer resistance following LPS treatment, suggesting that the increase of transcellular permeability occurs first and may be required for the increase in paracellular permeability.
Overexpression of Cav-1 in endothelial cells was able to induce an increase in the transcytosis of albumin.7 LPS treatment for 4 h increases the expression of Cav-1 mRNA and protein in human lung microvascular endothelial cells, resulting in increased caveolae number, thereby contributing to increased transendothelial albumin permeability.10 Overexpression of Cav-1 also enhances the LPS-induced inflammatory response in mouse lung alveolar type-1 cells.18 Cav-1-knockout markedly attenuates LPS-induced microvascular barrier breakdown and edema formation.19 Phosphorylation of Cav-1 on Tyr-14 also plays an important role in regulating the pulmonary vascular permeability via paracellular and transcellular pathways.4 Phosphorylation of Cav-1 may lead to the dissociation of vascular endothelial cadherin/beta-catenin complexes and resultant endothelial barrier disruption.4 In the isolated lung, enhanced phosphorylation of Cav-1 also increases transendothelial albumin permeability through the enhancement of caveolae-mediated albumin uptake and transport induced by isoflurane.20 In the present study, we showed that LPS treatment rapidly induced the phosphorylation of Cav-1 on Tyr-14 in rat PMVECs before total Cav-1 levels were changed. LPS treatment also induced the enrichment of Cav-1 in triton-insoluble region, suggesting redistribution from the cell membrane to the cytoplasm. However, the total Cav-1 protein levels were not changed by LPS up to 60 min. These observations suggest that LPS is able to induce the phosphorylation of Cav-1 before increasing Cav-1 expression. Furthermore, LPS treatment also increased the endocytosis of albumin and transport of EBA across PMVEC monolayers, suggesting that LPS treatment increases transcellular albumin permeability.
Toll-like receptor 4 signaling is coupled to Src family tyrosine kinase activation.21 After being recognized by Toll-like receptor 4, LPS is able to activate the Src family kinases including c-SRC, FYN, and YES, and then phosphorylate zonula adherens proteins to open the endothelial paracellular pathway.21 Therefore, we used a Src inhibitor to determine whether LPS-induced phosphorylation of Cav-1 was Src dependent. Our results showed that phosphorylation of Cav-1 was blocked by PP2, indicating LPS-induced Cav-1 phosphorylation is Src dependent, consistent with previous reports. Transcytosis of albumin was shown to play an important role in the increase of microvascular permeability induced by ischemic shock.22 In vitro experiments showed that VEGF induces an increase of caveolae density in endothelial cells, increasing the endocytosis and transcytosis of albumin and resulting in an increase in endothelial permeability,23 indicating that caveolae-mediated transcytosis of albumin plays an important role in the increase in microvascular endothelial permeability and tissue injury in pathological conditions. In this study, when the phosphorylation of Cav-1 was blocked by PP2 or the caveolae were disrupted by MβCD, LPS-induced increase of transcytosis of albumin was significantly suppressed, implicating that the increase of PMVEC permeability induced by LPS depends on the phosphorylation of Cav-1 and the integrity of caveolae. However, PP2 only partially inhibited LPS-induced Cav-1 phosphorylation and increase of albumin transcytosis, indicating there may be Src-independent pathways of LPS-induced Cav-1 phosphorylation.24,25 Previously, many studies have investigated the mechanisms of LPS-induced PMVEC hyperpermeability via the paracellular pathway.21,26,27 LPS treatment causes stress fiber formation, centripetal retraction of the cells, and substantial gap formation in the monolayer resulting in disruption of monolayer integrity.27 However, although extensive interstitial and alveolar edema have been shown in many patients with acute respiratory distress syndrome induced by sepsis, the lung microvascular endothelial cell–cell junctions are not destructed.28,29 In the present study, our results showed that LPS treatment induced a fast increase in the endocytosis and transcytosis of albumin (within 15 min), which occurred prior to the decrease of monolayer electrical resistance, consistent with previous results that the increase in transcellular permeability preceded the increase in paracellular permeability.17 These results suggest that caveolae-mediated transcytosis of albumin contributes to the increase of microvascular permeability in the early stage of acute lung injury.
In conclusion, our study provides a new prospective in the understanding of LPS-induced pulmonary microvascular hyperpermeability, especially in the early stage of acute lung injury. To date, since the nature of vascular leakage is only partially understood, pharmacological approaches for its management are limited. LPS-induced Cav-1 phosphorylation may be a therapeutic target for the treatment of diseases associated with pulmonary microvascular hyperpermeability.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No 81370170 and 81070054), the College Scientific Research Project of Anhui Province (KJ2011Z185) and E (Rui Yi) emergency medical research special fund (R2014003).
Disclosure
The authors report no conflicts of interest in this work.
References
Kelly JJ, Moore TM, Babal P, Diwan AH, Stevens T, Thompson WJ. Pulmonary microvascular and macrovascular endothelial cells: differential regulation of Ca2+ and permeability. Am J Physiol. 1998;274(5 Pt 1):L810–L819. | ||
van Zyl JM, Smith J. Surgactant treatment before first breath for respiratory distress syndrome in preterm lambs: comparison of peptide-containing lung surfactant with porcine-derived surfactant. Drug Des Devel Ther. 2013;7:905–916. | ||
Tsushima K, King LS, Aggarwal NR, De Gorordo A, D’Alessio FR, Kubo K. Acute lung injury review. Intern Med. 2009;48(9):621–630. | ||
Sun Y, Hu G, Zhang X, Minshall RD. Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ Res. 2009;105(7):676–685. | ||
Miyoshi S, Ito R, Katayama H, et al. Combination therapy with sivelestat and recombinant human soluble thrombomodulin for ARDS and DIC patients. Drug Des Devel Ther. 2014;8:1211–1219. | ||
Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. | ||
Pascariu M, Bendayan M, Ghitescu L. Correlated endothelial caveolin overexpression and increased transcytosis in experimental diabetes. J Histochem Cytochem. 2004;52(1):65–76. | ||
Lantz RC, Keller GE, Burrell R. The role of platelet-activating factor in the pulmonary response to inhaled bacterial endotoxin. Am Rev Respir Dis. 1991;144(1):167–172. | ||
Zhao B, Bowden RA, Stavchansky SA, Bowman PD. Human endothelial cell response to gram-negative lipopolysaccharide assessed with cDNA microarrays. Am J Physiol Cell Physiol. 2001;281(5):C1587–C1595. | ||
Tiruppathi C, Shimizu J, Miyawaki-Shimizu K, et al. Role of NF-kappaB-dependent caveolin-1 expression in the mechanism of increased endothelial permeability induced by lipopolysaccharide. J Biol Chem. 2008;283(7):4210–4218. | ||
Lei MG, Morrison DC. Differential expression of caveolin-1 in lipopolysaccharide-activated murine macrophages. Infect Immun. 2000;68:5084–5089. | ||
Choi YH, Kim GY, Lee HH. Anti-inflammatory effects of cordycepin in lipopolysaccharide-stimulated RAW 264.7 macrophages through Toll-like 4-mediated suppression of mitogen-activated protein kinases and NF-kB signaling pathways. Drug Des Devel Ther. 2014;8:1941–1953. | ||
Zhang H, Sun GY. LPS induces permeability injury in lung microvascular endothelium via AT(1) receptor. Arch Biochem Biophys. 2005;441(1):75–83. | ||
Yumoto Y, Nishikawa H, Okamoto M, Katayama H, Nagai J, Takano M. Clathrin-mediated endocytosis of FITC-albumin in alveolar type II epithelial cell line RLE-6TN. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L946–L955. | ||
Sun SX, Ge BX, Miao CH. Effects of preconditioning with sevoflurane on TNF-alpha-induced permeability and activation of p38 MAPK in rat pulmonary microvascular endothelial cells. Cell Biochem Biophys. 2011;61(1):123–129. | ||
Anderson RG. The caveolae membrane system. Annu Rev Biochem. 1998;67(1):199–225. | ||
Hu C, Place AT, Minshall RD. Regulation of endothelial permeability by Src kinase signaling: vascular leakage versus transcellular transport of drugs and macromolecules. Chem Biol Interact. 2008;171(2):177–189. | ||
Lv XJ, Li YY, Zhang YJ, Mao M, Qian GS. Over-expression of caveolin-1 aggravate LPS-induced inflammatory response in AT-1 cells via up-regulation of cPLA2/p38 MAPK. Inflamm Res. 2010;59(7):531–541. | ||
Garrean S, Gao XP, Brovkovych V, et al. Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J Immunol. 2006;177(7):4853–4860. | ||
Hu G, Schwartz DE, Shajahan AN, et al. Isoflurane, but not sevoflurane, increases transendothelial albumin permeability in the isolated rat lung: role for enhanced phosphorylation of caveolin-1. Anesthesiology. 2006;104(4):777–785. | ||
Gong P, Angelini DJ, Yang S, et al. TLR4 signaling is coupled to SRC family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. J Biol Chem. 2008;283(19):13437–13449. | ||
Childs EW, Udobi KF, Hunter FA, Dhevan V. Evidence of transcellular albumin transport after hemorrhagic shock. Shock. 2005;23(6):565–570. | ||
Chen J, Braet F, Brodsky S, et al. VEGF-induced mobilization of caveolae and increase in permeability of endothelial cells. Am J Physiol Cell Physiol. 2002;282(5):C1053–C1063. | ||
Galvagni F, Anselmi F, Salameh A, Orlandini M, Rocchigiani M, Oliviero S. Vascular endothelial growth factor receptor-3 activity is modulated by its association with caveolin-1 on endothelial membrane. Biochemistry. 2007;46(13):3998–4005. | ||
Singleton PA, Chatchavalvanich S, Fu P, et al. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res. 2009;104(8):978–986. | ||
Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 2008;1123(1):134–145. | ||
Bogatcheva NV, Zemskova MV, Kovalenkov Y, Poirier C, Verin AD. Molecular mechanisms mediating protective effect of cAMP on lipopolysaccharide (LPS)-induced human lung microvascular endothelial cells (HLMVEC) hyperpermeability. J Cell Physiol. 2009;221(3):750–759. | ||
Hu G, Vogel SM, Schwartz DE, Malik AB, Minshall RD. Intercellular adhesion molecule-1-dependent neutrophil adhesion to endothelial cells induces caveolae-mediated pulmonary vascular hyperpermeability. Circ Res. 2008;102(12):e120–e131. | ||
Hohenforst-Schmidt W, Petermann A, Visouli A, et al. Successful application extracorporeal membrane oxygenation due to pulmonary hemorrhage secondary to granulomatosis with polyangiitis. Drug Des Devel Ther. 2013;7:627–633. |
Supplementary materials
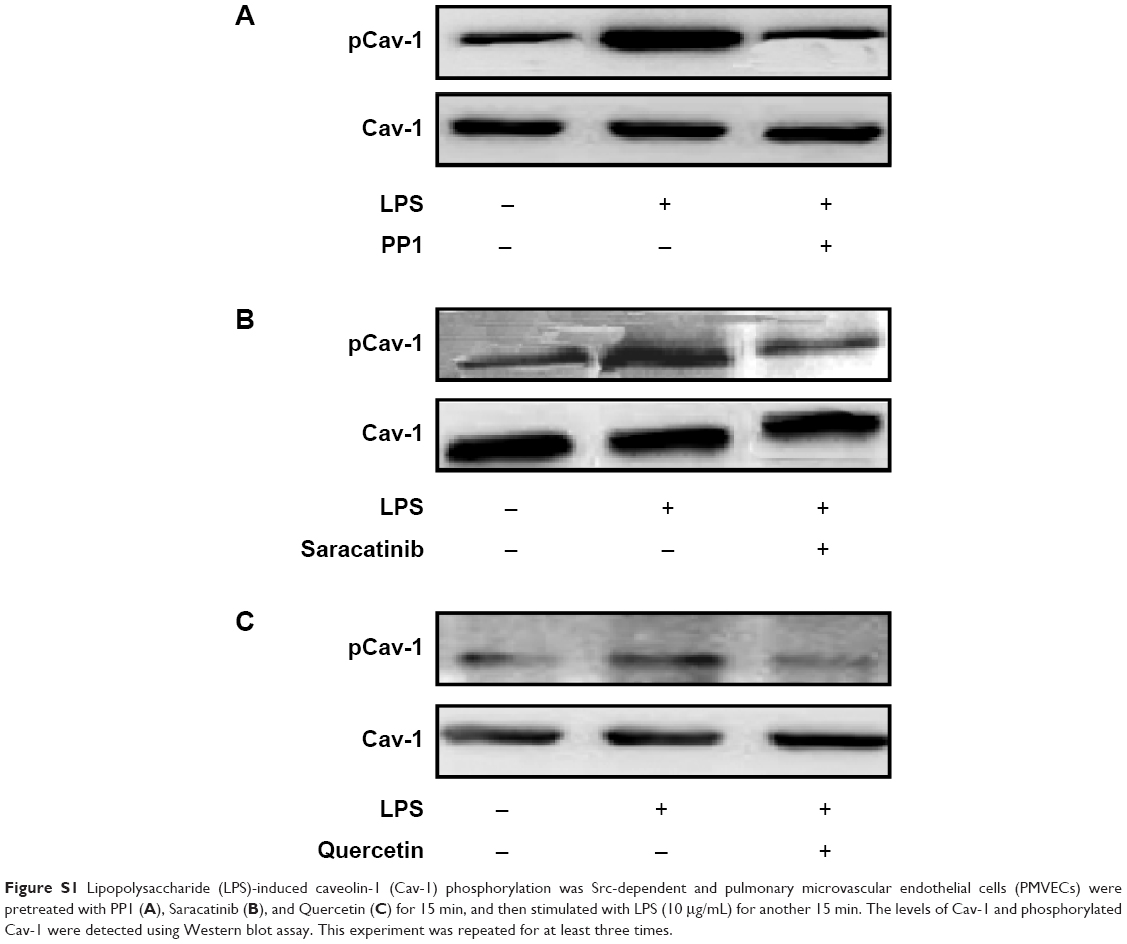 | Figure S1 Lipopolysaccharide (LPS)-induced caveolin-1 (Cav-1) phosphorylation was Src-dependent and pulmonary microvascular endothelial cells (PMVECs) were pretreated with PP1 (A), Saracatinib (B), and Quercetin (C) for 15 min, and then stimulated with LPS (10 μg/mL) for another 15 min. The levels of Cav-1 and phosphorylated Cav-1 were detected using Western blot assay. This experiment was repeated for at least three times. |
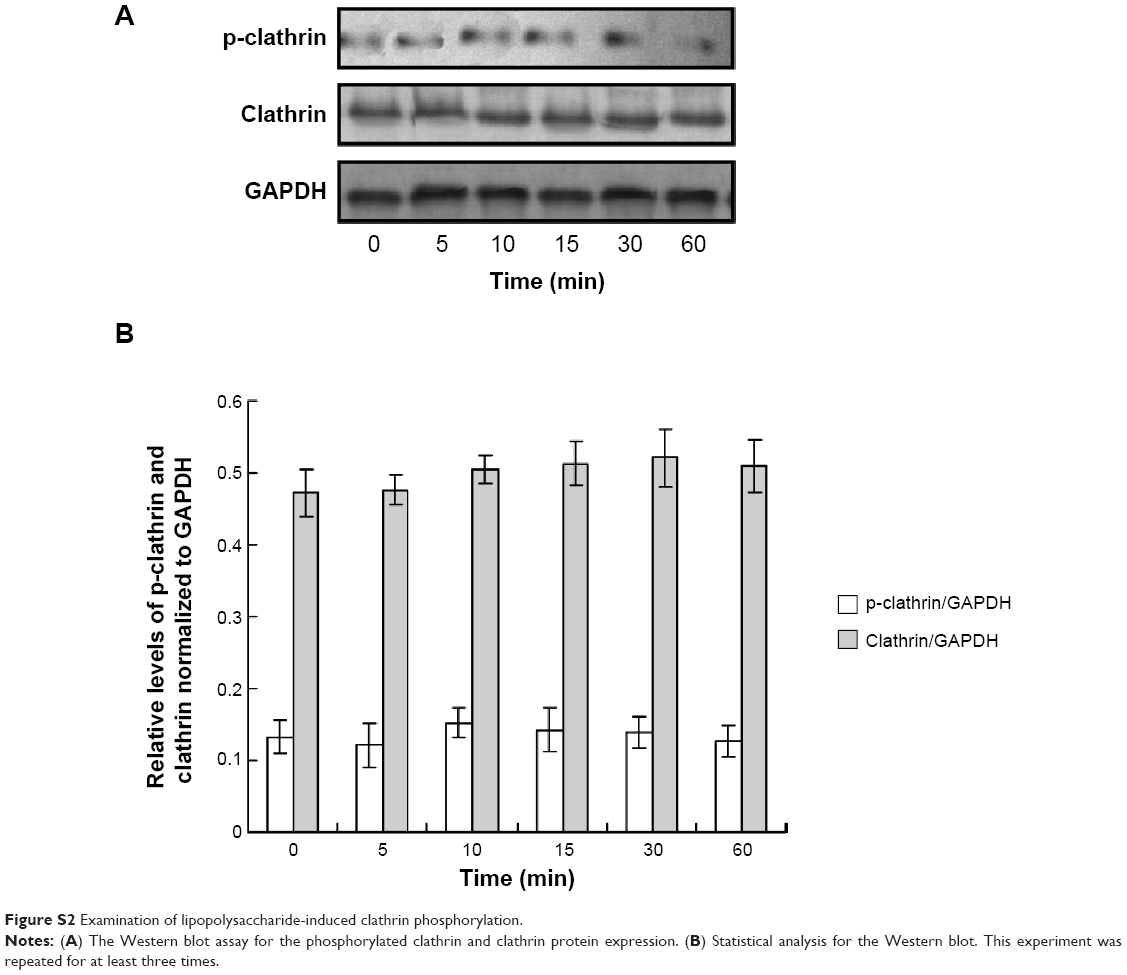 | Figure S2 Examination of lipopolysaccharide-induced clathrin phosphorylation. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.