")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 11
Lipid Metabolism as a Potential Target of Liver Cancer
Received 17 November 2023
Accepted for publication 25 January 2024
Published 14 February 2024 Volume 2024:11 Pages 327—346
DOI https://doi.org/10.2147/JHC.S450423
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David Gerber
Kangze Wu, Feizhuan Lin
Department of Hepatobiliary Surgery, Shaoxing People’s Hospital, Shaoxing, People’s Republic of China
Correspondence: Feizhuan Lin, Department of Hepatobiliary Surgery, Shaoxing People’s Hospital, Shaoxing, People’s Republic of China, Email [email protected]
Abstract: Hepatocellular carcinoma (HCC) stands as a severe malignant tumor with a profound impact on overall health, often accompanied by an unfavorable prognosis. Despite some advancements in the diagnosis and treatment of this disease, improving the prognosis of HCC remains a formidable challenge. It is noteworthy that lipid metabolism plays a pivotal role in the onset, development, and progression of tumor cells. Existing research indicates the potential application of targeting lipid metabolism in the treatment of HCC. This review aims to thoroughly explore the alterations in lipid metabolism in HCC, offering a detailed account of the potential advantages associated with innovative therapeutic strategies targeting lipid metabolism. Targeting lipid metabolism holds promise for potentially enhancing the prognosis of HCC.
Keywords: cholesterol, fatty acid, hepatocellular carcinoma, lipid uptake, lipid catabolism, lipid synthesis
Introduction
Liver cancer significantly impacts human health, ranking as the sixth most common malignant tumor and the third leading cause of cancer-related deaths globally. Hepatocellular carcinoma (HCC) constitutes roughly 75%-85% of liver cancer cases. Multiple factors contribute to the development of HCC, including aflatoxin exposure, alcohol consumption, hepatitis B, hepatitis C, obesity, and other related elements. In high-risk regions like China, Korea, and sub-Saharan Africa, chronic HBV infection and aflatoxin exposure are predominant causes.1
Over time, primary risk factors for HCC have shifted. In several regions, the proportion of HCC cases linked to obesity and diabetes has progressively risen.2 Western countries are witnessing an increase in HCC incidence due to non-alcoholic fatty liver disease, particularly non-alcoholic steatohepatitis (NASH), associated with metabolic syndrome or diabetes.3 While advancements have been made in comprehending the pathophysiology of HCC, it remains a formidable disease.3 Typically diagnosed at advanced stages, HCC often presents a poor prognosis.
In the United States, HCC patients have alarming survival rates. The average one-year survival rate is less than 50%, and the average five-year survival rate is less than 10%.4 This underlines the urgency to enhance early detection and more effective treatments to improve the outlook for individuals diagnosed with HCC.
Numerous conventional treatments are available for HCC, selected according to the patient’s health status. Early-stage HCC patients are typically recommended for resection, transplantation, or local ablation. Intermediate-stage patients often opt for transarterial chemoembolization (TACE), while those in the advanced stage generally receive systemic treatment as the primary approach.5–7 Currently, surgical intervention remains the primary treatment for HCC.5,8 However, a notable challenge post-surgery is the high recurrence rate, which can be as high as 70% even in patients with a single tumor of 2cm or less.9
In the last decade, sorafenib has stood out as a classic systemic drug with therapeutic effectiveness against HCC.10 Advanced understanding of HCC development mechanisms has led to the establishment of anti-PD-L1 antibody atezolizumab and anti-vascular endothelial growth factor antibody bevacizumab as standard treatments for untreated advanced HCC, demonstrating superior effectiveness to sorafenib.11,12 Yet, current treatments have limitations, stimulating researchers to actively search for new therapeutic targets. Among these, lipid metabolism has emerged as a notably promising focus of interest.13–17
The liver, a vital organ, performs various physiological functions, including the intricate processing of lipids.18 Lipids, comprising fats and lipoids, are diverse nutrients widely distributed in cellular organelles and serve as fundamental components of cellular membranes.19 Central to the synthesis, storage, and breakdown of lipids, the liver plays a critical role in maintaining lipid balance within the body.
One notable trait of cancer metabolism involves the promotion of de novo lipid synthesis, a pivotal pathway responsible for the generation of fatty acids (FAs).20 This metabolic shift is essential for rapidly dividing tumor cells to acquire necessary lipids, crucial for membrane construction, energy supply, and post-translational modifications.21 Relevant studies on lipid metabolism in HCC have already been published. Studies indicate that non-coding RNA can influence the progression of HCC by modulating the reprogramming of FAs metabolism.22,23 It has been reported that selective splicing of RNA can regulate lipid metabolism, thereby preventing the onset of HCC.24 Additionally, research has identified lipid metabolism-related gene markers that can be utilized to predict the prognosis of HCC.25 Given the significance of lipid metabolism in malignant tumors, a comprehensive understanding of how lipid metabolism is reprogrammed in HCC could provide valuable insights into targeting metabolic networks within HCC.
This review aims to summarize and analyze the influence of lipid metabolism on the genesis and advancement of HCC, introducing potential targets for therapeutic strategies based on lipid metabolism in the context of HCC.
Alteration of FAs Metabolism in HCC
In the physiological milieu of individuals with HCC, alterations in lipid metabolism manifest due to the heightened metabolic requirements of the tumor cells. Studies indicate that lipid biosynthesis and desaturation play crucial roles in the onset, sustenance, and advancement of HCC (Figure 1).26–28
 |
Figure 1 Aberrant FAs metabolism in HCC. In HCC, enzymes associated with fatty acid synthesis, such as ACLY, ACC, FASN, and SCD, are typically upregulated, and their increased expression is linked to a poor prognosis in HCC. However, ACSS stands out as relatively unique, as both upregulation and downregulation of its expression may be associated with adverse outcomes in HCC. In the FAs catabolism of HCC, there is typically an observed enhancement in FAO and LDs, while ferroptosis induced by FAs peroxidation tends to decrease. This is often associated with a poor prognosis in HCC. Abbreviations: TCA, Tricarboxylic acid; FAs, fatty acids; ACLY, ATP-citrate lyase; CoA, coenzyme A; ACC, acetyl-CoA carboxylase; AT, acyltransferase; LPA, lysophosphatidic acid;ACSS, acetyl-CoA synthetase; FASN, fatty acid synthase, SCD, Stearoyl-Coenzyme A Desaturase-1; PA, phosphatidic acid; DAG, diacylglycerol; DGAT, DAG acyltransferase; TAG, triacylglycerol; SFA, saturated fatty acid; MUFA, monounsaturated FA; FAO, fatty acid oxidation; LDS, lipid droplets. |
There remain unexplored facets of lipid metabolism in HCC that warrant further investigation in current research.
Aberrant FAs Uptake
Cells have the capacity to absorb FAs and cholesterol from their external surroundings.29 This uptake of FA necessitates the involvement of diverse membrane-associated transport proteins, such as fatty acid transport proteins (FATPs), fatty acid translocase (CD36), and FA-binding proteins (FABPs) (Figure 2).30
 |
Figure 2 Lipid uptake. Cells uptake exogenous fatty acids through CD36, FABPs, and FATPs, and cholesterol transported by LDL is taken up through LDLR. Abbreviations: CD36, Fatty acid translocase; FABPs, FA-binding proteins; FATPs, fatty acid transport proteins; LDL, low-density lipoprotein; LDLR, low-density lipoprotein receptor. |
CD36, a complete transmembrane glycoprotein, is expressed in various tissues, functioning as a scavenger receptor involved in immune recognition, inflammation, molecular adhesion, apoptosis, and lipid uptake.31–33 Research has revealed its pivotal role in regulating proliferation, metastasis, and angiogenesis in various tumor types.34–36 For instance, in esophageal squamous cell carcinoma, high CD36 expression significantly influences the reliance on FAs as a primary energy source, making it a critical regulator in this particular cancer.34 Additionally, studies indicate a positive correlation between baseline CD36 expression levels and migration, invasion, as well as the expression of epithelial-mesenchymal transition (EMT) markers in gastric cancer cell lines. Furthermore, research has demonstrated that in gastric cancer, CD36 promotes migration by activating serine/threonine kinase phosphorylation and inhibiting glycogen synthase kinase 3/β-catenin degradation, thereby facilitating the EMT.35 Intriguingly, in pancreatic cancer, low CD36 expression is associated with tumor growth and reduced survival rates.37
In the context of HCC, CD36’s role is intricately linked to the tumor microenvironment, where it functions in mediating the progression of HCC by reprogramming tumor metabolism. Experimental evidence indicates that the upregulation of CD36 in HCC significantly enhances both proliferation and metastatic potential, both in in vitro and in vivo settings.38 CD36, as a receptor for FAs, when deficient, leads to a reduction in FAs uptake across various human and mouse tissues.39 Studies have revealed that the absence of CD36 notably decreases phospholipids, triglycerides, and neutral lipids in HCC cells. This deficiency also impacts the expression of key enzymes involved in lipid metabolism, such as fatty acid synthase (FASN), acetyl-coenzyme A(CoA) carboxylase1 (ACC1), and FABP5. Furthermore, research has shown that CD36 plays a role in regulating the proliferation and migration of HCC cells by influencing their FAs uptake.38 Given these findings, CD36 emerges as a potential target for therapeutic intervention in HCC.
De Novo FAs Synthesis
In addition to the uptake of esters, de novo lipid synthesis in the body stands as a vital source of lipids. Primarily, de novo FAs synthesis takes place within the cytoplasm of liver cells or adipocytes, commencing with glucose conversion to pyruvate via glycolysis.40 Pyruvate then enters the mitochondria, transforming into citrate through the citric acid cycle. Exiting the mitochondria, citrate, catalyzed by ATP-citrate lyase (ACLY), converts into acetyl-CoA and oxaloacetate. Acetyl-CoA, further broken down into pyruvate and NADPH, along with previously generated acetyl-CoA, enters the fatty acid synthesis pathway.41 Acetyl-CoA is converted to malonyl-CoA by ACC. Malonyl-CoA, in conjunction with FASN, facilitates the formation of saturated fatty acids (SFAs) like palmitoyl-CoA and stearoyl-CoA.42 Stearoyl-CoA Desaturase-1 (SCD) catalyzes the conversion of saturated FAs to monounsaturated FA (MUFA) palmitoyl-CoA and oleoyl-CoA (Figure 3).43
 |
Figure 3 FAs synthesis. The citrate produced in the tricarboxylic acid cycle generates acetyl-CoA through ACLY. Acetyl-CoA is then converted to malonyl-CoA by ACC. Malonyl-CoA and acetyl-CoA combine through FASN to aid in the formation of SFA. Under the catalysis of SCD, SFAs are transformed into MUFA. G-3-P combines with activated fatty acids through AT to form LPA, which further converts to DAG. Subsequently, DGAT transforms DAG into TAG. Additionally, acetate can also be converted to acetyl-CoA through ACSS. Abbreviations: TCA, Tricarboxylic acid; FAs, fatty acids; ACLY, ATP-citrate lyase; CoA, coenzyme A; ACC, acetyl-CoA carboxylase; AT, acyltransferase; LPA, lysophosphatidic acid; ACSS, acetyl-CoA synthetase; FASN, fatty acid synthase; SCD, Stearoyl-Coenzyme A Desaturase-1; PA, phosphatidic acid; DAG, diacylglycerol; DGAT, DAG acyltransferase; TAG, triacylglycerol; SFA, saturated fatty acid; MUFA, monounsaturated FA; G-3-P, Glycerol-3-phosphate. |
Once FAs are produced, the liver begins to store them. Glucose, in the liver, is transformed into glycerol-3-phosphate (G3P) via glycolysis. G3P, combined with activated FAs by acyltransferase (AT), generates lysophosphatidic acid (LPA). AT further adds another activated FA to LPA, forming phosphatidic acid, which is then converted into diacylglycerol (DAG) by phosphatase. DAG acyltransferase (DGAT) then converts DAG into triacylglycerol (TAG).42 Moreover, acetate can be converted into acetyl-CoA via acetyl-CoA synthetase (ACSS) (Figure 3).44
Enzymes of Producing Acetyl-CoA in HCC
ACLY is an enzyme responsible for catalyzing the conversion of citrate and coenzyme A into acetyl-coenzyme A and oxaloacetate.45 The upregulation of ACLY is notably observed in various cancer cells. For instance, in breast cancer, research indicates that in HER2+/PIK3CAmut cells, mTORC2 stimulates the phosphorylation of ACLY, promoting the production of acetyl-CoA and enhancing de novo FAs synthesis, thereby facilitating tumor growth. Conversely, reduced ACLY exerts inhibitory effects on the growth of breast cancer cells.46 Immunohistochemical (IHC) analyses of gastric cancer patients have revealed a significant increase in ACLY in tumor tissues compared to surrounding normal tissues. Elevated ACLY levels are also positively associated with late-stage lymph node metastasis and shorter survival times in cancer.47
In the context of HCC, ACLY plays a crucial role in disease occurrence and development.48,49 Studies have shown that downregulating ACLY can reduce lipid synthesis, inflammation, and the incidence of HCC. ACLY’s role in promotingHCC has been elucidated by research.49
Some studies report that HCA, an ACLY inhibitor, can redirect the flow of acetyl-L-carnitine towards lipids while decreasing glucose flux towards lipids. This redirection suggests that in HepG2 HCC cells, acetyl-L-carnitine bypasses the citric acid cycle and utilizes cytoplasmic acetyl-CoA.50
Ning and colleagues’ research has identified that USP22 deubiquitinates and stabilizes Peroxisome Proliferator-Activated Receptor-γ (PPARγ), consequently upregulating ACLY expression, promoting lipid accumulation, and fostering tumor development in HCC cells.51
As ACLY mediates acetyl-CoA production, which is pivotal in tumor development, targeting ACLY could be an effective approach in cancer treatment.
The ACSS enzyme family, including ACSS1, ACSS2, and ACSS3, is involved in acetyl-CoA synthesis. In conditions of hypoxia and lipid depletion, ACSS2 is upregulated to produce acetyl-CoA.52–54 ACSS1, a mitochondrial protein, plays a crucial role in the growth of low glycolytic phenotype HCC cells, with inhibiting ACSS1 reducing acetate uptake and cell viability in this cell line.55
ACSS3, found in both the cytoplasm and nucleus, can be utilized for subtyping HCC.53,54,56 ACSS2, transcriptionally upregulated by sterol regulatory element-binding proteins (SREBPs), is identified through functional genomics as critical for cancer cell survival in hypoxic or low serum conditions.52 Inhibiting ACSS2 can suppress tumor growth, potentially linked to increased acetate consumption during tumor growth.57–59
In HCC, ACSS2 contributes acetyl groups for the acetylation of hypoxia-inducible factor (HIF)-2α. Under hypoxic conditions, silencing ACSS2 leads to decreased acetylation of HIF-2α, enhancing HIF-2α activity, increasing invasion, migration capabilities of HCC cells, and promoting EMT. Reduced expression of ACSS2 in a cohort of HCC patients is associated with advanced stages and poorer overall survival and disease-free survival rates, suggesting potential implications for the prognosis of HCC by targeting ACSS.60
FA Biosynthesis Enzymes in HCC
ACC serves as the rate-limiting enzyme in fatty acid biosynthesis61,62 and exists in two isoforms in humans: cytosolic ACC1, involved in metabolism, and outer mitochondrial membrane-anchored ACC2, responsible for regulating FAs beta-oxidation.62 Current research highlights that ACC gene expression is regulated by various transcription factors including SREBP1a, SREBP1c and carbohydrate-responsive element-binding protein (ChREBP).63
In various human cancer cells, ACC1 is notably highly expressed, showing relevance to cancer growth.64–66 In HCC, studies have identified ND-654, a liver-specific ACC inhibitor that mimics ACC phosphorylation, effectively inhibiting hepatic de novo lipogenesis and suppressing HCC development. Dysregulation of de novo lipogenesis and AMP-activated protein kinase (AMPK)-mediated ACC phosphorylation play pivotal roles in accelerating HCC.67
Zinc fingers and homeoboxes protein 2 (ZHX2) have been found to inhibit HCC tumor growth by significantly inhibiting de novo lipogenesis in HCC cells and reducing ACC1 expression.68 Serine/threonine protein kinase 25 (STK25), highly expressed in HCC patients, promotes HCC progression through the STRN/AMPK/ACC1 pathway.69
In laryngeal cancer, ACC2 exhibits high expression positively correlated with clinical tumor staging and negatively correlated with the 5-year survival rate of patients.70 miRNA-122’s role in mediating nonalcoholic fatty liver disease (NAFLD) development involves reducing ACC2 expression.71
However, research on ACC2’s role in HCC and its potential as a therapeutic target is still limited, requiring further investigation to expand our understanding in this area.
FASN is pivotal in de novo FAs synthesis, catalyzing the production of palmitate from acetyl-CoA, malonyl-CoA, and NADPH. Palmitate is crucial in more complex FA synthesis, contributes to cell membrane structure, and plays a significant role in post-translational protein acylation.44 Tumor-associated FASN is commonly regulated by SREBP1.72 Research across various cancers, including breast, prostate, and lung cancers, has consistently linked heightened FASN activity and overexpression to poor prognosis.44
In HCC, investigations have revealed intriguing findings. ZHX2 significantly reduces FASN expression, resulting in decreased de novo lipid synthesis in HCC cells, thereby inhibiting HCC development.68 Moreover, Wu and colleagues’ research highlighted that mitochondrial fission can elevate the acetylation level of SREBP1, subsequently upregulating FASN, promoting proliferation and metastasis in HCC.73
Additionally, upregulation of FASN has been associated with counteracting ferroptosis mediated by SLC7A11, leading to enhanced resistance to sorafenib. Combining FASN inhibitors with sorafenib has demonstrated synergistic anti-tumor effects in both in vitro and in vivo experiments, improving resistance to sorafenib in HCC.74
Studies have identified that ACAT1 can inhibit FASN degradation, promoting lipid synthesis and thereby stimulating HCC growth.75
SCD is an integral membrane protein situated in the endoplasmic reticulum. Its primary function involves catalyzing the production of MUFA, such as oleic acid and palmitoleic acid. SCD’s activity is regulated by SREBP. In animals, the SCD gene presents five genotypes, whereas in humans, the predominant genotypes are SCD1 and SCD5.44 SCD1 is notably overexpressed in various cancers, including pancreatic cancer.76–78 It has been observed to protect ovarian cancer cells from ferroptosis, and combining SCD1 inhibitors with ferroptosis inducers significantly reduces ovarian tumor volume in mouse models.44,79
In the context of HCC, Ma and colleagues found upregulation of SCD1 in HCC-initiating cells and sorafenib-resistant cells. Their research highlights that SCD1 regulates endoplasmic reticulum stress, inhibiting tumor self-renewal, migration, invasion, and resistance to sorafenib.80
A study by Liu and colleagues demonstrated that SCD1 reprograms the lipid metabolism of HCC cells, altering the cell’s lipid composition and impairing cytoplasmic membrane fluidity, consequently inhibiting invasion and metastasis of HCC cells. It was suggested that the damage to cytoplasmic membrane fluidity by SCD1 may be due to reduced levels of its primary product, oleic acid.81 Furthermore, research has shown that miR-4310 inhibits HCC cell proliferation, migration, and invasion in vitro and suppresses HCC growth and metastasis in vivo by targeting SCD1 and thereby inhibiting lipid synthesis.82 SCD5’s role in HCC remains less explored, warranting further research.
Collectively, ACC, FASN, and SCD pathways have shown significance in HCC. Investigating these pathways further offers potential therapeutic avenues for HCC treatment.
Abnormal FAs Catabolism
Fatty Acid Oxidation
Fatty acid oxidation (FAO), also known as β-oxidation, is the mitochondrial process that breaks down long-chain FAs into acetyl-CoA, NADH, and FADH2.83 The sequence of reactions in FAO involves multiple steps, starting with the activation of FAs catalyzed by fatty acyl-CoA synthetase. This step produces fatty acyl-CoA.
Further in the process, on the outer mitochondrial membrane, carnitine palmitoyltransferase 1 (CPT1), consisting of subtypes CPT1A, CPT1B, and CPT1C, converts fatty acyl-CoA to acylcarnitine. Subsequently, acylcarnitine is transported into the mitochondrial matrix by the carnitine-acylcarnitine translocase (CACT). Once in the matrix, CPT2 converts acylcarnitine back to fatty acyl-CoA. The fatty acyl-CoA undergoes a cyclic series of four steps inside the mitochondria, leading to the generation of acetyl-CoA. The resulting acetyl-CoA is further utilized in the tricarboxylic acid cycle to produce ATP.44,84
Research indicates that FAO is linked to various aspects of cancer, including tumor growth, metastasis, immune evasion, and chemotherapy resistance.85–88
In different cancers, such as breast cancer,85 glioblastoma,86 and acute myeloid leukemia,87 components of the FAO pathway have been associated with cancer development and progression.
In breast cancer, blocking CPT1B, a component of the FAO process, by inhibiting the JAK/STAT3 pathway has shown potential in suppressing tumor stem cells and resensitizing cancer cells to chemotherapy.85 Similarly, in glioblastoma, FAO has been suggested to facilitate immune evasion through CD47, contributing to invasive growth and radioresistance.86
In HCC, FAO has been shown to enhance chemoresistance by providing ATP for cell proliferation.89 Inhibition of FAO can reverse immune-suppressive activities in tumor-associated macrophages and inhibit HCC tumor progression. Additionally, overexpression of FAO-related genes in HCC cells with CTNNB1 mutations suggests a role in the progression of this cancer.90
The involvement of FAO in cancer development and progression highlights its potential as a therapeutic target. Further investigation and research on FAO might offer promising pathways for novel treatments in HCC.
Lipid Peroxidation and Cell Death
Lipids, particularly polyunsaturated fatty acids (PUFAs), are highly susceptible to lipid peroxidation. This process can be categorized into non-enzymatic and enzymatic-mediated forms.44,91 Non-enzymatic lipid peroxidation, also known as autoxidation of lipids, is a chain reaction triggered by reactive oxygen species (ROS), initiating the oxidation of PUFAs.44 Meanwhile, enzymatic lipid peroxidation is driven by the lipoxygenase (LOX) family, catalyzing the deoxygenation of free and esterified PUFAs, leading to the creation of various lipid peroxidation products.44
Ferroptosis, an iron-dependent form of cell death, has garnered significant attention in the realm of cancer.92 This non-apoptotic process can be activated via both exogenous and endogenous pathways. The exogenous pathway involves modulating transport proteins, such as inhibiting the amino acid reverse transport system xc− or activating iron transport proteins transferrin and lactotransferrin. Conversely, the endogenous pathway primarily triggers ferroptosis by hindering intracellular antioxidant enzymes, such as glutathione peroxidase 4 (GPX4). Moreover, various stressors, including extreme temperatures, hypoxia, and radiation, can also induce ferroptotic cell death.93
Many traditional cancer treatment methods induce ferroptosis, and augmenting the induction of this process through these methods can enhance their therapeutic efficacy.94 Research indicates that under the influence of radiation therapy, cancer cells adapt by upregulating the expression of SLC7A11 or GPX4 to counteract ferroptosis triggered by radiation therapy.95 In pancreatic cancer, the chemotherapy drug gemcitabine’s effect of inducing GPX4 expression and activity might contribute to cancer cells developing resistance to chemotherapy.96
In liver cancer, it has been observed that cu and copper metabolism murr1 domain 10 (COMMD10) can effectively boost ferroptosis both in laboratory settings and in live subjects, thus enhancing sensitivity to radiotherapy.97 Likewise, investigations have revealed that the suppressor of cytokine signaling 2 (SOCS2) can trigger the ubiquitination degradation of SLC7A11, promoting ferroptosis and increasing radio sensitivity in HCC.98 Concurrently, a study found that reducing LCN2, an iron-consuming factor, amplified sorafenib-induced ferroptosis and its anticancer impact on xenograft tumors in liver cancer patients exhibiting low LIFR expression and high LCN2 expression.99
Sorafenib, a common treatment for HCC, has shown resistance in specific cases. Research demonstrates that the transcription factors YAP/TAZ impede sorafenib-induced ferroptosis, leading to resistance to sorafenib in HCC.100 Lipid peroxidation has been identified as a contributing factor to various types of cell death, such as apoptosis, necrosis, ferroptosis, and alkaliptosis.44 Focusing on these cell death pathways associated with lipid peroxidation might offer innovative targets for the treatment of liver cancer.
Lipid Droplets
Lipid droplets (LDs) are composed of TAG and cholesteryl esters (CE), acting as storage organelles for lipid and energy homeostasis.101 Within tumors, intracellular LDs are often mobilized and increased, correlating with tumor invasiveness and resistance to treatment.102 For instance, in ovarian cancer, studies have demonstrated that impeding the assembly and accumulation of LDs can work in conjunction with bevacizumab to hinder tumor growth and proliferation.103 In the context of glioblastoma, researchers have shown that LDs accumulate in glioblastoma cells under hypoxic conditions. Inhibiting LD generation reduced cell survival during in vitro hypoxia reoxygenation and significantly hampered tumor development in vivo.104
Studies in HCC have revealed that BNIP3, a mitochondrial cargo receptor, can impede the growth of HCC by accelerating the turnover of LDs in lysosomes.105 Additionally, ACSL4 has been implicated in promoting the buildup of cellular LDs, furthering the progression of HCC.106
There remains a need for expanded research on LDs in the context of HCC. Given the synergistic effects of LDs in other tumors, investigating this aspect might present a promising area to explore in the treatment of HCC as well.
Alteration of Cholesterol Metabolism in HCC
In HCC, changes occur in cholesterol metabolism, including uptake, synthesis, and catabolism, which, in turn, impact the prognosis of HCC (Figure 4).
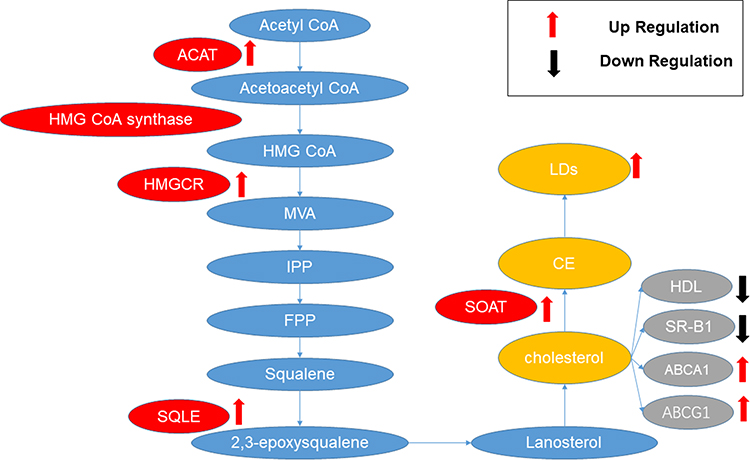 |
Figure 4 Aberrant cholesterol metabolism in HCC. In HCC, enzymes related to cholesterol synthesis, such as ACAT, HMGCR, SQLE, and SOAT, are typically upregulated, and their increased expression is associated with the occurrence and development of HCC. In the cholesterol catabolism of HCC, an observed enhancement in LDs is typically noted. The cholesterol efflux is characterized by a reduction in HDL and SR-B1, along with an increase in ABCA1 and ABCG1. This is associated with a poor prognosis in hepatocellular carcinoma (HCC). Abbreviations: ACAT, Acetyl-CoA acetyltransferase; HMG-CoA, 3-hydroxy-3-methyl glutaryl CoA; HMGCR, HMG-CoA reductase; MVA, mevalonate; IPP, isopentenyl pyrophosphate; FPP, farnesyl pyrophosphate; SQLE, squalene monooxygenase; LDs, lipid droplets; HDL, high-density lipoprotein; ABCA1, ATP-binding cassette transporters A1; ABCG1ATP-binding cassette transporter G1. |
Aberrant Cholesterol Uptake
Cholesterol plays a critical role in preserving membrane integrity, fluidity, and the creation of membrane microstructures.107 Cells produce cholesterol through de novo biosynthesis or by absorbing cholesterol transported by low-density lipoprotein (LDL) (Figure 2).108 Intracellular cholesterol balance is upheld through biosynthesis, uptake, excretion, and esterification processes.109,110 Several studies have highlighted dysregulated cholesterol metabolism in cancer cells.111–113 For instance, elevated levels of cholesterol, particularly 27-hydroxycholesterol (27HC), have been implicated as a significant risk factor for breast cancer incidence and recurrence. 27HC’s action on bone marrow cells, including macrophages, in a liver X receptor (LXR)-dependent manner hampers T-cell proliferation and cytotoxic function.111
In the context of HCC, research indicates that mice with high cholesterol exhibit fewer and smaller tumors when injected with HCC cells or exposed to carcinogens. Cholesterol accumulation in natural killer (NK) cells activates their effector functions against HCC cells.114 Studies by Chen and colleagues have unveiled that the downregulation of the LDL receptor (LDLR) is often linked to a poorer prognosis in HCC. Despite reducing LDL uptake, LDLR can partially stimulate cholesterol synthesis in HCC by activating the MEK/ERK pathway, thereby accelerating HCC proliferation and metastasis.115
While the specific mechanisms linking LDLR to HCC initiation, development, and prognosis are not yet fully understood, targeting LDLR remains a valuable avenue for research and potential intervention in HCC.
De Novo Cholesterol Synthesis
Cholesterol synthesis begins with acetyl-CoA. Two molecules of acetyl-CoA react with acetyl-CoA acetyltransferase (ACAT) to produce acetoacetyl-CoA. Under the catalysis of 3-hydroxy-3-methyl glutaryl CoA (HMG-CoA) synthase (HMGCS), acetoacetyl-CoA and another molecule of acetyl-CoA combine to produce HMG-CoA. HMG-CoA reductase (HMGCR), the major rate-limiting enzyme, catalyzes the reduction of HMG-CoA to mevalonate (MVA) while consuming two molecules of NADPH. MVA undergoes a three-step enzyme-catalyzed reaction to generate isopentenyl pyrophosphate (IPP), which further converts into farnesyl pyrophosphate (FPP) via a series of cytoplasmic enzymatic reactions. Condensation of two FPP molecules with pyrophosphate, under the action of squalene synthase, produces squalene. This squalene is oxidized to 2,3-oxidosqualene by squalene monooxygenase (SQLE), leading to cyclization in the endoplasmic reticulum, forming lanosterol. Lanosterol undergoes a complex series of reactions to synthesize cholesterol. Newly synthesized cholesterol in the endoplasmic reticulum is transported directly to the cell membrane or indirectly via the Golgi apparatus (Figure 5).116
 |
Figure 5 Cholesterol synthesis. Acetyl-CoA forms acetoacetyl-CoA catalyzed by ACAT, which then, through HMG CoA synthase, generates HMG-CoA. HMG-CoA, catalyzed by HMGCR, forms MVA, leading to the production of squalene. Squalene is oxidized by SQLE to 2,3-epoxysqualene, ultimately synthesizing cholesterol. Cholesterol, with the assistance of SOAT, is converted into CEs. Abbreviations: ACAT, Acetyl-CoA acetyltransferase; HMG-CoA, 3-hydroxy-3-methyl glutaryl CoA; HMGCR, HMG-CoA reductase; MVA, mevalonate; IPP, isopentenyl pyrophosphate; FPP, farnesyl pyrophosphate; SQLE, squalene monooxygenase; Ces, cholesteryl esters. |
In contrast to normal physiological responses, cancer cells activate lipid synthesis even in the presence of abundant exogenous lipids.117,118
Enzymes of Producing Acetyl-CoA in HCC
Acetyl-CoA is a pivotal node in both FAs and cholesterol synthesis, making it a critical factor in lipid metabolism. Cancer cells can upregulate acetyl-CoA synthesis through various pathways. For example, under conditions of hypoxia and lipid depletion, ACSS2 is upregulated to produce acetyl-CoA.52
ACLY and ACSS play a role in regulating cholesterol metabolism by modulating the production of acetyl-CoA. It has been reported that ACLY promotes cholesterol synthesis in patients with HCC. Meanwhile, researchers have discovered that the combined blockade of ACLY and immune checkpoints may potentially enhance the prognosis of HCC.119
ACSS is classified into three phenotypes: ACSS1, ACSS2, and ACSS3. In gastric cancer, knocking out ACSS3 can reduce cholesterol synthesis and improve the prognosis of gastric cancer.120 In HCC, ACSS1 has been found to be significantly upregulated in HCC tumors and is associated with tumor growth.121 The upregulation of ACSS promotes cholesterol production.
Cholesterol and Cholesterol Ester Biosynthesis Enzymes in HCC
ACAT, also known as acetoacetyl-CoA thiolase, primarily participates in the synthesis of acetoacetyl-CoA. ACAT has two isoforms: ACAT1 and ACAT2. Studies suggest that ACAT2 is downregulated in clear cell renal cell carcinoma, and this downregulation might correlate with a poorer prognosis in this type of cancer.122
In the context of HCC, Gu et al ‘s research unveiled that ACAT1 stabilizes the structure of GNPAT by acetylating the K128 site, consequently impeding the degradation of GNPAT. This stabilization suppresses TRIM21-mediated FASN degradation, ultimately promoting lipid synthesis. Simultaneously, it enhances xenograft tumor growth and exacerbates DEN/CCl4-induced mouse liver cancer development. The combined application of an ACAT1 inhibitor and sorafenib has demonstrated the capability to suppress DEN/CCl4-induced mouse HCC.75
HMGCR serves as the rate-limiting enzyme in the cholesterol synthesis pathway.116 Statins, known as competitive inhibitors of HMGCR, are widely employed to lower cholesterol levels.123 Existing research has highlighted the upregulation of HMGCR in various cancers.44 Additionally, studies have demonstrated that statin drugs inhibit cell proliferation and induce apoptosis.123
In gastric cancer, HMGCR expression is heightened in gastric cancer tissues. Its overexpression promotes the growth and migration of gastric cancer cells. Conversely, downregulation of HMGCR expression can inhibit the growth, migration, and tumorigenesis of gastric cancer cells.124
In HCC, HMGCR expression is also increased.125 A meta-analysis discovered that users of statin drugs have a reduced likelihood of developing HCC compared to non-users. Furthermore, the use of statin drugs is associated with a decreased risk of HCC.126 Reports suggest that ASPP2, a p53-activating factor, can restrain the activity of enzymes, including HMGCR, thus inhibiting tumor growth. Patients displaying high ASPP2 and low HMGCR tend to exhibit a better prognosis.127
Therefore, HMGCR stands as a highly promising target for therapeutic intervention.
SQLE is an essential enzyme in the cholesterol synthesis pathway responsible for converting squalene to 2.3-epoxysqualene. Research indicates the pivotal role of SQLE in various cancers, including colorectal, pancreatic, and prostate cancers, among others.128–131
In colorectal cancer, cholesterol accumulation leads to reduced SQLE levels, contributing to the progression of the disease.128 Conversely, in prostate cancer, SQLE is typically overexpressed, correlating with poorer survival rates. Inhibition of SQLE effectively impedes the growth of mouse orthotopic tumors.131
Regarding HCC, SQLE is often overexpressed in patients with NAFLD-HCC, linking this overexpression to a poorer prognosis. Inhibiting SQLE, particularly with terbinafine, significantly suppresses the growth of NAFLD-HCC and HCC cells. Increased SQLE levels in mice have been shown to promote CE biosynthesis, inducing NAFLD-HCC growth. Terbinafine inhibition of SQLE also mitigates tumor development in xenograft models and SQLE transgenic mice.132 However, some studies suggest that terbinafine’s anti-tumor effects may be unrelated to SQLE.133 Additionally, overexpression of SQLE in HCC promotes its growth, EMT, and metastasis in both in vitro and in vivo settings. Inhibiting SQLE exhibits potential in suppressing the growth and development of HCC.134
The exact role of SQLE in the onset, progression, and prognosis of HCC is currently ambiguous. Nonetheless, the available data suggest that targeting SQLE might hold promise in aiding drug development and treatment strategies for HCC.
Sterol O-acyltransferase (SOAT), also recognized as acyl-CoA cholesterol acyltransferase, belongs to the membrane-bound O-acyltransferase (MBOAT) family, primarily facilitating the transfer of acyl groups from acyl-CoA to cholesterol, generating CE on the endoplasmic reticulum (ER) membrane. In mammals, two primary subtypes exist: SOAT1 and SOAT2.44
SOAT1 is broadly expressed in most cells and has shown some tumor-suppressive effects when targeted in pancreatic cancer,135 prostate cancer,136 and glioblastoma.137 In HCC, research highlights significantly increased SOAT1 protein expression in tumor tissues compared to adjacent ones. Additionally, specific SOAT1 gene variants are associated with HCC susceptibility.138 High SOAT1 expression in liver cancers contributes to altered cellular cholesterol distribution, effectively inhibiting HCC proliferation and migration. In mouse models, SOAT1 inhibitors significantly reduce tumor size when SOAT1 expression is high.139
Furthermore, studies indicate that in HCC patients with p53 deficiency, knocking down SOAT1 significantly reduces cholesterol esterification and the incidence of HCC.140 SOAT2 is primarily distributed in hepatocytes and intestinal epithelial cells, participating in intracellular cholesterol storage and lipoprotein assembly.141 In the context of leptin-induced breast cancer cells, silencing SOAT2 significantly diminishes cancer cell proliferation, migration, and invasion.142 In HCC, inhibiting SOAT2 leads to the accumulation of non-esterified oxidized sterols intracellularly and suppresses the growth of HCC cell lines and their xenograft tumors.143
However, the specific mechanisms of SOAT in HCC necessitate further exploration.
Abnormal Cholesterol Catabolism
Lipid Droplets
For lipid and energy homeostasis, LDs act as storage organelles and are composed of TAG and CE.101 Within tumors, intracellular LDs are often mobilized and increased, correlating with tumor invasiveness and resistance to treatment.102 It is reported that elevated cholesterol levels lead to the formation of cholesterol crystals on the membrane of LDs, subsequently inducing the development of NASH, and may potentially contribute to HCC.144
Cholesterol Efflux
Cholesterol is excreted through four primary pathways: 1) Passive diffusion into mature high-density lipoprotein (HDL) particles. 2) Facilitated diffusion mediated by scavenger receptor class B type 1 (SR-B1). 3) Excretion through ATP-binding cassette transporter A1 (ABCA1) into lipid-poor apolipoprotein A1. 4) Excretion into lipid-rich mature HDL mediated by ATP-binding cassette transporter G1 (ABCG1).145
HDL exhibits anti-tumor activity and has demonstrated inhibitory effects on ovarian, colon, breast, and metastatic lung cancers in preclinical models.146 SR-B1 is notably overexpressed in various cancers, including breast,147 prostate,148 and nasopharyngeal cancers.149 In breast cancer, researchers have observed a correlation between SR-B1 overexpression and poor prognosis.150
ABCA1 is considered an anticancer protein. Loss of function, mutations, or abnormal expression of ABCA1 may contribute to cancer development.145 Some studies show a negative correlation between ABCA1 expression and breast cancer invasiveness.151 Conversely, the loss of ABCA1 function in colorectal cancer has been associated with increased cancer survival rates.152 However, certain studies have found that overexpression of ABCA1 in colorectal cancer enhances tumor migration and invasion capabilities.153
ABCG1 has been suggested to be associated with lower survival rates in a study on non-small cell lung cancer.154 Research indicates its enhanced expression in prostate cancer, showing a negative correlation with overall survival.155
In HCC, researchers have discovered that the HDL-binding protein is clinically linked to tumor metastasis. In vitro and in vivo experiments have demonstrated that the downregulation and overexpression of HDL-binding proteins significantly hinder and enhance the migration, invasion, and epithelial-mesenchymal transition of liver cancer cells, respectively.156
There has been a reported upregulation of ABCA1 in tumor monocytes/macrophages in HCC, resulting in an increased production of immature and immunosuppressive macrophages. The rise in ABCA1+ macrophages within HCC tumors is associated with a poorer prognosis.157
Furthermore, in HCC, researchers found that saracatinib might cause an overexpression of the gene encoding ABCG1, leading to oxaliplatin resistance. Interfering with ABCG1 expression can reverse oxaliplatin resistance in HCC patients.158
The presence of abnormal cholesterol efflux in HCC indicates that targeting cholesterol efflux might be beneficial for the treatment of this disease.
Transcriptional Regulation of Lipid Metabolism
SREBPs
SREBPs comprise a transcription factor family, which includes SREBP1a and SREBP1c encoded by the SREBF1 gene, as well as SREBP2 encoded by the SREBF2 gene. SREBP1 mainly governs genes related to fatty acid synthesis and the expression of LDL receptor (LDLR), while SREBP2 primarily regulates genes involved in cholesterol biosynthesis.159 These SREBPs are often activated in cancer cells.44
In HCC, studies have uncovered that ZHX2 can impede tumor growth. These studies demonstrated an inverse relationship between ZHX2 and the master regulator of de novo lipogenesis, SREBP1c, in HCC cell lines and human specimens.68 Additionally, research by Cheng et al unveiled that the knockdown of SLC25A47 modulates hepatic lipid metabolism via the AMPKα-SREBPs signaling pathway, promoting lipid synthesis and contributing to the onset and progression of HCC.160
Researchers have also observed in hepatic cells that SREBP2 can induce excessive cholesterol production, leading to the accumulation of lipid peroxides and functional impairment of natural killer T cells. This impairment further diminishes the immune system’s tumor surveillance function, thereby promoting the development of HCC.161
Furthermore, researchers have uncovered the pivotal role of SREBP2-mediated cholesterol biosynthesis in enhancing liver cancer stem cells (CSCs). The proliferation of CSCs, in turn, gives rise to drug resistance in tumor cells. Targeting SREBP2 may thus represent a potential strategy to overcome this drug resistance.162
LXRs
The LXRs are a class of nuclear receptors crucial for the transcriptional regulation of lipid metabolism, encompassing LXRα and LXRβ. These receptors govern fatty acid metabolism by controlling SREBP1c and are involved in various aspects of cholesterol function, including absorption, transport, efflux, and excretion through their capacity to regulate pertinent genes.163
In ovarian cancer, LXR ligands have been observed to induce the expression of P27, contributing to apoptosis in cancer cells.164 Studies have identified increased LXRs in pancreatic cancer.165 However, an alternate study reported significantly reduced LXR expression in tumor tissues compared to adjacent normal tissue in human pancreatic cancer patients.166
Regarding liver cancer, research has highlighted the capacity of LXRα to induce lipotoxicity and inhibit HCC.167 It has been noted that LXRα agonists can potentially reverse resistance to sorafenib treatment in sorafenib-resistant cells.168
Targeting LXR might hold promise in improving the prognosis of HCC.
PPARs
PPAR is a ligand-activated transcription factor belonging to the nuclear receptor superfamily, playing a crucial role in the transcriptional regulation of lipid metabolism.169 This family encompasses three subtypes: PPARα, primarily expressed in liver, heart, and kidneys, closely associated with FAO and energy metabolism;170 PPARβ/δ, mainly expressed in skeletal muscles with lesser expression in adipose tissue and skin, influencing FAO metabolism and energy uncoupling processes;171 and PPARγ, highly expressed in adipose tissue, regulating fat cell differentiation and energy storage in adipocytes.172 PPARγ is also found in the liver, kidneys, lungs, and colon, and its overexpression can lead to lipid accumulation in these tissues.173–177
It is reported that CD147 has been identified as downregulating PPARα, inhibiting FAs β-oxidation, and promoting the proliferation and metastasis of HCC cells.178
Research indicates a significant upregulation of PPARβ/δ in HCC tissues and cell lines, correlating with unfavorable clinical staging and prognosis in HCC.179
In HCC, tumor cells have been observed to evade immune checkpoint-targeted therapy through PPARγ/VEGF-A-mediated immune suppression in the tumor microenvironment (TME). Targeting PPARγ is suggested as a potential strategy to overcome immunotherapy resistance in HCC.180
As previously mentioned, the study by Ning et al highlights that USP22 stabilizes PPARγ through deubiquitination, leading to the upregulation of ACLY expression and promoting lipid accumulation in HCC cells, contributing to tumor development.51
Overall, PPAR emerges as a highly promising target for addressing HCC.
Therapeutically Exploring Lipid Metabolism in HCC Treatment
Targeting FA Synthesis
Although there is substantial evidence indicating the potential effectiveness of targeting FAs synthesis for treating HCC, there is a lack of clinical trials focusing on this area at present (Table 1). Specifically, there is a scarcity of studies exploring the anticancer effects of ACLY and SCD. In contrast, for FASN, a FASN inhibitor known as TVB-2640 has entered clinical trials.181
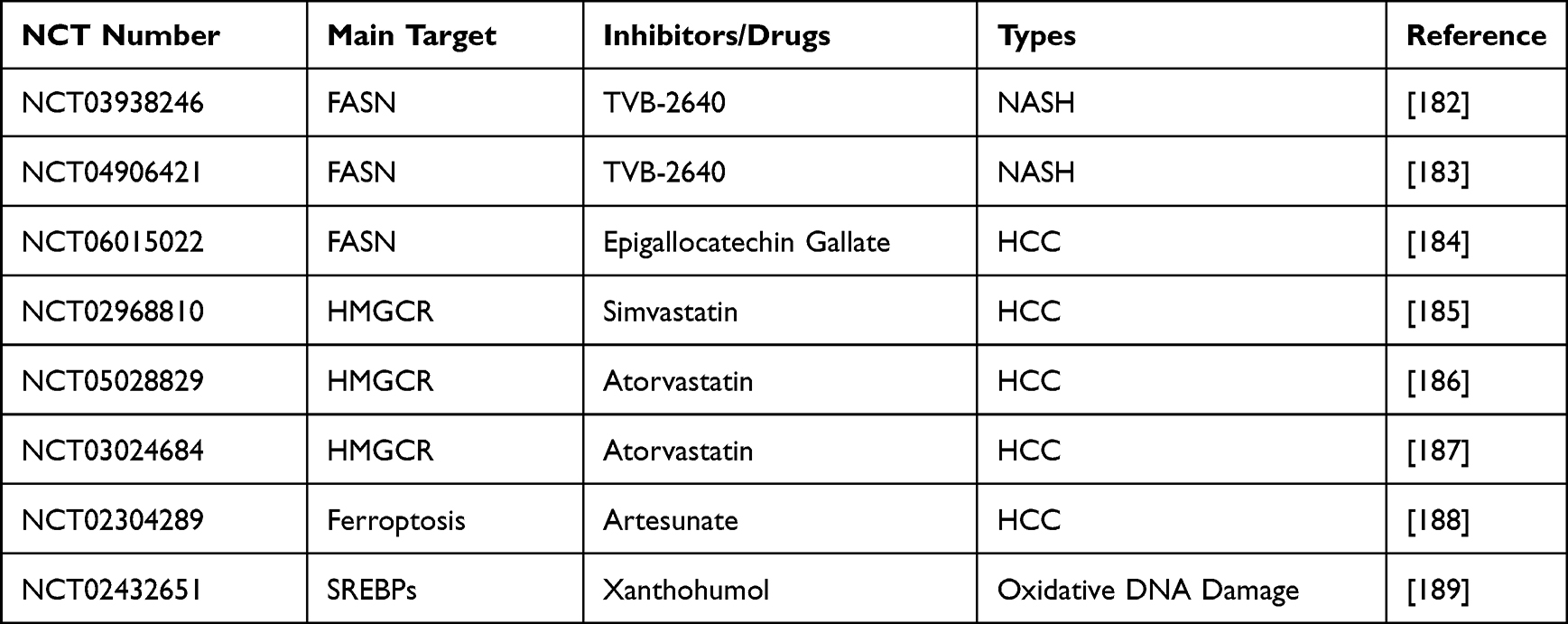 |
Table 1 Clinical Trials Targeting Lipid Metabolism |
The ongoing studies have primarily investigated the anticancer effects of FASN inhibition in glioblastoma, colon cancer, non-small cell lung cancer, and prostate cancer.190–193 However, there is currently no research focused on its effects specifically in HCC. On a related note, clinical trials are evaluating its role in NASH,182,183 which is a form of NAFLD considered a leading cause of HCC in many regions.194
Furthermore, a Phase II clinical trial aiming at FASN is currently recruiting participants for HCC. Researchers are using epigallocatechin gallate as an intervention to assess its effectiveness and safety in preventing the occurrence of HCC in patients with cirrhosis.184
Targeting Cholesterol Synthesis
Targeting cholesterol has emerged as a promising therapeutic approach in various cancers. Presently, there is a focus on targeting HMGCR, an enzyme involved in cholesterol production. Ongoing clinical research indicates promising results, demonstrating that the use of statins in metastatic prostate cancer patients receiving first-line chemotherapy is associated with improved overall survival.195
In the context of HCC, there is an ongoing Phase II clinical trial examining the efficacy of simvastatin in preventing HCC in patients with liver cirrhosis, with an expected completion this year.185 Simultaneously, another prospective, randomized, multicenter, double-blind, placebo-controlled trial is actively recruiting participants. This trial aims to evaluate the potential chemopreventive effect of atorvastatin in high-risk individuals with liver cirrhosis and fibrosis against the development of HCC.186 Furthermore, a multicenter, double-blind, randomized, placebo-controlled trial is recruiting patients to explore the preventive effects of atorvastatin on the risk of HCC recurrence after treatment (Table 1).187
Targeting Lipid Catabolism
Lipid catabolism plays a crucial role in providing energy for cancer cell survival and growth, making the targeting of lipid metabolism a critical focus in cancer treatment. Ferroptosis, an iron-dependent form of lipid peroxidation, characterizes a non-apoptotic type of cell death.92
In an attempt to explore new treatments for advanced HCC, a single-center Phase I dose escalation study aimed to evaluate the safety and pharmacokinetics of oral Artemisinin (Table 1).188 Unfortunately, the study was canceled due to slow patient recruitment. Despite this setback, further research is eagerly anticipated to confirm the clinical significance of lipid metabolism in the context of HCC.
Targeting Transcriptional Regulators of Lipid Metabolism
FA and cholesterol synthesis, which depend on gene transcription, are regulated by the membrane-bound transcription factor SREBPs. This makes SREBPs a potential target for cancer therapy.44 Xanthohumol, an SREBPs inactivator, reduces the re-synthesis of FAs and cholesterol, potentially offering benefits in conditions such as obesity and fatty liver.196 Xanthohumol has been clinically studied to showcase its ability to prevent DNA oxidative damage, potentially slowing down or impeding processes that can lead to cancer (Table 1).
Unfortunately, there are currently no published clinical studies on its effects specifically related to HCC. Further research in this area is eagerly anticipated to unveil its potential impact on HCC.
Conclusions
Undoubtedly, lipid metabolism significantly influences the development and prognosis of HCC. Changes in FAs and cholesterol intake have been correlated with the prognosis of HCC. Both FAs and cholesterol synthesis are key pathways, and their enhancement in HCC has been associated with its prognosis. Similarly, lipid catabolism, a crucial aspect of lipid metabolism, also contributes to the adverse prognosis of HCC. Current preclinical studies have demonstrated the benefits of targeting lipid metabolism in improving the prognosis of HCC, reducing the occurrence and progression of HCC, showcasing substantial potential.
Lipid metabolism is a complex network involving triglyceride intake, synthesis, breakdown, and regulatory factors, all impacting HCC growth. Current research predominantly concentrates on FAs and cholesterol synthesis and iron-induced cell death via lipid peroxidation. However, further studies investigating lipid intake, oxidation, and cholesterol excretion are needed. While existing clinical trials in HCC have not yet provided ideal outcomes, foundational research in related fields underscores the potential of targeting lipid metabolism as a promising avenue.
Currently, targets such as FASN, HMGCR, SREBPs, have been incorporated into clinical trials. These studies emphasize the de novo synthesis pathways of FAs and cholesterol, which may soon find successful applications in a clinical setting. Conversely, pathways related to lipid catabolism have not been extensively studied. However, it is evident that lipid catabolism significantly influences the treatment of HCC. Targeting lipid catabolism might lead to new and promising outcomes.
In summary, lipid metabolism is involved in the entire process of HCC occurrence and development. Targeting lipid metabolism is beneficial for preventing the occurrence of HCC and improving the prognosis. Therefore, targeting lipid metabolism provides a novel approach for the treatment of HCC. Further research in this field may open up new avenues for more effective treatment strategies for HCC.
Abbreviations
27HC, 27-hydroxycholesterol; ABCA1, ATP-binding cassette transporter A1; ABCG1, ATP-binding cassette transporter G1; ACAT1, acetyl-CoA acetyltransferase 1; ACC, acetyl-CoA carboxylase; ACLY, ATP-citrate lyase; ACSS, acetyl-CoA synthetase; AMPK, AMP-activated protein kinase; AT, acyltransferase; CACT, carnitine-acylcarnitine translocase; CD36, fatty acid translocase; CEs, cholesteryl esters; ChREBP, carbohydrate-responsive element-binding protein; COA, coenzyme A; COMMD10, cu and copper metabolism murr1 domain 10; CPT, carnitine palmitoyltransferase; DAG, diacylglycerol; DGAT, DAG acyltransferase; EMT, epithelial-mesenchymal transition; ER, endoplasmic reticulum; FABPs, FA-binding proteins; FAO, Fatty acid oxidation; FAs, fatty acids; FASN, fatty acid synthase; FATPs, fatty acid transport proteins; FPP, farnesyl pyrophosphate; G-3-P, glycerol-3-phosphate; GPX4, glutathione peroxidase 4; HCC, Hepatocellular carcinoma; HDL, high-density lipoprotein; HIF, Hypoxia-Inducible Factor; HMG-CoA, 3-hydroxy-3-methyl glutaryl CoA; HMGCR, HMG-CoA reductase; HMGCS, HMG-CoA synthase; IHC, Immunohistochemical; IPP, isopentenyl pyrophosphate; LDL, low-density lipoprotein; LDLR, LDL receptor; LDs, Lipid droplets; LOX, lipoxygenase; LPA, lysophosphatidic acid; LXR, liver X receptor; MBOAT, membrane-bound O-acyltransferase; MUFA, monounsaturated fatty acids; MVA, mevalonate; NAFLD, nonalcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; NK, natural killer; PUFAs, polyunsaturated fatty acids; ROS, reactive oxygen species; SCD, Stearoyl-Coenzyme A Desaturase; SFAs, saturated fatty acids; SOAT, Sterol O-acyltransferase; SOCS2, suppressor of cytokine signaling 2; SQLE, squalene monooxygenase; SR-B1, scavenger receptor class B type 1; SREBPs, sterol regulatory element-binding proteins; STK25, Serine/threonine protein kinase 25; TACE, transarterial chemoembolization; TAG, triacylglycerol; TCA, tricarboxylic acid; TME, tumor microenvironment.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Marengo A, Rosso C, Bugianesi E. Liver Cancer: connections with Obesity, Fatty Liver, and Cirrhosis. Annu Rev Med. 2016;67(1):103–117. doi:10.1146/annurev-med-090514-013832
3. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67(1):123–133.
4. McGlynn KA, London WT. The global epidemiology of hepatocellular carcinoma: present and future. Clin Liver Dis. 2011;15(2):2. doi:10.1016/j.cld.2011.03.006
5. EASL Clinical Practice. Guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236. doi:10.1016/j.jhep.2018.03.019
6. Llovet JM, Villanueva A, Marrero JA, et al. Trial Design and Endpoints in Hepatocellular Carcinoma: AASLD Consensus Conference. Hepatology. 2021;73(Suppl 1):158–191. doi:10.1002/hep.31327
7. Llovet JM, Brú C, Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis. 1999;19(3):329–338. doi:10.1055/s-2007-1007122
8. Marrero JA, Kulik LM, Sirlin CB, et al. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology. 2018;68(2):723–750. doi:10.1002/hep.29913
9. Roayaie S, Obeidat K, Sposito C, et al. Resection of hepatocellular cancer ≤2 cm: results from two Western centers. Hepatology. 2013;57(4):1426–1435. doi:10.1002/hep.25832
10. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi:10.1056/NEJMoa0708857
11. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894–1905. doi:10.1056/NEJMoa1915745
12. Lee MS, Ryoo B-Y, Hsu C-H, et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, Phase 1b study. Lancet Oncol. 2020;21(6):808–820. doi:10.1016/S1470-2045(20)30156-X
13. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279(15):2610–2623. doi:10.1111/j.1742-4658.2012.08644.x
14. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–777. doi:10.1038/nrc2222
15. Abramson HN. The lipogenesis pathway as a cancer target. J Med Chem. 2011;54(16):5615–5638. doi:10.1021/jm2005805
16. Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol. 2014;24(8):472–478. doi:10.1016/j.tcb.2014.06.001
17. von Roemeling CA, Marlow LA, Wei JJ, et al. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin Cancer Res. 2013;19(9):2368–2380. doi:10.1158/1078-0432.CCR-12-3249
18. Watt MJ, Miotto PM, De Nardo W, Montgomery MK. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr Rev. 2019;40(5):1367–1393.
19. Luo X, Cheng C, Tan Z, et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16(1):76.
20. Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res. 2013;52(4):585–589. doi:10.1016/j.plipres.2013.08.005
21. Satriano L, Lewinska M, Rodrigues PM, Banales JM, Andersen JB. Metabolic rearrangements in primary liver cancers: cause and consequences. Nat Rev Gastroenterol Hepatol. 2019;16(12):748–766.
22. Xu K, Xia P, Gongye X, et al. A novel lncRNA RP11-386G11.10 reprograms lipid metabolism to promote hepatocellular carcinoma progression. Mol Metabol. 2022;63:101540. doi:10.1016/j.molmet.2022.101540
23. Xu K, Xia P, Chen X, Ma W, Yuan Y. ncRNA-mediated fatty acid metabolism reprogramming in HCC. Trend Endocrinol Metabol. 2023;34(5):278–291. doi:10.1016/j.tem.2023.02.007
24. Xu K, Wu T, Xia P, Chen X, Yuan Y. Alternative splicing: a bridge connecting NAFLD and HCC. Trends Mol Med. 2023;29(10):859–872.
25. Xu K, Xia P, Liu P, Zhang X. A six lipid metabolism related gene signature for predicting the prognosis of hepatocellular carcinoma. Sci Rep. 2022;12(1):20781. doi:10.1038/s41598-022-25356-2
26. Muir K, Hazim A, He Y, et al. Proteomic and lipidomic signatures of lipid metabolism in NASH-associated hepatocellular carcinoma. Cancer Res. 2013;73(15):4722–4731. doi:10.1158/0008-5472.CAN-12-3797
27. Nelson GM, Ahlborn GJ, Allen JW, et al. Transcriptional changes associated with reduced spontaneous liver tumor incidence in mice chronically exposed to high dose arsenic. Toxicology. 2009;266(1–3):1–3. doi:10.1016/j.tox.2009.10.004
28. Falvella FS, Pascale RM, Gariboldi M, et al. Stearoyl-CoA desaturase 1 (Scd1) gene overexpression is associated with genetic predisposition to hepatocarcinogenesis in mice and rats. Carcinogenesis. 2002;23(11):1933–1936. doi:10.1093/carcin/23.11.1933
29. Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med. 2021;218:1.
30. Matsushita Y, Nakagawa H, Koike K. Lipid Metabolism in Oncology: why It Matters, How to Research, and How to Treat. Cancers. 2021;13(3):3. doi:10.3390/cancers13030474
31. Wang J, Li Y. CD36 tango in cancer: signaling pathways and functions. Theranostics. 2019;9(17):4893–4908. doi:10.7150/thno.36037
32. Yang X, Okamura DM, Lu X, et al. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat Rev Nephrol. 2017;13(12):769–781.
33. DeFilippis RA, Chang H, Dumont N, et al. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discov. 2012;2(9):826–839. doi:10.1158/2159-8290.CD-12-0107
34. Yoshida T, Yokobori T, Saito H, et al. CD36 Expression Is Associated with Cancer Aggressiveness and Energy Source in Esophageal Squamous Cell Carcinoma. Ann Surg Oncol. 2021;28(2):1217–1227.
35. Wang J, Wen T, Li Z, et al. CD36 upregulates DEK transcription and promotes cell migration and invasion via GSK-3β/β-catenin-mediated epithelial-to-mesenchymal transition in gastric cancer. Aging. 2020;13(2):1883–1897. doi:10.18632/aging.103985
36. Panda D, Chatterjee G, Sardana R, et al. Utility of CD36 as a novel addition to the immunophenotypic signature of RAM-phenotype acute myeloid leukemia and study of its clinicopathological characteristics. Cytometry B Clin Cytom. 2021;100(2):206–217. doi:10.1002/cyto.b.21943
37. Jia S, Zhou L, Shen T, Zhou S, Ding G, Cao L. Down-expression of CD36 in pancreatic adenocarcinoma and its correlation with clinicopathological features and prognosis. J Cancer. 2018;9(3):578–583. doi:10.7150/jca.21046
38. Tao L, Ding X, Yan L, et al. CD36 accelerates the progression of hepatocellular carcinoma by promoting FAs absorption. Med Oncol. 2022;39(12):202. doi:10.1007/s12032-022-01808-7
39. Son N-H, Basu D, Samovski D, et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J Clin Invest. 2018;128(10):4329–4342. doi:10.1172/JCI99315
40. Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. 2012;56(4):952–964. doi:10.1016/j.jhep.2011.08.025
41. Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24(41):6314–6322. doi:10.1038/sj.onc.1208773
42. Pope ED, Kimbrough EO, Vemireddy LP, Surapaneni PK, Copland JA, Mody K. Aberrant lipid metabolism as a therapeutic target in liver cancer. Expert Opin Ther Targets. 2019;23(6):473–483. doi:10.1080/14728222.2019.1615883
43. Miyazaki M, Kim HJ, Man WC, Ntambi JM. Oleoyl-CoA is the major de novo product of stearoyl-CoA desaturase 1 gene isoform and substrate for the biosynthesis of the Harderian gland 1-alkyl-2,3-diacylglycerol. J Biol Chem. 2001;276(42):39455–39461. doi:10.1074/jbc.M106442200
44. Yin X, Xu R, Song J, et al. Lipid metabolism in pancreatic cancer: emerging roles and potential targets. Cancer Commun. 2022;42(12):1234–1256. doi:10.1002/cac2.12360
45. Bempedoic GP. Acid to Lower LDL Cholesterol - Safety and Efficacy. N Engl J Med. 2020;383(7):e49.
46. Chen Y, Qian J, He Q, et al. mTOR complex-2 stimulates acetyl-CoA and de novo lipogenesis through ATP citrate lyase in HER2/PIK3CA-hyperactive breast cancer. Oncotarget. 2016;7(18):25224–25240. doi:10.18632/oncotarget.8279
47. Qian X, Hu J, Zhao J, Chen H. ATP citrate lyase expression is associated with advanced stage and prognosis in gastric adenocarcinoma. Int J Clin Exp Med. 2015;8(5):7855–7860.
48. Khwairakpam AD, Banik K, Girisa S, et al. The vital role of ATP citrate lyase in chronic diseases. J Mol Med. 2020;98(1):71–95. doi:10.1007/s00109-019-01863-0
49. Gu L, Zhu Y, Lin X, et al. The IKKβ-USP30-ACLY Axis Controls Lipogenesis and Tumorigenesis. Hepatology. 2021;73(1):160–174. doi:10.1002/hep.31249
50. Lligona-Trulla L, Arduini A, Aldaghlas TA, Calvani M, Kelleher JK. Acetyl-L-carnitine flux to lipids in cells estimated using isotopomer spectral analysis. J Lipid Res. 1997;38(7):1454–1462. doi:10.1016/S0022-2275(20)37427-7
51. Ning Z, Guo X, Liu X, et al. USP22 regulates lipidome accumulation by stabilizing PPARγ in hepatocellular carcinoma. Nat Commun. 2022;13(1):2187. doi:10.1038/s41467-022-29846-9
52. Schug ZT, Peck B, Jones DT, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27(1):57–71. doi:10.1016/j.ccell.2014.12.002
53. Li X, Yu W, Qian X, et al. Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol Cell. 2017;66(5):5. doi:10.1016/j.molcel.2017.04.026
54. Li Z, Liu H, He J, et al. Acetyl-CoA Synthetase 2: a Critical Linkage in Obesity-Induced Tumorigenesis in Myeloma. Cell Metab. 2021;33(1):1. doi:10.1016/j.cmet.2020.12.011
55. Yun M, Bang S-H, Kim JW, Park JY, Kim KS, Lee JD. The importance of acetyl coenzyme A synthetase for 11C-acetate uptake and cell survival in hepatocellular carcinoma. J Nucl Med. 2009;50(8):1222–1228. doi:10.2967/jnumed.109.062703
56. Bidkhori G, Benfeitas R, Klevstig M, et al. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc Natl Acad Sci U S A. 2018;115(50):E11874–E11883. doi:10.1073/pnas.1807305115
57. Moffett JR, Puthillathu N, Vengilote R, Jaworski DM, Namboodiri AM. Acetate Revisited: a Key Biomolecule at the Nexus of Metabolism, Epigenetics and Oncogenesis-Part 1: acetyl-CoA, Acetogenesis and Acyl-CoA Short-Chain Synthetases. Front Physiol. 2020;11:580167. doi:10.3389/fphys.2020.580167
58. Moffett JR, Puthillathu N, Vengilote R, Jaworski DM, Namboodiri AM. Acetate Revisited: a Key Biomolecule at the Nexus of Metabolism, Epigenetics, and Oncogenesis - Part 2: acetate and ACSS2 in Health and Disease. Front Physiol. 2020;11:580171. doi:10.3389/fphys.2020.580171
59. Xu H, Luo J, Ma G, et al. Acyl-CoA synthetase short-chain family member 2 (ACSS2) is regulated by SREBP-1 and plays a role in fatty acid synthesis in caprine mammary epithelial cells. J Cell Physiol. 2018;233(2):1005–1016. doi:10.1002/jcp.25954
60. Sun L, Kong Y, Cao M, et al. Decreased expression of acetyl-CoA synthase 2 promotes metastasis and predicts poor prognosis in hepatocellular carcinoma. Cancer Sci. 2017;108(7):1338–1346. doi:10.1111/cas.13252
61. Tong L. Structure and function of biotin-dependent carboxylases. Cell Mol Life Sci. 2013;70(5):863–891. doi:10.1007/s00018-012-1096-0
62. Hunkeler M, Hagmann A, Stuttfeld E, et al. Structural basis for regulation of human acetyl-CoA carboxylase. Nature. 2018;558(7710):470–474. doi:10.1038/s41586-018-0201-4
63. Chen L, Duan Y, Wei H, et al. Acetyl-CoA carboxylase (ACC) as a therapeutic target for metabolic syndrome and recent developments in ACC1/2 inhibitors. Expert Opin Investig Drugs. 2019;28(10):917–930. doi:10.1080/13543784.2019.1657825
64. Rios Garcia M, Steinbauer B, Srivastava K, et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017;26(6):6. doi:10.1016/j.cmet.2017.09.018
65. Ueda K, Nakatsu Y, Yamamotoya T, et al. Prolyl isomerase Pin1 binds to and stabilizes acetyl CoA carboxylase 1 protein, thereby supporting cancer cell proliferation. Oncotarget. 2019;10(17):1637–1648. doi:10.18632/oncotarget.26691
66. Fang W, Cui H, Yu D, Chen Y, Wang J, Yu G. Increased expression of phospho-acetyl-CoA carboxylase protein is an independent prognostic factor for human gastric cancer without lymph node metastasis. Med Oncol. 2014;31(7):15. doi:10.1007/s12032-014-0015-7
67. Ghoshal S. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019;29(1):174–182.e5. doi:10.1016/j.cmet.2018.08.020
68. Yu X, Lin Q, Wu Z, et al. ZHX2 inhibits SREBP1c-mediated de novo lipogenesis in hepatocellular carcinoma via miR-24-3p. J Pathol. 2020;252(4):358–370. doi:10.1002/path.5530
69. Zhang Y, Xu J, Qiu Z, et al. STK25 enhances hepatocellular carcinoma progression through the STRN/AMPK/ACC1 pathway. Cancer Cell Int. 2022;22(1):4. doi:10.1186/s12935-021-02421-w
70. Li K, Zhang C, Chen L, et al. The role of acetyl-coA carboxylase2 in head and neck squamous cell carcinoma. PeerJ. 2019;7:e7037. doi:10.7717/peerj.7037
71. Banerjee P. Genetic and Epigenetic Culprits in the Pathogenesis of Nonalcoholic Fatty Liver Disease. J Clin Exp Hepatol. 2018;8(4):390–402. doi:10.1016/j.jceh.2018.04.001
72. Rawson R. The SREBP pathway--insights from Insigs and insects. Nat Rev Mol Cell Biol. 2003;4(8):631–640. doi:10.1038/nrm1174
73. Wu D, Yang Y, Hou Y, et al. Increased mitochondrial fission drives the reprogramming of fatty acid metabolism in hepatocellular carcinoma cells through suppression of Sirtuin 1. Cancer Commun. 2022;42(1):37–55. doi:10.1002/cac2.12247
74. Li Y, Yang W, Zheng Y, et al. Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J Exp Clin Cancer Res. 2023;42(1):6. doi:10.1186/s13046-022-02567-z
75. Gu L, Zhu Y, Lin X, Tan X, Lu B, Li Y. Stabilization of FASN by ACAT1-mediated GNPAT acetylation promotes lipid metabolism and hepatocarcinogenesis. Oncogene. 2020;39(11):2437–2449. doi:10.1038/s41388-020-1156-0
76. Castro LFC, Wilson JM, Gonçalves O, Galante-Oliveira S, Rocha E, Cunha I. The evolutionary history of the stearoyl-CoA desaturase gene family in vertebrates. BMC Evol Biol. 2011;11:132. doi:10.1186/1471-2148-11-132
77. Wang J, Yu L, Schmidt RE, et al. Characterization of HSCD5, a novel human stearoyl-CoA desaturase unique to primates. Biochem Biophys Res Commun. 2005;332(3):735–742. doi:10.1016/j.bbrc.2005.05.013
78. Bené H, Lasky D, Ntambi JM. Cloning and characterization of the human stearoyl-CoA desaturase gene promoter: transcriptional activation by sterol regulatory element binding protein and repression by polyunsaturated fatty acids and cholesterol. Biochem Biophys Res Commun. 2001;284(5):1194–1198. doi:10.1006/bbrc.2001.5102
79. Carbone M, Melino G. Stearoyl CoA Desaturase Regulates Ferroptosis in Ovarian Cancer Offering New Therapeutic Perspectives. Cancer Res. 2019;79(20):5149–5150. doi:10.1158/0008-5472.CAN-19-2453
80. Mkf M, Lau EYT, Leung DHW, et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J Hepatol. 2017;67(5):979–990. doi:10.1016/j.jhep.2017.06.015
81. Liu -H-H, Xu Y, C-J L, et al. An SCD1-dependent mechanoresponsive pathway promotes HCC invasion and metastasis through lipid metabolic reprogramming. Mol Ther. 2022;30(7):2554–2567. doi:10.1016/j.ymthe.2022.03.015
82. Li H, Chen Z, Zhang Y, et al. MiR-4310 regulates hepatocellular carcinoma growth and metastasis through lipid synthesis. Cancer Lett. 2021;519:161–171. doi:10.1016/j.canlet.2021.07.029
83. Lim GB. Inhibiting fatty acid oxidation promotes cardiomyocyte proliferation. Nat Rev Cardiol. 2020;17(5):266–267. doi:10.1038/s41569-020-0361-4
84. Houten SM, Violante S, Ventura FV, Wanders RJA. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu Rev Physiol. 2016;78:23–44. doi:10.1146/annurev-physiol-021115-105045
85. Wang T, Fahrmann JF, Lee H, et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018;27(1):136–150.e5. doi:10.1016/j.cmet.2017.11.001
86. Jiang N, Xie B, Xiao W, et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47-mediated immune evasion. Nat Commun. 2022;13(1):1511. doi:10.1038/s41467-022-29137-3
87. Shi J, Fu H, Jia Z, He K, Fu L, Wang W. High Expression of CPT1A Predicts Adverse Outcomes: a Potential Therapeutic Target for Acute Myeloid Leukemia. EBioMedicine. 2016;14:55–64. doi:10.1016/j.ebiom.2016.11.025
88. Shao H, Mohamed EM, Xu GG, et al. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget. 2016;7(4):3832–3846. doi:10.18632/oncotarget.6757
89. Wu T, Luo G, Lian Q, et al. Discovery of a Carbamoyl Phosphate Synthetase 1-Deficient HCC Subtype With Therapeutic Potential Through Integrative Genomic and Experimental Analysis. Hepatology. 2021;74(6):3249–3268. doi:10.1002/hep.32088
90. Senni N, Savall M, Cabrerizo Granados D, et al. β-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut. 2019;68(2):322–334. doi:10.1136/gutjnl-2017-315448
91. Zielinski ZAM, Pratt DA. Lipid Peroxidation: kinetics, Mechanisms, and Products. J Org Chem. 2017;82(6):2817–2825. doi:10.1021/acs.joc.7b00152
92. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
93. Tang D, Kroemer G. Ferroptosis. Curr Biol. 2020;30(21):R1292–R1297. doi:10.1016/j.cub.2020.09.068
94. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22(7):381–396. doi:10.1038/s41568-022-00459-0
95. Lei G, Zhang Y, Koppula P, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30(2):146–162. doi:10.1038/s41422-019-0263-3
96. Zhu S, Zhang Q, Sun X, et al. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017;77(8):2064–2077. doi:10.1158/0008-5472.CAN-16-1979
97. Yang M, Wu X, Hu J, et al. COMMD10 inhibits HIF1α/CP loop to enhance ferroptosis and radiosensitivity by disrupting Cu-Fe balance in hepatocellular carcinoma. J Hepatol. 2022;76(5):1138–1150. doi:10.1016/j.jhep.2022.01.009
98. Chen Q, Zheng W, Guan J, et al. SOCS2-enhanced ubiquitination of SLC7A11 promotes ferroptosis and radiosensitization in hepatocellular carcinoma. Cell Death Differ. 2023;30(1):137–151. doi:10.1038/s41418-022-01051-7
99. Yao F, Deng Y, Zhao Y, et al. A targetable LIFR-NF-κB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis. Nat Commun. 2021;12(1):7333. doi:10.1038/s41467-021-27452-9
100. Gao R, Kalathur RKR, Coto-Llerena M, et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol Med. 2021;13(12):e14351. doi:10.15252/emmm.202114351
101. Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20(3):137–155. doi:10.1038/s41580-018-0085-z
102. Bacci M, Lorito N, Smiriglia A, Morandi A. Fat and Furious: lipid Metabolism in Antitumoral Therapy Response and Resistance. Trends Cancer. 2021;7(3):198–213. doi:10.1016/j.trecan.2020.10.004
103. Curtarello M, Tognon M, Venturoli C, et al. Rewiring of Lipid Metabolism and Storage in Ovarian Cancer Cells after Anti-VEGF Therapy. Cells. 2019;8(12):1601. doi:10.3390/cells8121601
104. Bensaad K, Favaro E, Lewis CA, et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014;9(1):349–365. doi:10.1016/j.celrep.2014.08.056
105. Berardi DE, Bock-Hughes A, Terry AR, Drake LE, Bozek G, Macleod KF. Lipid droplet turnover at the lysosome inhibits growth of hepatocellular carcinoma in a BNIP3-dependent manner. Sci Adv. 2022;8(41):eabo2510. doi:10.1126/sciadv.abo2510
106. Chen J, Ding C, Chen Y, et al. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer Lett. 2021;502:154–165. doi:10.1016/j.canlet.2020.12.019
107. Pellerin L, Carrié L, Dufau C, et al. Lipid metabolic Reprogramming: role in Melanoma Progression and Therapeutic Perspectives. Cancers. 2020;12(11):3147. doi:10.3390/cancers12113147
108. Luo J, Yang H, Song B-L. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. 2020;21(4):225–245.
109. Huang B, Song B-L, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab. 2020;2(2):132–141. doi:10.1038/s42255-020-0174-0
110. Xu H, Zhou S, Tang Q, Xia H, Bi F. Cholesterol metabolism: new functions and therapeutic approaches in cancer. Biochim Biophys Acta Rev Cancer. 2020;1874(1):188394. doi:10.1016/j.bbcan.2020.188394
111. Ma L, Wang L, Nelson AT, et al. 27-Hydroxycholesterol acts on myeloid immune cells to induce T cell dysfunction, promoting breast cancer progression. Cancer Lett. 2020;493:266–283. doi:10.1016/j.canlet.2020.08.020
112. Garcia-Bermudez J, Baudrier L, Bayraktar EC, et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature. 2019;567(7746):118–122. doi:10.1038/s41586-019-0945-5
113. Guillaumond F, Bidaut G, Ouaissi M, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2015;112(8):2473–2478. doi:10.1073/pnas.1421601112
114. Qin W-H, Yang Z-S, Li M, et al. High Serum Levels of Cholesterol Increase Antitumor Functions of Nature Killer Cells and Reduce Growth of Liver Tumors in Mice. Gastroenterology. 2020;158(6):1713–1727. doi:10.1053/j.gastro.2020.01.028
115. Chen Z, Chen L, Sun B, et al. LDLR inhibition promotes hepatocellular carcinoma proliferation and metastasis by elevating intracellular cholesterol synthesis through the MEK/ERK signaling pathway. Mol Metab. 2021;51:101230. doi:10.1016/j.molmet.2021.101230
116. Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9(2):125–138. doi:10.1038/nrm2336
117. Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. 2016;16(11):732–749. doi:10.1038/nrc.2016.89
118. Corn KC, Windham MA, Rafat M. Lipids in the tumor microenvironment: from cancer progression to treatment. Prog Lipid Res. 2020;80:101055. doi:10.1016/j.plipres.2020.101055
119. Xu Y, Zhang Z, Xu D, Yang X, Zhou L, Zhu Y. Identification and integrative analysis of ACLY and related gene panels associated with immune microenvironment reveal prognostic significance in hepatocellular carcinoma. Can Cell Inter. 2021;21(1):409. doi:10.1186/s12935-021-02108-2
120. Chang WC, Cheng WC, Cheng BH, et al. Mitochondrial Acetyl-CoA Synthetase 3 is Biosignature of Gastric Cancer Progression. Cancer Med. 2018;7(4):1240–1252. doi:10.1002/cam4.1295
121. Björnson E, Mukhopadhyay B, Asplund A, et al. Stratification of Hepatocellular Carcinoma Patients Based on Acetate Utilization. Cell Rep. 2015;13(9):2014–2026. doi:10.1016/j.celrep.2015.10.045
122. Zhao Z, Lu J, Han L, Wang X, Man Q, Liu S. Prognostic significance of two lipid metabolism enzymes, HADHA and ACAT2, in clear cell renal cell carcinoma. Tumour Biol. 2016;37(6):8121–8130. doi:10.1007/s13277-015-4720-4
123. Mo H, Jeter R, Bachmann A, Yount ST, Shen C-L, Yeganehjoo H. The Potential of Isoprenoids in Adjuvant Cancer Therapy to Reduce Adverse Effects of Statins. Front Pharmacol. 2018;9:1515. doi:10.3389/fphar.2018.01515
124. Chushi L, Wei W, Kangkang X, Yongzeng F, Ning X, Xiaolei C. HMGCR is up-regulated in gastric cancer and promotes the growth and migration of the cancer cells. Gene. 2016;587(1):42–47. doi:10.1016/j.gene.2016.04.029
125. Sohda T, Iwata K, Hirano G, et al. 3-Hydroxyl-3-methylglutaryl-coenzyme A reductase is up regulated in hepatocellular carcinoma associated with paraneoplastic hypercholesterolemia. Med Mol Morphol. 2013;46(4):239–242. doi:10.1007/s00795-013-0042-z
126. Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. 2013;144(2):323–332. doi:10.1053/j.gastro.2012.10.005
127. Liang B, Chen R, Song S, et al. ASPP2 inhibits tumor growth by repressing the mevalonate pathway in hepatocellular carcinoma. Cell Death Dis. 2019;10(11):830. doi:10.1038/s41419-019-2054-7
128. Jun SY, Brown AJ, Chua NK, et al. Reduction of Squalene Epoxidase by Cholesterol Accumulation Accelerates Colorectal Cancer Progression and Metastasis. Gastroenterology. 2021;160(4):1194–1207.e28. doi:10.1053/j.gastro.2020.09.009
129. Li C, Wang Y, Liu D, et al. Squalene epoxidase drives cancer cell proliferation and promotes gut dysbiosis to accelerate colorectal carcinogenesis. Gut. 2022;71(11):2253–2265. doi:10.1136/gutjnl-2021-325851
130. You W, Ke J, Chen Y, et al. SQLE, A Key Enzyme in Cholesterol Metabolism, Correlates With Tumor Immune Infiltration and Immunotherapy Outcome of Pancreatic Adenocarcinoma. Front Immunol. 2022;13:864244. doi:10.3389/fimmu.2022.864244
131. Kalogirou C, Linxweiler J, Schmucker P, et al. MiR-205-driven downregulation of cholesterol biosynthesis through SQLE-inhibition identifies therapeutic vulnerability in aggressive prostate cancer. Nat Commun. 2021;12(1):5066. doi:10.1038/s41467-021-25325-9
132. Liu D, Wong CC, Fu L, et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci Transl Med. 2018;10(437). doi:10.1126/scitranslmed.aap9840
133. Zhang E-B, Zhang X, Wang K, et al. Antifungal agent Terbinafine restrains tumor growth in preclinical models of hepatocellular carcinoma via AMPK-mTOR axis. Oncogene. 2021;40(34):5302–5313. doi:10.1038/s41388-021-01934-y
134. Zhang Z, Wu W, Jiao H, et al. Squalene epoxidase promotes hepatocellular carcinoma development by activating STRAP transcription and TGF-β/SMAD signalling. Br J Pharmacol. 2023;180(12):1562–1581. doi:10.1111/bph.16024
135. Oni TE, Biffi G, Baker LA, et al. SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. J Exp Med. 2020;217(9). doi:10.1084/jem.20192389
136. Yue S, Li J, Lee S-Y, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014;19(3):393–406. doi:10.1016/j.cmet.2014.01.019
137. Geng F, Cheng X, Wu X, et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin Cancer Res. 2016;22(21):5337–5348. doi:10.1158/1078-0432.CCR-15-2973
138. Chen Y, Yang X, Chen Y, et al. Impacts of the SOAT1 genetic variants and protein expression on HBV-related hepatocellular carcinoma. BMC Cancer. 2021;21(1):615. doi:10.1186/s12885-021-08245-1
139. Jiang Y, Sun A, Zhao Y, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567(7747):257–261. doi:10.1038/s41586-019-0987-8
140. Zhu Y, Gu L, Lin X, et al. P53 deficiency affects cholesterol esterification to exacerbate hepatocarcinogenesis. Hepatology. 2023;77(5):1499–1511. doi:10.1002/hep.32518
141. Chang CC, Sakashita N, Ornvold K, et al. Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine. J Biol Chem. 2000;275(36):28083–28092. doi:10.1074/jbc.M003927200
142. Huang Y, Jin Q, Su M, et al. Leptin promotes the migration and invasion of breast cancer cells by upregulating ACAT2. Cell Oncol Dordr. 2017;40(6):537–547. doi:10.1007/s13402-017-0342-8
143. Lu M, X-H H, Li Q, et al. A specific cholesterol metabolic pathway is established in a subset of HCCs for tumor growth. J Mol Cell Biol. 2013;5(6):404–415. doi:10.1093/jmcb/mjt039
144. Ioannou GN, Subramanian S, Chait A, et al. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J Lipid Res. 2017;58(6):1067–1079. doi:10.1194/jlr.M072454
145. Sharma B, Agnihotri N. Role of cholesterol homeostasis and its efflux pathways in cancer progression. J Steroid Biochem Mol Biol. 2019;191:105377. doi:10.1016/j.jsbmb.2019.105377
146. Delk SC, Chattopadhyay A, Escola-Gil JC, Fogelman AM, Reddy ST. Apolipoprotein mimetics in cancer. Semin Cancer Biol. 2021;73:158–168. doi:10.1016/j.semcancer.2020.11.002
147. Cao WM, Murao K, Imachi H, et al. A mutant high-density lipoprotein receptor inhibits proliferation of human breast cancer cells. Cancer Res. 2004;64(4):1515–1521. doi:10.1158/0008-5472.CAN-03-0675
148. Schörghofer D, Kinslechner K, Preitschopf A, et al. The HDL receptor SR-BI is associated with human prostate cancer progression and plays a possible role in establishing androgen Independence. Reprod Biol Endocrinol. 2015;13:88. doi:10.1186/s12958-015-0087-z
149. Zheng Y, Liu Y, Jin H, et al. Scavenger receptor B1 is a potential biomarker of human nasopharyngeal carcinoma and its growth is inhibited by HDL-mimetic nanoparticles. Theranostics. 2013;3(7):477–486. doi:10.7150/thno.6617
150. Yuan B, Wu C, Wang X, et al. High scavenger receptor class B type I expression is related to tumor aggressiveness and poor prognosis in breast cancer. Tumour Biol. 2016;37(3):3581–3588. doi:10.1007/s13277-015-4141-4
151. Schimanski S, Wild PJ, Treeck O, et al. Expression of the lipid transporters ABCA3 and ABCA1 is diminished in human breast cancer tissue. Horm Metab Res. 2010;42(2):102–109. doi:10.1055/s-0029-1241859
152. Smith B, Land H. Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Rep. 2012;2(3):580–590. doi:10.1016/j.celrep.2012.08.011
153. Aguirre-Portolés C, Feliu J, Reglero G, Ramírez de Molina A. ABCA1 overexpression worsens colorectal cancer prognosis by facilitating tumour growth and caveolin-1-dependent invasiveness, and these effects can be ameliorated using the BET inhibitor apabetalone. Mol Oncol. 2018;12(10):1735–1752. doi:10.1002/1878-0261.12367
154. Wang Y, Liu H, Ready NE, et al. Genetic variants in ABCG1 are associated with survival of nonsmall-cell lung cancer patients. Int J Cancer. 2016;138(11):2592–2601. doi:10.1002/ijc.29991
155. Demidenko R, Razanauskas D, Daniunaite K, Lazutka JR, Jankevicius F, Jarmalaite S. Frequent down-regulation of ABC transporter genes in prostate cancer. BMC Cancer. 2015;15:683. doi:10.1186/s12885-015-1689-8
156. Yuan J, Lv T, Yang J, et al. The lipid transporter HDLBP promotes hepatocellular carcinoma metastasis through BRAF-dependent epithelial-mesenchymal transition. Cancer Lett. 2022;549:215921. doi:10.1016/j.canlet.2022.215921
157. Li Z, Wang Y, Xing R, et al. Cholesterol Efflux Drives the Generation of Immunosuppressive Macrophages to Promote the Progression of Human Hepatocellular Carcinoma. Cancer Immunol Res. 2023;11(10):1400–1413. doi:10.1158/2326-6066.CIR-22-0907
158. Liao X, Song G, Xu Z, et al. Oxaliplatin resistance is enhanced by saracatinib via upregulation Wnt-ABCG1 signaling in hepatocellular carcinoma. BMC Cancer. 2020;20(1):31. doi:10.1186/s12885-019-6480-9
159. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. 2017;13(12):710–730. doi:10.1038/nrendo.2017.91
160. Cheng L, Deepak RNVK, Wang G, et al. Hepatic mitochondrial NAD + transporter SLC25A47 activates AMPKα mediating lipid metabolism and tumorigenesis. Hepatology. 2023;78(6):1828–1842. doi:10.1097/HEP.0000000000000314
161. Tang W, Zhou J, Yang W, et al. Aberrant cholesterol metabolic signaling impairs antitumor immunosurveillance through natural killer T cell dysfunction in obese liver. Cell Mol Immunol. 2022;19(7):834–847. doi:10.1038/s41423-022-00872-3
162. Mok EHK, Leung CON, Zhou L, et al. Caspase-3-Induced Activation of SREBP2 Drives Drug Resistance via Promotion of Cholesterol Biosynthesis in Hepatocellular Carcinoma. Cancer Res. 2022;82(17):3102–3115. doi:10.1158/0008-5472.CAN-21-2934
163. Wang Y, Viscarra J, Kim S-J, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol. 2015;16(11):678–689. doi:10.1038/nrm4074
164. Rough JJ, Monroy MA, Yerrum S, Daly JM. Anti-proliferative effect of LXR agonist T0901317 in ovarian carcinoma cells. J Ovarian Res. 2010;3:13. doi:10.1186/1757-2215-3-13
165. Srivastava S, Widmann S, Ho C, et al. Novel Liver X Receptor Ligand GAC0001E5 Disrupts Glutamine Metabolism and Induces Oxidative Stress in Pancreatic Cancer Cells. Int J Mol Sci. 2020;21(24):9622. doi:10.3390/ijms21249622
166. Yang B, Zhang B, Cao Z, et al. The lipogenic LXR-SREBF1 signaling pathway controls cancer cell DNA repair and apoptosis and is a vulnerable point of malignant tumors for cancer therapy. Cell Death Differ. 2020;27(8):2433–2450. doi:10.1038/s41418-020-0514-3
167. Rudalska R, Harbig J, Snaebjornsson MT, et al. LXRα activation and Raf inhibition trigger lethal lipotoxicity in liver cancer. Nat Cancer. 2021;2(2):201–217. doi:10.1038/s43018-020-00168-3
168. Lin Z, Xia S, Liang Y, et al. LXR activation potentiates sorafenib sensitivity in HCC by activating microRNA-378a transcription. Theranostics. 2020;10(19):8834–8850. doi:10.7150/thno.45158
169. Wagner N, Wagner KD. The Role of PPARs in Disease. Cells. 2020;9(11):2367. doi:10.3390/cells9112367
170. Pyper SR, Viswakarma N, Yu S, Reddy JK. PPARalpha: energy combustion, hypolipidemia, inflammation and cancer. Nuclear Receptor Signaling. 2010;8:e002. doi:10.1621/nrs.08002
171. Blunder S, Pavel P, Minzaghi D, Dubrac S. PPARdelta in Affected Atopic Dermatitis and Psoriasis: a Possible Role in Metabolic Reprograming. Int J Mol Sci. 2021;22(14):7354. doi:10.3390/ijms22147354
172. Kliewer SA, Xu HE, Lambert MH, Willson TM. Peroxisome proliferator-activated receptors: from genes to physiology. Recent Prog Horm Res. 2001;56:239–263. doi:10.1210/rp.56.1.239
173. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015;62(3):720–733. doi:10.1016/j.jhep.2014.10.039
174. Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. BBA. 2012;1821(5):809–818. doi:10.1016/j.bbalip.2011.10.016
175. Corton JC. Evaluation of the role of peroxisome proliferator-activated receptor alpha (PPARalpha) in mouse liver tumor induction by trichloroethylene and metabolites. Crit. Rev. Toxicol. 2008;38(10):857–875. doi:10.1080/10408440802209796
176. Fisher CD, Jackson JP, Lickteig AJ, Augustine LM, Cherrington NJ. Drug metabolizing enzyme induction pathways in experimental non-alcoholic steatohepatitis. Arch. Toxicol. 2008;82(12):959–964. doi:10.1007/s00204-008-0312-z
177. Takeuchi S, Matsuda T, Kobayashi S, Takahashi T, Kojima H. In vitro screening of 200 pesticides for agonistic activity via mouse peroxisome proliferator-activated receptor (PPAR)alpha and PPARgamma and quantitative analysis of in vivo induction pathway. Toxicol Appl Pharmacol. 2006;217(3):235–244. doi:10.1016/j.taap.2006.08.011
178. Li J, Huang Q, Long X, et al. CD147 reprograms fatty acid metabolism in hepatocellular carcinoma cells through Akt/mTOR/SREBP1c and P38/PPARα pathways. J Hepatol. 2015;63(6):1378–1389.
179. Han W, Wang N, Kong R, Bao W, Lu J. Ligand-activated PPARδ expression promotes hepatocellular carcinoma progression by regulating the PI3K-AKT signaling pathway. J Transl Med. 2022;20(1):86. doi:10.1186/s12967-022-03288-9
180. Xiong Z, Chan SL, Zhou J, et al. Targeting PPAR-gamma counteracts tumour adaptation to immune-checkpoint blockade in hepatocellular carcinoma. Gut. 2023;72(9):1758–1773. doi:10.1136/gutjnl-2022-328364
181. Inc. SB. A Phase 1, First-In-Human Study of Escalating Doses of Oral TVB-2640 in Patients With Solid Tumors; 2013. Available from: https://classic.clinicaltrials.gov/show/NCT02223247.
182. Inc. SB. Study of TVB 2640 in Subjects With Non-Alcoholic Steatohepatitis (NASH); 2019. Available from: https://classic.clinicaltrials.gov/show/NCT03938246;.
183. Inc. SB. Study of TVB-2640 in Subjects With Nonalcoholic Steatohepatitis (NASH); 2019. Available from: https://classic.clinicaltrials.gov/show/NCT04906421.
184. Center UoTSM. EGCG for Hepatocellular Carcinoma Chemoprevention; 2023. Available from: https://classic.clinicaltrials.gov/show/NCT06015022.
185. Institute NC. Simvastatin in Preventing Liver Cancer in Patients With Liver Cirrhosis; 2017. Available from: https://classic.clinicaltrials.gov/show/NCT02968810.
186. Chung R, Center Uo TSM, Hospital MG Safety and Efficacy of Atorvastatin v. Placebo on HCC Risk; 2023. Available from: https://classic.clinicaltrials.gov/show/NCT05028829;.
187. Hospital CC, Hospital E-D, Hospital NTU, et al. Statin for Preventing Hepatocellular Carcinoma Recurrence After Curative Treatment; 2017. Available from: https://classic.clinicaltrials.gov/show/NCT03024684;.
188. University Hospital G, Ghent U, Anticancer Fund B Dose-Escalation Study Evaluating the Safety and Pharmacokinetics of Artesunate in Patients With Hepatocellular Carcinoma; 2014. Available from: https://classic.clinicaltrials.gov/show/NCT02304289;.
189. University OS. Xanthohumol and Prevention of DNA Damage; 2015. Available from: https://classic.clinicaltrials.gov/show/NCT02432651;.
190. Antonio TUoTHSCaS. TVB- 2640 in Combination With Bevacizumab in Patients With First Relapse of High Grade Astrocytoma; 2017. Availablr from: https://classic.clinicaltrials.gov/show/NCT03032484.
191. Evers M, Institute NC, Kentucky U. TVB 2640 for Resectable Colon Cancer Other Resectable Cancers; a Window Trial. 2017. Available from: https://classic.clinicaltrials.gov/show/NCT02980029;.
192. Gerber DE, Sb I, Texas CPRIo, Center UoTSM. Phase 2 Study of TVB-2640 in KRAS Non-Small Cell Lung Carcinomas; 2019. Available from: https://classic.clinicaltrials.gov/show/NCT03808558.
193. University WMCoC, Inc. SB. Study of TVB-2640 in Men With Metastatic Castration-Resistant Prostate Cancer; 2023. Available from: https://classic.clinicaltrials.gov/show/NCT05743621.
194. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16(7):411–428. doi:10.1038/s41575-019-0145-7
195. Abdel-Rahman O. Statin treatment and outcomes of metastatic pancreatic cancer: a pooled analysis of two Phase III studies. Clin Transl Oncol. 2019;21(6):810–816. doi:10.1007/s12094-018-1992-3
196. Miyata S, Inoue J, Shimizu M, Sato R. Xanthohumol Improves Diet-induced Obesity and Fatty Liver by Suppressing Sterol Regulatory Element-binding Protein (SREBP) Activation. J Biol Chem. 2015;290(33):20565–20579.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.