")
Back to Journals » Journal of Inflammation Research » Volume 14
Interferon-Driven Immune Dysregulation in Down Syndrome: A Review of the Evidence
Authors Chung H , Green PH, Wang TC, Kong XF
Received 23 July 2021
Accepted for publication 22 September 2021
Published 7 October 2021 Volume 2021:14 Pages 5187—5200
DOI https://doi.org/10.2147/JIR.S280953
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Howard Chung,1,2 Peter HR Green,1,3 Timothy C Wang,1 Xiao-Fei Kong1,3
1Division of Digestive and Liver Diseases, Department of Medicine, Columbia University Irving Medical Center, New York, NY, 10032, USA; 2Department of Internal Medicine, Icahn School of Medicine at Mount Sinai/Queens (Queens Hospital Center), Jamaica, NY, 11432, USA; 3Celiac Disease Center, Department of Medicine, Columbia University Medical Center, New York, NY, 10032, USA
Correspondence: Xiao-Fei Kong
Division of Digestive and Liver Diseases, Department of Medicine, Columbia University Irving Medical Center, 622 West 168th ST, PH7W Suite 318, New York, NY, 10032, USA
Email [email protected]
Abstract: Down syndrome (DS) is a unique genetic disease caused by the presence of an extra copy of chromosome 21, which carries four of the six interferon receptor (IFN-R) genes on its long arm. Recent studies reporting higher levels of interferon-stimulated gene (ISG) expression in primary immune cells studied ex vivo have suggested that the additional copies of the IFN-R genes in DS result in mild interferonopathy. In this review, we analyze the potential clinical and immunological impacts of this interferonopathy in DS. We performed a literature review to explore the epidemiology and risks of celiac disease, type 1 diabetes, thyroid dysfunction, mucocutaneous manifestations, infectious diseases (including COVID-19), and Alzheimer’s disease in individuals with DS relative to the general population with or without iatrogenic exposure to interferons. We analyzed immunophenotyping data and the current experimental evidence concerning IFN-R expression, constitutive JAK-STAT activation, and ISG overexpression in DS. Despite the lack of direct evidence that implicating this mild interferonopathy directly in illnesses in individuals with DS, we highlight the challenges ahead and directions that could be taken to determine more clearly the biological impact of interferonopathy on various immune-related conditions in DS.
Keywords: down syndrome, interferon receptors, JAK-STAT, celiac disease, T cells, gene dosage effect
Introduction
Down syndrome (DS) is the most common chromosomal abnormality, causing a wide spectrum of illnesses affecting multiple organ systems. According to the Centers for Disease Control and Prevention (CDC),1 approximately 6000 babies are born annually with DS in the US, accounting for 1 in 691 live births, corresponding to a 30% increase over the last four decades.2 Collaboration between families, physicians, scientists, and federal agencies is required to improve the clinical care of individuals with DS. Physicians and families are frequently faced with medical problems and questions concerning individuals with DS, and there is, therefore, an urgent need for caregivers to resolve these uncertainties, and find solutions rapidly. The most common clinical phenotypes in DS include intellectual disability and developmental delays, the severity of which vary between individuals. DS also causes many other abnormalities, including congenital heart disorders and gastrointestinal defects.2–4 Immune disorders have frequently been reported in DS (Figure 1).5–7 However, it remains unclear why individuals with DS are more prone to celiac disease (CeD), for example. The basis of their higher risk of developing mucocutaneous infection and inflammations, thyroid abnormalities, type 1 diabetes, and leukemia also remains unknown, and the best way to prevent and manage these conditions in DS is also unclear.
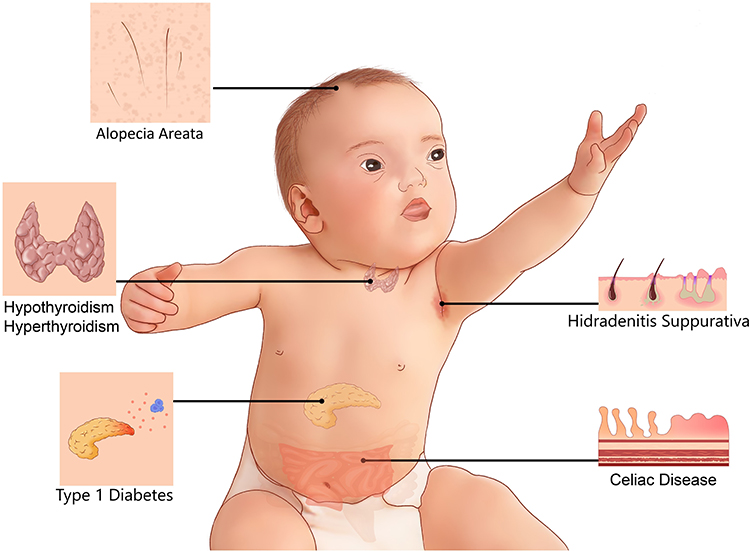 |
Figure 1 Common immune system-mediated disorders in DS. |
The pioneers in the field, Jerome Lejeune and his colleagues, discovered the genetic etiology of DS through their use of early cell culture technologies and karyotype analysis. In 1959, they reported the presence of an extra copy of human chromosome 21 (HSA21) in individuals with DS.8 They named this condition trisomy 21, and this discovery has served as the foundation of our understanding of DS pathophysiology for the last 60 years. It has been suggested that the wide spectrum of clinical phenotypes in DS is due to a dosage effect of genes on HSA21. The current working paradigm is that the clinical phenotype emerges once the cumulative levels of mRNA/protein corresponding to three alleles of genes on HSA21 surpass a critical level. However, individuals with DS vary considerably in terms of clinical phenotype, which can be unpredictable. It has been suggested that this variability is due to interindividual allelic differences and the indirect impact of non-HSA21 genes.2,4,9,10 Unraveling the precise biological mechanism underlying each clinical phenotype in DS is much more challenging than elucidating the mechanisms underlying monogenic Mendelian disorders, due to the possible involvement of multiple genetic factors.
The key to understanding the pathophysiology of DS is establishing a direct link between the clinical phenotypes and specific cellular and immunological alterations. Historically, DS patients have proved an important group for deciphering the molecular basis of human illness. Baruch S. Blumberg, a populational geneticist and Nobel Prize winner, studied the Australian antigen, which was initially thought to be a genetically encoded serological polymorphism. In a study of a population of DS patients in 1967, it became increasingly evident that the Australian antigen was responsible for hepatitis B, overcoming the existing uncertainty and ruling out the previously suspected association between the Australian antigen and leukemia, which was frequently observed in DS.11 In 1976, before the advent of the Human Genome Project, interferon-induced antiviral states were reported in human skin fibroblasts from individuals with DS.12,13 Cells with trisomy 21 were three to seven times more responsive to interferon, resulting in higher levels of viral protection than were observed in normal diploid fibroblasts or fibroblasts with trisomy 13 or 18. With further technological advances, it has become possible to measure immunological phenotypes precisely, by systemic approaches.14–16 The greater sensitivity to interferon observed in DS has now been confirmed in multiple studies from several independent laboratories (Figure 2).14,15,17 However, individuals with DS have a paradoxical susceptibility to certain viral infections, with higher mortality rates due to SARS-CoV2 and respiratory syncytial virus (RSV) infections.18–23 The mismatch between these immunological findings and the clinical phenotypes of individuals with DS has remained puzzling, rendering the development of new treatments more challenging.
 |
Figure 2 Mild interferonopathy in Down syndrome. Left: Diagram of the type I and type II IFN-activated JAK-STAT pathway. IFNAR1, IFNAR2, IFNGR2 are located on HSA21. ISGF3 is a DNA-binding complex formed by STAT1/STAT2/IRF9; GAF is a STAT1/STAT1 homodimer. Right: summary of observations for the testing of serum, monocytes, EBV-transformed B cells, and T cells from euploid healthy controls and individuals with DS. Abbreviation: ISGs, interferon-stimulated genes. |
We present here the results of a comprehensive review of the available clinical, immunological, and molecular evidence for interferonopathy in DS and highlight possible future directions for extracting the maximal benefit to the DS community from research findings.
Interferon- and Immune-Driven Clinical Phenotypes in DS
We conducted a review of the literature concerning celiac disease (CeD), type 1 diabetes, thyroid dysfunction, mucocutaneous problems, infectious diseases and other medical problems in individuals with DS, to improve the dissection of immune-mediated phenotypes and interferonopathy in DS. We include adverse effects of interferon treatment in euploid individuals in this analysis for comparison, to provide clinical insight into these manifestations (Table 1).
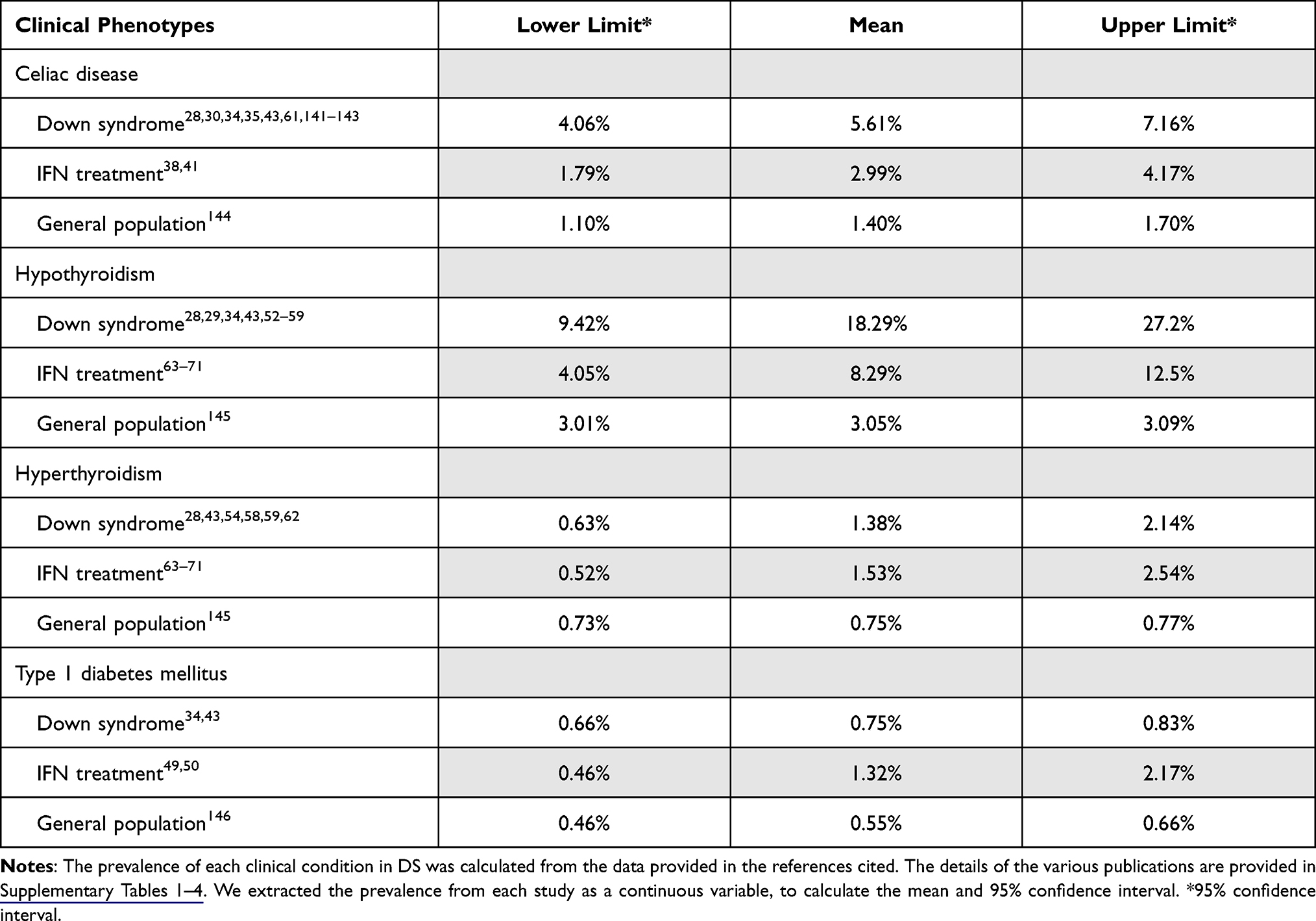 |
Table 1 The Prevalence of Celiac Disease, Hypothyroidism, Hyperthyroidism, and Type 1 Diabetes Mellitus in Individuals with Trisomy 21 and in the General Population with and without IFN Treatment |
Celiac Disease in DS
Celiac disease occurs at a significantly higher frequency among individuals with DS than in the general population.24–29 Based on the findings of 39 studies published in the past 30 years, 274 of the 7003 DS patients included were diagnosed with CeD, corresponding to an estimated prevalence of 5.61% (95% CI, 4.06–7.16%). Children and adolescents (age <15 years old) accounted for 25% of reported DS-CeD cases. The most common symptoms of DS-CeD are diarrhea, abdominal bloating, weight loss, anemia, failure to thrive, vomiting, and anemia. Four studies reported that a quarter of patients (11/40) were asymptomatic at the time of CeD diagnosis, CeD being diagnosed through screening.30–33 Thyroid diseases are reported as common coexisting conditions in 27.8% to 77.8% of DS-CeD patients.30,31,34 CeD was diagnosed by tissue biopsy in 32 of the 39 studies on DS-CeD. It remains a matter of debate whether positive serological tests are sufficient for CeD diagnosis in individuals with DS, given the broad range of gluten-related problems.27,35 Current guidelines recommend that initial testing for CeD should be based on serology, but that diagnosis requires a duodenal biopsy and histological analysis for confirmation.36 Three serological tests are widely used for CeD screening. They are based on anti-deamidated gliadin peptide antibodies (DGP), anti-endomysial antibodies (EMA), or anti-tissue transglutaminase antibodies (tTG). Anti-DGP IgA is more sensitive for CeD screening than IgG, however, but both anti-EMA and anti-tTG antibodies are more specific than Anti-DGP IgA.27,32,35 IgA deficiency is rare in individuals with DS, with only one case reported to date.26 In four studies including both DS patients and healthy control groups, the DS patients had significantly higher rates of abnormal CeD-related antibody levels than the control group.24–27 HLA DQA1*0501 /DQB1*0201 were the most frequent alleles reported in both DS-CeD patients and euploid patients with CeD.27,35,37 In karyotype analyses, only two patients with DS mosaicism have been reported to have CeD. Symptomatic DS-CeD patients display clinical and histological improvements, with the disappearance or decrease in levels of anti-DGP Abs, on a gluten-free diet.29,31,37 Interferon has been reported to play an important role in CeD pathogenesis.38–41 Clinical evidence in support of this notion is provided by the reported higher risk of developing CeD Ab and pathological features in patients treated with interferon, although only nine interferon-induced cases of CeD have been described.38,41 Additional studies are required to determine the relationship between interferon and DS-CeD.
Type 1 Diabetes (T1D) in DS
Type 1 diabetes occurs in about 0.7% of individuals with DS.34,42–45 A populational-based study reported a prevalence of T1D of 0.71% in DS patients, a value 4.2 times higher than that for the general population (0.17%), according to the data in the Danish national database.42 T1D tended to be diagnosed earlier in DS patients, with 22% of those affected diagnosed before the age of two years, versus only 7% in the general population.44 Concurrent T1D and thyroid dysfunction were reported in 29.9% of DS cases.46 In total, 27 of 117 (23%) individuals with DS and T1D were found to be positive for DR4-DQ8/DR3-DQ2, versus 39.8% of euploid patients with T1D.47,48 Patients on IFN treatments have been reported to have a higher rate of autoantibody (directed against GAD-65 and other proteins) production (4.5%) than untreated individuals, resulting in a 0.5% chance of developing T1D.49,50 Patients with IFN-related T1D patients tend to have a higher C-peptide level and a lower insulin requirement.51 IFN-related T1D should be differentiated from T1D in general. However, the mechanism underlying interferon-related T1D remains unclear.
Thyroid Dysfunction in DS
Hypothyroidism, encompassing subclinical hypothyroidism, congenital hypothyroidism, and acquired hypothyroidism, is the most common thyroid dysfunction in DS. In about four thousand individuals with DS from 12 different studies, 18.29% had been diagnosed with hypothyroidism.28,29,34,43,52–59 A retrospective study of 1453 DS patients indicated that these patients have a nine times higher risk of acquired hypothyroidism than individuals without DS.28 Subclinical hypothyroidism is a common manifestation of DS in young individuals.43,54 Individuals with DS who are positive for auto-antibodies (against microsome, TPO or thyroglobulin) have a higher risk of acquired hypothyroidism.54 Routine serum TSH determinations are recommended in young DS patients, because early treatment with thyroxine can effectively prevent abnormalities of mental development or growth.52,54,60,61 DS patients also have a higher risk of hyperthyroidism than euploid individuals, with a prevalence of 0.71% according to data from five retrospective studies.28,43,54,58,62 Most patients with hyperthyroidism tested positive for anti-TPO antibody and thyroid-stimulating immunoglobulin. In addition to these abnormal laboratory results, thyrotoxicosis, diagnosed on the basis of an increase in diffuse uptake on thyroid scans and clinical presentations, may also occur in individuals with DS. Interferon treatment-related thyroid dysfunction has been widely studied,63–71 with hypothyroidism detected in 7.5% and hyperthyroidism in 2.6% of patients treated with interferon.69 All these data suggest that IFN may play a unique role in the thyroid abnormalities observed in DS.
Infections and Mucocutaneous Abnormalities in DS
The prevalence of orofacial soft-tissue abnormalities is higher in individuals with DS than in the general population.72 Common lesions include cheilitis, fissured tongue, lingual papillary hypertrophy, angular cheilitis, and lip fissures.57,73,74 Most of the lip fissures concern the lower lip and the midline.57,72 Individuals with DS are also at higher risk of skin abnormalities, including onychomycosis (4.4–67.8%), syringoma (6–39.2%), seborrheic dermatitis (8–36%), folliculitis (10.3–26%), alopecia areata (1.4–20%), hidradenitis suppurativa (2.1–15%) livedo reticularis (2–12.6%) psoriasis (0.5–8%), keratosis pilaris (2.3–4%), and vitiligo (1.9–3%).74–76 Individuals with DS are also highly susceptible to candidiasis. The oral-mucosal lesions observed include erythema and white pseudomembrane in 22 (40%) individuals with DS in one study, but only one member of the control group.77 Positive culture results for candida are more frequent for isolates from lip lesions and/or angular cheilitis in individuals with DS than for isolates from individuals without oral lesions (77.27% vs 29.41%).73 In four separate studies, 140 Candida albicans-positive cultures were obtained following oral swab tests on 262 individuals with DS.73,77–79 Greater susceptibility to bacterial and viral infections, including streptococcal infections80 and RSV infection,22,81 has also been noted in individuals with DS. Nevertheless, there is still no sufficiently large cohort of individuals with DS to determine the overall prevalence of these infections.
Individuals with DS have been reported to be particularly susceptible to COVID-19. Retrospective studies have shown that individuals with DS are more likely to develop complications of COVID-19, including altered mental status, superinfection, and sepsis.19,23 Mechanical ventilation due to acute respiratory distress syndrome is frequently required in individuals with DS suffering from COVID-19 (41.7%), as shown by our study performed in New York City.23 Consistent with this greater severity of COVID-19 in individuals with DS, COVID-19-related mortality is also higher in these individuals, with death occurring at a younger age.20–22 Individuals with DS have been reported to have a four times higher risk of hospitalization and a 10 times higher risk of death from SARS-CoV-2 infection than individuals for the general population.20 DS patients were more frequently treated with systematic steroids, potentially due to the high levels of inflammatory markers found in DS patients with COVID-19 pneumonia.19 The mechanism underlying the poorer clinical outcomes of COVID-19 in DS patients remains unclear, and future evaluations are required to evaluate the protective effect of the vaccine in this particular population.
Arthropathy and Alzheimer’s Disease in DS
Arthropathy was reported in about 2 in 1000 of individuals with DS, with an onset at ages ranging from 0.3 to 20 years. Arthropathy of DS (A-DS) has been proposed as a term to differentiate this condition from juvenile idiopathic arthritis (JIA) in the general population.82,83 Individuals with A-DS present with more small joints involvement, but lower levels of inflammatory markers including C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR), than euploid individuals with JIA.83 Cases with interferon-induced arthritis have been reported, together with a type I IFN signature in rheumatoid arthritis,84,85 but the contribution of IFN to A-DS remains unclear. Early screening and interventions are the key preventive approaches for A-DS. Another inflammation-related neurological disorder, Alzheimer’s disease, is diagnosed in 30% of individuals with DS in their fifties and 50% of those in their 60s.86,87 The amyloid-beta protein (Aβ) gene is located on HSA21, and interferon and other neuroinflammatory cytokines may affect Aβ production and metabolism by regulating β- and γ-secretase activities.88 An ISG signature has been detected in the brain from multiple mouse models of Alzheimer’s disease. Nucleic acid-containing amyloid fibrils can lead to microglial activation, microgliosis, and synapse loss.89–91 Studies in the IFN-R knockout model have suggested that the IFN pathway plays a key role in the neuroinflammatory network in Alzheimer’s disease. Future studies to identify the potential clinical and biological risk factors, such as interferon score, for early-onset Alzheimer’s disease in DS will be beneficial not only to individuals with DS, but to society as a whole (Figure 3).
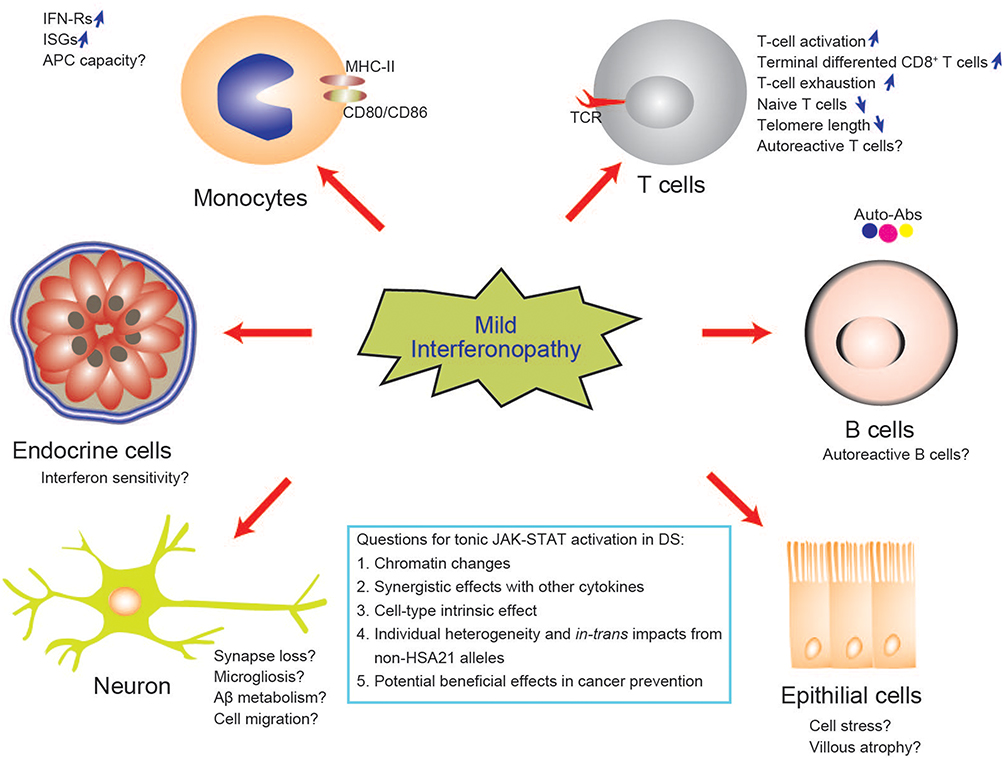 |
Figure 3 Biological implications and potential mechanisms of mild interferonopathy in DS. Interferons have pleiotropic effects on multiple organs and different effects on different cell types. IFN can drive T-cell activation and differentiation which is considered to be the third signal for T-cell development, in addition to the TCR and the costimulatory receptor. The tissue and temporal effects of interferonopathy in DS remain to be explored. Abbreviation: APC, antigen-presenting cells. |
Interferon Receptors Gene Dosage Effects of in DS
Four of the 225 genes on chromosome 21—IFNAR1, IFNAR2, IFNGR2, and IL-10RB—form a cluster on the long arm of the chromosome and are essential for JAK-STAT activation and cellular responses to all three types of interferon.3,17 Type I interferon, of which there are 13 different subtypes, requires IFN-αR1 and IFN-αR2 for activity.92 IFN-γR2 is the limiting factor for type II interferon or IFN-γ (Figure 2).93 Type III interferons, including IFNL1 (IL29), IFNL2 (IL28A), IFNL3 (IL28B), and IFNL4, require IL-10RB.94 The biological importance of these four genes and the phenotypes associated with them have been deciphered in studies of human autosomal recessive monogenic immunodeficiencies, with IFNAR1 and IFNAR2 deficiencies causing severe viral infection,95–98 IFNGR2 deficiency causing MSMD,99,100 and IL-10RB deficiency causing very early onset inflammatory bowel disease (VEO-IBD).101 The haploinsufficiency of the IFNGR2 gene provided information about the basis for understanding the gene dosage effect of IFN-R in trisomy 21. This haploinsufficiency is cell-type dependent, affecting naïve CD4+ T cells and B cells, but not monocytes or monocyte-derived macrophages, accounting for the low clinical penetrance of IFNGR2 haploinsufficiency.93 In determinations of the gene dosage effects of three copies of this cluster of interferon receptors on phenotypic manifestations, both transcriptional and post-transcriptional regulation must be taken into account, to unravel cell type-dependent impacts.
High Levels of IFN-Rs mRNA and Protein in DS
DS has been considered to be a disorder of gene transcription, with about 1.5 times the usual level of transcription for the genes located on HSA21.3 The transcriptomic analyses of DS cells have been revolutionized with the advances in mRNA quantification, from northern blotting, hybridization, and fluorescence-based quantitative PCR to next-generation sequencing techniques, including RNA-Seq and single-cell RNA-sequencing in particular, which are now widely used.102,103 There is growing evidence to suggest that the transcript levels of IFN-Rs, including IFNAR1, IFNAR2, IFNGR2 and IL-10RB, in the monocytes, T cells, whole blood, fibroblasts, EBV-transformed B lymphocytes (EBV-B), and induced pluripotent stem (iPS) cells of DS patients are significantly higher than those in euploid individuals.14,15,102 However, the difference in IFN-R mRNA levels between the DS and healthy controls may vary considerably, depending on the methods of transcriptome analysis used.102 One major concern is that the transcripts levels are not always predictive of protein levels and posttranscriptional modifications. As a means of circumventing this problem, we demonstrated, by FACS, that the levels of IFN-αR1, IFN-αR2, and IFN-γR2 proteins on the cell surface of EBV-B cells and monocytes from individuals with DS patients were higher than those in healthy controls, whereas IL-10RB protein levels were similar in individuals with DS and healthy controls.17 Nevertheless, not all cell types display IFN-R gene dosage effects. A Cyto-F analysis showed that 85 of 100 primary immune cell types from individuals with DS had significantly higher levels of IFN-αR1 expression at the cell surface than were observed in the corresponding cells from healthy controls.16 Nevertheless, determinations of IFN-R levels, either ex vivo or in vitro, are not sufficient for any firm conclusions to be drawn about biological implications. First, the biological effects of IFN-Rs require agonists, downstream Janus kinases including JAK1, JAK2, and Tyk2, and signal transduction and activation of transcription factors,92 including STAT1, STAT2, and STAT3, together with a JAK-STAT-independent pathway. Second, there is crosstalk between type I and type II interferon and synergistic effects between IFN-αR1 and IFN-γR2.104 Third, IFN-R proteins may be glycosylated,99 degraded via a pathway involving USP18 and ISG15,105,106 or distributed between different cytoplasmic regions.107 All these features may affect the response to interferons, suggesting that the assessment of interferon-driven activities in humans requires a systemic approach.
Mild Interferonopathy in DS
An interferon signature, revealing an upregulation of interferon-stimulated genes(ISGs), was first reported in patients with systemic lupus erythematosus (SLE) and was found to be associated with disease severity,108 with 15 ISGs identified as biomarkers of SLE severity. Such signatures have since been reported in many autoimmune conditions.109 Another related term, “Type I Interferonopathies”, was first proposed by Dr. Yanick J. Crow in 2011, based on his studies of patients with monogenic Aicardi-Goutières syndrome (AGS).110,111 The main clinical features of AGS include abnormal neurological findings, intracranial calcification, high level of liver enzymes, and thrombocytopenia. Crow’s group used six ISGs to generate an interferon score as a biomarker for screening for interferonopathy, and for determining the pathophysiological elements common to different genetic etiologies.112 The group of Dr. Joaquin M. Espinosa identified ISG signatures in individuals with DS in 2016, in RNA-Seq-based transcriptomic analyses of fibroblasts, EBV-B cells, monocytes, and T cells from several individuals with DS. Using a systemic approach, they clearly demonstrated that ISG mRNA levels were upregulated in these cell types from DS individuals, but in a cell-type specific manner. Whilst the author (X.F Kong) was working in Dr. Jean-Laurent Casanova’s group, we initiated a study on DS in 2011, after discovering human gain-of-function STAT1 disorders and IFNGR2 haploinsufficiency.93,113 The initial hypothesis tested was that the three copies of interferon receptors genes in DS patients might act as a phenocopy of STAT1 gain-of-function, predisposing DS patients to mucocutaneous candidiasis with impaired Th17 immunity. Monocytes are the major PBMC cell type in which the level of IFN-R expression are highest. We demonstrated high levels of monocyte ISG expression in 14 individuals with DS, and revealed the occurrence of an independent cluster with the interferon modular signal, intermediate between the values for healthy controls and patients with gain-of-function STAT1 mutations. We also observed that type I interferon levels were high in 12.5% of individuals with DS (Figure 2). However, neither serum type I interferon level nor the ISGs signature was associated with the clinical infectious phenotypes, such as mucocutaneous Candida albicans and Staphylococcus aureus infection.17 We concluded that interferon overactivation in DS is milder than that in patients with STAT1 gain of function, monogenic type I interferonopathies, or SLE.
Interferon-Related Immunological Phenotypes in DS
Immunological dysfunctions were first reported in individuals with DS more than 40 years ago,114 and several review articles have focused on innate and acquired immunity in DS.115,116 Here, we will focus on two main topics relating to interferon: T-cell development and immunosenescence in DS.
T-Cell Abnormalities May Be Related to IFN-Rs
Studies on human primary immunodeficiency disorders and mouse models have demonstrated the essential immunomodulatory role of interferons in T cells.117 The T-cell abnormalities most frequently observed in individuals with DS include a low thymic output and low levels of naïve T cells, an altered CD4/CD8 ratio, limited proliferation in response to PHA stimulation, high levels of expression for cell-surface markers related to T-cell activation and senescence, and high levels of IFN-γ production, and terminally differentiated CD8+ T cells (Figure 3).5,15–17,116 The T-cell phenotypes observed in DS mimic those observed in chronic viral infections with persistent interferon exposure.118 With the recent development of a systematic approach for immunophenotyping based on CyTOF, more details have emerged about each subset of T cells and other immune cells, including their cell-surface marker expression, and cytokine production profiles.15,16 Exhausted CD8+ T-cell phenotypes are also observed in other primary immunodeficiencies presenting with clinical manifestations due to EBV infection, such as DOCK8, STK4 and PIK3CD deficiencies.119 Overall, CyTOF profiling has provided no additional evidence to suggest that the gene dosage effect of IFN-Rs causes the T-cell phenotypes observed in DS. Intriguingly, none of the patients with inherited JAK-STAT overactivation or interferon hyperresponsive disorders, such as those with gain-of-function JAK1,120,121 STAT1,113 STAT2,122 or STAT3123 mutations, has been reported to display a similar immunophenotype. Additional studies are required to determine the association between these T-cell abnormalities and autoimmune phenotypes in DS, and to provide additional insight into the incomplete clinical penetrance of infectious and autoimmune diseases in DS.
Clinically, individuals with DS displayed no particular susceptibility to the common viral infections — with EBV, CMV, and HSV, for example, — frequently observed in patients with primary T-cell defects.124 The high levels of COVID-19-related morbidity and mortality in individuals with DS are intriguing, and several speculative hypotheses have been proposed.18 However, further investigation of the mechanisms involved will be essential to understand the reasons for the severity of COVID-19 in these individuals. A recent study on two DS children with COVID-19 revealed an atypical immune response, with high levels of STAT3 phosphorylation in various cell types.125 A mouse model would be useful for deciphering the contributions of IFN-Rs to T-cell abnormalities in DS. Several mouse models of DS, including Ts65Dn, Ts1Yey, Ts1Cje, and Tc1 have been widely used to study the pathophysiology of trisomy 21. However, all of these mouse models have genetic and phenotypical imperfections, as discussed in a recent review.2 A previous study revealed that mouse trisomy 16 lacking one functional IFNAR1 and an IFNGR1 allele had significantly faster growth and greater cortical neuron viability.126 In a mouse model (Dp16/Ts1Yey) of DS, TLR3 agonists can induce a lethal response with acute liver injury that can be attenuated by treatment with a JAK inhibitor.127 Mice with various numbers of functional IFN-R copies could be generated, to help to clarify whether the T-cell phenotypes observed are dependent on type I or type II interferon. Various in vitro methods have been used to investigate the cellular effects of differential levels of IFN-R expression, using shRNA, CRISPR, or even tetracycline-inducible systems for regulating IFNAR-1/2 expression in human fibroblasts. For example, indoleamine 2,3-dioxygenase (IDO1), an ISG that is crucial for tryptophan metabolism, has been reported to be sensitive to IFN-R dosage.128 Other rare scenarios, such as individuals with DS carrying one or two loss-of-function IFN-R alleles,129 might explain the causal relationship between IFN-R dosage effects and T-cell abnormalities. Further investigation, including genomic sequencing in DS patients with various chronic inflammatory phenotypes, is required to explore these possibilities.
The Immunological Enigma in DS
The complexity of the human immune system presents a challenge for both physicians and scientists. There is currently no single clinical test for evaluating the immunological status of any given individual, but technologies are improving.130 Clinicians use blood counts, T-cell counts, levels of immunoglobulins and interleukins, CRP, ESR, and other available markers to obtain a general impression of the patient’s immunological status, and to determine infection, inflammation, or autoimmune disease status. Systemic immunophenotyping has revealed a global dysregulation of the immune system in DS, with increases in the levels of fibrocytes, conventional CD1c+ dendritic cells, hyperactivated NK and cytotoxic T cells, and memory B cells.16 However, translating this information into a better clinical understanding of susceptibility to particular illnesses in DS remains a challenge. For example, 28.6% of individuals with DS tested positive rheumatoid factors (RF), but displayed no clinical evidence of rheumatic disease.131 This suggests that some immunological findings may be of little, if any, relevance to pathogenesis. Desensitization to interferon has also been proposed, based on the observation that prolonged interferon stimulation can lead to an expression of negative regulators of JAK-STAT, such as ISG15 or USP18,105,106 leading to a downregulation of IFN-αR2-dependent JAK1 activation.132,133 However, ex vivo analyses of primary immune cells from individuals with DS have shown that most of the cell types analyzed maintained hyperresponsive to IFN.15,16 It is unclear whether such phenomena occur in pathogenic states, such as acute SARS-CoV-2 infection and prolonged pneumonia and inflammation.
Another enigma concerns the nuclear transcription factors involved in modulating the transcriptional disorder in DS. Different cell types have different, specific patterns of chromatin binding and of expression for IFN-γR2 and STATs.134 CD4+ naïve T cells have the highest STAT1 level, whereas CD8+ TERMA T cells have the lowest levels of STAT1 expression, but the highest levels of T-bet and STAT4 expression of the various CD3+ T-cell subsets.15,17 Genes for two other important immune system-related transcription factors are also located on HSA21: AIRE and RUNX1. AIRE-deficient patients have APECED syndrome and are prone to autoimmune diseases due to the production of various antibodies. The impact of an additional copy of AIRE and RUNX1 on immune development and chromatin structure remains unclear. In addition, the interferon system is complicated by the synergistic effects of type I and type II interferons and other cytokines, and by temporal and cell type-specific effects.135 Given all these complexities, further experiments based on a reductionistic approach are required to decipher the mechanisms involved. Such an approach has already revealed that DS provides protection against colon cancer and polyposis in the APC mutant mouse model (Figure 3).136 This observation may pave the way for improvements in our understanding of protective immunity to gastrointestinal cancer.
Future Directions for Evaluating the Clinical Impact of Interferonopathy in DS
Following the observation of ISG signatures in DS, a clinical trial has been launched to assess the benefits of a JAK inhibitor, tofacitinib for treating alopecia and other skin disorders by targeting JAK-STAT pathway in individuals with DS.137 A few individuals with DS have been successfully treated with JAK inhibitor.138,139 However, a clear association between clinical phenotype and ISG signature in DS patients has yet to be established. Is the presence of alopecia or other immune-mediated clinical phenotypes in DS driven by JAK-STAT overactivation? Is there a simple way to measure the severity of interferonopathy in DS? Is the overexpression of particular ISGs phenotype-specific in DS? Which cell types should be tested to explore this association? We are still far from having a reliable biomarker for the use of ISG mRNAs to guide precise clinical care in DS patients. From a biological perspective, the key is understanding the mechanisms controlling ISG transcription in DS: tonic JAK-STAT signaling or delayed pulse-dependent transcription (Figure 3)?140 What impact do DNA methylation and histone modification have on transcriptional regulation? More collaborative multicenter studies are now required to increase power and shed light on the clinical utility of ISG signatures in DS, with a view to developing more effective future treatments, given that interferons may also have neurotoxic effects.
Acknowledgment
We thank Drs. Jean-Laurent Casanova, Benjamin Lebwohl, Benjamin Tycko, Beishi Zheng, Bing Chen for careful reading of the manuscript and suggestions. Dr. Xiao-Fei Kong has been supported by K08 DK128631 NIDDK, and grants from Jerome Lejeune Foundation and Alexander’s Angels Inc.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Centers for Disease Control and Prevention. Data and statistics on Down syndrome. Available from: https://www.cdc.gov/ncbddd/birthdefects/downsyndrome/data.html.
2. Antonarakis SE, Skotko BG, Rafii MS, et al. Down syndrome. Nat Rev Dis Primers. 2020;6(1):9. doi:10.1038/s41572-019-0143-7
3. Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. Chromosome 21 and Down syndrome: from genomics to pathophysiology. Nat Rev Genet. 2004;5:725–738. doi:10.1038/nrg1448
4. Letourneau A, Santoni F, Bonilla X, et al. Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature. 2014;508:345–350. doi:10.1038/nature13200
5. Ram G, Chinen J. Infections and immunodeficiency in Down syndrome. Clin Exp Immunol. 2011;164:9–16. doi:10.1111/j.1365-2249.2011.04335.x
6. Kusters MAA, Verstegen RHJ, Gemen EFA, de Vries E. Intrinsic defect of the immune system in children with Down syndrome: a review. Clin Exp Immunol. 2009;156:189–193. doi:10.1111/j.1365-2249.2009.03890.x
7. Levo Y, Green P. Down’s syndrome and autoimmunity. Am J Med Sci. 1977;273:95–100. doi:10.1097/00000441-197701000-00010
8. Lejeune J, Gautier M, Turpin R. A study of somatic chromosomes in nine infants with mongolism. CR Acad Sci. 1959;248:1721–1722.
9. Gardiner K, Costa ACS. The proteins of human chromosome 21. Am J Med Genet C Semin Med Genet. 2006;142:196–205. doi:10.1002/ajmg.c.30098
10. Lana-Elola E, Watson-Scales SD, Fisher EMC, Tybulewicz VLJ. Down syndrome: searching for the genetic culprits. Dis Model Mech. 2011;4:586–595. doi:10.1242/dmm.008078
11. Blumberg BS, Gerstley BJ, Hungerford DA, London WT, Sutnick AI. A serum antigen (Australia antigen) in Down’s syndrome, leukemia, and hepatitis. Ann Intern Med. 1967;66(5):924–931. doi:10.7326/0003-4819-66-5-924
12. Tan YH, Schneider EL, Tischfield J, Epstein CJ, Ruddle FH. Human chromosome 21 dosage: effect on the expression of the interferon induced antiviral state. Science. 1974;186(4158):61–63. doi:10.1126/science.186.4158.61
13. Cupples CG, Tan YH. Effect of human interferon preparations on lymphoblastogenesis in Down’s syndrome. Nature. 1977;267:165–167. doi:10.1038/267165a0
14. Sullivan KD, Lewis HC, Hill AA, et al. Trisomy 21 consistently activates the interferon response. eLife. 2016;5:e16220. doi:10.7554/eLife.16220
15. Araya P, Waugh KA, Sullivan KD, et al. Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc Natl Acad Sci. 2019;116:24231–24241. doi:10.1073/pnas.1908129116
16. Waugh KA, Araya P, Pandey A, et al. Mass cytometry reveals global immune remodeling with multi-lineage hypersensitivity to type I interferon in Down syndrome. Cell Rep. 2019;29(7):1893–1908 e4. doi:10.1016/j.celrep.2019.10.038
17. Kong XF, Worley L, Rinchai D, et al. Three copies of four interferon receptor genes underlie a mild type I interferonopathy in Down syndrome. J Clin Immunol. 2020;40(6):807–819. doi:10.1007/s10875-020-00803-9
18. Espinosa JM. Down syndrome and COVID-19: a perfect storm? Cell Rep Med. 2020;1(2):100019. doi:10.1016/j.xcrm.2020.100019
19. Villani ER, Carfì A, Di Paola A, et al. Clinical characteristics of individuals with Down syndrome deceased with CoVID-19 in Italy-A case series. Am J Med Genet A. 2020;182(12):2964–2970. doi:10.1002/ajmg.a.61867
20. Clift AK, Coupland CAC, Keogh RH, Hemingway H, Hippisley-Cox J. COVID-19 mortality risk in Down syndrome: results from a cohort study of 8 million adults. Ann Intern Med. 2021;174(4):572–576. doi:10.7326/m20-4986
21. Emami A, Javanmardi F, Akbari A, Asadi-Pooya AA. COVID-19 in patients with Down syndrome. Neurol Sci. 2021;42(5):1649–1652. doi:10.1007/s10072-021-05091-8
22. Hüls A, Costa ACS, Dierssen M, et al. Medical vulnerability of individuals with Down syndrome to severe COVID-19-data from the Trisomy 21 Research Society and the UK ISARIC4C survey. EClinicalMedicine. 2021;33:100769. doi:10.1016/j.eclinm.2021.100769
23. Malle L, Gao C, Hur C, et al. Individuals with Down syndrome hospitalized with COVID-19 have more severe disease. Genet Med. 2021;23(3):576–580. doi:10.1038/s41436-020-01004-w
24. Storm W. Prevalence and diagnostic significance of gliadin antibodies in children with Down syndrome. Eur J Pediatr. 1990;149(12):833–834. doi:10.1007/bf02072069
25. Castro M, Crinò A, Papadatou B, et al. Down’s syndrome and celiac disease: the prevalence of high IgA-antigliadin antibodies and HLA-DR and DQ antigens in trisomy 21. J Pediatr Gastroenterol Nutr. 1993;16(3):265–268. doi:10.1097/00005176-199304000-00007
26. Gale L, Wimalaratna H, Brotodiharjo A, Duggan JM. Down’s syndrome is strongly associated with coeliac disease. Gut. 1997;40(4):492–496. doi:10.1136/gut.40.4.492
27. Carlsson A, Axelsson I, Borulf S, et al. Prevalence of IgA-antigliadin antibodies and IgA-antiendomysium antibodies related to celiac disease in children with Down syndrome. Pediatrics. 1998;101(2):272–275. doi:10.1542/peds.101.2.272
28. Goldacre MJ, Wotton CJ, Seagroatt V, Yeates D. Cancers and immune related diseases associated with Down’s syndrome: a record linkage study. Arch Dis Child. 2004;89(11):1014–1017. doi:10.1136/adc.2003.046219
29. Hansson T, Dahlbom I, Rogberg S, et al. Antitissue transglutaminase and antithyroid autoantibodies in children with Down syndrome and celiac disease. J Pediatr Gastroenterol Nutr. 2005;40(2):
30. AlRuwaily F, Kattan HA, AlMehaidib AM, AlDekhail W. Prevalence of celiac disease in Saudi children with Down syndrome: a retrospective study. Int J Pediatr Adolesc Med. 2017;4(2):51–53. doi:10.1016/j.ijpam.2016.12.002
31. Carnicer J, Farré C, Varea V, Vilar P, Moreno J, Artigas J. Prevalence of coeliac disease in Down’s syndrome. Eur J Gastroenterol Hepatol. 2001;13(3):263–267. doi:10.1097/00042737-200103000-00008
32. George EK, Mearin ML, Bouquet J, et al. High frequency of celiac disease in Down syndrome. J Pediatr. 1996;128(4):555–557. doi:10.1016/s0022-3476(96)70369-4
33. Zachor DA, Mroczek-Musulman E, Brown P. Prevalence of celiac disease in Down syndrome in the United States. J Pediatr Gastroenterol Nutr. 2000;31(3):275–279. doi:10.1097/00005176-200009000-00014
34. Abdulrazzaq Y, El-Azzabi TI, Al Hamad SM, Attia S, Deeb A, Aburawi EH. Occurrence of hypothyroidism, diabetes mellitus, and celiac disease in Emirati children with Down’s syndrome. Oman Med J. 2018;33(5):387–392. doi:10.5001/omj.2018.72
35. Book L, Hart A, Black J, Feolo M, Zone JJ, Neuhausen SL. Prevalence and clinical characteristics of celiac disease in Downs syndrome in a US study. Am J Med Genet. 2001;98(1):70–74. doi:10.1002/1096-8628(20010101)98:1<70::AID-AJMG1002>3.0.CO;2-G
36. Rubio-Tapia A, Hill ID, Kelly CP, Calderwood AH, Murray JA. ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gastroenterol. 2013;108(5):
37. Uibo O, Teesalu K, Metskula K, et al. Screening for celiac disease in Down’s syndrome patients revealed cases of subtotal villous atrophy without typical for celiac disease HLA-DQ and tissue transglutaminase antibodies. World J Gastroenterol. 2006;12(9):1430–1434. doi:10.3748/wjg.v12.i9.1430
38. Durante-Mangoni E, Iardino P, Resse M, et al. Silent celiac disease in chronic hepatitis C: impact of interferon treatment on the disease onset and clinical outcome. J Clin Gastroenterol. 2004;38(10):901–905. doi:10.1097/00004836-200411000-00014
39. Martins EV, Gaburri AK. Celiac disease onset after pegylated interferon and ribavirin treatment of chronic hepatitis C. Arq Gastroenterol. 2004;41:132–133. doi:10.1590/S0004-28032004000200012
40. Hernandez L, Johnson TC, Naiyer AJ, et al. Chronic hepatitis C virus and celiac disease, is there an association? Dig Dis Sci. 2008;53:256–261. doi:10.1007/s10620-007-9851-z
41. Ruggeri C, La Masa AT, Rudi S, et al. Celiac disease and non-organ-specific autoantibodies in patients with chronic hepatitis C virus infection. Dig Dis Sci. 2008;53(8):2151–2155. doi:10.1007/s10620-007-0146-1
42. Bergholdt R, Eising S, Nerup J, Pociot F. Increased prevalence of Down’s syndrome in individuals with type 1 diabetes in Denmark: a nationwide population-based study. Diabetologia. 2006;49(6):1179–1182. doi:10.1007/s00125-006-0231-6
43. Pierce MJ, LaFranchi SH, Pinter JD. Characterization of thyroid abnormalities in a large cohort of children with Down syndrome. Horm Res Paediatr. 2017;87(3):170–178. doi:10.1159/000457952
44. Mortimer GL, Gillespie KM. Early onset of autoimmune diabetes in children with Down syndrome—two separate aetiologies or an immune system pre-programmed for autoimmunity? Curr Diab Rep. 2020;20(9). doi:10.1007/s11892-020-01318-8
45. Anwar AJ, Walker JD, Frier BM. Type 1 diabetes mellitus and Down’s syndrome: prevalence, management and diabetic complications. Diabetic Med. 1998;15(2):160–163. doi:10.1002/(sici)1096-9136(199802)15:2<160::Aid-dia537>3.0.Co;2-j
46. Zornitzki T. Interferon therapy in hepatitis C leading to chronic type 1 diabetes. World J Gastroenterol. 2015;21(1):233. doi:10.3748/wjg.v21.i1.233
47. Gillespie KM, Dix RJ, Williams AJ, et al. Islet autoimmunity in children with Down’s syndrome. Diabetes. 2006;55(11):3185–3188. doi:10.2337/db06-0856
48. Aitken RJ, Mehers KL, Williams AJ, et al. Early-onset, coexisting autoimmunity and decreased HLA-mediated susceptibility are the characteristics of diabetes in Down syndrome. Diabetes Care. 2013;36(5):1181–1185. doi:10.2337/dc12-1712
49. Fabris P, Floreani A, Tositti G, Vergani D, De Lalla F, Betterle C. Type 1 diabetes mellitus in patients with chronic hepatitis C before and after interferon therapy. Aliment Pharmacol Ther. 2003;18(6):549–558. doi:10.1046/j.1365-2036.2003.01681.x
50. Molleston JP, Mellman W, Narkewicz MR, et al. Autoantibodies and autoimmune disease during treatment of children with chronic hepatitis C. J Pediatr Gastroenterol Nutr. 2013;56(3):304–310. doi:10.1097/MPG.0b013e3182774cae
51. Nakanishi K, Saitoh S. Clinical and genetic characteristics of patients with type 1 diabetes associated with interferon therapy. Diabetes Care. 2011;34(2):471–473. doi:10.2337/dc10-1237
52. Fort P, Lifshitz F, Bellisario R, et al. Abnormalities of thyroid function in infants with Down syndrome. J Pediatr. 1984;104(4):545–549. doi:10.1016/s0022-3476(84)80544-2
53. Selikowitz M. Health problems and health checks in school-aged children with Down syndrome. J Paediatr Child Health. 1992;28(5):383–386. doi:10.1111/j.1440-1754.1992.tb02697.x
54. Karlsson B, Gustafsson J, Hedov G, Ivarsson SA, Annerén G. Thyroid dysfunction in Down’s syndrome: relation to age and thyroid autoimmunity. Arch Dis Child. 1998;79(3):242–245. doi:10.1136/adc.79.3.242
55. Tüysüz B, Beker DB. Thyroid dysfunction in children with Down’s syndrome. Acta paediatrica. 2001;90(12):1389–1393. doi:10.1080/08035250152708770
56. Shott SR, Joseph A, Heithaus D. Hearing loss in children with Down syndrome. Int J Pediatr Otorhinolaryngol. 2001;61(3):199–205. doi:10.1016/s0165-5876(01)00572-9
57. Schepis C, Barone C, Siragusa M, Pettinato R, Romano C. An updated survey on skin conditions in Down syndrome. Dermatology. 2002;205(3):234–238. doi:10.1159/000065859
58. Chen MH, Chen SJ, Su LY, Yang W. Thyroid dysfunction in patients with Down syndrome. Acta paediatrica Taiwanica. 2007;48(4):191–195.
59. Martínez E, Castañeda D, Jaramillo S, et al. Altered immune parameters correlate with infection-related hospitalizations in children with Down syndrome. Hum Immunol. 2016;77(7):594–599. doi:10.1016/j.humimm.2016.05.004
60. Noble SE, Leyland K, Findlay CA, et al. School based screening for hypothyroidism in Down’s syndrome by dried blood spot TSH measurement. Arch Dis Child. 2000;82(1):27–31. doi:10.1136/adc.82.1.27
61. van Trotsenburg AS, Vulsma T, van Rozenburg-marres SL, et al. The effect of thyroxine treatment started in the neonatal period on development and growth of two-year-old Down syndrome children: a randomized clinical trial. J Clin Endocrinol Metab. 2005;90(6):3304–3311. doi:10.1210/jc.2005-0130
62. Goday-Arno A, Cerda-Esteva M, Flores-le-roux JA, Chillaron-Jordan JJ, Corretger JM, Cano-Pérez JF. Hyperthyroidism in a population with Down syndrome (DS). Clin Endocrinol. 2009;71(1):110–114. doi:10.1111/j.1365-2265.2008.03419.x
63. Serranti D, Indolfi G, Nebbia G, et al. Transient hypothyroidism and autoimmune thyroiditis in children with chronic hepatitis C treated with pegylated-interferon-α-2b and ribavirin. Pediatr Infect Dis J. 2018;37(4):287–291. doi:10.1097/INF.0000000000001791
64. Hameed MA, Mehmood A, Farooq MA, Tayyab GUN, Haq Toor IU. Hypothyroidism in hepatitis C patients on pegylated interferon therapy. J Ayub Med Coll. 2016;28(4):706–708.
65. Vezali E, Elefsiniotis I, Mihas C, Konstantinou E, Saroglou G. Thyroid dysfunction in patients with chronic hepatitis C: virus- or therapy-related? J Gastroenterol Hepatol. 2009;24(6):1024–1029. doi:10.1111/j.1440-1746.2009.05812.x
66. Yan Z, Fan K, Fan Y, et al. Thyroid dysfunction in Chinese patients with chronic hepatitis C treated with interferon alpha: incidence, long-term outcome and predictive factors. Hepat Mon. 2012;12(9):e6390. doi:10.5812/hepatmon.6390
67. Bini EJ, Mehandru S. Incidence of thyroid dysfunction during interferon α-2b and ribavirin therapy in men with chronic hepatitis C: a prospective cohort study. Arch Intern Med. 2004;164(21):2371–2376. doi:10.1001/archinte.164.21.2371
68. Földes I, Dávid K, Horváth G, Osztrogonácz H, Jankovics K, Tolvaj G. [Thyroid dysfunctions in patients with viral hepatitis treated with interferon-alpha]. Orv Hetil. 2004;145(23):1211–1216. [Danish].
69. Durelli L, Ferrero B, Oggero A, et al. Liver and thyroid function and autoimmunity during interferon-beta 1b treatment for MS. Neurology. 2001;57(8):1363–1370. doi:10.1212/wnl.57.8.1363
70. Pavan MH, Pavin EJ, Gonçales FL, Zantut-Wittmann DE, Wittmann DE. Virus C genotype predisposes to primary hypothyroidism during interferon-α treatment for chronic hepatitis C. Braz J Infect Dis. 2011;15(5):449–456. doi:10.1016/s1413-8670(11)70226-4
71. Kozielewicz D, Zalesna A, Dybowska D. Can pegylated interferon alpha 2a cause development of thyroid disorders in patients with chronic hepatitis B? Expert Opin Drug Saf. 2014;13(8):1009–1014. doi:10.1517/14740338.2014.921156
72. Al-Maweri SA, Tarakji B, Al-Sufyani GA, Al-Shamiri HM, Gazal G. Lip and oral lesions in children with Down syndrome. A controlled study. J Clin Exp Dent. 2015;7(2):e284–8. doi:10.4317/jced.52283
73. Scully C, van Bruggen W, Diz Dios P, Casal B, Porter S, Davison M-F. Down syndrome: lip lesions (angular stomatitis and fissures) and Candida albicans. Br J Dermatol. 2002;147:37–40. doi:10.1046/j.1365-2133.2002.04741.x
74. Daneshpazhooh M, Nazemi TM, Bigdeloo L, Yoosefi M. Mucocutaneous findings in 100 children with Down syndrome. Pediatr Dermatol. 2007;24(3):317–320. doi:10.1111/j.1525-1470.2007.00412.x
75. Madan V, Williams J, Lear JT. Dermatological manifestations of Down’s syndrome. Clin Exp Dermatol. 2006;31(5):623–629. doi:10.1111/j.1365-2230.2006.02164.x
76. Giovanardi G, Chiricozzi A, Bianchi L, et al. Hidradenitis suppurativa associated with Down syndrome is characterized by early age at diagnosis. Dermatology. 2018;234(1–2):66–70. doi:10.1159/000487799
77. Carlstedt K, Krekmanova L, Dahllöf G, Ericsson B, Braathen G, Modéer T. Oral carriage of Candida species in children and adolescents with Down’s syndrome. Int J Paediatr Dent. 1996;6(2):95–100. doi:10.1111/j.1365-263x.1996.tb00219.x
78. Mohiddin G, Narayanaswamy AB, Masthan KM, Nagarajan A, Panda A, Behura SS. Oral Candidal and Streptococcal carriage in Down syndrome patients. J Nat Sci Biol Med. 2015;6(2):300–305. doi:10.4103/0976-9668.159983
79. Maranhão FC, Mendonça NM, Teixeira TC, et al. Molecular identification of Candida species in the oral microbiota of individuals with Down syndrome: a case-control study. Mycopathologia. 2020;185(3):537–543. doi:10.1007/s11046-020-00457-6
80. Megged O, Schlesinger Y. Down syndrome and streptococcus group A disease in hospitalized children. Acta paediatrica. 2010;99(6):796–797. doi:10.1111/j.1651-2227.2009.01640.x
81. Sánchez-Luna M, Medrano C, Lirio J. Down syndrome as risk factor for respiratory syncytial virus hospitalization: a prospective multicenter epidemiological study. Influenza Other Respi Viruses. 2017;11(2):157–164. doi:10.1111/irv.12431
82. Yancey CL, Zmijewski C, Athreya BH, Doughty RA. Arthropathy of Down’s syndrome. Arthritis Rheum. 1984;27(8):929–934. doi:10.1002/art.1780270813
83. Foley CM, Deely DA, MacDermott EJ, Killeen OG. Arthropathy of Down syndrome: an under-diagnosed inflammatory joint disease that warrants a name change. RMD Open. 2019;5(1):e000890. doi:10.1136/rmdopen-2018-000890
84. Passos de Souza E, Evangelista Segundo PT, Jose FF, Lemaire D, Santiago M. Rheumatoid arthritis induced by α-interferon therapy. Clin Rheumatol. 2001;20(4):297–299. doi:10.1007/pl00011206
85. Verweyen EL, Schulert GS. Interfering with Interferons: targeting the JAK-STAT pathway in complications of systemic juvenile idiopathic arthritis (SJIA). Rheumatology. 2021. doi:10.1093/rheumatology/keab673
86. Hartley D, Blumenthal T, Carrillo M, et al. Down syndrome and Alzheimer’s disease: common pathways, common goals. Alzheimers Dement. 2015;11(6):700–709. doi:10.1016/j.jalz.2014.10.007
87. Cohen AD, Head E, Lee JH. Alzheimer’s disease in aging Down syndrome. Dev Neurobiol. 2019;79(7):611–612. doi:10.1002/dneu.22717
88. Yamamoto M, Kiyota T, Horiba M, et al. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170(2):680–692. doi:10.2353/ajpath.2007.060378
89. Taylor JM, Moore Z, Minter MR, Crack PJ. Type-I interferon pathway in neuroinflammation and neurodegeneration: focus on Alzheimer’s disease. J Neural Transm. 2018;125(5):797–807. doi:10.1007/s00702-017-1745-4
90. Moore Z, Mobilio F, Walker FR, Taylor JM, Crack PJ. Abrogation of type-I interferon signalling alters the microglial response to Aβ1–42. Sci Rep. 2020;10(1):3153. doi:10.1038/s41598-020-59917-0
91. Roy ER, Wang B, Wan YW, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. 2020;130(4):1912–1930. doi:10.1172/JCI133737
92. Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity. 2012;36(4):503–514. doi:10.1016/j.immuni.2012.03.013
93. Kong XF, Vogt G, Itan Y, et al. Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease. Hum Mol Genet. 2013;22(4):769–781. doi:10.1093/hmg/dds484
94. Hemann EA, Gale M, Savan R. Interferon lambda genetics and biology in regulation of viral control. Front Immunol. 2017;8:1707. doi:10.3389/fimmu.2017.01707
95. Duncan CJ, Mohamad SM, Young DF, et al. Human IFNAR2 deficiency: lessons for antiviral immunity. Sci Transl Med. 2015;7(307):307ra154. doi:10.1126/scitranslmed.aac4227
96. Hernandez N, Bucciol G, Moens L, et al. Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J Exp Med. 2019;216(9):2057–2070. doi:10.1084/jem.20182295
97. Zhang Q, Bastard P, Liu Z, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370(6515):eabd4570. doi:10.1126/science.abd4570
98. Bastard P, Manry J, Chen J, et al. Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency. J Clin Invest. 2021;131(1). doi:10.1172/JCI139980
99. Vogt G, Bustamante J, Chapgier A, et al. Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med. 2008;205(8):1729–1737. doi:10.1084/jem.20071987
100. Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol. 2014;26(6):454–470. doi:10.1016/j.smim.2014.09.008
101. Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361(21):2033–2045. doi:10.1056/NEJMoa0907206
102. Pelleri MC, Cattani C, Vitale L, et al. Integrated quantitative transcriptome maps of human trisomy 21 tissues and cells. Front Genet. 2018;9:125. doi:10.3389/fgene.2018.00125
103. Prandini P, Deutsch S, Lyle R, et al. Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance. Am J Hum Genet. 2007;81:252–263. doi:10.1086/519248
104. Takaoka A, Mitani Y, Suemori H, et al. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science. 2000;288(5475):2357–2360. doi:10.1126/science.288.5475.2357
105. Meuwissen MEC, Schot R, Buta S, et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med. 2016;213(7):1163–1174. doi:10.1084/jem.20151529
106. Zhang X, Bogunovic D, Payelle-Brogard B, et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature. 2015;517(7532):89–93. doi:10.1038/nature13801
107. Blouin CM, Hamon Y, Gonnord P, et al. Glycosylation-dependent IFN-γR partitioning in lipid and actin nanodomains is critical for JAK activation. Cell. 2016;166(4):920–934. doi:10.1016/j.cell.2016.07.003
108. Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197(6):711–723. doi:10.1084/jem.20021553
109. Banchereau R, Cepika AM, Banchereau J, Pascual V. Understanding human autoimmunity and autoinflammation through transcriptomics. Annu Rev Immunol. 2017;35:337–370. doi:10.1146/annurev-immunol-051116-052225
110. Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. 2011;1238(1):91–98. doi:10.1111/j.1749-6632.2011.06220.x
111. Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med. 2016;213(12):2527–2538. doi:10.1084/jem.20161596
112. Rice GI, Forte GMA, Szynkiewicz M, et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013;12(12):1159–1169. doi:10.1016/S1474-4422(13)70258-8
113. Liu L, Okada S, Kong X-F, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011;208(8):1635–1648. doi:10.1084/jem.20110958
114. Ugazio AG, Maccario R, Notarangelo LD, Burgio GR. Immunology of Down syndrome: a review. Am J Med Genet Suppl. 1990;7:204–212. doi:10.1002/ajmg.1320370742
115. Malle L, Bogunovic D. Down syndrome and type I interferon: not so simple. Curr Opin Immunol. 2021;72:196–205. doi:10.1016/j.coi.2021.06.006
116. Verstegen RHJ, Kusters MAA. Inborn errors of adaptive immunity in Down syndrome. J Clin Immunol. 2020;40(6):791–806. doi:10.1007/s10875-020-00805-7
117. Fischer A. Recent advances in understanding the pathophysiology of primary T cell immunodeficiencies. Trends Mol Med. 2015;21:408–416. doi:10.1016/j.molmed.2015.04.002
118. Gil MP, Ploquin MJY, Watford WT, et al. Regulating type 1 IFN effects in CD8 T cells during viral infections: changing STAT4 and STAT1 expression for function. Blood. 2012;120(18):3718–3728. doi:10.1182/blood-2012-05-428672
119. Tangye SG. Genetic susceptibility to EBV infection: insights from inborn errors of immunity. Hum Genet. 2020;139(6–7):885–901. doi:10.1007/s00439-020-02145-3
120. Gruber CN, Calis JJA, Buta S, et al. Complex autoinflammatory syndrome unveils fundamental principles of JAK1 kinase transcriptional and biochemical function. Immunity. 2020;53(3):672–684.e11. doi:10.1016/j.immuni.2020.07.006
121. Del Bel KL, Ragotte RJ, Saferali A, et al. JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. J Allergy Clin Immunol. 2017;139(6):2016–2020e5. doi:10.1016/j.jaci.2016.12.957
122. Gruber C, Martin-Fernandez M, Ailal F, et al. Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy. J Exp Med. 2020;217(5). doi:10.1084/jem.20192319
123. Milner JD, Vogel TP, Forbes L, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood: Am Soc Hematol. 2015;125(4):591–599. doi:10.1182/blood-2014-09-602763
124. Casanova JL. Human genetic basis of interindividual variability in the course of infection. Proc Natl Acad Sci U S A. 2015;112(51):E7118–27. doi:10.1073/pnas.1521644112
125. Malle L, Bastard P, Martin-Nalda A, et al. Atypical inflammatory syndrome triggered by SARS-CoV-2 in infants with Down syndrome. J Clin Immunol. 2021;41(7):1457–1462. doi:10.1007/s10875-021-01078-4
126. Maroun LE, Heffernan TN, Hallam DM. Partial IFN-alpha/beta and IFN-gamma receptor knockout trisomy 16 mouse fetuses show improved growth and cultured neuron viability. J Interferon Cytokine Res. 2000;20(2):197–203. doi:10.1089/107999000312612
127. Tuttle KD, Waugh KA, Araya P, et al. JAK1 inhibition blocks lethal immune hypersensitivity in a mouse model of Down syndrome. Cell Rep. 2020;33(7):108407. doi:10.1016/j.celrep.2020.108407
128. Powers RK, Culp-Hill R, Ludwig MP, et al. Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat Commun. 2019;10(1):4766. doi:10.1038/s41467-019-12739-9
129. Charbit-Henrion F, Begue B, Sierra A, et al. Copy number variations and founder effect underlying complete IL-10Rβ deficiency in Portuguese kindreds. PLoS One. 2018;13(10):e0205826. doi:10.1371/journal.pone.0205826
130. Pulendran B, Davis MM. The science and medicine of human immunology. Science. 2020;369(6511):eaay4014. doi:10.1126/science.aay4014
131. da Rosa Utiyama SR, Nisihara RM, Nass FR, Oliveira NP, Fiedler PT, de Messias-reason IT. Autoantibodies in patients with Down syndrome: early senescence of the immune system or precocious markers for immunological diseases? J Paediatr Child Health. 2008;44(4):182–186. doi:10.1111/j.1440-1754.2007.01229.x
132. Malakhova OA, Kim KI, Luo JK, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006;25(11):2358–2367. doi:10.1038/sj.emboj.7601149
133. Jimenez Fernandez D, Hess S, Knobeloch KP. Strategies to target ISG15 and USP18 toward therapeutic applications. Front Chem. 2019;7:923. doi:10.3389/fchem.2019.00923
134. O’Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat Rev Immunol. 2011;11(4):239–250. doi:10.1038/nri2958
135. Gough DJ, Messina NL, Clarke CJ, et al. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity. 2012;36:166–174. doi:10.1016/j.immuni.2012.01.011
136. Sussan TE, Yang A, Li F, Ostrowski MC, Reeves RH. Trisomy represses Apc(Min)-mediated tumours in mouse models of Down’s syndrome. Nature. 2008;451(7174):73–75. doi:10.1038/nature06446
137. Clinicaltrials.gov. Tofacitinib for immune skin conditions in Down Syndrome. Available from: https://clinicaltrials.gov/ct2/show/NCT04246372.
138. Rachubinski AL, Estrada BE, Norris D, Dunnick CA, Boldrick JC, Espinosa JM. Janus kinase inhibition in Down syndrome: 2 cases of therapeutic benefit for alopecia areata. JAAD Case Rep. 2019;5(4):365–367. doi:10.1016/j.jdcr.2019.02.007
139. Pham AT, Rachubinski AL, Enriquez-Estrada B, Worek K, Griffith M, Espinosa JM. JAK inhibition for treatment of psoriatic arthritis in Down syndrome. Rheumatology. 2021;60(9):e309–e311. doi:10.1093/rheumatology/keab203
140. Yu YE, Xing Z, Do C, et al. Genetic and epigenetic pathways in Down syndrome: insights to the brain and immune system from humans and mouse models. Prog Brain Res. 2020;251:1–28. doi:10.1016/bs.pbr.2019.09.002
141. Du Y, Shan LF, Cao ZZ, Feng JC, Cheng Y. Prevalence of celiac disease in patients with Down syndrome: a meta-analysis. Oncotarget. 2018;9:5387–5396. doi:10.18632/oncotarget.23624
142. Agardh D, Nilsson A, Carlsson A, Kockum I, Lernmark A, Ivarsson SA. Tissue transglutaminase autoantibodies and human leucocyte antigen in Down’s syndrome patients with coeliac disease. Acta paediatrica. 2002;91(1):34–38. doi:10.1080/080352502753457914
143. Alanay Y, Boduroğlu K, Tunçbilek E. Celiac disease screening in 100 Turkish children with Down syndrome. Turk J Pediatr. 2005;47(2):138–140.
144. Singh P, Arora A, Strand TA, et al. Global prevalence of celiac disease: systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2018;16(6):823–836.e2. doi:10.1016/j.cgh.2017.06.037
145. Garmendia Madariaga A, Santos Palacios S, Guillén-Grima F, Galofré JC. The incidence and prevalence of thyroid dysfunction in Europe: a meta-analysis. J Clin Endocrinol Metab. 2014;99(3):923–931. doi:10.1210/jc.2013-2409
146. Bullard KM, Cowie CC, Lessem SE, et al. Prevalence of diagnosed diabetes in adults by diabetes type - United States, 2016. Morb Mortal Wkly Rep. 2018;67(12):359–361. doi:10.15585/mmwr.mm6712a2
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.