")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Inhibition of mitotic Aurora kinase A by alisertib induces apoptosis and autophagy of human gastric cancer AGS and NCI-N78 cells
Authors Yuan C, Zhou Z, Yang Y, He Z, Zhang X, Wang D, Yang T, Wang N, Zhao RJ, Zhou S
Received 11 September 2014
Accepted for publication 3 October 2014
Published 14 January 2015 Volume 2015:9 Pages 487—508
DOI https://doi.org/10.2147/DDDT.S74127
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Chun-Xiu Yuan,1,2 Zhi-Wei Zhou,2,3 Yin-Xue Yang,4 Zhi-Xu He,3 Xueji Zhang,5 Dong Wang,6 Tianxing Yang,7 Ning-Ju Wang,1 Ruan Jin Zhao,8 Shu-Feng Zhou2
1Department of Oncology, General Hospital Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 2Department of Pharmaceutical Science, College of Pharmacy, University of South Florida, Tampa, FL, USA; 3Guizhou Provincial Key Laboratory for Regenerative Medicine, Stem Cell and Tissue Engineering Research Center and Sino–US Joint Laboratory for Medical Sciences, Guiyang Medical University, Guiyang, Guizhou, People’s Republic of China; 4Department of Colorectal Surgery, General Hospital, Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 5Research Center for Bioengineering and Sensing Technology, University of Science and Technology Beijing, Beijing, People’s Republic of China; 6Cancer Center, Daping Hospital and Research Institute of Surgery, Third Military Medical University, Chongqing, People’s Republic of China; 7Department of Internal Medicine, University of Utah and Salt Lake Veterans Affairs Medical Center, Salt Lake City, UT, USA; 8Center for Traditional Chinese Medicine, Sarasota, FL, USA
Abstract: Gastric cancer is one of the most common cancers and responds poorly to current chemotherapy. Alisertib (ALS) is a second-generation, orally bioavailable, highly selective small-molecule inhibitor of the serine/threonine protein kinase Aurora kinase A (AURKA). ALS has been shown to have potent anticancer effects in preclinical and clinical studies, but its role in gastric cancer treatment is unclear. This study aimed to investigate the cancer cell-killing effect of ALS on gastric cancer cell lines AGS and NCI-N78, with a focus on cell proliferation, cell-cycle distribution, apoptosis, and autophagy and the mechanism of action. The results showed that ALS exhibited potent growth-inhibitory, proapoptotic, and proautophagic effects on AGS and NCI-N78 cells. ALS concentration-dependently inhibited cell proliferation and induced cell-cycle arrest at G2/M phase in both cell lines, with a downregulation of cyclin-dependent kinase 1 and cyclin B1 expression but upregulation of p21 Waf1/Cip1, p27 Kip1, and p53 expression. ALS induced mitochondria-mediated apoptosis and autophagy in both AGS and NCI-N78 cells. ALS induced the expression of proapoptotic proteins but inhibited the expression of antiapoptotic proteins, with a significant increase in the release of cytochrome c and the activation of caspase 9 and caspase 3 in both cell lines. ALS induced inhibition of phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) and p38 mitogen-activated protein kinase (MAPK) signaling pathways while activating the 5'-adenosine monophosphate-activated protein kinase (AMPK) signaling pathway as indicated by their altered phosphorylation, contributing to the proautophagic effects of ALS. SB202191 and wortmannin enhanced the autophagy-inducing effect of ALS in AGS and NCI-N78 cells. Notably, ALS treatment significantly decreased the ratio of phosphorylated AURKA over AURKA, which may contribute, at least in part, to the inducing effects of ALS on cell-cycle arrest and autophagy in AGS and NCI-N78 cells. Taken together, these results indicate that ALS exerts a potent inhibitory effect on cell proliferation but inducing effects on cell-cycle arrest, mitochondria-dependent apoptosis, and autophagy with the involvement of PI3K/Akt/mTOR, p38 MAPK, and AURKA-mediated signaling pathways in AGS and NCI-N78 cells.
Keywords: gastric cancer, alisertib, AURKA, apoptosis, autophagy
Introduction
According to Globocan 2012, there were about 952,000 new cases of gastric cancer in the world in 2012, accounting for 6.8% of all cancer diagnoses and making it the fifth-most-common malignancy globally after lung, breast, colorectum, and prostate cancer.1 The highest mortality rate for gastric cancer was in East Asia, at 24 per 100,000 in men and 9.8 per 100,000 in women; the lowest mortality rate was in North America.1 According to the Ministry of Health in the People’s Republic of China, the mortality rate for gastric cancer in that country was in third place behind cancers of the lung and liver, with 24.71 per 100,000 in 2004-2005.2 Gastric cancer accounts for 8.8% of the cancer-related deaths in the People’s Republic of China. Surgery is the primary therapy for gastric cancer, but the prognosis is still quite unsatisfactory, with a 5-year overall survival rate of less than 30%.3 Although recent studies have demonstrated that combined-modality therapy based on surgery contributes to ameliorating mortality, the optimal tailored treatment for high-risk patients remains elusive.4 At present, no effective treatment is available for gastric cancer. Therefore, there is an urgent need to develop mechanism-based, effective, pharmacologically safe agents, preferably of dietary origin, to prevent and treat gastric cancer.
The Aurora kinases (A, B, and C) belong to a family of oncogenic serine/threonine kinases that play a key role in proper mitotic execution.5 Aurora kinase A (AURKA), the most well-characterized member of Aurora kinase family, is essential for centrosome function and maturation, spindle assembly, chromosome segregation, and mitotic entry.6 Inhibition of AURKA by specific small-molecule inhibitors or knockdown of AURKA by small-RNA interference causes abnormal spindle formation, mitotic defects, senescence, and cell death.7,8 Abnormalities of the activities and expression of AURKA have been implicated in cancer development, progression, and metastasis.9 AURKA amplification and overexpression frequently occur in upper gastrointestinal adenocarcinomas as well as several other malignancies.10 AURKA acts as an oncogene resulting in genetic instability, dedifferentiated morphology, and a poor prognosis in patients with upper gastrointestinal adenocarcinoma.11 The overexpression of AURKA promotes cancer cell growth and resistance to chemotherapy by upregulating oncogenic signaling pathways and suppressing cell-death mechanisms.9 Several studies have shown that AURKA overexpression promotes drug resistance and tumor recurrence,12 and induces growth-promoting and survival-promoting oncogenic signaling pathways, such as the phosphoinsitide 3-kinase (PI3K)/protein kinase B (Akt) and β-catenin signaling pathways.9 This suggests that AURKA could serve as a therapeutic target for cancer treatment.
Alisertib (ALS, MLN8237, Figure 1A) is an investigational, orally available, and selective small-molecule AURKA inhibitor.13 ALS has the ability to selectively inhibit AURKA and thereby induces cell-cycle arrest, aneuploidy, polyploidy, mitotic catastrophe, and cell death.8,10 In preclinical studies, ALS exhibited potent AURKA inhibition and high antitumor activity in a wide range of tumor cells.14 However, there is a lack of evidence for the anticancer effect of ALS in gastric cancer. In this present study, in order to explore the anticancer effect of ALS in gastric cancer, we examined the proapoptotic and proautophagic effects of ALS on AGS and NCI-N78 cells and the potential mechanisms.
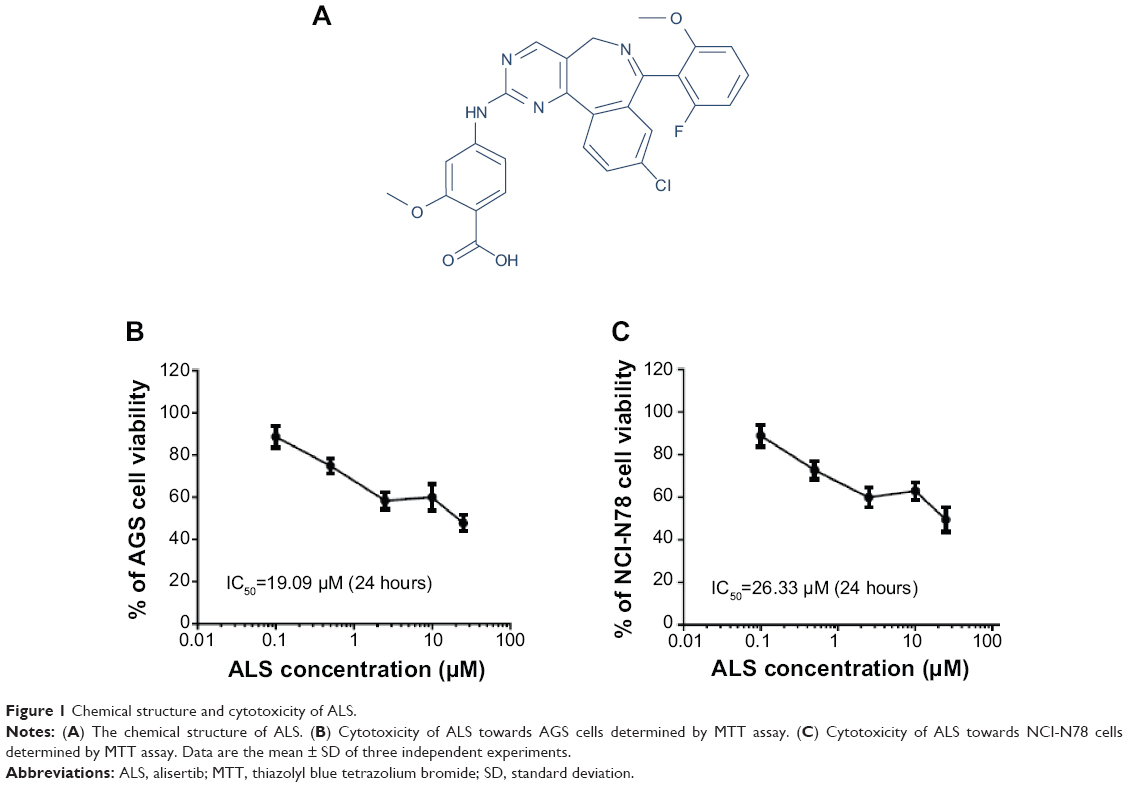 | Figure 1 Chemical structure and cytotoxicity of ALS. |
Materials and methods
Chemicals and reagents
Fetal bovine serum (FBS), Dulbecco’s phosphate buffered saline (PBS), thiazolyl blue tetrazolium bromide (MTT), RNase A, and propidium iodide (PI) were purchased from Sigma-Aldrich Inc (St Louis, MO, USA). Dulbecco’s Modified Eagle’s Medium (DMEM) and RPMI-1640 medium were obtained from Corning Cellgro Inc (Herndon, VA, USA). SB202190 (4-[4-fluorophenyl]-2-[4-hydroxyphenyl]-5-[4-pyridyl]1H-imidazole, a selective inhibitor of p38 mitogen-activated protein kinase [MAPK] used as an autophagy inducer) and wortmannin (WM, a potent, irreversible, and selective PI3K inhibitor and a blocker of autophagosome formation) were purchased from Invivogen Inc (San Diego, CA, USA). Phenol red-free culture medium and 4,6-diamidino-2-phenylindole were bought from Invitrogen (Carlsbad, CA, USA). The annexin V: phycoerythrin (PE) apoptosis detection kit was purchased from BD Biosciences (San Jose, CA, USA). The Cyto-ID® autophagy detection kit was obtained from Enzo Life Sciences Inc (Farmingdale, NY, USA). The Pierce bicinchoninic acid (BCA) protein assay kit, skim milk, and Western blot substrate were purchased from Thermo Scientific (Waltham, MA, USA). Polyvinylidene difluoride membrane was purchased from EMD Millipore (Bedford, MA, USA). Primary antibodies against human p21 Waf1/Cip1, p27 Kip1, p53, cyclin B1, cyclin-dependent kinase 1(CDK1/cell division cycle protein 2 homologue [CDC2]/CDKN1), the p53 upregulated modulator of apoptosis (PUMA), B-cell lymphoma 2 (Bcl-2), Bcl-2-like protein 4/Bcl-2-associated X protein (Bax), B-cell lymphoma-extra-large (Bcl-xl), cytochrome c, cleaved caspase 9, cleaved caspase 3, p38 MAPK, phosphorylated (p-) p38 MAPK at Thr180/Tyr182, 5′-adenosine monophosphate-activated protein kinase (AMPK), p-AMPK at Thr172, Akt, p-Akt at Ser473, mammalian target of rapamycin (mTOR), p-mTOR at Ser2448, PI3K, p-PI3K/p85 at Tyr458, AURKA, p-AURKA at Thr288, phosphatase and tensin homolog (PTEN), beclin1, microtubule-associated protein 1A/1B-light chain 3 (LC3)-I, and LC3-II were all purchased from Cell Signaling Technology Inc (Beverly, MA, USA). The antibody against human β-actin was obtained from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA).
Cell lines and cell culture
The AGS and NCI-N78 cell lines are the most common human gastric adenocarcinoma cell lines and are widely used for the investigation of cancer cell-killing effect and underlying mechanisms of new compounds in anti-gastric-cancer drug development. The AGS cell line was derived from the fragments of a tumor resected from a patient who had received no prior therapy. It is a hyperdiploid cell line. The modal chromosome number was 49, occurring in 60% of cells. The percentage of polyploidy was 3.6%.15 The NCI-N78 cell line was derived from metastatic site of liver. It is a near-diploid cell line and double minutes were present in 64% of cells examined.16 The two cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM (AGS cells) and RPMI-1640 medium (NCI-N78 cells) supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin. The cells were maintained in a 5% CO2/95% air humidified incubator at 37°C. ALS was dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 50 mM and was freshly diluted to the desired concentration with culture medium. The final concentration of DMSO was at 0.05% (v/v). The control cells received the vehicle only.
Cell viability assay
The MTT assay was performed to examine the effect of ALB on cell viability as previously described.17,18 Briefly, AGS and NCI-N78 cells were seeded into a 96-well culture plate at a density of 8,000 cells/well. After 24-hour incubation, the cells were treated with ALS at concentrations ranging from 0.1–25 μM for 24 hours at a volume of 100 μL medium. Following the drug treatment, 10 μL of MTT stock solution (5 mg/mL) was added to each well and incubated for 2–4 hours. Then, the medium was carefully removed and 100 μL DMSO was added into each well. The plate was placed on a rocker to mix the solution thoroughly for 30 seconds and incubated at 37°C for 10 minutes. The absorbance was measured using a Synergy H4 Hybrid microplate reader (BioTek Inc, Winooski, VT, USA) at a wavelength of 450 nm. The concentration required for 50% inhibition of growth was determined from concentration–response curves obtained in multireplicated experiments by nonlinear regression analysis using GraphPad Prism 6 software (San Diego, CA, USA). The experiment was performed at least three times.
Cell-cycle distribution analysis
The effect of ALS on cell-cycle distribution of AGS and NCI-N78 cells was examined using flow cytometry as previously described.18 Briefly, AGS and NCI-N78 cells were treated with ALS at concentrations of 0.1 μM, 1 μM, and 5 μM for 24 hours. In separate experiments, AGS and NCI-N78 cells were treated with 5 μM ALS for 4 hours, 8 hours, 12 hours, 24 hours, 48 hours, and 72 hours. After cells were treated with ALS, cells were trypsinized and fixed in 70% ethanol at −20°C overnight. Following the fixation, cells were collected and resuspended in 1 mL of PBS containing 1 mg/mL RNase A and 50 μM/mL PI. Then, cells were incubated in the dark for 30 minutes at room temperature. A total number of 1×104 cells were subject to cell-cycle analysis using a flow cytometer (Becton Dickinson Immunocytometry Systems; San Jose, CA, USA).
Quantification of cellular apoptosis
The effects of ALS on the apoptosis of AGS and NCI-N78 cells were quantitated using the annexin V: PE apoptosis detection kit according to the manufacturer’s instruction. Annexin V is a 35–36 kDa Ca2+-dependent phospholipid-binding protein that has a high affinity for negatively charged phospholipid phosphatidylserine and binds to cells that are actively undergoing apoptosis with exposed phospholipid phosphatidylserine.19 Cells were treated with ALS at concentrations of 0.1 μM, 1 μM, and 5 μM for 24 hours, and then cells were trypsinized and washed twice with cold PBS. Subsequently, cells were resuspended at a concentration of 1×106/mL cells in 1× binding buffer (BD Biosciences). A quota of cell suspension (100 μL) was transferred into a 5 mL clean tube and incubated with 5 μL PE annexin V and 5 μL 7-amino-actinomycin D (a vital nucleic acid dye) in the dark for 15 minutes at room temperature. A quota of 1× binding buffer (400 μL) was then added to each tube and the number of apoptotic cells was quantified using a flow cytometer within 1 hour. Cells that stain positive for PE and annexin V and negative for 7-amino-actinomycin D are undergoing apoptosis; cells that stain positive for PE, annexin V, and 7-amino-actinomycin D are either in the end stage of apoptosis, are undergoing necrosis, or are already dead; and cells that stain negative for PE annexin V and 7-amino-actinomycin D are alive and not undergoing measurable apoptosis.
Determination of cellular autophagy
To determine the effect of ALS on autophagy in AGS and NCI-N78 cells, the intracellular autophagy level was examined using flow cytometry as previous described.17,18 Briefly, the cells were treated with fresh medium alone, control vehicle alone (0.05% DMSO, v/v), or ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours. After 24 hours of incubation, the cells were trypsinized and collected with 1× assay buffer containing 5% FBS. Cells were resuspended in 250 μL of phenol red-free culture medium containing 5% FBS, and 250 μL of the diluted Cyto-ID® Green stain solution was added to each sample and mixed well. Cells were incubated for 30 minutes at 37°C in the dark. Following the incubation, cells were collected by centrifugation at 250× g for 3 minutes and washed with 1× assay buffer. Subsequently, the cells were resuspended in 500 μL fresh 1× assay buffer containing 5% FBS and subject to flow cytometric analysis within 1 hour. Cells were analyzed using the green (FL1) channel of a flow cytometer.
Confocal fluorescence microscopy
Confocal fluorescence microscopy was performed to further examine the cellular autophagy level and the mechanisms for ALS-induced autophagy in AGS and NCI-N78 cells using a Cyto-ID® autophagy detection kit according to the manufacturer’s instruction. The kit was used to measure cellular autophagic vacuoles and autophagic flux using a novel dye that selectively labels autophagic vacuoles. The assay provides a rapid and quantitative approach to monitoring autophagy in live cells without the need for cell transfection and allows the measurement and differentiation between autophagic flux and autophagolysosome accumulation.20 Briefly, AGS and NCI-N78 cells were seeded into an 8-well chamber slide at 30% confluence for 24 hours. Then, the cells were treated with ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours. In separate experiments, to investigate the mechanisms for ALS-induced autophagy, cells were pretreated with 10 μM WM (a PI3K inhibitor and autophagy blocker) or 10 μM SB202190 (a selective inhibitor of p38 MAPK used as an autophagy inducer), then co-treated with 5 μM ALS for a further 24 hours. After the ALS treatment, cells were washed by 1× assay buffer and incubated with 100 μL of microscopy dual detection reagent for 30 minutes at 37°C in the dark. Following the incubation, cells were washed with 1× assay buffer to remove detection reagent and maintained in 200 μL fresh 1× assay buffer containing 5% FBS. The cells were examined using a Leica TCS SP2 laser scanning confocal microscopy (Wetzlar, Germany) using a standard fluorescein isothiocyanate filter set for imaging the autophagic signal at wavelengths of 405/488 nm.
Western blot analysis
The expression levels of various cellular proteins related to cell cycle, apoptosis, and autophagy were determined using Western blot assays. AGS and NCI-N78 cells were washed with PBS after 24-hour treatment with ALS at 0.1 μM, 1 μM, and 5 μM, lysed on ice with lysis buffer – 50 mmol 4-(2-hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES) at pH 7.5, 150 mmol NaCl, 10% glycerol, 1.5 mmol MgCl2, 1% Triton-X 100, 1 mmol ethylenediaminetetraacetic acid at pH 8.0, 10 mmol sodium pyrophosphate, 10 mmol sodium fluoride, phosphatase inhibitor cocktail, and protease inhibitor cocktail – and centrifuged at 3,000× g for 10 minutes at 4°C. The supernatant was collected and the protein concentrations were measured using a Pierce BCA protein assay kit. An equal amount of protein sample (30 μg) was resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) sample loading buffer and electrophoresed on 7% or 12% SDS–PAGE minigel after thermal denaturation at 95°C for 5 minutes. Proteins were transferred onto polyvinylidene difluoride membrane at 200 mA for 3 hours at 4°C. Membranes were probed with indicated primary antibody overnight at 4°C and then blotted with appropriate horseradish peroxidase-conjugated secondary antimouse or antirabbit antibody. Visualization was performed using an enhanced chemiluminescence kit (Thermal Scientific) and the blots were analyzed using Image Lab 3.0 (BioRad, Hercules, CA, USA). Protein level was normalized to the matching densitometric value of the internal control β-actin.
Statistical analysis
Data are presented as mean ± standard deviation (SD). Comparisons of multiple groups were evaluated by one-way analysis of variance followed by Tukey’s multiple comparison procedure. Values of P<0.05 were considered statistically different. Assays were performed at least three times independently.
Results
ALS inhibits the proliferation of AGS and NCI-N78 cells
To examine the effect of ALS on the viability of gastric cancer cell lines AGS and NCI-N78, we first tested the cell proliferation using the MTT assay. There was a concentration-dependent inhibition in the growth of AGS and NCI-N78 cells when cells were treated with ALS at concentrations ranging from 0.1–25 μM for 24 hours (Figure 1B and C). When the cells were treated with ALS at concentrations of 0.1 μM, 0.5 μM, 2.5 μM, 10 μM, and 25 μM for 24 hours, the percentages of the viability of AGS cells were 88.7%, 75.0%, 58.4%, 60.0%, and 47.8%, respectively (Figure 1B) and the percentages of the viability of NCI-N78 cells were 89.0%, 72.7%, 59.9%, 63.0%, and 49.48%, respectively (Figure 1C). The 50% inhibition of growth values were 19.1 μM and 26.3 μM for AGS and NCI-N78 cells, respectively. These results demonstrate that ALS induces a concentration-dependent inhibition in the growth of AGS and NCI-N78 cells.
ALS downregulates the phosphorylation level of AURKA in AGS and NCI-N78 cells
ALS is a selective inhibitor of AURKA that plays a critical role the cell mitosis. Since we examined the inhibitory effects of ALS on the viability of AGS and NCI-N78 cells, we further examined the effect of ALS on the phosphorylation of AURKA in AGS and NCI-N78 cells. Both cell lines were treated with ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours. As shown in Figure 2A and B, the phosphorylation level of AURKA was significantly decreased in both cells. In comparison to the control cells, treatment of AGS cells with ALS at 0.1 μM, 1 μM, and 5 μM resulted in 76%, 67%, and 81% decreases in the ratio of p-AURKA/AURKA, respectively. There was an 83% reduction in the ratio of p-AURKA/AURKA in NCI-N78 cells treated with 1 μM ALS, compared to the control cells (Figure 2A and B). The results showed that downregulation of the phosphorylation level of AURKA may contribute to cell-growth inhibition in these two cell lines.
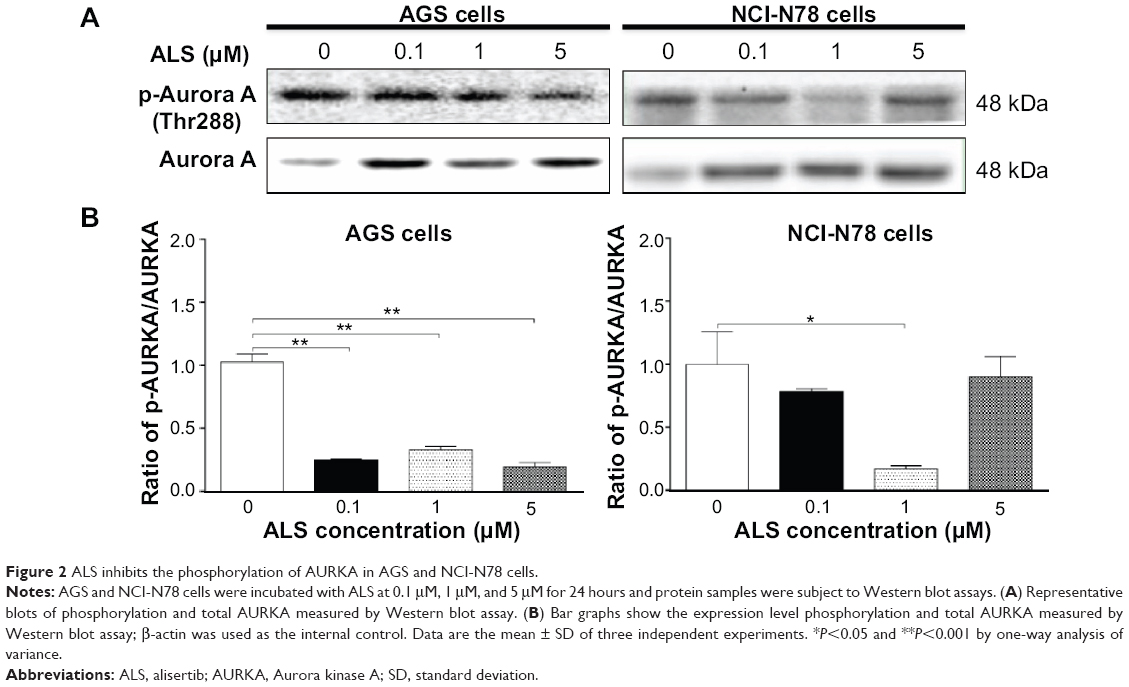 | Figure 2 ALS inhibits the phosphorylation of AURKA in AGS and NCI-N78 cells. |
ALS alters the cell-cycle distribution of AGS and NCI-N78 cells
We next examined the cell-cycle distribution of AGS and NCI-N78 cells treated with ALS using a flow cytometer. The data showed that ALS significantly induced cell-cycle arrest in G2/M phase in both cell lines (Figure 3A and B). The percentages of AGS and NCI-N78 cells in G2/M phase were 31.8% and 19.7%, respectively, at the basal level. Compared to control cells, a concentration-dependent increase in the cell number in G2/M phase was observed after ALS treatment at 0.1 μM, 1 μM, and 5 μM for 24 hours. The percentages of AGS cells arrested in G2/M phase were 51.6%, 87%, and 86.4% when cells were treated with ALS at 0.1 μM, 1 μM, and 5 μM, respectively (Figure 3A and B); the percentages of NCI-N78 cells arrested in G2/M phase were 71.4%, 79.3%, and 74.4% when treated with ALS at 0.1 μM, 1 μM, and 5 μM, respectively (Figure 3A and B). ALS treatment also decreased the percentages of cells in G1 phase in both cell lines compared to the control cells. The basal levels of cells in G1 phase for AGS and NCI-N78 cell lines were 56.5% and 74%, respectively. After the treatment of cells with ALS at 0.1 μM, 1 μM, and 5 μM, the percentages of AGS cells in G1 phase were 41.0%, 9.9%, and 10.4%, respectively, while the percentages of NCI-N78 cells in G1 phase were 21.4%, 15.4%, and 19.7%, respectively (Figure 3A and B). There was no significant difference in the number of cells in S phase in both cell lines treated with ALS. Intriguingly, exposure of NCI-N78 cells to ALS at 0.1 μM, 1 μM, and 5 μM resulted in an accumulation of aneuploidy (Figure 3A), which may be due to the interruption of the spindle assembly, chromosome segregation, and mitotic entry.
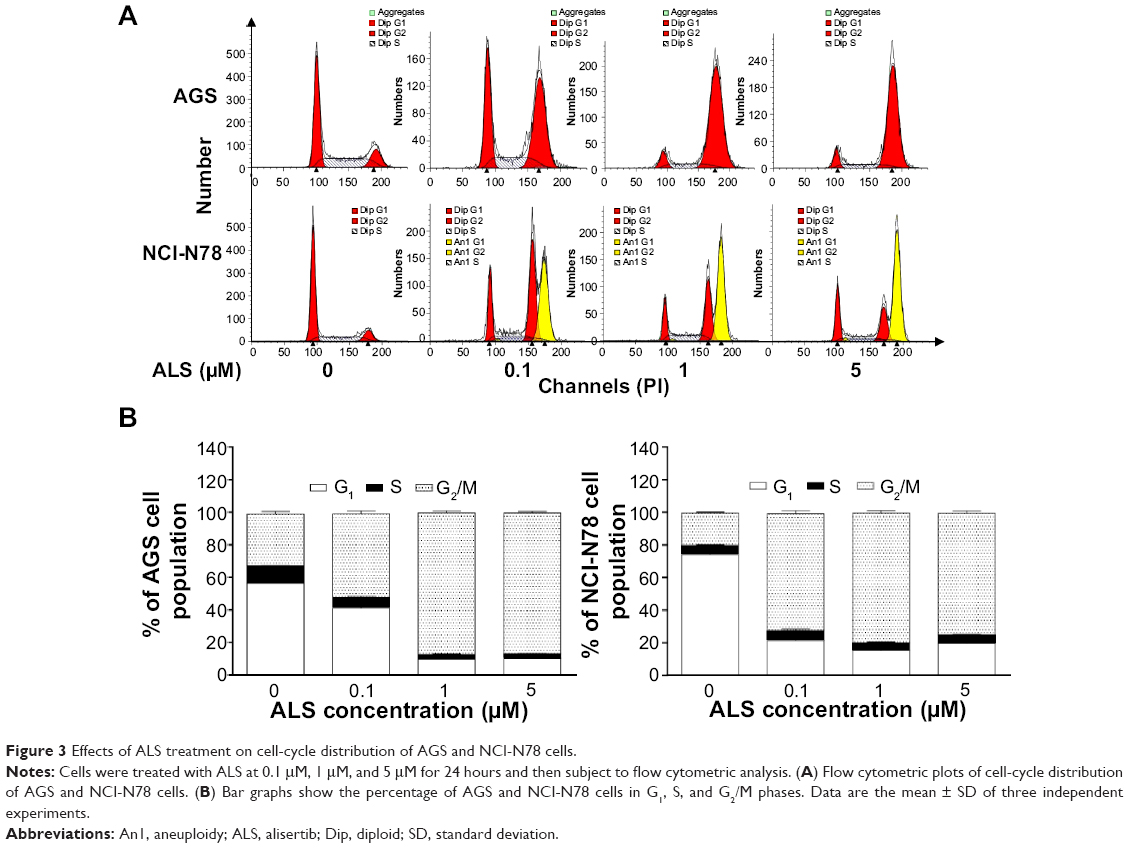 | Figure 3 Effects of ALS treatment on cell-cycle distribution of AGS and NCI-N78 cells. |
We did further experiments to evaluate the effect of ALS treatment at 1 μM on cell-cycle distribution in both cell lines over 72 hours (Figure 4). For AGS cells, the percentages of cells at G2/M phase went from 27.7% at the basal level to 44.8%, 57.6%, 68.9%, 80.7%, 88.9%, and 85.4% after 4 hours, 8 hours, 12 hours, 24 hours, 48 hours, and 72 hours, respectively (Figure 4A and B). Compared to the control cells, the percentages of NCI-N78 cells at G2/M phase increased from 21.8% at the basal level to 41.5%, 69.9%, 69.4%, and 67.1% after treatments for 12 hours, 24 hours, 48 hours, and 72 hours, respectively (Figure 4A and B). The percentages of AGS cells in G1/M phase went from 62.3% at the basal level to 39.6%, 31%, 21.2%, 10.9%, 7.53%, and 5.4% after 4 hours, 8 hours, 12 hours, 24 hours, 48 hours, and 72 hours, respectively. There was a decrease in the percentages of NCI-N78 cells in G1 phase from 60.0% at the basal level to 36.1%, 16.7%, 17.0%, and 16.2% after treatments for 12 hours, 24 hours, 48 hours, and 72 hours, respectively (Figure 4). These results indicate that ALS inhibits the growth of AGS and NCI-N78 cells.
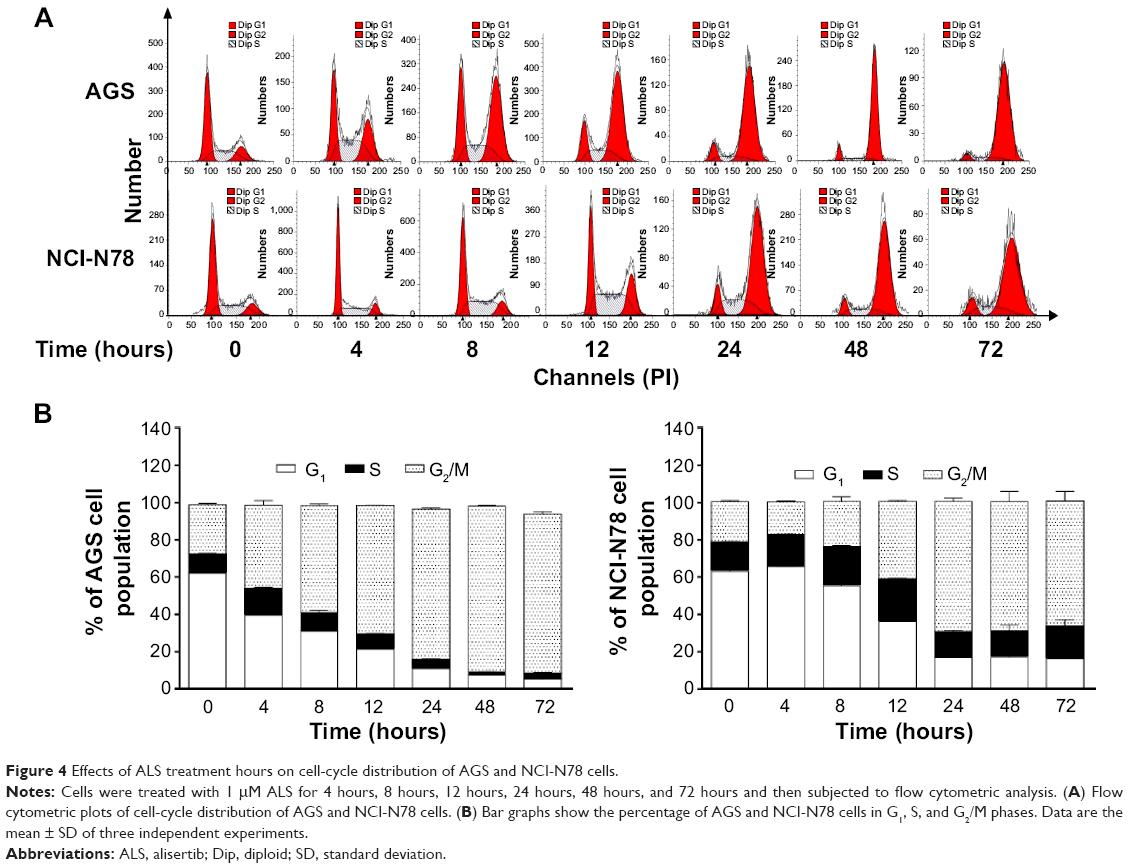 | Figure 4 Effects of ALS treatment hours on cell-cycle distribution of AGS and NCI-N78 cells. |
ALS upregulates the expression of p53, p21 Waf1/Cip1, and p27 Kip1 and downregulates the expression of cyclin B1 and CDC2 in AGS and NCI-N78 cells
To study the mechanism for ALS-induced cell-cycle arrest in AGS and NCI-N78 cells, we examined the expression level of key regulators responsible for G2/M-phase checkpoints using the Western blot assay. Both cells were treated with ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours. In comparison to the control cells, the expression level of CDC2 was reduced 1.8-fold, 1.5-fold, and 1.6-fold when treated with ALS at concentrations of 0.1 μM, 1 μM, and 5 μM for 24 hours, respectively, and the expression level of cyclin B1 was reduced 1.8-fold with 5 μM ALS for 24 hours in AGS cells (Figure 5A and B). Similarly, the inhibitory effect on expression of CDC2 and cyclin B1 was observed in NCI-N78 cells. Compared to the control cells, the expression levels of CDC2 and cyclin B1 were significantly decreased 3.2-fold and 1.8-fold, respectively, in NCI-N78 cells treated with ALS at 0.1 μM for 24 hours (Figure 5A and B). These results suggest that the inhibitory effect of ALS on cell growth could be partially ascribed to the downregulation of the expression of CDC2 and cyclin B1.
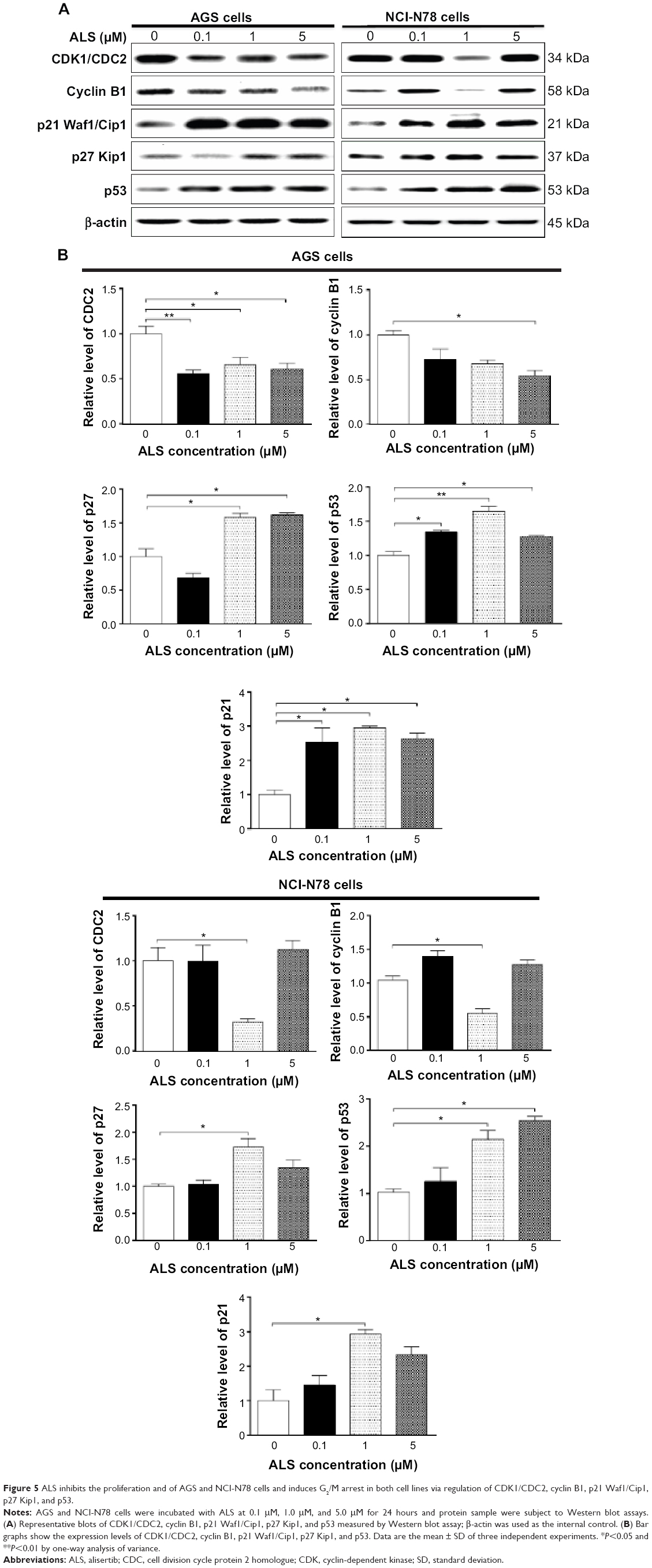 | Figure 5 ALS inhibits the proliferation and of AGS and NCI-N78 cells and induces G2/M arrest in both cell lines via regulation of CDK1/CDC2, cyclin B1, p21 Waf1/Cip1, p27 Kip1, and p53. |
To further explore the molecular mechanism for the inducing effect of ALS on cell-cycle arrest, the expression levels of p21 Waf1/Cip1, p27 Kip1, and p53 in AGS and NCI-N78 cells treated with ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours were evaluated using Western blot assay (Figure 5A and B). The tumor-suppressor protein p21 Waf1/Cip1 acts as an inhibitor of cell-cycle progression, and it serves to inhibit kinase activity and block progression through G1/S in association with CDK2 complexes.21 p53 is a tumor-suppressor protein that plays a major role in cellular response to DNA damage and other genomic aberrations. p53 is phosphorylated and upregulates p21 Waf1/Cip1 transcription via a p53-responsive element during cell-cycle stages.22 p27 Kip1 is a member of the Cip/Kip family of cyclin-dependent kinase inhibitors and enforces the G1 restriction point via its inhibitory binding to CDK2/cyclin E and other CDK/cyclin complexes.23 In comparison to control cells, the expression levels of p21 Waf1/Cip1 and p53 were increased in AGS cells when treated with ALS at 0.1 μM, 1 μM, and 5 μM. There were 2.5-fold, 2.9-fold, and 2.6-fold increases in the expression level of p21 Waf1/Cip1 and 1.3-fold, 1.6-fold, and 1.2-fold increases in the expression of p53. Incubation of AGS cells with ALS at 1 μM and 5 μM led to a 1.6-fold increase in the expression level of p27 (P<0.05%, Figure 5A and B). For NCI-N78 cells, there were 2.9-fold and 1.7-fold increases in the expression of p21 Waf1/Cip1 and p27 in NCI-N78 cells when treated with ALS at 1 μM and 5 μM, respectively, for 24 hours. The expression of p53 was also increased 2.1-fold and 2.5-fold when cells were treated with ALS at 1 μM and 5 μM, respectively, for 24 hours (Figure 5A and B). These results suggest that ALS can upregulate the expression of p21 Waf1/Cip1, p27 Kip1, and p53 in both cell lines.
ALS induces the apoptosis of AGS and NCI-N78 cells via activation of mitochondria-dependent pathway
To test the killing effects of ALS on gastric cancer cells, we observed the apoptosis-inducing effect of ALS on AGS and NCI-N78 cells when the number of apoptotic cells was quantified by flow cytometry. Cells were incubated with ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours; results are shown in Figure 6A and B. The total percentage of apoptotic AGS cells (early + late apoptosis) was 4.5% at the basal level. When AGS cells were treated with ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours, the total percentages of apoptotic cells were 6.3%, 7.5%, and 10.9%, respectively. Compared to the control cells, there was a 2.4-fold increase in the total percentage of apoptotic AGS cells when treated 5 μM ALS (P<0.01; Figure 6A and B). The number of apoptotic NCI-N78 cells at the basal level was 5.4%. When NCI-N78 cells were treated with ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours, the total percentages of apoptotic cells (early + late apoptosis) were 6.2%, 7.7%, and 10.6 %, respectively. In comparison to the control cells, there were 1.4-fold and 2.0-fold increases in the total percentage of apoptotic NCI-N78 cells when treated with ALS at 1 μM and 5 μM, respectively (P<0.05 or P<0.001; Figure 6A and B). The results clearly show that ALS induces apoptosis of both AGS and NCI-N78 cells in a concentration-dependent manner.
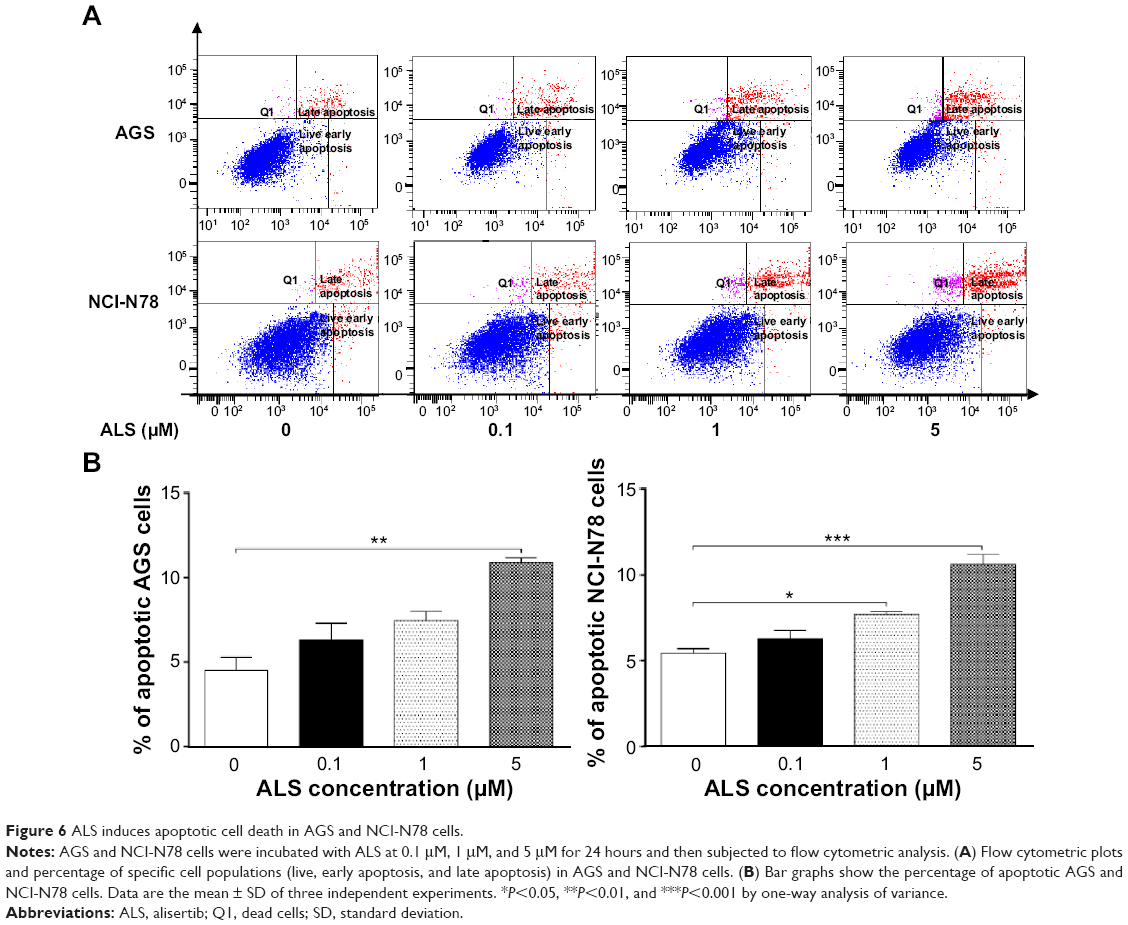 | Figure 6 ALS induces apoptotic cell death in AGS and NCI-N78 cells. |
In order to reveal the possible mechanisms for the proapoptotic effect of ALS on AGS and NCI-N78 cells, we tested the effect of ALS treatment in these two cell lines by measuring the expression levels of the proapoptotic protein Bax and the antiapoptotic proteins Bcl-2 and Bcl-xl (Figure 7A and B). AGS and NCI-N78 cells were treated with ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours. The expression level of Bax was increased in the two cell lines (P<0.05; Figure 7A and B). In comparison to the control cells, incubation of AGS and NCI-N78 cells with 5 μM ALS significantly increased Bax expression levels 1.3-fold and 1.8-fold, respectively (P<0.05 or P<0.001). However, there was a remarkable decline in the expression of antiapoptotic proteins Bcl-xl and Bcl-2 when AGS and NCI-N78 cells were treated with ALS. Compared to the control cells, the expression level of Bcl-xl was reduced 33.3% in AGS cells and 38.3% in NCI-N78 cells when cells were treated with 1 μM ALS for 24 hours. The expression level of Bcl-2 was decreased 55.9% in AGS cells and 47.8% in NCI-N78 cells when treated with 5 μM ALS (Figure 7A and B).
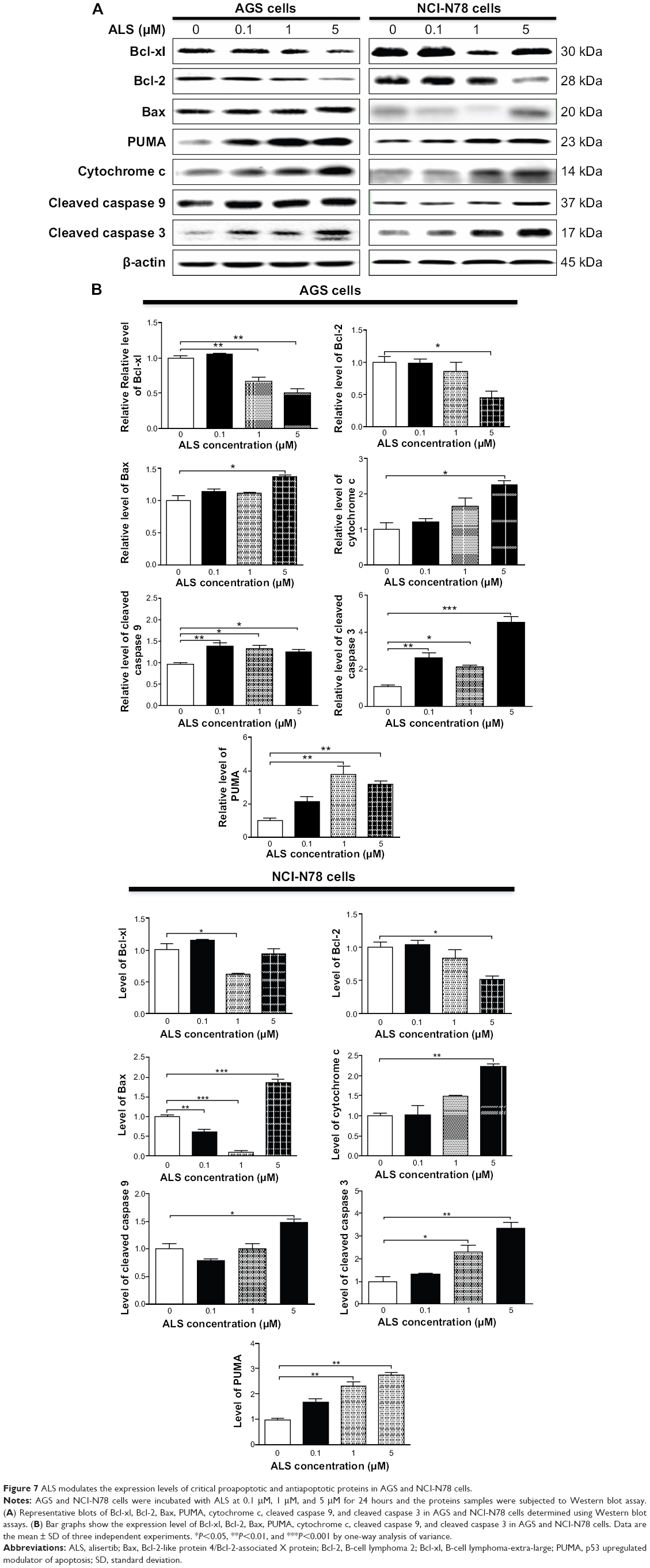 | Figure 7 ALS modulates the expression levels of critical proapoptotic and antiapoptotic proteins in AGS and NCI-N78 cells. |
We observed a marked increase in the expression level of Bax but decrease in the expression levels of Bcl-xl and Bcl-2, resulting in a rise in the ratio of proapoptotic protein/antiapoptotic protein and shifting the balance to apoptotic status. Thus, we investigated the expression of other critical proapoptotic and antiapoptotic proteins in AGS and NCI-N78 cells. We examined the effect of ALS on the expression of PUMA, which plays an important role in the regulation of antiapoptotic proteins. The expression level of PUMA was significantly increased 3.8-fold and 3.2-fold in AGS cells when treated with 1 μM and 5 μM ALS, respectively, for 24 hours. Incubation of NCI-N78 cells with ALS at 1 μM and 5 μM increased the expression level of PUMA 2.3-fold and 2.7-fold, respectively (Figure 7A and B).
The effect of ALS treatment on the mitochondria-related apoptotic pathway was further examined in AGS and NCI-N78 cells. The release of cytochrome c from mitochondria to cytosol initiates the caspase-dependent apoptotic cascade.24 The release of cytochrome c from the mitochondrial intermembrane space denotes the early event during apoptotic cell death. As such, we observed the effect of ALS treatment on the release of cytochrome c in both AGS and NCI-N78 cells. Treatment of AGS and NCI-N78 cells with ALS for 24 hours significantly increased the release of cytochrome c from mitochondria in concentration-dependent manner (Figure 7A and B). Subsequently, we observed a significant increase in the activation of caspases 9 and 3, which are two critical proteins in mitochondria-mediated apoptosis. Compared to the control cells, treatment of AGS and NCI-N78 cells with ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours significantly increased the level of cleaved caspases 3 and 9 (Figure 7A and B). These results indicate that ALS induces a significant activation of caspases 9 and 3 and eventually leads to apoptosis of both AGS and NCI-N78 cells.
ALS induces the autophagy of AGS and NCI-N78 cells in via inhibition of the PI3K/Akt/mTOR pathway
Since we observed ALS-induced apoptosis in AGS and NCI-N78 cells, we next examined the effects of ALS on the autophagy of AGS and NCI-N78 cells using flow cytometric analysis and confocal microscopic examination. The percentages of autophagic cells at the basal level were 1.4% and 4.7% for AGS and NCI-N78 cells, respectively (Figure 8A and B). Incubation of both cell lines with ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours significantly increased the percentages of autophagic cells in a concentration-dependent manner. Compared to the control cells, treatment of AGS cells with 0.1 μM, 1 μM, and 5 μM ALS for 24 hours resulted in 5.8-fold, 10-fold, and 11.1-fold increases, respectively, in the percentage of autophagic cells. For NCI-N78 cells, there were 2.8-fold, 4.2-fold, and 4.4-fold increases in the percentage of autophagic cells when treated with ALS at 0.1 μM, 1 μM, and 5 μM, respectively, compared to the control cells (P<0.05; Figure 8A and B).
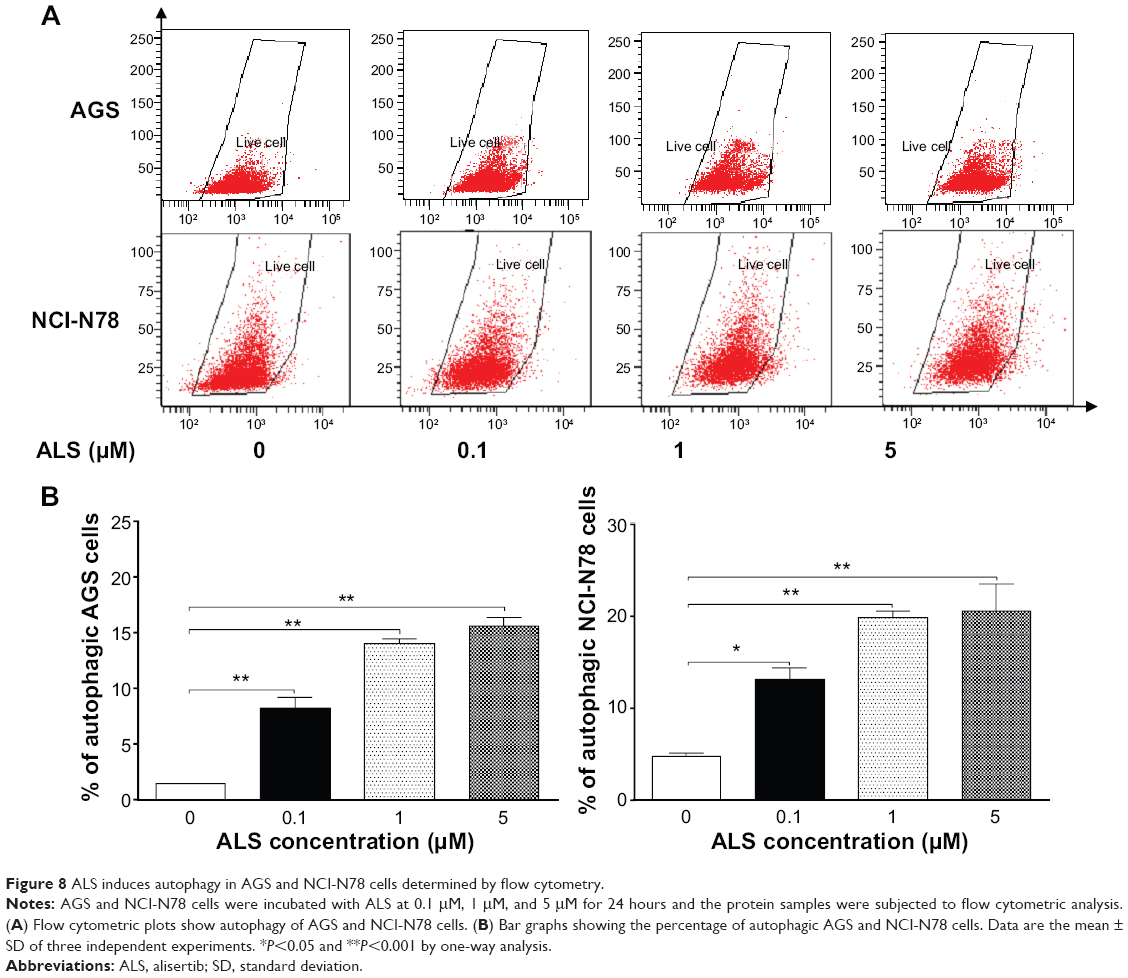 | Figure 8 ALS induces autophagy in AGS and NCI-N78 cells determined by flow cytometry. |
We further observed the effect of ALS on autophagy in AGS and NCI-N78 cells using confocal microscopic examination. In comparison to the control cells, ALS treatment caused a significant increase in autophagy of AGS and NCI-N78 cells in a concentration-dependent manner (Figure 9A and B). For AGS cells, there were 1.1-fold, 1.5-fold, and 1.7-fold increases in autophagy when treated with ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours, respectively (Figure 9A and B). For NCI-N78 cells, there were 1.9-fold, 3.0-fold, and 3.2-fold increases in autophagic death when treated with ALS at 0.1 μM, 1 μM, and 5 μM, respectively, for 24 hours (Figure 9A and B). These results show that ALS induces autophagy in both AGS and NCI-N78 cells.
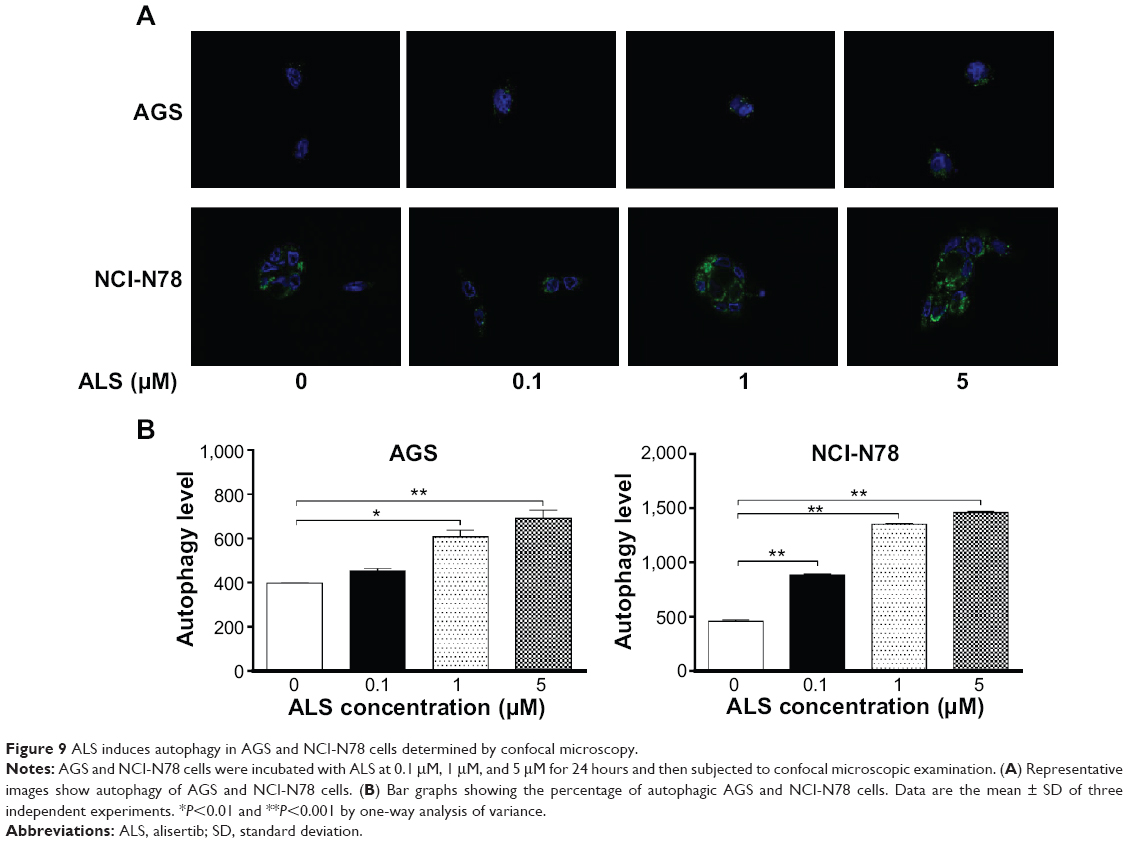 | Figure 9 ALS induces autophagy in AGS and NCI-N78 cells determined by confocal microscopy. |
Next, we investigated the mechanisms for the autophagy-inducing effect of ALS in AGS and NCI-N78 cells. First, we examined the phosphorylation levels of PI3K at Tyr199, AMPK at Thr172, and p38 MAPK at Thr180/Tyr182, which are upstream signaling molecules of the Akt/mTOR pathway and play important roles in the regulation of cell survival and cell death.25,26 In this study, ALS significantly inhibited the phosphorylation of PI3K at Tyr199 in both cell lines, compared to the control cells. The ratio of p-PI3K/PI3K was significantly decreased by ALS treatment in AGS and NCI-N78 cells compared to the control cells. The p-PI3K/PI3K ratios were decreased 48% and 51% when AGS cells were treated with ALS at 1 μM and 5 μM, respectively (P<0.05; Figure 10A and B). In NCI-N78 cells, 1 μM ALS decreased the ratio of p-PI3K/PI3K 55% compared to the control cells (P<0.05; Figure 10A and B).
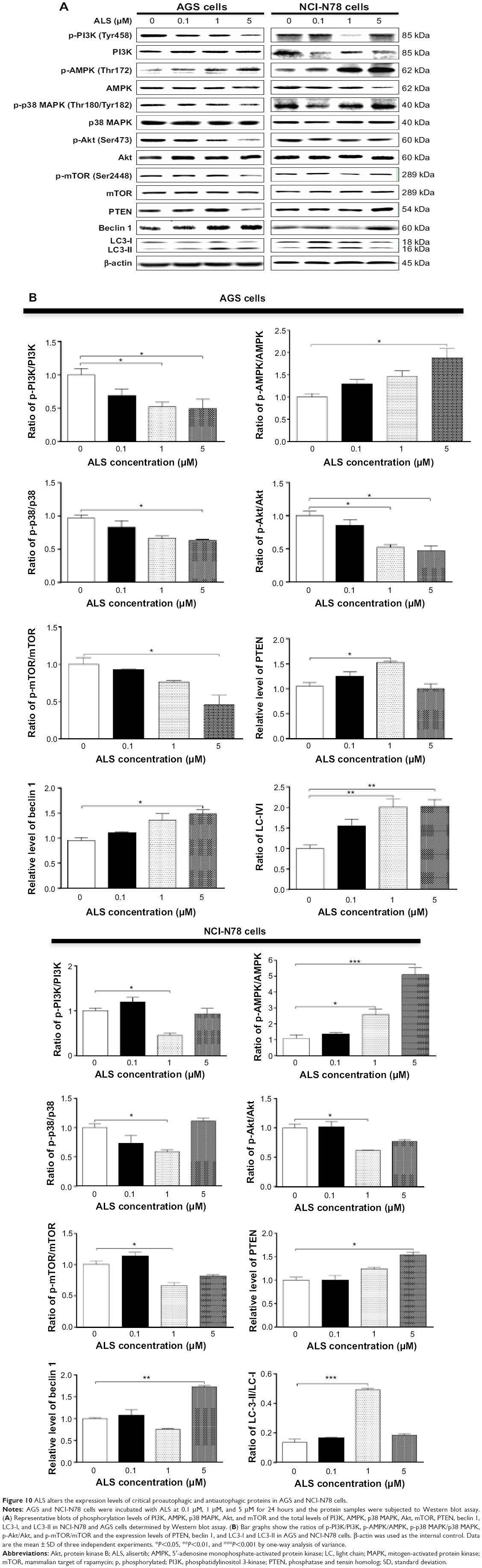 | Figure 10 ALS alters the expression levels of critical proautophagic and antiautophagic proteins in AGS and NCI-N78 cells. |
We further evaluated the effect of ALS on the phosphorylation of Akt at Ser473 and mTOR at Ser2448 and the expression of PTEN in AGS and NCI-N78 cells (Figure 10A and B). Akt is involved in the regulation of various downstream signaling pathways, including metabolism, cell proliferation, survival, growth, and angiogenesis.27 As a downstream effector of PI3K, Akt can activate mTOR, while mTORC2 phosphorylates Akt at Ser473, which stimulates Akt phosphorylation at Thr308 by 3-phosphoinositide–dependent protein kinase-1 and leads to full Akt activation.27 mTOR plays a key role in cell growth, autophagic cell death, and homeostasis.28 mTOR is phosphorylated at Ser2448 via the PI3K/Akt signaling pathway and autophosphorylated at Ser2481.28 mTOR inhibition promotes dissociation of mTOR from the complex of autophagy-related protein (Atg)13 with unc-51–like kinase (ULK)1 and ULK2, thus releasing ULK1/2 to activate focal adhesion kinase family interacting protein of 200 kD, a protein critical for autophagosome formation and autophagy initiation.28 PTEN, a dual-specificity phosphatase and tumor suppressor gene, inhibits Akt/mTOR and MAPK signaling, leading to cell death.29 In comparison to the control cells, the phosphorylation level of Akt at Ser473 was decreased in AGS and NCI-N78 cells with the treatment of ALS at 0.1 μM, 1 μM, and 5 μM for 24 hours (Figure 10A and B). There was no significant alteration in the expression of total Akt in both cell lines. The ratio of p-Akt/Akt was significantly decreased in both cell lines treated with ALS. In AGS cells, the ratio of p-Akt/Akt was decreased 48% and 53% with the treatment of AGS at 1 μM and 5 μM, respectively (Figure 10A and B). In NCI-N78 cells, the ratio of p-Akt/Akt was decreased 39% when cells were treated with ALS at 1 μM for 24 hours, compared to controls.
In addition, the expression level of PTEN was significantly increased when AGS and NCI-N78 cells were treated with ALS for 24 hours (P<0.05; Figure 10A and B). Exposure of AGS and NCI-N78 cells to ALS at 0.1 μM, 1 μM, and 5 μM resulted in a decrease in the phosphorylation level of mTOR at Ser2448. There was no significant change in the expression of total mTOR in both cell lines when treated with ALS for 24 hours. However, a significant decrease in the ratio of p-mTOR/mTOR was observed in both cell lines. In AGS cells, the ratio of p-mTOR/mTOR was decreased by 55% with treatment of ALS at 5 μM (Figure 10A and B). In NCI-N78 cells, the ratio of p-mTOR/mTOR was decreased by 34% when treated with 1 μM ALS (Figure 10A and B).
AMPK is a heterotrimeric complex composed of a catalytic α subunit and regulatory β and γ subunits, playing a key role in the regulation of energy homeostasis, cell survival, and cell death.30 Under certain circumstances, AMPK promotes autophagy by directly activating ULK1, a homologue of yeast Atg1, through phosphorylation of Ser317 and Ser777,31 and high mTOR activity prevents ULK1 activation by phosphorylating ULK1 Ser757 and disrupting the interaction between ULK1 and AMPK.31 As shown in Figure 10A and B, ALS exhibited a promoting effect on the phosphorylation of AMPK at Thr172 in both cell lines. In comparison to controls, treatment of AGS cells with 5 μM ALS resulted in a 1.9-fold increase in the ratio of p-AMPK/AMPK (P<0.05; Figure 10A and B). Similarly, treating NCI-N78 cells with ALS at 1 μM and 5 μM for 24 hours led to 2.6-fold and 5.1-fold increases, respectively, in the ratio of p-AMPK/AMPK (P<0.05 or P<0.01; Figure 10A and B).
p38 MAPK regulates cellular responses to cytokines and stress, and thus controls cell differentiation, cell death, cell migration, and invasion.32 p38 MAPK plays a dual role as a regulator of cell death, in that it can mediate either cell survival or cell death depending on the type of stimulus and in a cell-type–specific manner.33 In contrast to the promoting effects on AMPK phosphorylation at Thr172 by ALS exposure, we observed an inhibitory effect of ALS on the activation of p38 MAPK at Thr180/Tyr182 in both AGS and NCI-N78 cells (Figure 10A and B). Incubation of AGS cells with ALS at 5 μM resulted in a 38% decrease in the ratio of p-p38 MAPK/p38 MAPK (P<0.05; Figure 10A and B). In comparison to the control cells, the ratio of p-p38 MAPK/p38 MAPK was decreased 42% when NCI-N78 cells were treated with ALS at 1 μM (P<0.05; Figure 10A and B). These findings demonstrate that ALS inhibits the phosphorylation of PI3K Tyr199 and p38 MAPK Thr180/Tyr182 but enhances the phosphorylation of AMPK Thr172 in AGS and NCI-N78 cells, contributing to its gastric cancer cell-killing effect.
Finally, we evaluated the effect of ALS on the expression levels of beclin 1 and LC3-I/II. Autophagy is tightly regulated by beclin 1, which forms a complex with vacuolar sorting protein 34 (also called class III PI3K), and serves as a platform for recruitment of other Atgs that are critical for autophagosome formation.34,35 Upon autophagy initiation, LC3 is cleaved at the C-terminus by Atg4 to form the cytosolic LC3-I,36 and LC3-I is consequently proteolytically cleaved and lipidated by Atg3 and Atg7 to form LC3-II, which localizes to the autophagosome membrane. Treatment of AGS and NCI-N78 cells with ALS for 24 hours significantly increased the expression of beclin 1 (Figure 10A and B). There was a 1.3-fold increase in the expression of beclin 1 in NCI-N78 cells treated with 5 μM ALS for 24 hours (P<0.05; Figure 10A and B). Our Western blot analysis found two clear bands of LC3-I and LC3-II in AGS and NCI-N78 cells (Figure 10A and B). After 24-hour treatment with ALS, there was an increase in the expression of LC3-II in both AGS and NCI-N78 cells (Figure 10A and B). In comparison to the control cells, 1 μM and 5 μM ALS treatment resulted in a 2-fold increase in the expression level of LC3-II in AGS cells, and there was a 2-fold increase in the expression level of LC3-II in NCI-N78 cells when treated with ALS at 1 μM for 24 hours (Figure 10A and B). Taken together, these results suggest that ALS exhibits a strong autophagy-inducing effect on AGS and NCI-N78 cells via modulation of PI3K/Akt/mTOR, AMPK, and p38 MAPK-mediated signaling pathways.
SB202190 and WM enhance ALS-induced autophagy in AGS and NCI-N78 cells
Since, we observed that ALS induces significant autophagy in both AGS and NCI-N78 cells involving the PI3K/Akt/mTOR pathway, we used SB202190 and WM to further evaluate the possible mechanisms for the autophagy-inducing effect of ALS. SB202190 is a selective p38 MAPK inhibitor that can induce autophagic vacuole formation through cross-inhibition of the PI3K/mTOR pathway.37 WM is a well-known PI3K inhibitor. In both AGS and NCI-N78 cells, coincubation of SB202190 or WM at 10 μM significantly enhanced ALS-induced autophagy compared to the control cells receiving ALS alone (Figure 11A and B). In AGS cells, SB202190 and WM at 10 μM significantly increased ALS-induced autophagy 37.5% and 22.5%, respectively, compared to the control cells receiving ALS alone (Figure 11A and B). Similarly, in NCI-N78 cells, both SB202190 and WM at 10 μM significantly increased ALS-induced autophagy 22.2% and 10.6%, respectively, compared to the control cells receiving ALS alone (Figure 11A and B). The results show that ALS induces autophagic cell death via PI3K-mediated and p38 MAPK-mediated signaling pathways.
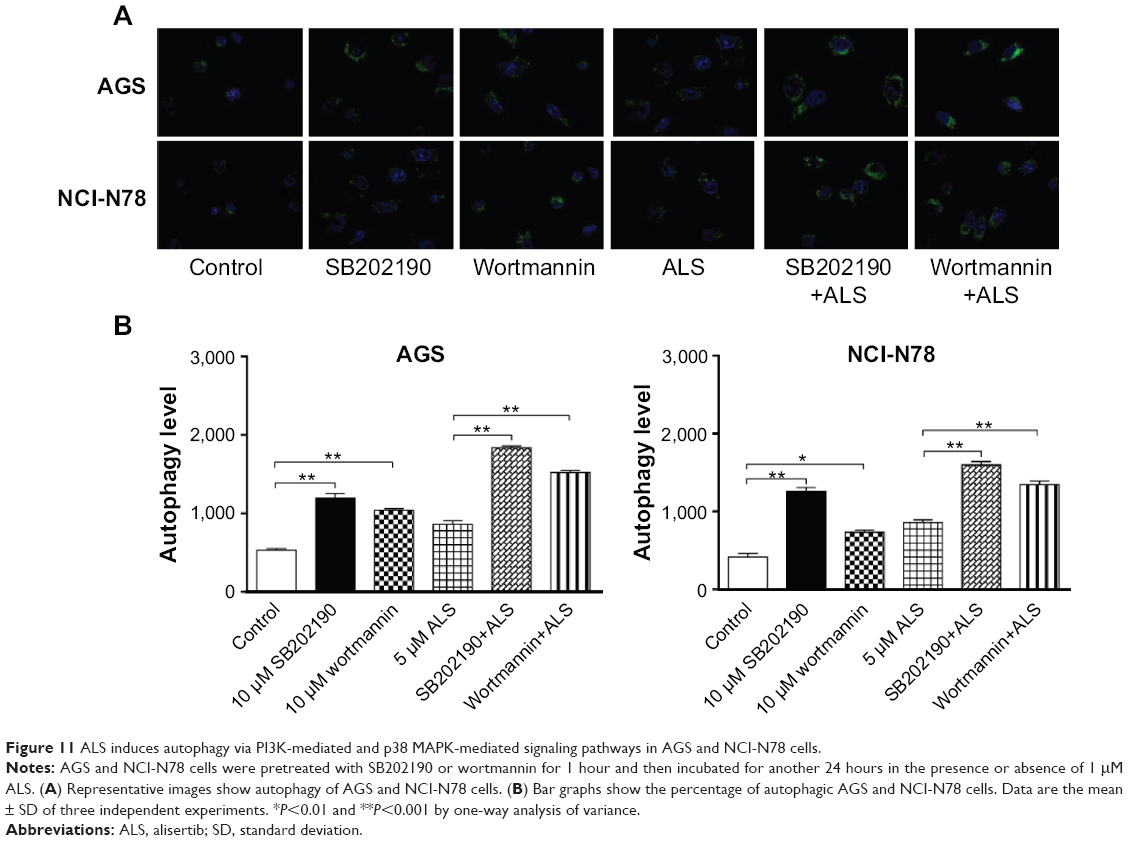 | Figure 11 ALS induces autophagy via PI3K-mediated and p38 MAPK-mediated signaling pathways in AGS and NCI-N78 cells. |
Discussion
Gastric cancer is one of the most common cancers and it responds poorly to current chemotherapy, with unfavorable clinical outcomes.38 Despite novel therapeutic advancements, improvement in the survival rate of patients with gastric cancer is marginal, which suggests the presence of unique, active, intrinsic mechanisms that render resistance to current chemotherapy in gastric cancer.39,40 AURKA has a strong association with cancer initiation, development, and progression.41 It has been reported that abnormalities in the expression and activity of AURKA have been implicated in the pathogenesis of cancer.41 Accumulating evidence suggests that overexpression of AURKA induces chemotherapeutic resistance and regulates several key signaling pathways associated with cell proliferation, cell survival, cell migration and invasion, and cell death in various types of cancer cells.9 Targeting AURKA has been proposed to be a promising therapeutic strategy for cancer treatment, and a number of Aurora kinase inhibitors that have been developed.42–44 ALS is an investigational small-molecule inhibitor that selectively inhibits AURKA. It can induce cell-cycle arrest, polyploidy, and mitotic catastrophe in various types of tumor cells and can induce tumor regression in vivo.8,45,46 In our benchmarking study, we have observed a potent inhibitory effect of ALS on cell proliferation of AGS and NCI-N78 cells. ALS induces gastric cancer cell arrest in G2/M phase and promotes mitochondria-dependent apoptosis and autophagy with the involvement of PI3K/Akt/mTOR, p38 MAPK, and AMPK-mediated signaling pathways.
During a normal cell cycle, the progression of cells in G2 phase to M phase is triggered by the activation of the cyclin-B1–dependent CDC2 kinase,47 which is regulated by a series of phosphorylation and dephosphorylation events and protein–protein interactions involving a network of signaling pathways.48 In general, a cell with suppressed cyclin B1/CDC2 activity would tend to be arrested in the G2/M phase, whereas a cell with elevated cyclin B1/CDC2 activity would be favored to enter mitosis.49 In the present study, we found that ALS concentration-dependently arrested AGS and NCI-N78 cells in G2/M phase. It has been reported that AURKA regulates G2 phase to M phase transitions and its inhibition results in G2/M-phase arrest.50 Our results show that treatment of gastric cancer cells with the AURKA inhibitor ALS is able to inhibit cell proliferation and arrest cell cycle in G2/M phase.
CDC2 and cyclin B1 are two key regulators in cell-cycle checkpoints, and the CDC2–cyclin B1 complex is pivotal in regulating the G2/M-phase transition and mitosis. We have observed a significant decrease in the expression levels of cyclin B1 and CDC2 in AGS and NCI-N78 cells when treated with ALS, which provides an explanation for the inducing effect of ALS on G2/M-phase arrest in both gastric cancer cell lines. Furthermore, it has been reported that p21 Waf1/Cip1, a cyclin-dependent kinase inhibitor regulated by p53, has a negative modulating effect on the CDK1/CDC2–cyclin B1 complex, thereby inducing cell-cycle arrest. We have observed that the expression of p53, p27 Kip1 and p21 Waf1/Cip1 were increased in AGS and NCI-N78 cells treated with ALS, which probably contributes to the inhibitory effect of ALS on the cell proliferation and the inducing effect on cell-cycle arrest. The results indicate that upregulation of p53, p21 Waf1/Cip1, and p27 Kip1 expression by ALS may result in the G2/M-phase arrest in AGS and NCI-N78 cells.
Notably, AURKA has a strong capability of regulating a number of important functional proteins and their associated signaling pathways that regulate cell cycle, cell survival, and cell death in gastric cancer, such as p53,51,52 nuclear factor-κB,53 and signal transducer and activator of transcription 3 (STAT3).54 Sehdev et al52 showed that AURKA promoted gastric tumor growth and cell survival via the regulation of human double-minute-2–induced ubiquitination and inhibition of p53. It also has been reported that overexpression of AURKA promotes inflammation and tumorigenesis in mice via upregulation of the nuclear factor-κB signaling pathway.53 In addition, it has been showed that AURKA increases STAT3 activity through modulation of the expression and phosphorylation levels of STAT3 upstream of nonreceptor tyrosine kinase Janus kinase 2 in gastric cancer cells.54 In the present study, treatment of AGS and NCI-N78 cells with ALS resulted in a significant decrease in the ratio of p-AURKA/AURKA, suggesting that the inhibition of phosphorylation of AURKA contributes, at least in part, to the inhibitory effect of ALS on cell proliferation and the inducing effect on cell-cycle arrest, with the involvement of p53 in AGS and NCI-N78 cells.
Recently, increasing evidence supports the notion that programmed cell death, which includes modulation of apoptosis and autophagy, is a promising strategy to fight against cancer.55–57 Apoptosis is executed by members of the caspase family of cysteine proteases. There are two main pathways, namely the extrinsic death receptor pathway and the intrinsic mitochondria/cytochrome-c–mediated pathway.24,58 In the present study, our findings have shown that treatment of ALS significantly induces apoptotic cell death in AGS and NCI-N78 cells. It is well known that mitochondrial disruption and the subsequent release of cytochrome c trigger the process of apoptosis,24 with proapoptotic members of the Bcl-2 family being promoted and antiapoptotic members of this family being antagonized. Furthermore, antiapoptotic members of Bcl-2 can be inhibited by posttranslational modification and/or increased expression of PUMA, which is an essential regulator of p53-mediated cell apoptosis.24 We have found that the cytosolic level of cytochrome c is significantly increased after ALS treatment, which subsequently activates caspase 9. Activated caspase 9 in turn activates caspase 3, and activated caspase 3 ultimately induces apoptosis with a decrease in Bcl-2 levels. Moreover, we noted a concentration-dependent increase in the expression of PUMA in AGS and NCI-N78 cells. These results indicate that ALS induces mitochondria-dependent apoptosis in both AGS and NCI-N78 cells.
Autophagy is also known as a type II programed cell death. Targeting autophagy for cancer treatment is promising but controversial, due to its multifaceted roles in the regulation of cell survival and cell death.56 Under certain circumstances, autophagy has been known to promote cellular survival during nutrient depletion and is essential for maintaining cellular hemostasis by degrading damaged organelles and proteins.25,26,59 The PI3K/Akt/mTOR signaling pathway is a central pathway involved in autophagy through the regulation of cell growth, motility, protein synthesis, cell metabolism, cell survival, and cell death in response to various stimuli.24,58,60 PI3K activates the serine/threonine kinase Akt, which in turn through a cascade of regulators results in the phosphorylation and activation of the serine/threonine kinase mTOR.60 In this study, ALS induced autophagy in both AGS and NCI-N78 cell lines, which may contribute to its anticancer effect. ALS treatment resulted in a significant inhibition of PI3K/Akt/mTOR signaling, contributing to the autophagy-inducing effect of ALS in AGS and NCI-N78 cells. Of note, it has been reported that AURKA promotes cancer cell survival through the activation of Akt.61,62 Thus, inhibition of AURKA may result in cancer cell death via inactivation of Akt. Our findings showed that ALS treatment inhibited the phosphorylation of AURKA, which may contribute, at least in part, to the inactivation of Akt in AGS and NCI-N78 cells.
In addition, there was a significant activation of AMPK with ALS treatment in the present study. AMPK is an important regulator of cell death under various conditions through activation of c-Jun N-terminal kinase and p53 and inhibition of mTOR.26 Thus, activation of AMPK may contribute to the inhibitory effect of ALS on the mTOR signaling pathway. Furthermore, our findings showed that ALS significantly suppressed the phosphorylation of p38 MAPK. SB202190, a p38/MAPK inhibitor, remarkably increased autophagic cell death in both cell lines. It has been reported that p38 MAPK is responsive to a variety of stimuli, such as cytokines, ultraviolet irradiation, heat shock, and osmotic shock, and regulates cell differentiation, apoptosis, and autophagy.24,26 Taken together, both AMPK and p38 MAPK play an important role in ALS-induced autophagy of gastric cancer cells. Notably, there are differential responses to ALS treatment with regard to the regulation of p38 MAPK in AGS and NCI-N78 cells. This may be due to the differences in the origin of cell lines, cytogenetics, and other related factors. For AGS cells, it is a hyperdiploid cell line and the percentage of polyploidy was 3.6%. The modal chromosome number was 49, occurring in 60% of AGS cells.15 For NCI-N78 cells, it is a near diploid cell line and the double minutes were present in 64% of cells examined.16
In conclusion, ALS inhibited cell proliferation, induced cell-cycle arrest, activated the mitochondria-dependent apoptotic pathway, and promoted autophagy in human gastric cancer AGS and NCI-N78 cells. ALS-induced inhibition of PI3K/Akt/mTOR and p38 MAPK pathways and activation of AMPK contribute to the autophagy-inducing effect of ALS. Inhibition of AURKA may play an important role in the gastric cancer cell-killing effect of ALS. In the future, more functional and mechanistic studies are warranted to elucidate the role of ALS in the treatment of gastric cancer.
Acknowledgments
The authors appreciate the financial support from the Startup Fund of the College of Pharmacy, University of South Florida, Tampa, FL, USA. Dr Zhi-Wei Zhou is a holder of a postdoctoral scholarship from the College of Pharmacy, University of South Florida, Tampa, FL, USA. The authors appreciate the help from Mr Jeffrey L Edelman from University of South Florida (Tampa, FL) for manuscript proofreading.
Disclosure
The authors report no conflicts of interest in this work.
References
GLOBOCAN 2012: Cancer Incidence and Mortality Worldwide in 2012 [webpage on the Internet]. Lyon: International Agency for Research on Cancer, World Health Organization; 2014. Available from: http://globocan.iarc.fr. Accessed November 10, 2014. | ||
Wang J, Yang S, Zhang H, et al. Aurora-A as an independent molecular prognostic marker in gastric cancer. OncolRep. 2011;26(1):23–32. | ||
Hohenberger P, Gretschel S. Gastric cancer. Lancet. 2003;362(9380):305–315. | ||
Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med. 2006;355(1):11–20. | ||
Bolanos-Garcia VM. Aurora kinases. Int J Biochem Cell Biol. 2005;37(8):1572–1577. | ||
Barr AR, Gergely F. Aurora-A: the maker and breaker of spindle poles. J Cell Sci. 2007;120(Pt 17):2987–2996. | ||
Huck JJ, Zhang M, McDonald A, et al. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol Cancer Res. 2010;8(3):373–384. | ||
Görgün G, Calabrese E, Hideshima T, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010;115(25):5202–5213. | ||
Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora kinase inhibitors – rising stars in cancer therapeutics? Mol Cancer Ther. 2010;9(2):268–278. | ||
Sehdev V, Peng D, Soutto M, et al. The aurora kinase A inhibitor MLN8237 enhances cisplatin-induced cell death in esophageal adenocarcinoma cells. Mol Cancer Ther. 2012;11(3):763–774. | ||
Rugge M, Fassan M, Zaninotto G, et al. Aurora kinase A in Barrett’s carcinogenesis. Hum Pathol. 2010;41(10):1380–1386. | ||
Cammareri P, Scopelliti A, Todaro M, et al. Aurora-a is essential for the tumorigenic capacity and chemoresistance of colorectal cancer stem cells. Cancer Res. 2010;70(11):4655–4665. | ||
Manfredi MG, Ecsedy JA, Chakravarty A, et al. Characterization of alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res. 2011;17(24):7614–7624. | ||
Carol H, Boehm I, Reynolds CP, et al. Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer Chemother Pharmacol. 2011;68(5):1291–1304. | ||
Barranco SC, Townsend CM Jr, Quraishi MA. Heterogeneous responses of an in vitro model of human stomach cancer to anticancer drugs. Invest New Drugs. 1983;1(2):117–127. | ||
Park JG, Frucht H, LaRocca RV. Characteristics of cell lines established from human gastric carcinoma. Cancer Res. 1990;50(9):2773–2780. | ||
Li YC, He SM, He ZX, et al. Plumbagin induces apoptotic and autophagic cell death through inhibition of the PI3K/Akt/mTOR pathway in human non-small cell lung cancer cells. Cancer Lett. 2014;344(2):239–259. | ||
Qiu JY, Zhou ZW, He ZX, et al. Plumbagin elicits differential proteomic responses mainly involving cell cycle, apoptosis, autophagy, and epithelial to mesenchymal transition pathways in human prostate cancer PC-3 and DU145 cells. Drug Des Devel Ther. In press. | ||
Stupack DG. Caspase-8 as a therapeutic target in cancer. Cancer Lett. 2013;332(2):133–140. | ||
Balan M, Pal S. A novel CXCR3-B chemokine receptor-induced growth-inhibitory signal in cancer cells is mediated through the regulation of Bach-1 protein and Nrf2 protein nuclear translocation. J Biol Chem. 2014;289(6):3126–3137. | ||
Warfel NA, El-Deiry WS. p21WAF1 and tumourigenesis: 20 years after. Curr Opin Oncol. 2013;25(1):52–58. | ||
Carvajal LA, Manfredi JJ. Another fork in the road – life or death decisions by the tumour suppressor p53. EMBO Rep. 2013;14(5):414–421. | ||
Yoon MK, Mitrea DM, Ou L, Kriwacki RW. Cell cycle regulation by the intrinsically disordered proteins p21 and p27. Biochem Soc Trans. 2012;40(5):981–988. | ||
Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–241. | ||
Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. | ||
Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19(1):87–95. | ||
Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813(11):1978–1986. | ||
Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344–1348. | ||
Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet. 2013;163C(2):114–121. | ||
Dunlop EA, Tee AR. The kinase triad, AMPK, mTORC1 and ULK1, maintains energy and nutrient homoeostasis. Biochem Soc Trans. 2013;41(4):939–943. | ||
Kim J, Guan KL. Regulation of the autophagy initiating kinase ULK1 by nutrients: roles of mTORC1 and AMPK. Cell Cycle. 2011;10(9):1337–1338. | ||
Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13(9):679–692. | ||
Koul HK, Pal M, Koul S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer. 2013;4(9–10):342–359. | ||
Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. | ||
Maiuri MC, Criollo A, Kroemer G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010;29(3):515–516. | ||
Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. | ||
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. | ||
Dar AA, Belkhiri A, El-Rifai W. The aurora kinase A regulates GSK-3beta in gastric cancer cells. Oncogene. 2009;28(6):866–875. | ||
Kubota E, Kataoka H, Tanaka M, et al. ERas enhances resistance to CPT-11 in gastric cancer. Anticancer Res. 2011;31(10):3353–3360. | ||
Hildebrandt MA, Yang H, Hung MC, et al. Genetic variations in the PI3K/PTEN/AKT/mTOR pathway are associated with clinical outcomes in esophageal cancer patients treated with chemoradiotherapy. J Clin Oncol. 2009;27(6):857–871. | ||
Carvajal RD, Tse A, Schwartz GK. Aurora kinases: new targets for cancer therapy. Clin Cancer Res. 2006;12(23):6869-6875. | ||
Hilton JF, Shapiro GI. Aurora kinase inhibition as an anticancer strategy. J Clin Oncol. 2014;32(1):57–59. | ||
Mountzios G, Terpos E, Dimopoulos MA. Aurora kinases as targets for cancer therapy. Cancer Treat Rev. 2008;34(2):175–182. | ||
Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, Hajduch M. Aurora kinase inhibitors: progress towards the clinic. Invest New Drugs. 2012;30(6):2411–2432. | ||
Maris JM, Morton CL, Gorlick R, et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr Blood Cancer. 2010;55(1):26–34. | ||
Tomita M, Mori N. Aurora A selective inhibitor MLN8237 suppresses the growth and survival of HTLV-1-infected T-cells in vitro. Cancer Sci. 2010;101(5):1204–1211. | ||
Baldin V, Ducommun B. Subcellular localisation of human wee1 kinase is regulated during the cell cycle. J Cell Sci. 1995;108(Pt 6):2425–2432. | ||
Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277(5331):1501–1505. | ||
Wang TH, Wang HS, Soong YK. Paclitaxel-induced cell death: where the cell cycle and apoptosis come together. Cancer. 2000;88(11):2619–2628. | ||
Qin L, Tong T, Song Y, Xue L, Fan F, Zhan Q. Aurora-A interacts with Cyclin B1 and enhances its stability. Cancer Lett. 2009;275(1):77–85. | ||
Dar AA, Zaika A, Piazuelo MB, et al. Frequent overexpression of Aurora Kinase A in upper gastrointestinal adenocarcinomas correlates with potent antiapoptotic functions. Cancer. 2008;112(8):1688–1698. | ||
Sehdev V, Katsha A, Arras J, et al. HDM2 regulation by AURKA promotes cell survival in gastric cancer. Clin Cancer Res. 2014;20(1):76–86. | ||
Katsha A, Soutto M, Sehdev V, et al. Aurora kinase A promotes inflammation and tumorigenesis in mice and human gastric neoplasia. Gastroenterology. 2013;145(6):1312–1322.e1–8. | ||
Katsha A, Arras J, Soutto M, Belkhiri A, El-Rifai W. AURKA regulates JAK2-STAT3 activity in human gastric and esophageal cancers. Mol Oncol. Epub 2014 Jun 3. | ||
Ouyang L, Shi Z, Zhao S, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45(6):487–498. | ||
Reyjal J, Cormier K, Turcotte S. Autophagy and cell death to target cancer cells: exploiting synthetic lethality as cancer therapies. Adv Exp Med Biol. 2014;772:167–188. | ||
Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int. 2014;2014:150845. | ||
Estaquier J, Vallette F, Vayssiere JL, Mignotte B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol. 2012;942:157–183. | ||
Chen Y, Yu L. Autophagic lysosome reformation. Exp Cell Res. 2013;319(2):142–146. | ||
Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10(3):143–153. | ||
Yao JE, Yan M, Guan Z, et al. Aurora-A down-regulates IkappaBalpha via Akt activation and interacts with insulin-like growth factor-1 induced phosphatidylinositol 3-kinase pathway for cancer cell survival. Mol Cancer. 2009;8:95. | ||
Long M, Hao M, Dong K, et al. AEG-1 overexpression is essential for maintenance of malignant state in human AML cells via up-regulation of Akt1 mediated by AURKA activation. Cell Signal. 2013;25(6):1438–1446. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.