")
Back to Journals » Drug Design, Development and Therapy » Volume 9
In silico approach for the discovery of new PPARγ modulators among plant-derived polyphenols
Authors Encinar JA , Fernández-Ballester GJ, Galiano-Ibarra V , Micol V
Received 30 July 2015
Accepted for publication 23 September 2015
Published 4 November 2015 Volume 2015:9 Pages 5877—5895
DOI https://doi.org/10.2147/DDDT.S93449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
José Antonio Encinar,1 Gregorio Fernández-Ballester,1 Vicente Galiano-Ibarra,2 Vicente Micol1,3
1Molecular and Cell Biology Institute, 2Physics and Computer Architecture Department, Miguel Hernández University, Elche, Spain; 3CIBER: CB12/03/30038 Physiopathology of Obesity and Nutrition, CIBERobn, Instituto de Salud Carlos III, Palma de Mallorca, Spain
Abstract: Peroxisome proliferator-activated receptor gamma (PPARγ) is a well-characterized member of the PPAR family that is predominantly expressed in adipose tissue and plays a significant role in lipid metabolism, adipogenesis, glucose homeostasis, and insulin sensitization. Full agonists of synthetic thiazolidinediones (TZDs) have been therapeutically used in clinical practice to treat type 2 diabetes for many years. Although it can effectively lower blood glucose levels and improve insulin sensitivity, the administration of TZDs has been associated with severe side effects. Based on recent evidence obtained with plant-derived polyphenols, the present in silico study aimed at finding new selective human PPARγ (hPPARγ) modulators that are able to improve glucose homeostasis with reduced side effects compared with TZDs. Docking experiments have been used to select compounds with strong binding affinity (ΔG values ranging from -10.0±0.9 to -11.4±0.9 kcal/mol) by docking against the binding site of several X-ray structures of hPPARγ. These putative modulators present several molecular interactions with the binding site of the protein. Additionally, most of the selected compounds have favorable druggability and good ADMET properties. These results aim to pave the way for further bench-scale analysis for the discovery of new modulators of hPPARγ that do not induce any side effects.
Keywords: virtual screening, molecular docking, high-throughput computing, TZDs, human PPARγ, AutoDock/Vina, ADMET, phenolic compounds
Introduction
Human peroxisome proliferator-activated receptors (hPPARs) are nuclear soluble proteins that function as ligand-dependent transcription factors belonging to the thyroid/retinoid nuclear receptor family.1 After binding to different types of ligands, PPARs form heterodimers with the retinoic X receptor (RXR), and the resulting PPAR/RXR heterodimer recruits different transcriptional cofactors that bind to the promoter region of the respective target gene and initiate transcription.2 To date, three PPAR proteins have been identified:3,4 PPARα (UniProt: Q07869-PPARA_HUMAN), PPARδ/β (UniProt: Q03181-PPARD_HUMAN), and PPARγ (UniProt: P37231-PPARG_HUMAN). All three of these proteins coordinate pathways involved in lipid and glucose metabolism. Despite the similarities in their primary and secondary structures, PPAR isoforms present marked differences in their tissue distribution, ligands, and physiological role.5 PPARα regulates the expression of genes involved in lipid metabolism and has a higher presence in the heart, liver, and brown adipose tissue.6 The PPARδ/β isoform is expressed ubiquitously in all tissues, particularly in tissues involved in lipid metabolism, such as adipose, liver, kidney, and muscle tissues. Based on the information currently available, it appears that PPARδ/β plays a role mainly in three areas: 1) regulation of energy metabolism, 2) cell proliferation and differentiation, and 3) protection under stress conditions, such as oxidative stress and inflammation.7 PPARγ is the best-characterized member of the PPAR family; it is predominantly expressed in adipose tissue and plays a significant role in lipid metabolism, adipogenesis, glucose homeostasis, and insulin sensitization.8,9 PPARγ presents two isoforms, PPARγ1 and PPARγ2, as a result of differential promoter usage and alternative splicing which results in PPARγ1 having 28 additional amino acids at the N-terminus. PPARγ1 is abundantly expressed in adipose tissue, the large intestine, and hematopoietic cells and to a lower degree in the kidneys, liver, muscles, pancreas, and small intestine. PPARγ2 is restricted to white and brown adipose tissue under physiological conditions.10,11
Various molecules have been suggested as biological ligands of PPARγ: polyunsaturated fatty acids, prostanoids, eicosanoids, components of oxidized low-density lipoproteins, and oxidized alkyl phospholipids.12 These molecules can activate PPARγ and lead to increased expression of PPARγ-target genes. However, the in vivo concentration of many of these molecules is not sufficient to activate PPARγ. Therefore, further studies are needed to determine specific endogenous ligands of PPARγ. As synthetic ligands, thiazolidinediones (TZDs) are full agonists that have been used in clinical practice to treat type 2 diabetes for many years and they effectively lower blood glucose levels and improve insulin sensitivity.13 However, the administration of TZDs has been associated with severe side effects such as fluid retention, weight gain, cardiac hypertrophy, bone fractures, and hepatotoxicity.14 Troglitazone was withdrawn from the market due to liver toxicity; farglitazar failed to pass Phase III clinical trials due to the emergence of peripheral edema; rosiglitazone was removed from the European market due to its association with excessive cardiovascular risk. Pioglitazone is currently in clinical practice even though it has also been linked to controversial side effects, including an increased risk of cardiovascular-related death.15 Considering these facts, it appears evident that the search for safer new agonists is an important goal in the fight against obesity-related pathologies.
The use of natural products has re-emerged in the field of drug discovery.16 Plant-derived molecules possess a high chemical scaffold diversity, which leads to molecular promiscuity,17 and are evolutionarily optimized to serve different biological functions, conferring them a high drug-likeness and making them an excellent source for the identification of new drug leads.18 Several PPARγ ligands were identified in plants that are common food sources, including the tea plant, soybeans, palm oil, ginger, grapes, and wine, and a number of culinary herbs and spices1 (eg, Origanum vulgare, Rosmarinus officinalis, Salvia officinalis, and Thymus vulgaris). The use of these compounds, which are often weak PPARγ agonists, may become an alternative for the prevention of obesity through dietary intervention. Among the natural products that have been well characterized to serve as PPARγ ligands, various phenolic compounds have been identified.1 We recently reported19 that the capacities of lemon verbena polyphenolic extract and its major compound verbascoside to ameliorate high glucose-induced metabolic disturbances are mediated by the PPARγ-dependent transcriptional upregulation of adiponectin. Therefore, the role of polyphenols as partial PPARγ agonists based on selective receptor–cofactor interactions and target gene regulation may deserve intensive research.20
The diet of Western populations is rich in phenolic compounds, which are primarily found in fruits, vegetables, and beverages, such as tea, coffee, wine, and fruit juices. However, precise knowledge of the effects of each phenolic compound on health and disease, as well as the most accurate possible assessment of polyphenol intake, still requires extensive scientific research.
In this context, we performed an in silico study to find potential efficient hPPARγ agonists from the phenolic compounds recorded in the Phenol Explorer database21 (~924 compounds) together with other chemical libraries with molecules presenting 70% structural similarity to scutellarin (~10,437 compounds), a flavone that has been shown to bind PPAR in computational studies and has been validated in cellular assays.22 The best scoring compounds were compared with several phytochemicals found in the literature as well-characterized PPARγ ligands. These compounds were also evaluated for their pharmacodynamic and pharmacokinetic properties. Our in silico approach aims to establish a basis for the further evaluation of potential hPPARγ modulators.
Materials and methods
Protein structures for hPPARγ and chemical libraries
To date, 130 crystal structures of the hPPARγ protein have been solved and deposited into the Protein Data Bank, many of them with their potential inhibitors, including phenolic compounds. Only the X-ray-derived PPARγ structures in complex with modulators were taken from the Brookhaven Protein Data Bank, and these have the following codes: 4PRG, 4JL4, 4JAZ, 4HEE, 4FGY, 4F9M, 4EMA, 4EM9, 4E4Q, 4E4K, 4A4W, 4A4V, 3VSP, 3VSO, 3VSP, 3VN2, 3VJI, 3V9V, and 3V9T. Altogether, these structures represent a wide variety of sample configurations of the receptor.23 The residues forming the binding site for the ligand were investigated.24,25
The two-dimensional (2D) structures of a total of 924 compounds belonging to different classes of phenolic compounds were downloaded in spatial data file (SDF) format from the Phenol Explorer 3.6 database. Additionally, the 2D structure of 10,457 scutellarin-related compounds were download in SDF format from PubChem.26 To handle the large number of ligand structures, we developed a Python (http://www.python.org) script to convert 2D SDFs into individual three-dimensional structures in mol2 format using Marvin Suite 6.0 from ChemAxon (http://www.chemaxon.com).
Docking procedure
Prior to initiating the docking procedure, the protein (receptor) and ligand structures should be prepared. Each PDB file receptor (PPARγ) was edited using PyMOL27 to select a single polypeptide chain and remove all water molecules and cocrystallized ligands from the binding site. All of the selected protein structures were then subjected to geometry optimization using the repair function of the FoldX algorithm.28,29 To perform docking with AutoDock/Vina, the receptor and ligand structures were transformed to the pdbqt file format, which includes atomic charges, atom-type definitions and, for ligands, topological information (rotatable bonds).30 These file preparations were performed using the AutoDock/Vina plugin with scripts from the AutoDock Tools package.31 The ligands used for subsequent docking runs can be prepared either individually through PyMOL selections or by specifying a directory containing a library of ligands to be docked.31 A grid with dimensions of 22×22×22 points was centered to ensure coverage of the binding site of the structure. The files generated for each ligand (“ligand.pdbqt” and “ligand.vina_config.txt”) contain the path for “receptor.pdbqt” and “ligand.pdbqt”, the coordinates of the grid center, the size of the grid in points, the path and name for one output file with all of the best calculated poses, and the path and name for another output file with the ΔG (kcal/mol) for each pose. AutoDock/Vina was set up on a Linux cluster under the ROCKS 6.1 distribution (http://www.rocksclusters.org/) with Condor Roll (http://www.rocksclusters.org/roll-documentation/condor/) to distribute all AutoDock/Vina jobs to the nodes of the cluster. AutoDock/Vina can run on a Windows OS, but should be run on a Linux OS to achieve high performance. Once the calculation is completed, two files are generated per ligand, “ligand.docked.pdbqt” and “ligand.vina.log”, which contain the coordinates of the atoms for each pose (maximum of 20 poses) of a given ligand and the ΔG (kcal/mol) for each pose, respectively. When analyzing a large number of files we used a Python script to automate the reading and extraction of data from ligand.vina.log files. Compounds with minor calculated free energy variations (ie, the best theoretical binding energy) were selected as putative modulators.
In silico analysis of drug likeness and ADMET properties
The drug likeness of the screened compounds was calculated using Marvin Suite 6.0 from ChemAxon (http://www.chemaxon.com). Again, the analysis of large amounts of data was automated with a Python script to obtain up to 80 physicochemical properties of each compound. The in silico absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of the selected compounds were calculated as an alternative approach to the expensive experimental evaluation of ADMET profiles.32 For ADMET assessment, we used admetSAR32 and OSIRIS Property Explorer (http://www.organic-chemistry.org/prog/peo/).
Results and discussion
Cavity binding site analysis
hPPARs, particularly the hPPARγ protein, have been widely studied by crystallographers, as evidenced by the 130 X-ray structures that have been deposited in the Protein Data Bank and recorded in the UniProt database (http://www.uniprot.org/uniprot/P37231). The structural alignment of different PPARγ entries (backbone atoms of amino acids 223–505) showed root-mean-square deviation values below 0.5 Å (0.488±0.109 Å), indicating small conformational variability. All of the structures cocrystallized with a ligand present at the binding site cavity located in the area surrounding α-helix 304–330 (Figure 1A). Measurements of the volume and area of this internal cavity show a direct linear relationship with the size of the ligand, particularly the nonpolar area of the ligand (Figure 1B and Supplementary material 1). The analysis of the cavity binding site indicates that the size of the cavity is apparently adapted to the cocrystallized ligand. Because several amino acids were found to be involved in the interactions with hPPARγ ligands (modulators), the set of interacting residues should be ligand-dependent. As an example, in 3V9V.pdb, a derivative of cercosporamide,33 which acts as a partial agonist, was cocrystalized. This ligand provides several interactions with multiple residues of the protein, including Arg280 (polar contacts)24 and Ile262, Ile341, Ser342, Met348, Ile281, Leu353, Met343, Leu330, Tyr327, Met364, Lys367, His449, Ser289, Cys285, Arg288, Gly284, and Phe287 (nonpolar contacts).24 In this example, the measured34 volume and area are 971.8 Å3 and 1025.3 Å2, respectively. For the remaining 130 hPPARγ structures, the size of the deep groove encompassing the binding pocket was found to be highly variable (Figure 1B and Supplementary material 1). Thus, the binding of different natural phenolic compounds and other drugs to different sites within this large internal cavity can be assumed to modulate the activity of hPPARγ, and it appears to be a promising mode of action for the design of drug candidates35 against hPPARγ.
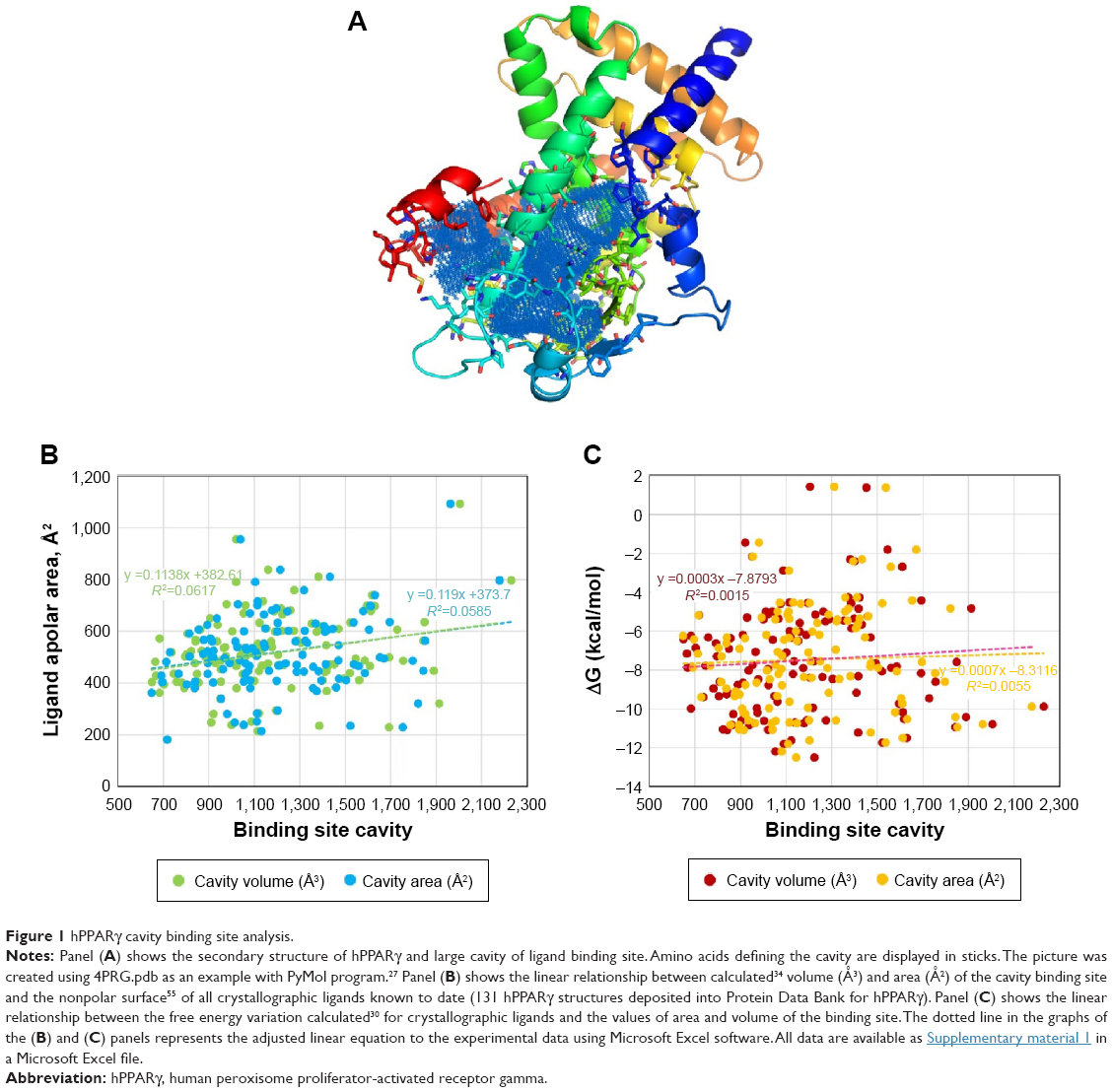 | Figure 1 hPPARγ cavity binding site analysis. |
The variation in the free energy for the binding of ligands cocrystallized with the 123 structures of hPPARγ was measured via AutoDock/Vina (Figure 1C). Binding energies are representative of how precisely the ligand (phenolic natural products or drug candidates) binds to the target protein and were thus considered a set of reference comparisons for selection of the compounds in the library to be screened.35 For the above-described example (3V9V.pdb), the ligand binding ΔG value measured with AutoDock/Vina was −10 kcal/mol, and only 23% of the ligands cocrystallized with hPPARγ have a ΔG≤–10 kcal/mol (Figure 1C). The calculation of the dissociation constant KD36 as a function of ΔG (KD = expΔG/RT) indicated that ΔG values ≤−10 kcal/mol imply the KD is in the nanomolar or subnanomolar range, which was used as a threshold to filter the docking results.
hPPARγ – ligand molecular docking analysis
Progress in high-performance computing allows the virtual screening of a large number of compounds in a short time and the selection of one of the compounds to be tested experimentally.37 The molecular docking of a protein target and small ligand compounds predicts the best interaction mode for a defined binding site.38,39 In the current study, hPPARγ was docked with two sets of compounds: the first contained 924 molecules registered in the Phenol Explorer database21 as phenolic compounds, and the second set includes 10,437 compounds with structural similarity to scutellarin22 (http://goo.gl/1O3O07), of which 1,085 comply with Lipinski’s Rule of Five.40
X-ray crystallography techniques are commonly used to obtain conformations of proteins with high resolution and usually provide snapshots of one or some of the conformations of the proteins.41 However, in the case of hPPARγ, there are a tremendous number of crystallographic structures that may show a large conformational space with some similarity to the native conditions of the protein. During ligand binding, the receptor protein undergoes conformational changes that are essential for its function.42 AutoDock/Vina has been implemented to ensure flexibility of the side chains of the amino acids constituting the ligand binding site. However, performing docking while considering the flexibility of a few amino acid binding sites requires a high computational cost that cannot be assumed during the analysis of the high number of compounds used in this study. For this reason, we chose a sample of 19 hPPARγ structures (see “Materials and methods” section) for docking, and we did not consider the flexibility of the side chains of the amino acids involved in the binding sites.
Tables 1 and 2 show the docking scores obtained for phenolic compounds from the Phenol Explorer database and scutellarin-related compounds, respectively; these tables show hydrogen bonds, direct contacts based on van der Waals radii, and the ΔG calculated using AutoDocK/Vina. The free energy variation is a representative value of the number and intensity of the atomic interactions between the receptor (protein) and the ligand and can thus be considered a baseline comparison for the selection of lead compounds in the process of drug design.35
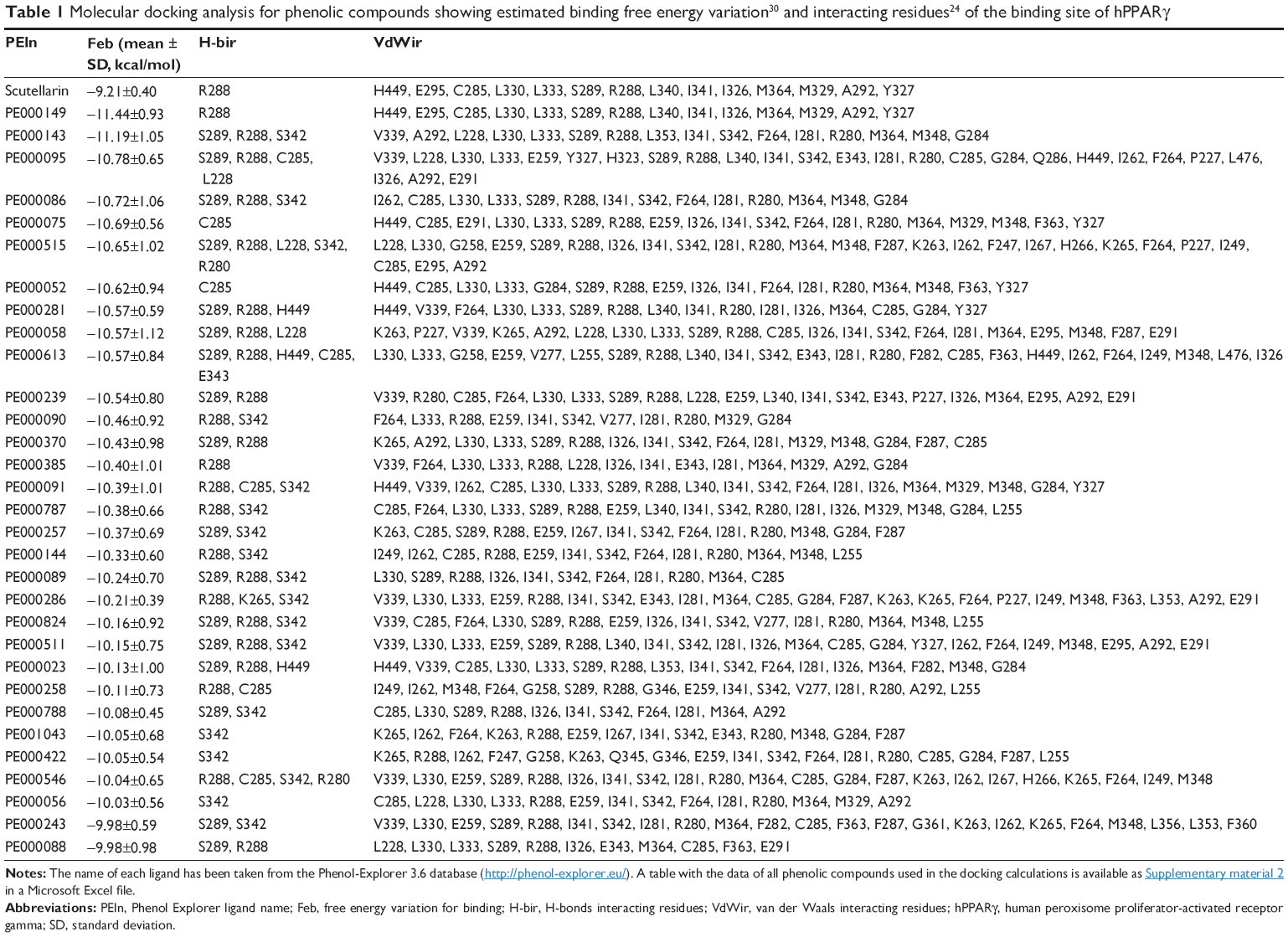 | Table 1 Molecular docking analysis for phenolic compounds showing estimated binding free energy variation30 and interacting residues24 of the binding site of hPPARγ |
 | Table 2 Molecular docking analysis for scutellarin-related compounds showing estimated binding free energy variation30 and interacting residues24 of potential modulators compounds in the binding site of hPPARγ |
Table 1 includes 32 phenolic compounds with free energy variation ranging from −10.0±0.9 to −11.4±0.9 kcal/mol, which are stronger compared with the calculated ΔG values for many phenolic compounds with experimentally demonstrated affinity for hPPARγ1 that were used as the reference set. The phenolic compounds included in Table 2 of the paper published by Wang et al1 bind to purified hPPARγ in the micromolar range, and our calculated ΔG values for these compounds ranged from −8.8±0.7 (PE000404 – genistein) to −7.5±0.4 kcal/mol (PE000592 – resveratrol). The ΔG values of other polyphenols that were within this range are the following: −8.2±0.5 (PE000229 – luteolin), −8.0±0.5 (PE000291 – quercetin), −8.0±0.5 (PE000290 – kaempferol), −7.7±0.6 (PE000124 – catechin), −7.8±0.3 (PE000104 – 2′-OH-chalcone), −8.2±0.5 (PE000397 – biochanin A), −7.8±0.5 (PE000905 – 6-OH-daidzein), and −7.5±0.2 (PE000848 – 6′-OH-O-desmethylangolensin). Interestingly, the other 175 tested polyphenols were found to have ΔG values ranging from −9.0±0.4 to −9.9±0.7 kcal/mol (full table in Supplementary material 2), indicating that these may represent a potential source for new hPPARγ modulators. This second set of polyphenols includes scutellarin (−9.2±0.4 kcal/mol), a phenolic compound that presents antiadipogenic activity through the modulation of PPARγ in 3T3-L1 preadipocytes22 and a reference compound that can be used to construct a structure-related chemical library that can be used in the docking experiments performed in this study.
Table 2 includes 83 scutellarin structure-related compounds (full table in Supplementary material 3) with free energy variations ranging from −10.0±0.6 to −11.0±0.6 kcal/mol; additionally, 2,772 compounds presented variations in the range of −9.0±0.6 to −9.9±0.5 kcal/mol. Among these, 179 satisfied Lipinski’s Rule of Five in evaluating drug-likeness in the PubChem database (http://goo.gl/NnQ4UR), 14 compounds have been tested for biological properties (http://goo.gl/xbkcEk), and 110 compounds can be obtained from a commercial source (http://goo.gl/LTZLQ1).
Prediction of ADMET profiles
Both for the development of drugs and for the environmental risk assessment of drug candidates, it is necessary to know the ADMET properties32 of pesticides and chemicals used in industrial chemistry. Because the experimental evaluation of ADMET is very costly and time-consuming, the application of computational techniques to predict ADMET profiles is a useful solution. The prediction of good ADMET profiles of drug candidates can help eliminate compounds with unacceptable side effects. We calculated ADMET profiles using free online applications32 (http://lmmd.ecust.edu.cn:8000/predict/). A limitation to the use of these applications is that the user must manually enter the chemical formula of each compound. To avoid this problem, we developed a Python script that uses the SMILE code of the compound as input and automatically obtains the calculated ADMET profile. Positive ADMET profiles for the compounds with the best calculated free energy variations (the first selection criterion) constitute a second selection criterion for the final proposed candidates of PPARγ modulators.
For ease of understanding, we present the ADMET profiles as ADME values (Tables 3 and 4 show the predicted molecular pharmacokinetic properties of the selected phenolic compounds of the Phenol Explorer database and scutellarin-related compounds, respectively) and toxicity profiles (Tables 5 and 6 show the results of the toxicity assessment of selected compounds of the Phenol Explorer database and scutellarin-related compounds, respectively). A full table with the ADME and toxicity profiles of all of the compounds used in the docking experiments is available as supplemental material (Supplementary material 2 and Supplementary material 3).
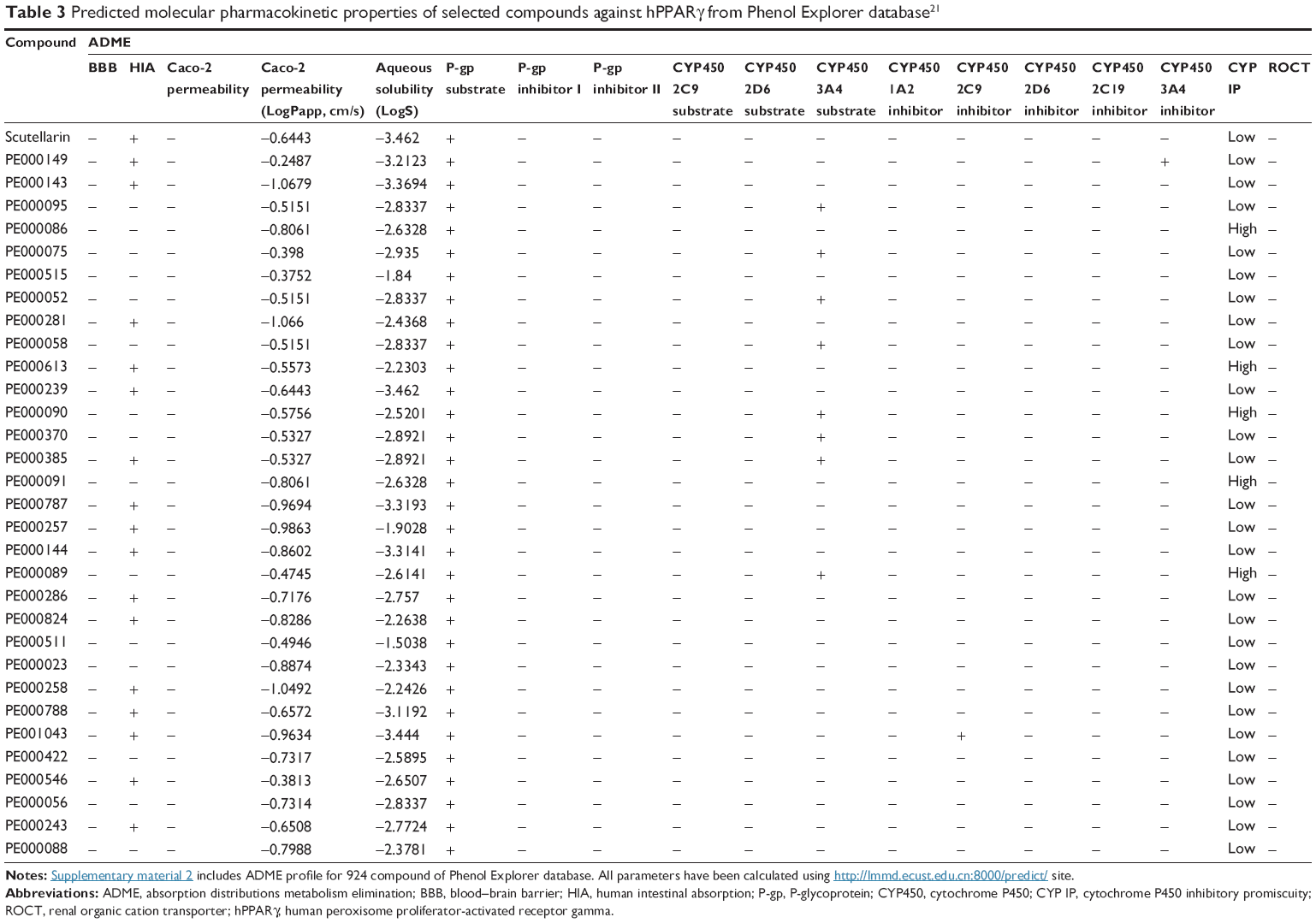 | Table 3 Predicted molecular pharmacokinetic properties of selected compounds against hPPARγ from Phenol Explorer database21 |
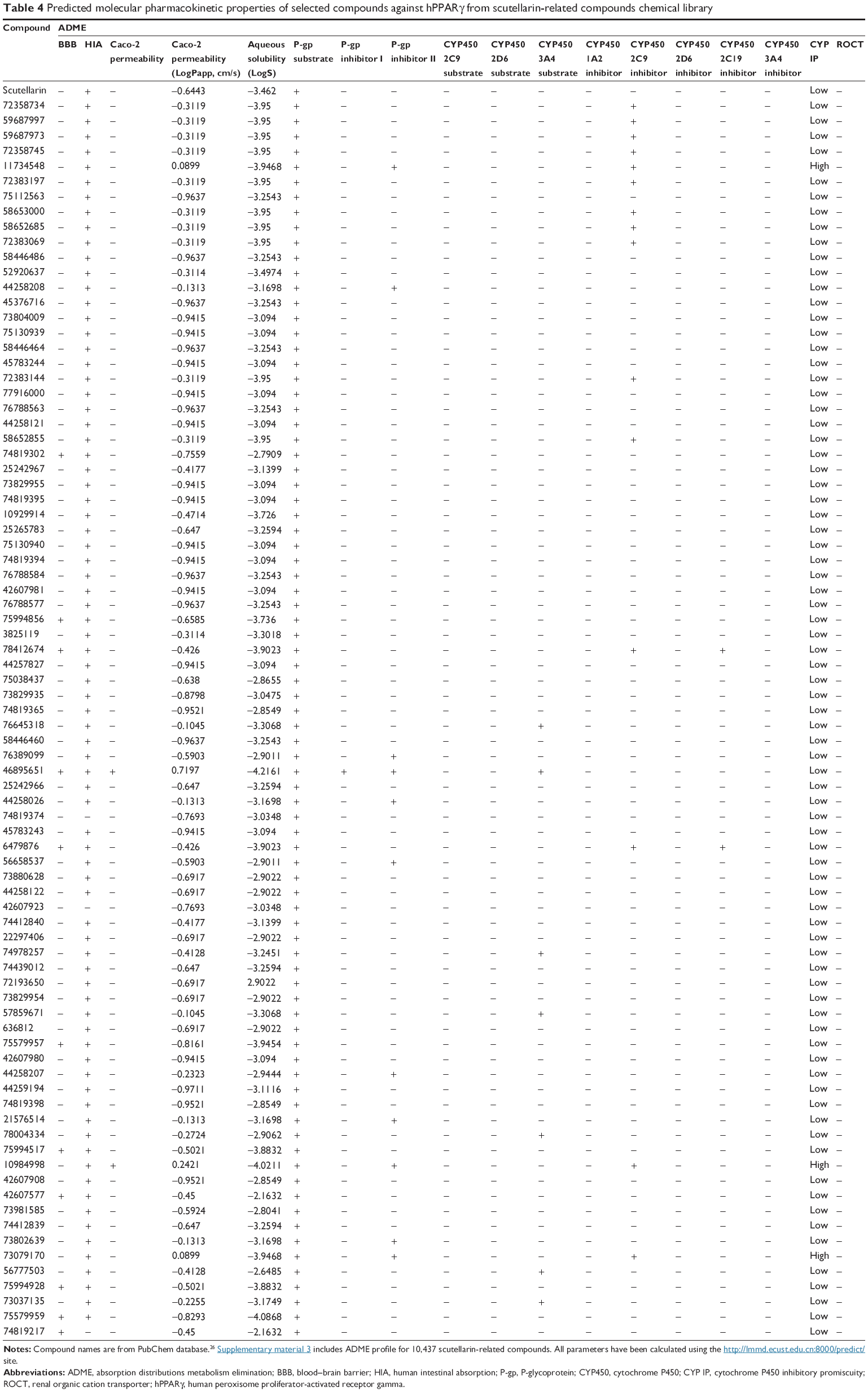 | Table 4 Predicted molecular pharmacokinetic properties of selected compounds against hPPARγ from scutellarin-related compounds chemical library |
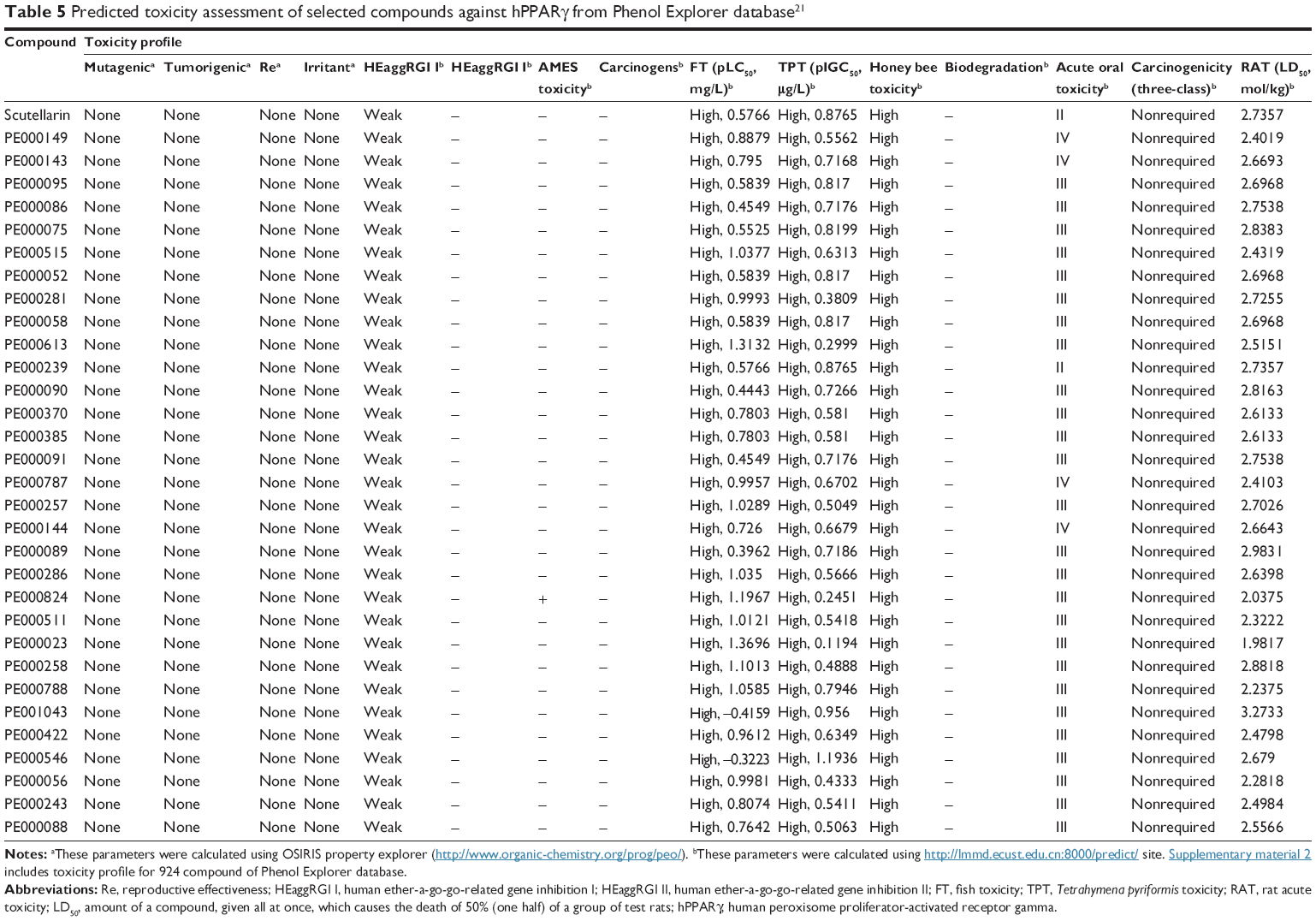 | Table 5 Predicted toxicity assessment of selected compounds against hPPARγ from Phenol Explorer database21 |
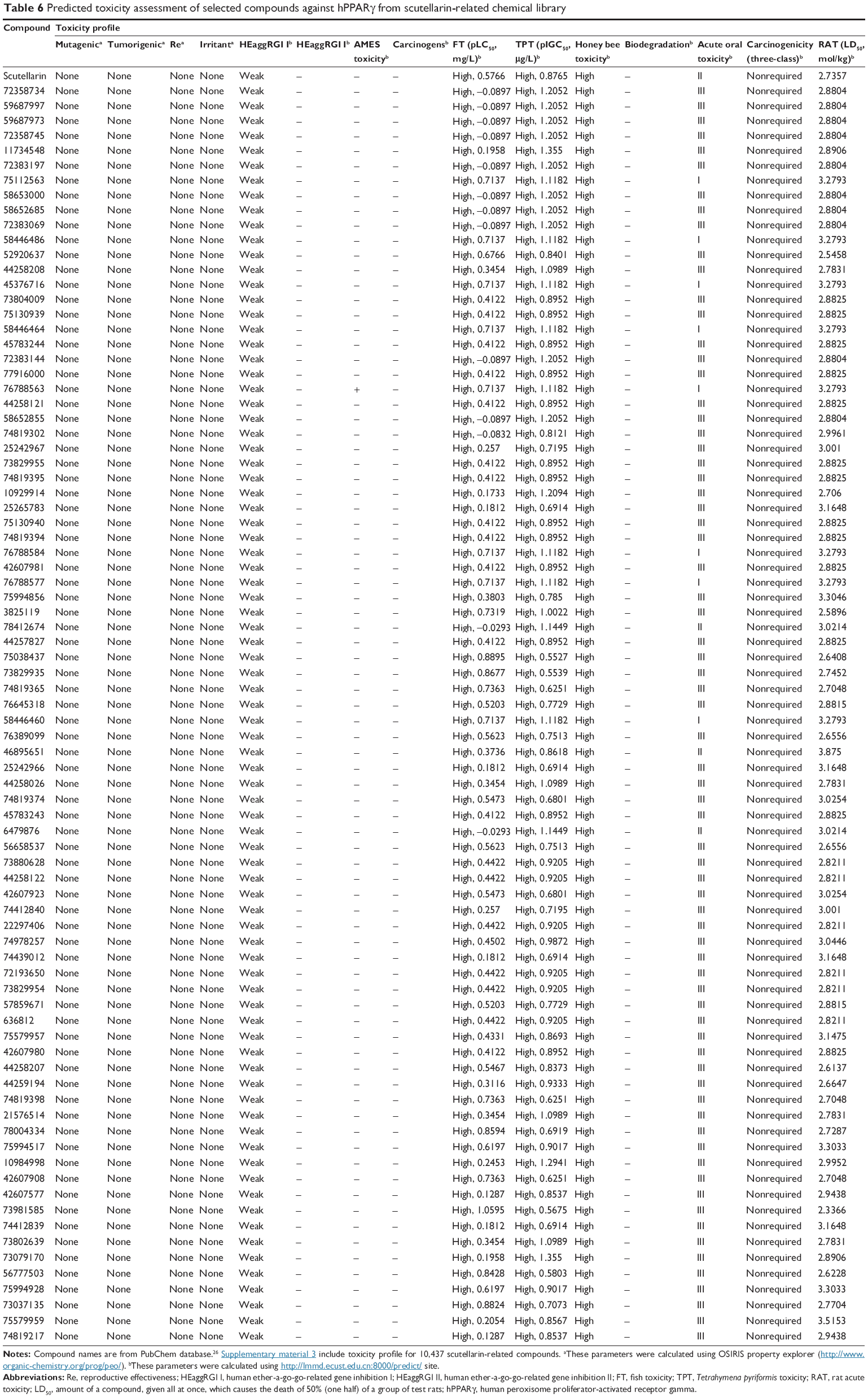 | Table 6 Predicted toxicity assessment of selected compounds against hPPARγ from scutellarin-related chemical library |
An ADME analysis includes the analysis of various properties such as ability to penetrate blood–brain barrier,43 capability of human intestinal absorption,43 Caco-2 permeability,44 and abilities to function as a P-glycoprotein (P-gp) substrate45 and inhibitor,46,47 renal organic cation transporter,48 and cytochrome P450 substrate49 and inhibitor.50
Almost all of the selected compounds showed positive results for human intestinal absorption but negative results for blood–brain barrier and Caco-2. The selected compounds showed no inhibitory side effects in terms of renal cation transport. Analysis of the ability of the compounds to serve as P-gp substrates showed that all of the selected ligands showed positive results and were identified as noninhibitors of P-gp. This predicted behavior for the selected compounds is similar to that of other compounds that have been proven to be hPPARγ activators, such as luteolin (PE000229), quercetin (PE000291), (+)-catechin (PE000124), 2′-OH-chalcone (PE000104), biochanin A (PE000397), genistein (PE000404), and 6-OH daidzein (PE000848). Cytochromes P450 are part of a ubiquitous superfamily of hemoproteins that are involved in various metabolic pathways in humans. In this context, we focused on the potential ability of some phenolic compounds to inhibit the capacity of Cyt P450 to catalyze the oxidation of drugs and other xenobiotics.51 These enzymes are predominantly expressed in the liver, but are also found in the small intestine (reducing drug bioavailability), lungs, placenta, and kidneys. Five isoforms, 1A2, 2C9, 2C19, 2D6, and 3A4, are considered the most important in xenobiotic metabolism.52 The heme group conducts reactions that are often oxidation reactions, such as aliphatic and aromatic oxidations, heteroatom oxidations and N- and O-dealkylations. These reactions will mainly generate more soluble compounds that are more easily excreted.52 Most of the selected ligands (Tables 3 and 4) did not serve as substrates for all cytochrome substrates (2C9, 2D6, and 3A4), whereas some compounds (PE000095, PE000075, PE000052, PE000058, PE000090, PE000370, and PE000385) were found to act as substrates for cytochrome P450 3A4. Inhibitors of cytochrome P450 decrease the enzymatic activity of one or more cytochrome enzymes in a dose-dependent manner and promote the accumulation of drugs to toxic levels.53 It is therefore desirable that the selected compounds do not serve as inhibitors of cytochrome P450. Most of the compounds included in Tables 3 and 4 satisfy this condition.
In the study of the prediction of toxicity profiles, several qualitative classification models, including mutagenicity,54 tumorigenicity,54 reproductive effectiveness,54 irritancy,54 human ether-a-go-go-related gene inhibition,32 Ames toxicity,32 carcinogenicity,32 fish toxicity,32 Tetrahymena pyriformis toxicity,32 honey bee toxicity,32 biodegradation,32 acute oral toxicity category,32 and acute rat toxicity32 were used. Tables 5 and 6 show the toxicity profiles of the selected compounds, and Supplementary materials 2 and Supplementary materials 3 present information for all of the studied compounds. The toxicity profiles of the selected compounds revealed that most of the compounds were not mutagenic, carcinogenic, or tumorigenic. Similarly, the selected compounds were negative for Ames toxicity, weak inhibitors of human ether-a-go-go-related genes, and exhibit no properties that exert significant toxicity in humans. On the contrary, all of the selected compounds were found to present high toxicity for fish, T. pyriformis, and honey bees.
Conclusion
This in silico study shows that a variety of plant-derived polyphenols found in dietary sources may modulate the activity of hPPARγ more strongly than other compounds reported in the literature.1 The compounds described in this study showed strong theoretical binding affinity (free energy variations ranging from −10.0±0.9 to −11.4±0.9 kcal/mol), as determined by docking against the binding site of several X-ray structures of hPPARγ. These putative modulators presented several molecular interactions (Tables 1 and 2) with the binding site of the protein. Additionally, most of the selected compounds present favorable druggability and good ADMET properties.
Taken together, the results of this computational study suggest that numerous plant-derived phenolic compounds, as well as other scutellarin-related compounds, can modulate the activity of hPPARγ. Although further cellular and in vivo investigations are required to confirm the physiological relevance of these results, these data highlight the potential of several phenolic compounds to become selective hPPARγ modulators able to alleviate obesity-related pathologies with reduced side effects compared with TZDs.
Acknowledgments
We thank Dr Javier Manuel Gozalvez-Sempere for allowing the use of facilities in the Linux cluster illice.umh.es. This work was partly supported by grants BFU2011-25920, AGL2014-51773-C3-1-R, and AGL2015-67995-C3-1-R from the Spanish MICINN, PROMETEO/2012/007 and ACOMP/2013/093 grants from Generalitat Valenciana, and CIBER (CB12/03/30038, Fisiopatología de la Obesidad y la Nutrición, CIBERobn, Instituto de Salud Carlos III).
Disclosure
The authors report no conflicts of interest in this work.
References
Wang L, Waltenberger B, Pferschy-Wenzig EM, et al. Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARgamma): a review. Biochem Pharmacol. 2014;92(1):73–89. | ||
Yu S, Reddy JK. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim Biophys Acta. 2007;1771(8):936–951. | ||
Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650. | ||
Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68(5):879–887. | ||
Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10(4):355–361. | ||
Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr. 2001;21:193–230. | ||
Attianese GMPG, Desvergne B. Integrative and systemic approaches for evaluating PPARbeta/delta (PPARD) function. Nucl Recept Signal. 2015;13:e001. | ||
Semple RK, Chatterjee VK, O’Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest. 2006;116(3):581–589. | ||
Higgins LS, Mantzoros CS. The development of INT131 as a selective PPARgamma modulator: approach to a safer insulin sensitizer. PPAR Res. 2008;2008:936906. | ||
Auboeuf D, Rieusset J, Fajas L, et al. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-alpha in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes. 1997;46(8):1319–1327. | ||
Medina-Gomez G, Gray SL, Yetukuri L, et al. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet. 2007;3(4):e64. | ||
Choi SS, Park J, Choi JH. Revisiting PPARgamma as a target for the treatment of metabolic disorders. BMB Rep. 2014;47(11):599–608. | ||
Elte JW, Blickle JF. Thiazolidinediones for the treatment of type 2 diabetes. Eur J Int Med. 2007;18(1):18–25. | ||
Ahmadian M, Suh JM, Hah N, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19(5):557–566. | ||
Petersen RK, Christensen KB, Assimopoulou AN, et al. Pharmacophore-driven identification of PPARgamma agonists from natural sources. J Comput Aided Mol Des. 2011;25(2):107–116. | ||
Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. 2015;14(2):111–129. | ||
Barrajon-Catalan E, Herranz-Lopez M, Joven J, et al. Molecular promiscuity of plant polyphenols in the management of age-related diseases: far beyond their antioxidant properties. Adv Exp Med Biol. 2014;824:141–159. | ||
Cragg GM, Newman DJ. Natural products: a continuing source of novel drug leads. Biochim Biophys Acta. 2013;1830(6):3670–3695. | ||
Herranz-Lopez M, Barrajon-Catalan E, Segura-Carretero A, Menendez JA, Joven J, Micol V. Lemon verbena (Lippia citriodora) polyphenols alleviate obesity-related disturbances in hypertrophic adipocytes through AMPK-dependent mechanisms. Phytomedicine. 2015;22(6):605–614. | ||
Higgins LS, Depaoli AM. Selective peroxisome proliferator-activated receptor gamma (PPARgamma) modulation as a strategy for safer therapeutic PPARgamma activation. Am J Clin Nutr. 2010;91(1):267S–272S. | ||
Rothwell JA, Perez-Jimenez J, Neveu V, et al. Phenol-Explorer 3.0: a major update of the Phenol-Explorer database to incorporate data on the effects of food processing on polyphenol content. Database. 2013;2013:bat070. | ||
Lu K, Han M, Ting HL, Liu Z, Zhang D. Scutellarin from Scutellaria baicalensis suppresses adipogenesis by upregulating PPARalpha in 3T3-L1 cells. J Nat Prod. 2013;76(4):672–678. | ||
Bottegoni G, Rocchia W, Rueda M, Abagyan R, Cavalli A. Systematic exploitation of multiple receptor conformations for virtual ligand screening. PLoS One. 2011;6(5):e18845. | ||
Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. | ||
Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015;43(W1):W443–W447. | ||
Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37:W623–W633. | ||
The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC [computer program]. 2015. | ||
Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320(2):369–387. | ||
Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24(5):417–422. | ||
Cheng F, Li W, Zhou Y, et al. AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11):3099–3105. | ||
Furukawa A, Arita T, Fukuzaki T, et al. Substituents at the naphthalene C3 position of (-)-cercosporamide derivatives significantly affect the maximal efficacy as PPARgamma partial agonists. Bioorg Med Chem Lett. 2012;22(3):1348–1351. | ||
Rother K, Preissner R, Goede A, Frommel C. Inhomogeneous molecular density: reference packing densities and distribution of cavities within proteins. Bioinformatics. 2003;19(16):2112–2121. | ||
Mirza MU, Ghori NU, Ikram N, Adil AR, Manzoor S. Pharmacoinformatics approach for investigation of alternative potential hepatitis C virus nonstructural protein 5B inhibitors. Drug Des Devel Ther. 2015;9:1825–1841. | ||
Borea PA, Varani K, Gessi S, Gilli P, Dalpiaz A. Receptor binding thermodynamics as a tool for linking drug efficacy and affinity. Farmaco. 1998;53(4):249–254. | ||
Yuriev E. Challenges and advances in structure-based virtual screening. Future Med Chem. 2014;6(1):5–7. | ||
Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–949. | ||
Ripphausen P, Stumpfe D, Bajorath J. Analysis of structure-based virtual screening studies and characterization of identified active compounds. Future Med Chem. 2012;4(5):603–613. | ||
Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today. 2004;1(4):337–341. | ||
Danishuddin M, Khan AU. Structure based virtual screening to discover putative drug candidates: necessary considerations and successful case studies. Methods. 2015;71:135–145. | ||
Shirai NC, Kikuchi M. Structural flexibility of intrinsically disordered proteins induces stepwise target recognition. J Chem Phys. 2013;139(22):225103. | ||
Shen J, Cheng F, Xu Y, Li W, Tang Y. Estimation of ADME properties with substructure pattern recognition. J Chem Inf Model. 2010;50(6):1034–1041. | ||
Pham-The H, González-Álvarez I, Bermejo M, et al. In silico prediction of Caco-2 Cell permeability by a classification QSAR approach. Mol Inf. 2011;30(4):376–385. | ||
Wang Z, Chen Y, Liang H, Bender A, Glen RC, Yan A. P-glycoprotein substrate models using support vector machines based on a comprehensive data set. J Chem Inf Model. 2011;51(6):1447–1456. | ||
Chen L, Li Y, Zhao Q, Peng H, Hou T. ADME evaluation in drug discovery. 10. Predictions of P-glycoprotein inhibitors using recursive partitioning and naive Bayesian classification techniques. Mol Pharm. 2011;8(3):889–900. | ||
Broccatelli F, Carosati E, Neri A, et al. A novel approach for predicting P-glycoprotein (ABCB1) inhibition using molecular interaction fields. J Med Chem. 2011;54(6):1740–1751. | ||
Kido Y, Matsson P, Giacomini KM. Profiling of a prescription drug library for potential renal drug-drug interactions mediated by the organic cation transporter 2. J Med Chem. 2011;54(13):4548–4558. | ||
Carbon-Mangels M, Hutter MC. Selecting relevant descriptors for classification by Bayesian estimates: a comparison with decision trees and support vector machines approaches for disparate data sets. Mol Inf. 2011;30(10):885–895. | ||
Cheng F, Yu Y, Shen J, et al. Classification of cytochrome P450 inhibitors and noninhibitors using combined classifiers. J Chem Inf Model. 2011;51(5):996–1011. | ||
Anzenbacher P, Anzenbacherova E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci. 2001;58(5–6):737–747. | ||
Olsen L, Oostenbrink C, Jorgensen FS. Prediction of cytochrome P450 mediated metabolism. Adv Drug Deliv Rev. 2015;86:61–71. | ||
Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76(3):391–396. | ||
OSIRIS Property Explorer. Available from: http://www.organic-chemistry.org/prog/peo/. Accessed June 29, 2015. | ||
Pedretti A, Villa L, Vistoli G. VEGA: a versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J Mol Graph Model. 2002;21(1):47–49. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.