")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Identification of covalent active site inhibitors of dengue virus protease
Authors Koh-Stenta X, Joy J, Wang SF, Kwek P, Wee J, Wan KF, Gayen S, Chen A, Kang C, Lee MA, Poulsen A, Vasudevan S, Hill J, Nacro K
Received 11 August 2015
Accepted for publication 9 October 2015
Published 10 December 2015 Volume 2015:9 Pages 6389—6399
DOI https://doi.org/10.2147/DDDT.S94207
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Xiaoying Koh-Stenta,1 Joma Joy,1 Si Fang Wang,1 Perlyn Zekui Kwek,1 John Liang Kuan Wee,1 Kah Fei Wan,2 Shovanlal Gayen,1 Angela Shuyi Chen,1 CongBao Kang,1 May Ann Lee,1 Anders Poulsen,1 Subhash G Vasudevan,3 Jeffrey Hill,1 Kassoum Nacro1
1Experimental Therapeutics Centre, Agency for Science, Technology and Research (A*STAR), Singapore; 2Novartis Institute for Tropical Diseases, Singapore; 3Program in Emerging Infectious Diseases, Duke-NUS Graduate Medical School, Singapore
Abstract: Dengue virus (DENV) protease is an attractive target for drug development; however, no compounds have reached clinical development to date. In this study, we utilized a potent West Nile virus protease inhibitor of the pyrazole ester derivative class as a chemical starting point for DENV protease drug development. Compound potency and selectivity for DENV protease were improved through structure-guided small molecule optimization, and protease-inhibitor binding interactions were validated biophysically using nuclear magnetic resonance. Our work strongly suggests that this class of compounds inhibits flavivirus protease through targeted covalent modification of active site serine, contrary to an allosteric binding mechanism as previously described.
Keywords: flavivirus protease, small molecule optimization, covalent inhibitor, active site binding, pyrazole ester derivatives
A Letter to the Editor has been received and published for this article.
Introduction
Dengue virus (DENV) is a mosquito-borne pathogen belonging to the Flaviviridae family, which includes West Nile virus (WNV), yellow fever virus, Murray Valley encephalitis virus, and Japanese encephalitis virus.1 Global incidence and geographical spread of the four known dengue serotypes (DENV-1 to 4) has increased significantly in recent years – half of the world’s population is currently at risk of infection.2–4 Dengue infection can develop into severe and potentially lethal conditions, in particular dengue hemorrhagic fever and dengue shock syndrome.5,6 Rising endemicity of multiple DENV serotypes in tropical countries today is a cause for concern, especially as secondary heterologous infection is a major risk factor for dengue hemorrhagic fever and dengue shock syndrome.4,7,8 There are currently no clinically-approved anti-viral drugs available for treatment of dengue infection.6,9 Hence, finding new and effective therapeutic measures for dengue remains a top global health priority.
The single-stranded, positive-sense RNA genome of DENV encodes for a polyprotein that is processed into three structural (capsid, envelope, and membrane) and seven non-structural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins. Virus-encoded NS3 serine protease, in complex with cofactor NS2B, plays a critical role alongside host cell proteases in the proteolytic processing of viral polyprotein and ultimately, in viral replication.9 DENV NS2B/NS3 protease is thus an attractive target for anti-dengue drug development. Recent successes of human immunodeficiency virus and hepatitis C virus protease inhibitors in the clinic have also renewed focus on viral protease inhibition as a proven strategy for anti-viral therapy.10–13
Several approaches have been taken to develop DENV protease inhibitors, including peptidic substrate-based inhibition,14,15 high-throughput screening (HTS),9,16,17 and in silico docking.18 No compounds have reached clinical development to date, mainly due to multiple challenges encountered during lead optimization, including poor cellular activity, selectivity, and compound stability.9 With the paucity of promising dengue compounds and a need for high-quality lead candidates in mind, we describe in this study an approach whereby a potent WNV NS2B/NS3 compound was utilized as a chemical starting point for structure-based DENV protease inhibitor discovery. WNV and DENV proteases share high sequence identity, particularly in the active sites.19 Moreover, it has been demonstrated through alignment of peptide-bound X-ray crystal structures that many protein-ligand interactions are conserved between both proteases.20 A recent study identified several substrate-competitive inhibitors that were active against WNV and DENV2 proteases, among them one with low micromolar potency.21 Similarly, DENV2 protease inhibitors from an HTS study had shown comparable activities against WNV protease.16
We focused our investigation on a class of pyrazole ester derivatives, which were previously identified as potent inhibitors of WNV protease in an HTS study of the National Institutes of Health compound library (>65,000 compounds).22 To our knowledge, this class of compounds has not been explored for dengue. Our results show that a particular compound with nanomolar potency against WNV was able to inhibit the DENV2 protease, albeit at micromolar potency. To gain insight into the mode of inhibition, we performed molecular modeling and molecular characterization of DENV2 and WNV proteases in the presence of compound. This was followed-up by nuclear magnetic resonance (NMR) spectroscopy as a biophysical method to validate protease-inhibitor binding interactions. Our work provides strong evidence that the pyrazole ester derivatives inhibit flavivirus protease through targeted covalent modification of the active site serine, contrary to an allosteric binding mechanism as previously described.22,23 Additionally, we have utilized a structure-guided approach for small molecule optimization to improve compound potency and selectivity for DENV2 protease.
Materials and methods
Expression and purification of DENV2 and WNV proteases
A WNV fusion construct encoding fragments from NS2B and NS3, fused by a G4-S-G3 linker, was codon optimized for expression in Escherichia coli and synthesized (GenScript). A synonymous DENV2 construct was generated after aligning the sequences of WNV and DENV2. Active site mutants, DENV2-S135A and WNV-S135A, were generated by site-directed mutagenesis using the QuikChange Kit (Stratagene Agilent Technologies, Santa Clara, CA, USA) and mutagenesis was confirmed by DNA sequencing. Recombinant WNV and DENV2 proteases were expressed and purified as previously described for Murray Valley encephalitis virus protease.24 Briefly, the proteins were expressed with an N-terminal 6-His tag, purified by immobilized metal affinity chromatography, and desalted on the Profinia Protein Purification System (Bio-Rad Laboratories Inc., Hercules, CA, USA). Finally, they were concentrated to 1.0 mg/mL and stored at −80°C.
DENV2 and WNV protease assays
Compounds were assayed for inhibitory activity at a final volume of 30 μL (384-well format) in 10 mM Tris-HCl (pH 8.5), 20% glycerol and 1 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate.25 Protease (10 nM and 40 nM for DENV2 and WNV respectively) was pre-incubated with compounds at room temperature for 30 minutes at a final dimethyl sulfoxide (DMSO) concentration of 0.2%. The reaction was initiated by addition of 20 μM fluorophore-tagged substrate: Bz-nKRR-AMC for DENV2, and Bz-RTKR-AMC for WNV. Reaction progress was followed by monitoring of fluorescence levels (excitation 380 nm, emission 450 nm) using a Tecan Safire 2 (Männedorf, Switzerland) microplate reader. Relative fluorescence values were determined after 90 minutes. Half maximal inhibitory concentration (IC50) values were determined by non-linear regression using GraphPad prism (GraphPad Software, Inc., La Jolla, CA, USA). All IC50 values reported were from a minimum of two experiments.
Electron spray ionization time-of-flight mass spectrometry (ESI-TOF MS)
Protein size determination was performed in 10 mM Tris buffer (pH 8.0) containing 20% (v/v) glycerol and 1 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate. Aliquots containing 5 μM of DENV2 or WNV protease were incubated with 20 μM of compound for 1 hour at 25°C. Control reactions were prepared by incubating the enzyme with 2% DMSO. Samples were analyzed on a quadrupole time-of-flight mass spectrometry and protein molecular mass determined by liquid chromatography time-of-flight mass spectrometry using an Agilent 6224 mass spectrometer coupled to an Agilent 1200 capillary high-performance liquid chromatography. Briefly, protein was loaded onto a C18 reversed-phase column and elution was carried out with a linear gradient from 10% acetonitrile and 0.1% formic acid, to 90% acetonitrile and 0.1% formic acid. The system was controlled by MassHunter Acquisition software (Version 3.03, Agilent Technologies) and deconvolution of the recorded m/z values was carried out using MassHunter Quantitative Analysis software (Version 3.03).
NMR spectroscopy
WNV and DENV2 proteases were expressed by induction in an M9 medium containing 1 g/L of 15NH4Cl and purified as previously described.26 Briefly, cells were harvested at OD600 0.8 by centrifugation and cell pellets were resuspended in lysis buffer. Protease was purified using Ni2+-NTA resin followed by gel filtration using a Superdex 200 column. Pooled fractions were then buffer exchanged into an NMR buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 2 mM dithiothreitol and 10% D2O) for structural analysis. Compounds were dissolved in deuterated-DMSO and titrated into a 0.4 mM 15N-labeled protease solution. All NMR spectra were acquired at 298 K on a Bruker Avance II 700 MHz spectrometer (Bruker Corporation, Billerica, MA, USA) equipped with a cryoprobe, and data were processed using NMRPipe and visualized with NMRView.
Molecular modeling
The WNV protease (Protein Data Bank [PDB] ID 3E90)27 and DENV protease (PDB ID 3U1I)20 X-ray structures were downloaded and prepared in Maestro 9.3 using standard settings. This included addition of hydrogen atoms, bond assignment, optimization of the hydrogen bond network, and restrained minimization using the optimized potential for liquid simulations-2005 force field.28 The co-crystallized inhibitors were deleted from both structures, and the hydroxyl group of S135 was deprotonated. Compounds 1 and 2 were manually docked into the binding sites by placing the phenyl or bi-phenyl of the phenyl-ester in the P1 site, the carbonyl of the ester in the oxyanion hole, the amine in the P1’ site and the phenyl of the phenylsulfonyl in the P2’ site. The inhibitor-protein complex was finally minimized using Macromodel 9. All residues more than 7Å from the ligand were constrained before the complex was subjected to 500 steps of Polak-Ribiere-Conjugate-Gradient minimization using the optimized potential for liquid simulations-2005 force field and GB/SA continuum solvation method. In all the minimized protein-ligand complexes, the distance between the deprotonated serine oxygen and the carbonyl carbon of the inhibitor was 3.2–3.3 Å.
Results and discussion
We selected from the literature a nanomolar potency inhibitor (compound 1) of WNV NS2B/NS3 as a starting point for our investigation. Compound 1, previously identified by Johnston et al through HTS of the National Institutes of Health small molecules library,22 consisted of a 5-amino-1-(phenyl)sulfonyl-pyrazol-3-yl core linked through the 3 position to a phenyl ester (Figure 1). We synthesized compound 1 as previously described29 and verified inhibition of WNV NS2B/NS3 in a low-volume, fluorescence-based biochemical assay (Figure 2). An IC50 of 0.16 μM was determined for compound 1 against the WNV protease (Table 1), comparable to that previously reported (0.105 μM).22,23 The biochemical assay was optimized for evaluation of compound inhibition of DENV2 NS2B/NS3 (Figure 2B). Compound 1 showed clear inhibition against the DENV2 protease, albeit at a weaker IC50 of 8.5 μM as compared with WNV (Table 1).
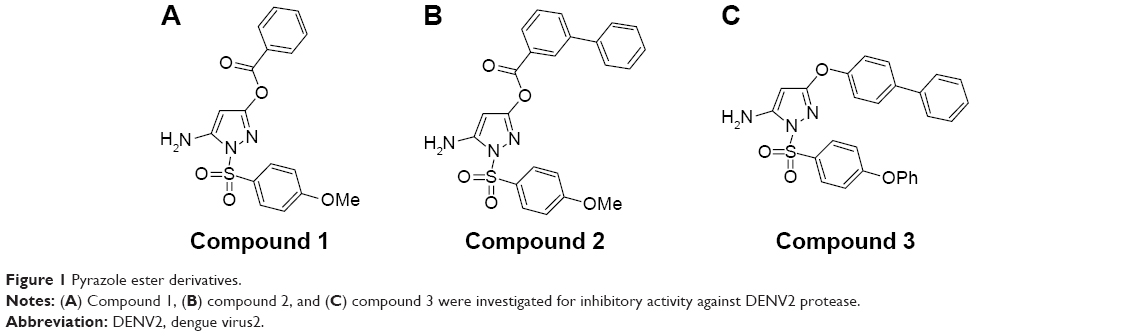 | Figure 1 Pyrazole ester derivatives. |
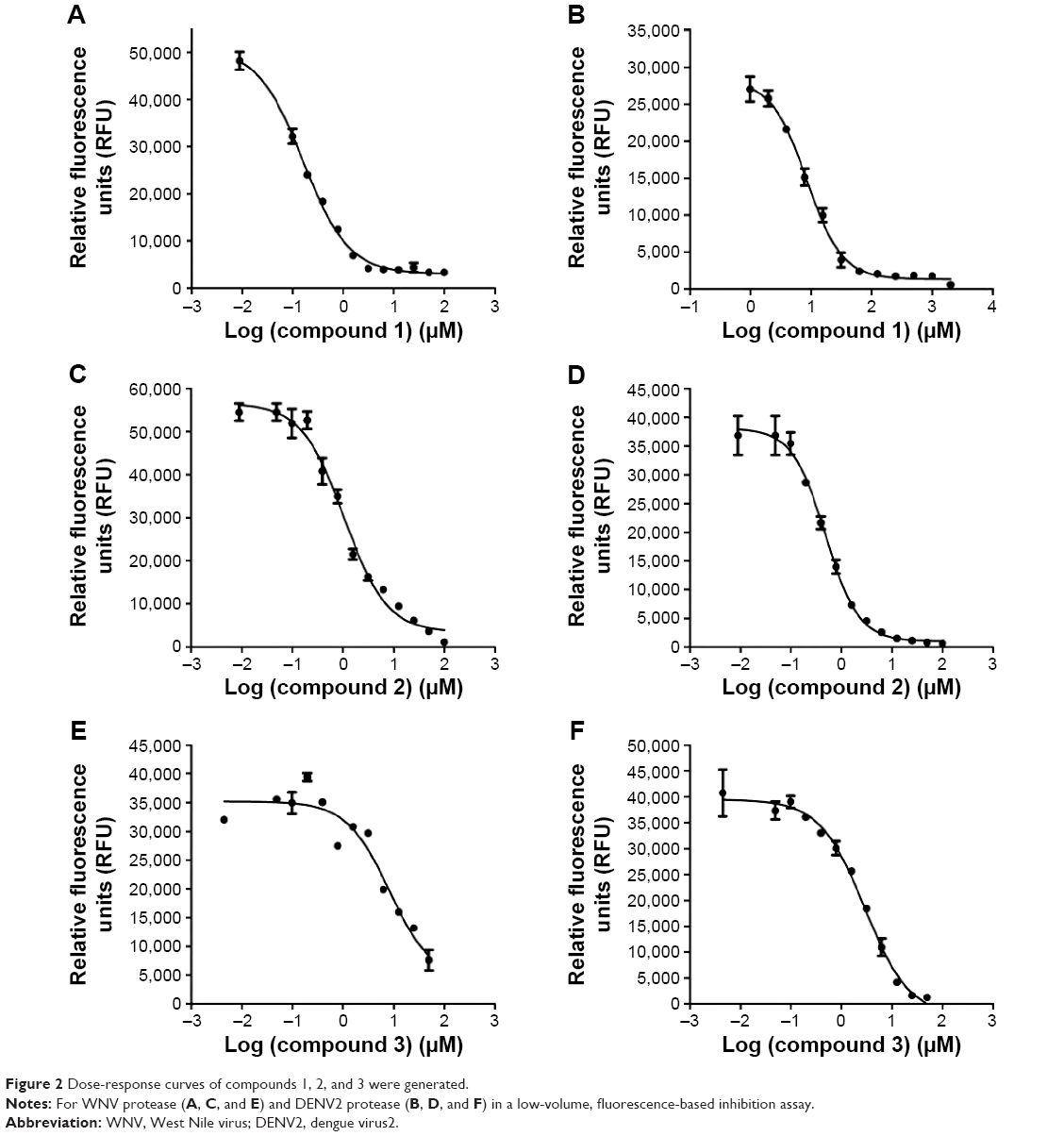 | Figure 2 Dose-response curves of compounds 1, 2, and 3 were generated. |
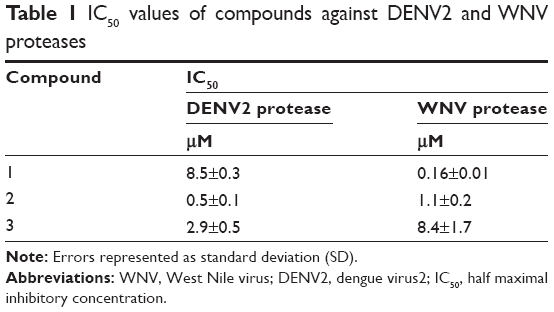 | Table 1 IC50 values of compounds against DENV2 and WNV proteases |
Subsequent analysis of DENV2 protease molecular weight by ESI-TOF MS yielded a surprising result – DENV2 protease that had been treated with compound 1 consistently displayed a 104 Da molecular weight increase over the control protease in DMSO solution (Figure 3). Significantly, this increase in molecular weight corresponded well with benzoylation of DENV2 protease by compound 1 (Table 2). Similar analysis was performed on a DENV2 protease active site mutant (DENV2-S135A) whereby the catalytic serine of the NS3 domain had been replaced with alanine (Figure 3B). Unlike the wild type protease, no change in molecular weight was observed for DENV2-S135A upon treatment with compound 1 (Table 3). The above data provided evidence of a covalent reaction between the catalytic serine of DENV2 protease and compound 1, and strongly suggested that compound binding took place within the active site. In the absence of the catalytic serine, covalent transfer of the benzoyl moiety to the protease did not take place.
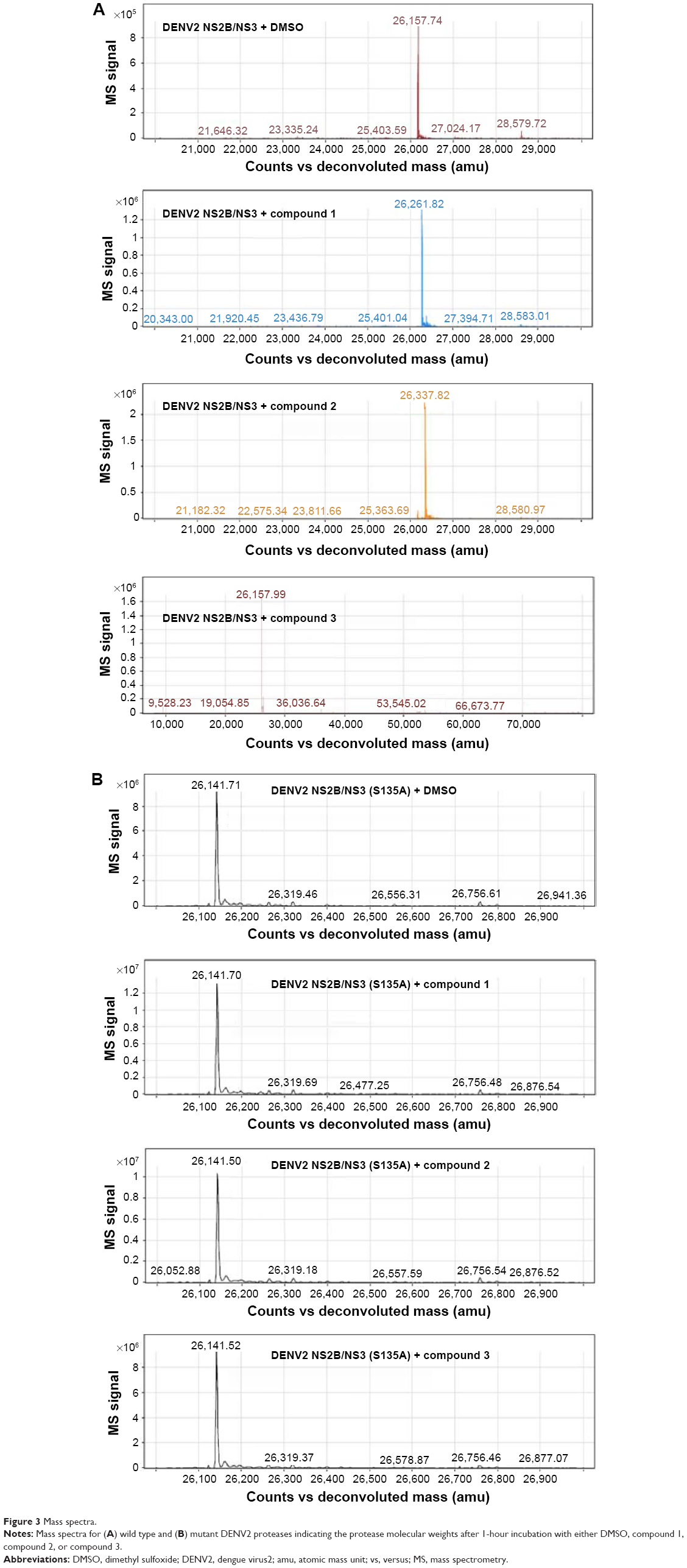 | Figure 3 Mass spectra. |
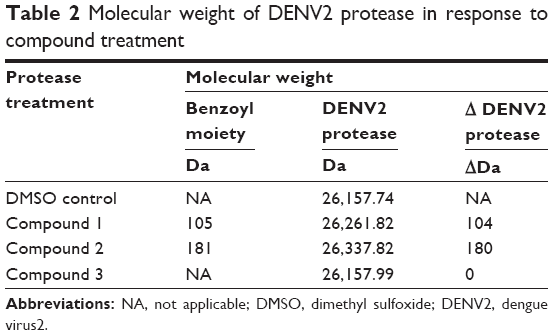 | Table 2 Molecular weight of DENV2 protease in response to compound treatment |
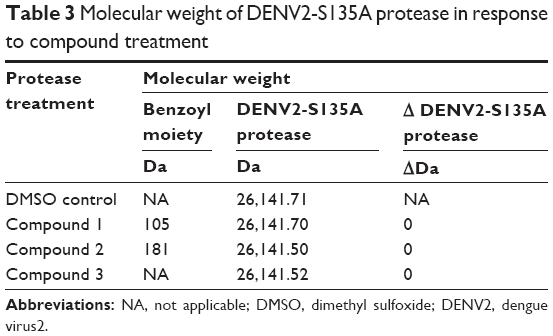 | Table 3 Molecular weight of DENV2-S135A protease in response to compound treatment |
NMR analyses were performed to investigate DENV2 protease-inhibitor binding interactions. In the absence of inhibitor, the 1H-15N-HSQC (heteronuclear single quantum coherence) spectrum of DENV2 protease exhibited peak overlap (Figure 4A, black). This could be explained by conformational dynamics between residues from NS2B and NS3 leading to a disappearance of NMR signal.26,30 In the presence of compound 1, the spectrum of DENV2 protease-inhibitor complex showed significant improvement over that of protease alone – cross peaks were well-dispersed and better defined (Figure 4, red). Similar improvement in spectrum quality was observed for WNV when mixed with compound 1 (Figure 4). In addition, WNV spectra obtained for compound 1 and the tripeptide inhibitor 2-naphthoyl-KKRH26,27 displayed high similarity, indicating that compound 1 interacted with similar residues as the peptide inhibitor (Figure 4F). Overall, these results reflected protease stabilization upon compound 1 binding in the active site, and provided biophysical validation of protease-inhibitor interactions.
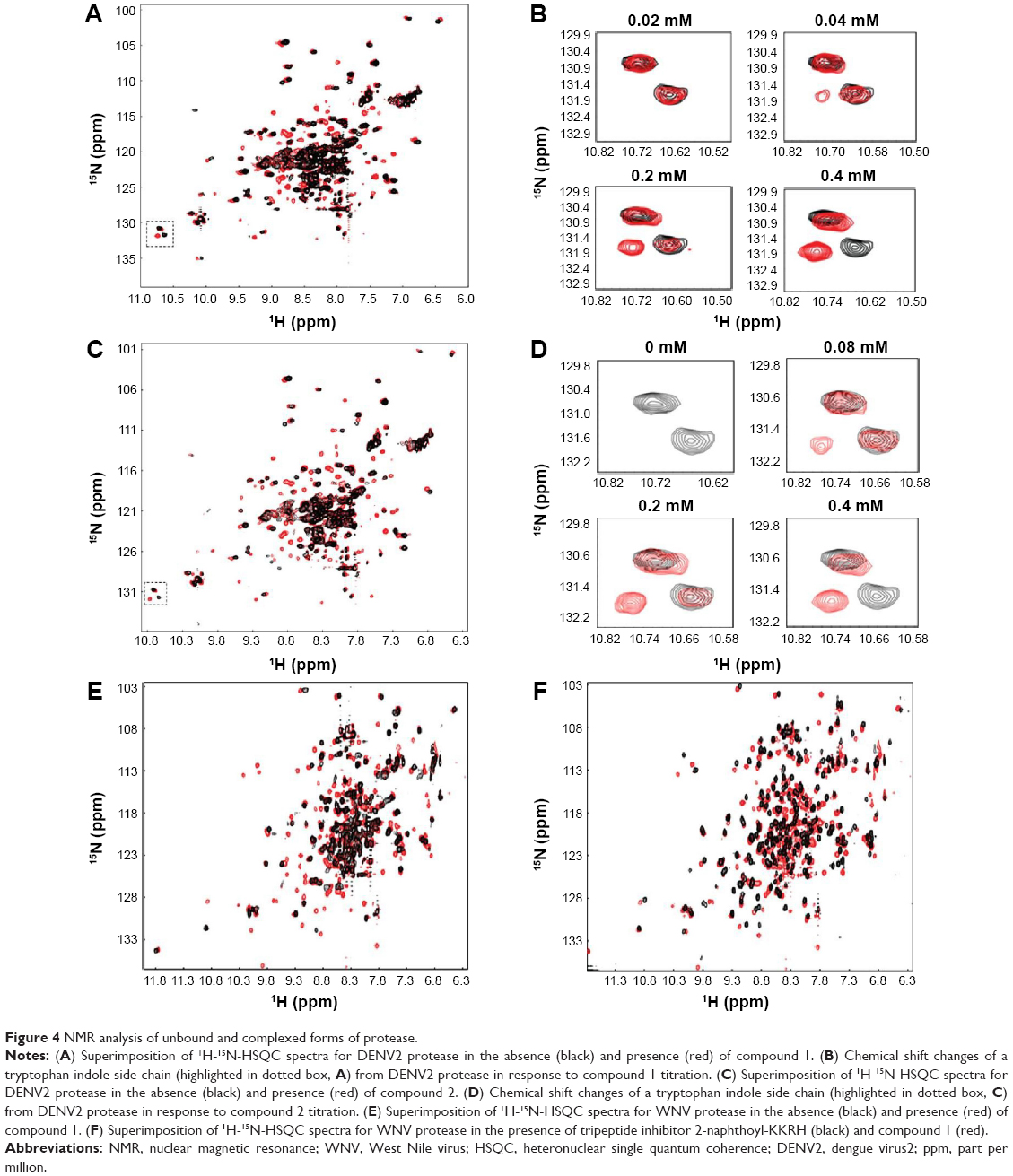 | Figure 4 NMR analysis of unbound and complexed forms of protease. |
To examine protease-inhibitor molecular interactions in further detail, compound 1 was titrated against DENV2 protease and chemical shift perturbations were analyzed (Figure 4B). The titration experiment, which yielded a series of 1H-15N-HSQC spectra with gradual cross peak shifts, demonstrated that the protease was in slow exchange between its bound and free states. One example could be observed from the indole side chain of a tryptophan residue (not assigned) of DENV2 protease (Figure 4B). With the addition of 0.04 mM of compound 1, a cross peak attributing to the protease-inhibitor complex was discernible, and became more prominent at 0.2 mM of compound 1. The protease became fully bound only upon addition of 0.4 mM of compound 1, at a protease-inhibitor ratio of 1:1. This indicated strong, monovalent binding between DENV2 protease and inhibitor forming a tight and stable complex, and corresponded well with findings from ESI-TOF MS.
To further our understanding on what protease-inhibitor interactions may take place in the active site, we performed molecular modeling based on an existing X-ray crystal structure of DENV3 protease in complex with a peptide inhibitor (PDB ID 3U1I).20 DENV2 and DENV3 proteases were highly similar, hence the selected structure provided a useful representation of the catalytically active and closed conformation of the DENV protease.20 Compound 1 was docked into the protease catalytic domain of the structure and the inhibitor-protease complex was subjected to constrained energy minimization. Docking of compound 1 resulted in a good fit with the protease active site (Figure 5A). The structure model showed that the sulfonamide-phenyl of compound 1 occupied the shallow P2’ subpocket, and formed van der Waals contacts with residues T34, V36, P102, and G133. Additionally, the pyrazole-amino group formed a hydrogen bond with the carbonyl backbone of V36 in the P1’ site. The ester-phenyl group of compound 1 occupied the deeper P1 site sandwiched between residues P132 and Y161. The proximity of this phenyl group to Y161 created an ideal environment for aromatic-aromatic stacking interactions. Overall, data from molecular modeling corroborated well with an active site binding mechanism for compound 1 to the DENV protease.
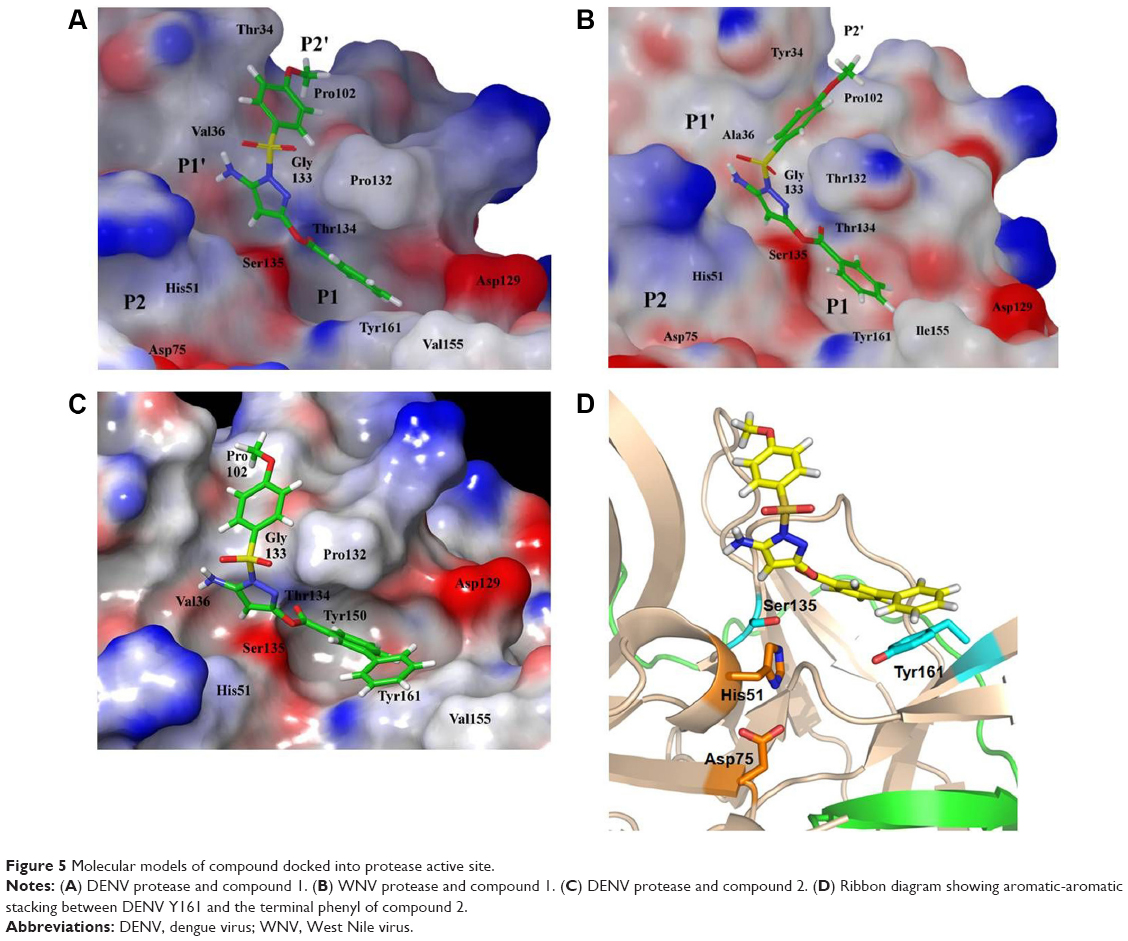 | Figure 5 Molecular models of compound docked into protease active site. |
The change in potencies for compound 1 against DENV2 (8.5 μM) and WNV proteases (0.16 μM) could be understood from structural differences observed in the respective docked models. Compound 1 was docked into an X-ray crystal structure of WNV protease (PDB ID 3E90) in a similar manner as for the DENV protease (Figure 5B). The compound binding mode in WNV was found to be similar to that in DENV, with the exception of a shallower P1 site surrounding the compound in WNV. In WNV protease, the P1 site consisted of I155 (Figure 5B) which came into close contact with the ester-phenyl group of compound 1 and contributed to the formation of van der Waals interactions with the inhibitor. The homologous position in DENV contained a less bulky residue, V155, which was slightly further away from the compound (Figure 5A). The resulting weaker van der Waals interactions with the ester-phenyl group in DENV could account for a drop in potency compared to WNV (Table 1). This insight into molecular interactions at the P1 site paved the way for us to perform small molecule optimization to improve compound potency. A second inhibitor, compound 2 (Figure 1B), was designed comprising an ester-biphenyl rather than ester-phenyl group to enable a closer fit in the DENV P1 pocket (Figure 5C). It was predicted that the additional phenyl group of compound 2 would be in a favorable position to establish stronger aromatic-aromatic stacking interactions with Y161 (Figure 5D).
Compound 2 was synthesized and tested for protease inhibition as earlier described (Figure 2). The IC50 for compound 2 against DENV2 protease was determined to be 0.5 μM, more than 15-fold improvement over that for compound 1 (Table 1). The opposite was true for WNV protease – here the additional phenyl group of compound 2 resulted in an IC50 increase from 0.16 to 1.1 μM (Table 1). The changes in potency corresponded well with predictions from molecular modeling. Molecular weights of wild type DENV2 and DENV2-S135A proteases upon treatment with compound 2 were analyzed by ESI-TOF MS (Figure 3). It was observed that the wild type protease had increased in size by 180 Da in response to compound 2 treatment (Table 2), whereas the size of the mutant protease remained unchanged (Table 3). The observed increase in molecular weight was consistent with covalent transfer of the 3-phenyl benzoyl group of compound 2 to the catalytic serine. NMR analysis of DENV2 protease and compound 2 binding resulted in similar chemical shift perturbations as those observed for compound 1 (Figure 4C). Titration of compound 2 against DENV2 protease revealed a slow exchange between the bound and free states, indicative of tight inhibitor binding (Figure 4D).
We have described key steps of the transesterification reaction between DENV2 protease and compound 2 in a mechanistic model (Figure 6). First, the carbonyl oxygen of the compound ester group formed hydrogen bonds with the backbone of residues T134 and G133 in the oxyanion hole (Figure 6A). The carbonyl carbon underwent nucleophilic attack from the side chain oxygen of S135, a member of the catalytic triad. This led to formation of a transition state, similar to tetrahedral intermediates formed during typical substrate cleavage reactions (Figure 6B). In the final step, the pyrazole-phenylsulfonamide was cleaved off, leaving the 3-phenyl benzoyl covalently bound to S135 (Figure 6C). To demonstrate the importance of the compound ester group in the reaction, we designed an analog of compound 2 in which the ester had been replaced by an ether group (compound 3) (Figure 1C). In contrast to compounds 1 and 2, treatment of wild type DENV2 protease with compound 3 did not result in a change of protease molecular weight (Figure 3A). Through this analysis, we have confirmed that the ester group was a functional moiety essential for covalent modification of the protease active site serine.
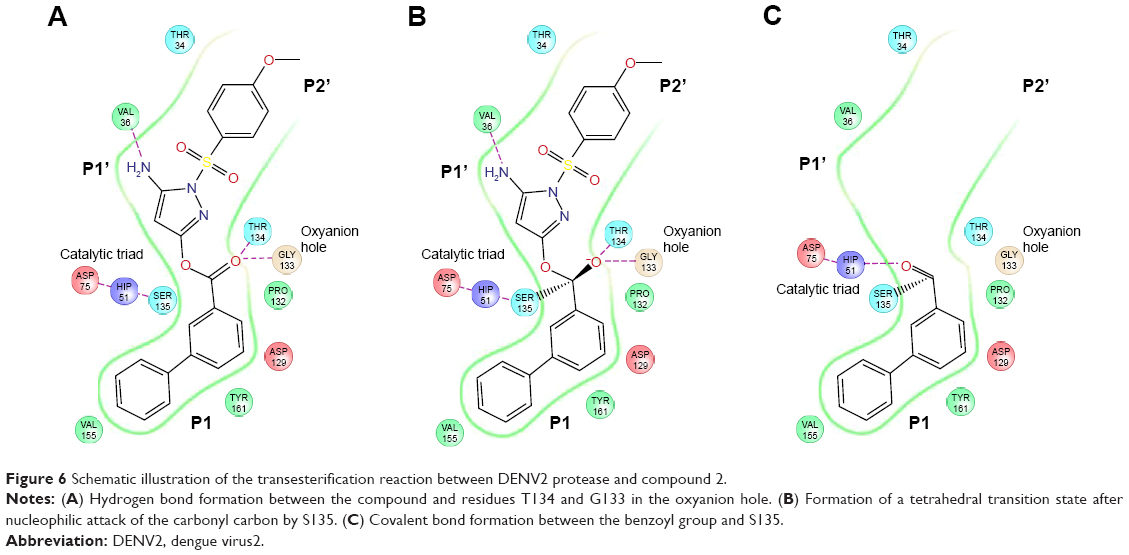 | Figure 6 Schematic illustration of the transesterification reaction between DENV2 protease and compound 2. |
In summary, we have described an approach for DENV protease drug development whereby a known potent inhibitor against a different flavivirus was used as a chemical starting point from which to perform small molecule optimization. Through molecular characterization and modeling, the selectivity and potency of a pyrazole ester-derived compound were improved for DENV2 protease. Protease-inhibitor binding interactions were validated biophysically using NMR. Notably, our systematic analyses using wild type and mutant proteases provide strong evidence that this class of compounds inhibits flavivirus protease through active site binding and covalent modification of catalytic serine.
Acknowledgment
This work was supported by the Agency for Science, Technology and Research (A*STAR), Singapore.
Disclosure
The authors report no conflicts of interest in this work.
References
Kuno G, Chang GJ, Tsuchiya KR, Karabatsos N, Cropp CB. Phylogeny of the genus Flavivirus. J Virol. 1998;72(1):73–83. | ||
World Health Organization [homepage on the Internet]. Dengue and severe dengue. WHO Fact sheet No 117 [updated May 2015]. Available from: http://www.who.int/mediacentre/factsheets/fs117/en/. Accessed October 29, 2015. | ||
Guzman MG, Halstead SB, Artsob H, et al. Dengue: a continuing global threat. Nat Rev Microbiol. 2010;8(12 Suppl):S7–S16. | ||
Guzman MG, Harris E. Dengue. Lancet. 2015;385(9966):453–465. | ||
Rigau–Perez JG, Clark GG, Gubler DJ, Reiter P, Sanders EJ, Vorndam AV. Dengue and dengue haemorrhagic fever. Lancet. 1998;352(9132):971–977. | ||
Nitsche C, Holloway S, Schirmeister T, Klein CD. Biochemistry and Medicinal Chemistry of the Dengue Virus Protease. Chem Rev. Epub 2014 Sep 30. | ||
Kyle JL, Harris E. Global spread and persistence of dengue. Annu Rev Microbiol. 2008;62:71–92. | ||
Guzman MG, Alvarez M, Halstead SB. Secondary infection as a risk factor for dengue hemorrhagic fever/dengue shock syndrome: an historical perspective and role of antibody-dependent enhancement of infection. Arch Virol. 2013;158(7):1445–1459. | ||
Lim SP, Wang QY, Noble CG, et al. Ten years of dengue drug discovery: progress and prospects. Antiviral Res. 2013;100(2):500–519. | ||
Wyles DL. Antiviral resistance and the future landscape of hepatitis C virus infection therapy. J Infect Dis. 2013;207 Suppl 1:S33–S39. | ||
De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Agents. 2009;33(4):307–320. | ||
Brecher M, Zhang J, Li H. The flavivirus protease as a target for drug discovery. Virol Sin. 2013;28(6):326–336. | ||
Menendez-Arias L. Molecular basis of human immunodeficiency virus drug resistance: an update. Antiviral Res. 2010;85(1):210–231. | ||
Nitsche C, Behnam MA, Steuer C, Klein CD. Retro peptide-hybrids as selective inhibitors of the Dengue virus NS2B-NS3 protease. Antiviral Res. 2012;94(1):72–79. | ||
Yin Z, Patel SJ, Wang WL, et al. Peptide inhibitors of Dengue virus NS3 protease. Part 1: Warhead. Bioorg Med Chem Lett. 2006;16(1):36–39. | ||
Tomlinson SM, Watowich SJ. Use of parallel validation high-throughput screens to reduce false positives and identify novel dengue NS2B-NS3 protease inhibitors. Antiviral Res. 2012;93(2):245–252. | ||
Yang CC, Hsieh YC, Lee SJ, et al. Novel dengue virus-specific NS2B/NS3 protease inhibitor, BP2109, discovered by a high-throughput screening assay. Antimicrob Agents Chemother. 2011;55(1):229–238. | ||
Deng J, Li N, Liu H, et al. Discovery of novel small molecule inhibitors of dengue viral NS2B-NS3 protease using virtual screening and scaffold hopping. J Med Chem. 2012;55(14):6278–6293. | ||
Erbel P, Schiering N, D’Arcy A, et al. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat Struct Mol Biol. 2006;13(4):372–373. | ||
Noble CG, Seh CC, Chao AT, Shi PY. Ligand-bound structures of the dengue virus protease reveal the active conformation. J Virol. 2012;86(1):438–446. | ||
Lai H, Dou D, Aravapalli S, et al. Design, synthesis and characterization of novel 1,2-benzisothiazol-3(2H)-one and 1,3,4-oxadiazole hybrid derivatives: potent inhibitors of Dengue and West Nile virus NS2B/NS3 proteases. Bioorg Med Chem. 2013;21(1):102–113. | ||
Johnston PA, Phillips J, Shun TY, et al. HTS identifies novel and specific uncompetitive inhibitors of the two-component NS2B-NS3 proteinase of West Nile virus. Assay Drug Dev Technol. 2007;5(6):737–750. | ||
Sidique S, Shiryaev SA, Ratnikov BI, et al. Structure-activity relationship and improved hydrolytic stability of pyrazole derivatives that are allosteric inhibitors of West Nile Virus NS2B-NS3 proteinase. Bioorg Med Chem Lett. 2009;19(19):5773–5777. | ||
Joy J, Mee NF, Kuan WL, Perlyn KZ, Wen TS, Hill J. Biochemical characterisation of Murray Valley encephalitis virus proteinase. FEBS Lett. 2010;584(14):3149–3152. | ||
Li J, Lim SP, Beer D, et al. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J Biol Chem. 2005;280(31):28766–28774. | ||
Kang C, Gayen S, Wang W, et al. Exploring the binding of peptidic West Nile virus NS2B-NS3 protease inhibitors by NMR. Antiviral Res. 2013;97(2):137–144. | ||
Robin G, Chappell K, Stoermer MJ, et al. Structure of West Nile virus NS3 protease: ligand stabilization of the catalytic conformation. J Mol Biol. 2009;385(5):1568–1577. | ||
Kaminski GA, Stern HA, Berne BJ, et al. Development of a polarizable force field for proteins via ab initio quantum chemistry: first generation model and gas phase tests. J Comput Chem. 2002;23(16):1515–1531. | ||
Myers MC, Napper AD, Motlekar N, et al. Identification and characterization of 3-substituted pyrazolyl esters as alternate substrates for cathepsin B: the confounding effects of DTT and cysteine in biological assays. Bioorg Med Chem Lett. 2007;17(17):4761–4766. | ||
de la Cruz L, Nguyen TH, Ozawa K, et al. Binding of low molecular weight inhibitors promotes large conformational changes in the dengue virus NS2B-NS3 protease: fold analysis by pseudocontact shifts. J Am Chem Soc. 2011;133(47):19205–19215. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.