")
Back to Journals » Journal of Inflammation Research » Volume 14
HMGB1/TLR4 Signaling Affects Regulatory T Cells in Acute Lung Injury
Authors Zhou M, Zhang Y, Tang R, Liu H, Du M, Gao Z, Ji Z, Fang H
Received 21 January 2021
Accepted for publication 31 March 2021
Published 19 April 2021 Volume 2021:14 Pages 1551—1561
DOI https://doi.org/10.2147/JIR.S302967
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Min Zhou,1,* Yadi Zhang,2,* Rui Tang,1 Haiyan Liu,1 Min Du,1 Zhi Gao,1 Zongshu Ji,1 Haoshu Fang3
1Neurocritical Care Unit, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui, 230001, People’s Republic of China; 2Department of Respiratory Medicine, The Second People’s Hospital of Hefei and Hefei Hospital Affiliated with Anhui Medical University, Hefei, Anhui, 230011, People’s Republic of China; 3Department of Pathophysiology, School of Basic Medical Sciences, Anhui Medical University, Hefei, Anhui, 230032, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Haoshu Fang
Department of Pathophysiology, School of Basic Medical Sciences, Anhui Medical University, 81 Meishan Road, Hefei, Anhui, 230032, People’s Republic of China
Tel +86 551-65161129
Email [email protected]
Background: High-mobility group box-1 protein (HMGB1) serves as the prototypic damage-associated molecular pattern molecule, and TLR4 is considered a receptor for HMGB1. Regulatory T cells (Tregs) play a crucial role in infectious diseases. The role of HMGB1 in the modulation of Tregs is of great interest.
Methods: Serum HMGB1 and Treg proportions were detected in 58 patients with acute lung injury (ALI) and 36 healthy volunteers. The correlations of these parameters with disease severity were analyzed. The WT and TLR4-/- mice were administered HMGB1 by intratracheal injection. After 48 h, the mice were sacrificed. The morphological changes and wet/dry ratio of the lung were measured. Spleen CD4+CD25+ Tregs were sorted from spleen cells, the expression of FOXP3 and CTLA-4, and releasing of cytokines was detected. CD4+CD25+ Tregs were cocultured with effector T cells, the inhibitory effect, and release of cytokines was detected.
Results: Significantly increased plasma levels of HMGB1 and reduced CD4+CD25+CD127low Tregs were detected in ALI patients. In the mouse model, lung injury was significantly increased after HMGB1 instillation in the WT and TLR4-/- groups compared with control group. The lung wet/dry ratio and the TNF-α and IL-1β contents in BALF were significantly increased, and the severity of WT mice was higher than that of TLR4-/- mice. The expression of FOXP3 and CTLA-4 in TLR4-/- mice was significantly increased compared with that in WT mice and was associated with a similar trend of IL-10 and TGF-β levels (p< 0.05). In coculture with effector T cells, Tregs isolated from TLR4-/- mice exhibited decreased IL-2 and IFN-γ and increased IL-4 levels compared with Tregs from WT mice. Increased polarization of TLR4-/- CD4+CD25+ Treg cells to Th2 cells was observed.
Conclusion: In HMGB1-induced lung injury, HMGB1 affects the expression of FOXP3 and CTLA-4 through TLR4, thus reducing the immunosuppressive function of Treg cells.
Keywords: HMGB1, TLR4, Treg, acute lung injury
Background
Acute respiratory distress syndrome (ARDS) is severe lung dysfunction occurring in critically ill patients and is caused by alveolar injury secondary to an inflammatory process.1 One important characteristic of ARDS is lung injury during the innate immune response to pathogens, which is mediated by inflammatory cytokines.
High-mobility group box 1 (HMGB1) is a nuclear protein that also functions as an inflammatory cytokine. HMGB1 is released in the late phase of the inflammatory response, and the release of HMGB1 from macrophages is induced by LPS, TNFα, and IL-1β stimulation.2 HMGB1 contributes to acute lung injury by activating the NF–κB signaling pathway and inducing the production of inflammatory cytokines.3–5 Based on the evidence, HMGB1-induced cytokine release occurs through a TLR4-dependent pathway.6,7 TLR4 is a key receptor involved in innate immunity. TLR4 activation induces an inflammatory response through the NF-κB signaling pathway.8 Notably, the activation of TLR4 by LPS leads to the induction of proinflammatory Th17 cells and a reduction in the number of anti-inflammatory T regulatory lymphocytes (Tregs).9 Stimulation with HMGB1 decreases the expression of CTLA-4, FOXP3, IL-4, and IL-10 and increases IL-2 and IFN-γ levels in splenic Treg cells.10 Elevated levels of HMGB1 depress Tregs and promote Th17 cell activity by modulating the TLR4-IL-6 axis in patients with liver failure and chronic HBV infection.11,12 Tregs have been shown to exert a protective effect on transfusion-associated acute lung injury (TRALI), and a depletion of Tregs increases susceptibility to antibody-mediated TRALI in a mouse model.13,14 In seeking to address the gaps in our understanding of the mechanism of HMGB1-associated acute lung injury, a better understanding of the effect of HMGB1-TLR4 activation on the immune properties of T cell polarization may help to enhance cell therapy for acute lung injury.
We hypothesize that the development of acute lung injury requires a TLR4-mediated reduction in Tregs, and thus strategies designed to increase Tregs may reduce the extent of HMGB1-induced lung injury. In the present study, the relationship between serum HMGB1 levels and the Treg population was analyzed in patients with sepsis and septic shock, and the role of TLR4 was investigated in HMGB1-induced acute lung injury models using TLR4 knockout mice.
Methods
Study Approval
The human study was approved by the Ethical Review Board of Anhui Medical University in China (No: 2013005). The healthy controls provided informed consent, and this study was conducted in accordance with the Declaration of Helsinki. The animal study was performed in strict accordance with the the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. The study protocol was approved by the Institutional Animal Care and Use Committee of Anhui Medical University (No: LLSC2013007).
Human Subjects
Fifty-eight adult patients (33 men and 25 women, 22 to 68 years old) were prospectively identified as suffering from ALI associated with severe sepsis or septic shock and recruited into this study. Thirty-six healthy individuals (21 men and 15 women, 26 to 70 years old) with normal lung function and free of any respiratory disease served as controls. The study was conducted at the hospitals of Anhui Medical University between June 2012 and January 2014. Informed consent was obtained from the next of kin of all participants. The diagnosis of severe sepsis and septic shock was defined by the criteria of ACCP/SCCM Consensus Conference.15 ALI/ARDS was diagnosed based on the definitions of the American European Consensus Conference on ARDS (E1), including patients who met consensus conference oxygenation and radiographic criteria.16 All patients with sepsis were required to have developed ALI onset within the preceding 24 hours (h) to be eligible for the study. Mechanical ventilation followed the low-tidal-volume strategy described in the ARDS Net study.17 Patients were excluded if they had immunodeficiency or were receiving any form of immune-modulating treatment, had an autoimmune or hypersensitivity disease, were undergoing dialysis or had a severe underlying disease. Patients treated with hydrocortisone for refractory hypotension were withdrawn from the study. The lung injury score and the criteria of the ACCP/SCCM Consensus Conference Committee for sepsis syndrome were used. The characteristics of the patients are summarized in Table 1.
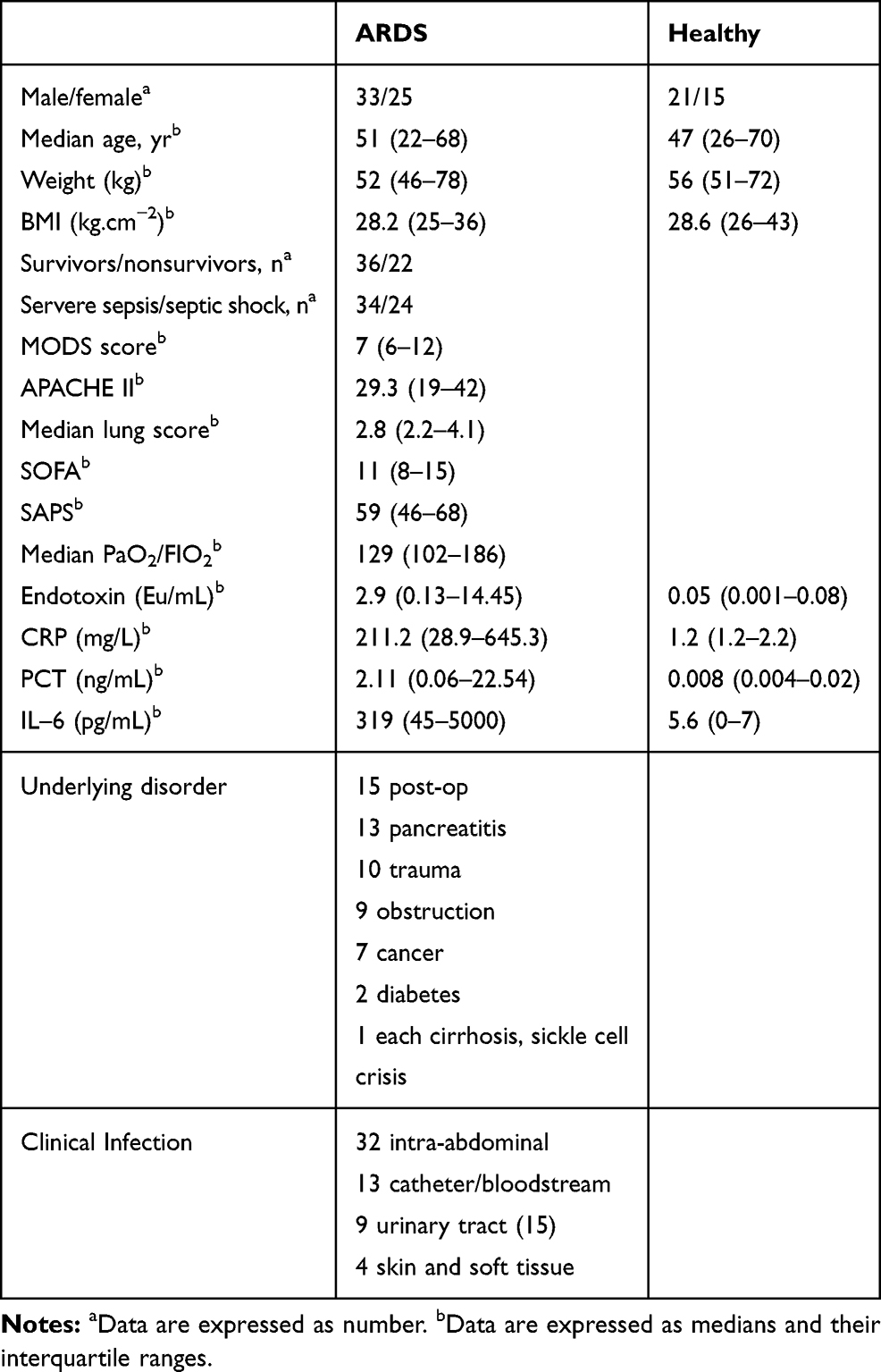 |
Table 1 Characteristics of Healthy and ARDS Subjects |
Human CD4+CD25+CD127lowTregs and Flow Cytometry Analysis
Peripheral blood mononuclear cells were isolated using a Ficoll-Paque Plus gradient (Amersham Biosciences, Uppsala, Sweden).18 The ratio of CD4+CD25+CD127low cells was measured using flow cytometry with anti-human CD4-FITC, CD25-PE-Cyanine5, and CD127-PE antibodies (eBioscience, San Diego, CA).
Measurements of Human HMGB1 Levels
ELISA kits were used to measure the levels of HMGB1 (Shino-TEST Co, Tokyo, Japan) in serum samples according to the manufacturer’s instructions.
Animal Models
C57BL/10J wild-type (WT) and C57BL/10Sc TLR4 global knockout (TLR4-/-) mice (8–10 weeks) were purchased from Nanjing University Model Animal Research Center. All animals were housed in a facility with standard animal care conditions and had free access to water and food.
The mouse acute lung injury model was approved by the Institute Research Ethics Committee of Anhui Medical University. Mice were distributed to three groups. In control group (control), WT mice were used, and treated with saline; in wild type group (WT), WT mice were used, and treated with HMGB1; in TLR4-/- group (TLR4-/-), TLR4-/- mice were used, and treated with HGMB1. Ten mice per group were anesthetized with an i.p. injection of ketamine (150 mg/kg). Mice were placed in a supine position head up on a board tilted at a 50-degree angle. A midline incision was made in the neck, and the trachea was exposed. A catheter was inserted into the trachea, and mice received an intratracheal injection of HMGB1 (Sigma-Aldrich, St. Louis, MO, USA) at a dose of 20 μg in 50 μL of PBS. Control mice received an intratracheal injection of 50 μL of saline. After the intratracheal injection, the mice were placed in a vertical position and rotated for 1 min to distribute the instillation evenly within the lungs. Forty-eight hours after the intratracheal injection, the animals were sacrificed. Blood and tissue samples were harvested.
Preparation of Bronchoalveolar Lavage Fluid (BALF)
BALF isolation was performed as described previously.18 Briefly, 48 hours after HMGB1 challenge in the mouse model, bronchoalveolar lavage (BAL) was performed three times by instilling 0.5 mL of PBS through the tracheal cannula after ligation of the left bronchus. The fluid was centrifuged for 10 min at 1200 rpm, and the supernatant was collected and stored at −80 °C until further detection.
Wet to-Dry (W/D) Lung Weight Ratio
We used the W/D lung weight ratio to assess lung edema and lung inflammation. The left lung was ligated, and the weight was measured. The lung tissue was incubated at 60°C for 3 days, and the dry weight was recorded. The W/D weight ratio was calculated by dividing the wet weight by the dry weight.
Histopathology
The lung tissue was fixed with 4.5% buffered formalin. Paraffin embedding was performed using standard techniques. Sections (5 μm) were stained with hematoxylin and eosin. The tissue damage was evaluated. The lung injury was quantified according to the criteria for assessment of acute lung injury reported by Matute-Bello et al.19
Isolation and Purification of Splenic CD4+CD25+Tregs
Spleens were harvested, and cell suspensions were prepared by mincing tissues. Cells were treated with ammonium chloride solution and washed twice before suspension. CD4+CD25+ and CD4+CD25− T cells were purified using regulatory T cell magnetic cell sorting kits (Miltenyi Biotec, Germany). The purity of CD4+CD25+ and CD4+CD25− T cells obtained using this approach was consistently >90%. Cell purity was assessed by staining cells with Cy5-labeled anti-CD4 Ab and PE-labeled anti-CD25 Ab followed by an analysis using a FACSCalibur Instrument (BD Biosciences).
Treg Culture and Cytokine Measurements
All cells were cultured at 37 °C with 5% CO2 in RPMI 1640 supplemented with 10% FCS. CD4+CD25+ Tregs isolated from the mouse spleens with magnetic beads were seeded on 96-well (5 × 106 cells/well) cell culture plates coated with anti-CD3 (5 mg/L) and soluble anti-CD28 (1 mg/L) antibodies. After 24 h, Tregs were collected and incubated with the APC-labeled CTLA-4 Ab and FITC-labeled FOXP3 Ab followed by an analysis using a FACSCalibur Instrument (BD Biosciences).
In vitro Assessment of Cytokine Production
Purified CD4+CD25+ Treg cells were cultured in 96-well plates at a density of 1×105 cells/well. After 24 h of culture, the supernatants were harvested and stored at −80°C. Purified CD4+CD25+ Treg cells were cultured in 96-well plates at a density of 1×105 cells/well, and 100 ng/mL HMGB1 was added to each well to assess the effect of HMGB1 on Tregs. After 48 h of stimulation, culture supernatants were harvested and stored at −80°C. The cytokines IL-10 and TGF-β were measured using cytokine-specific ELISAs (ExCell Bio, China) according to the manufacturer’s instructions. The ELISA results were analyzed using an ELISA plate reader (DUTM-800, Beckman Coulter, USA) and the associated softMas pro software. Cytokine levels in culture supernatants were calculated based on cytokine standards included in each assay plate.
The concentrations of secreted IL-10 and TGF-β1 in the supernatants of CD4+CD25+ Tregs were determined using commercially available double-antibody sandwich ELISA kits (ExCell Bio, China). The color reaction was terminated by adding 100 μL of ortho-phosphoric acid. Plates were read in a microplate reader (Spectra MR, Dynex, Richfield, MN). All samples were run in quadruplicate.
The CD4+CD25+ and CD4+CD25− T Cell Cocultures
Purified CD4+CD25+ and CD4+CD25− T cells (1:1) were seeded on 96-well (2 × 105 cells/well) cell culture plates coated with anti-CD3 (1 μg/mL) and soluble anti-CD28 (1 μg/mL) antibodies. Seventy-two hours after coculture, 100 μL of supernatant were collected and stored at −80°C until the ELISA measurement. Ten microliters of CCK-8 reagent were added, and after 4 h of culture, the absorption value was measured at 450 nm (DUTM-800, Beckman Coulter, USA).
Statistical Analysis
Data are presented as the mean±SD. The clinical data were analyzed using the permutation t-test and Pearson’s correlation analysis. For each comparison, two-sided p values less than 0.05 were considered statistically significant. These analyses were performed using SAS/STAT software, version 9.4. Tukey’s test was performed for multiple comparisons in the animal experiment and in vitro experiment. Data from two groups were compared using t-tests. A p-value less than 0.05 was considered statistically significant. These analyses were performed using SPSS software.
Results
Elevated HMGB1 Levels and Reduced Tregs in Patients with ALI
We first measured HMGB1 levels in patients with sepsis or septic shock to determine whether HMGB1 levels in patients correlated with the severity of sepsis. Indeed, HMGB1 levels were increased in patients with sepsis (n=34) compared with healthy controls (n=36, Figures 1A, 105.3±45.6 ng/mL vs 8.3±3.62 ng/mL, p<0.01). The HMGB1 levels were further increased in patients with septic shock (n=24) compared to patients with sepsis (Figures 1A, 167.5±78.5 ng/mL, p<0.05). Next, we measured the HMGB1 levels in patients from the surviving ARDS (n=36) and nonsurviving ARDS (n=22) groups. HMGB1 levels were significantly increased in the nonsurviving ARDS group compared with the surviving ARDS group (Figures 1B, 108.3±67.9 ng/mL vs 67.4±42.6 ng/mL, p<0.05). We analyzed the frequency of CD4+CD25+CD127low Tregs in patients with ARDS (including surviving and nonsurviving patients with ARDS) to determine whether HMGB1 affects Treg production during sepsis-induced ALI. Indeed, decreased percentages of CD4+CD25+CD127low Tregs were observed in patients with sepsis-induced ARDS (4.2±2.56, n=58) compared to healthy controls (7.9±1.31, n=36, p<0.01) (Figure 1C and D). The correlation was further analyzed between the percentage of CD4+CD25+CD127low Tregs and HMGB1 levels in patients with ARDS and shown to be significant (Figure 1E, R2=0.535, n=58, p<0.001). Based on these data, HMGB1 levels may be correlated with Treg accumulation in patients with sepsis-induced lung injury.
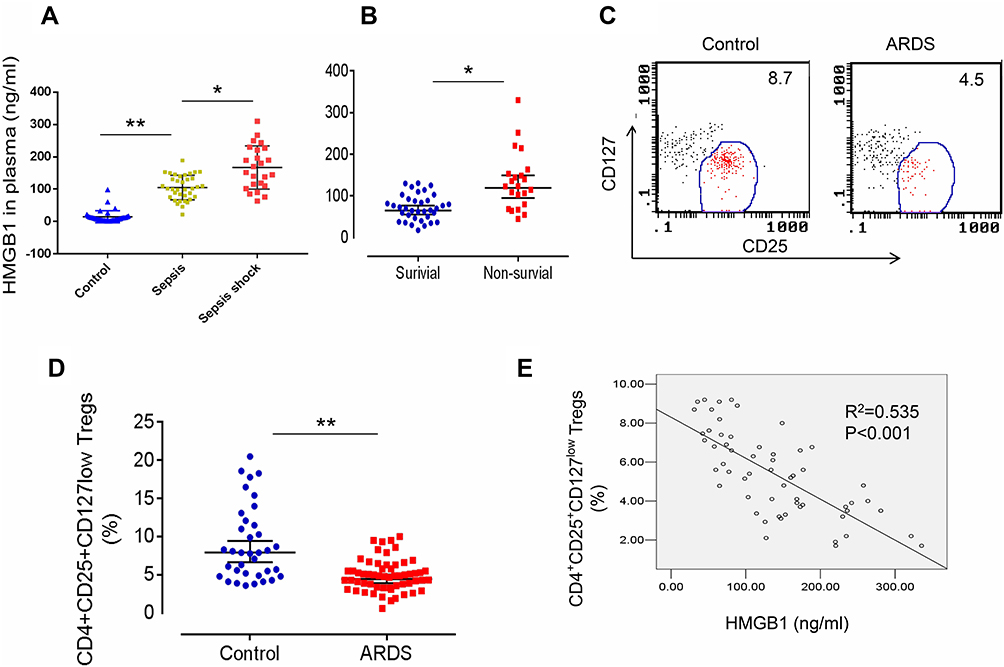 |
Figure 1 Elevated HMGB1 levels and reduced Treg proportions in patients with sepsis-induced ALI. (A) Circulating HMGB1 levels in plasma were measured in patients with sepsis (n=34), septic shock (n=24), and healthy controls (n=36) using an ELISA. **p<0.01. (B) Plasma HMGB1 levels in surviving (n=36) and nonsurviving (n=22) patients with ARDS. *p<0.05 and **p<0.01. (C) Representative two-dimensional scatter diagrams of CD4+CD25+CD127low Tregs in the peripheral blood of patients with ARDS and healthy controls were analyzed using flow cytometry. (D) The percentage of CD4+CD25+CD127low Tregs in the peripheral blood of patients with ARDS (n=58) and healthy controls (n=36); means±SD, **p<0.01. (E) Correlation between the percentage of CD4+CD25+CD127low Tregs and HMGB1 levels in patients with ARDS; R2=0.535, p<0.001. The analyses in this figure were performed using permutation t-tests (A, B, and D) and Pearson’s correlation analyses (E). |
TLR4 Deletion Accelerates Lung Injury and the Local Inflammatory Response Induced by the HMGB1 Instillation
HMGB1 has been shown to induce an inflammatory response and cause tissue damage. The intratracheal instillation of HMGB1 caused morphological changes in the lung tissue, including interstitial edema, alveolar thickening, and severe leukocyte infiltration (Figure 2A), the quantified lung injury score was listed in Figure 2B. Deletion of TLR4 ameliorated the injury caused by HMGB1, as indicated by reduced morphological changes in the lung tissue. Treatment with HMGB1 caused an increase in the W/D weight ratio of lung tissue, which was decreased by TLR4-/- deletion (5.53±0.32 vs 4.82±0.22, p<0.05, Figure 2C). The HMGB1 instillation induced local expression of TNF-α and IL-1β, which was associated with increased serum cytokine levels (p<0.05, Figure 2D–G). The increases in local and systemic TNF-α and IL-1β levels were blocked by the deletion of TLR4-.
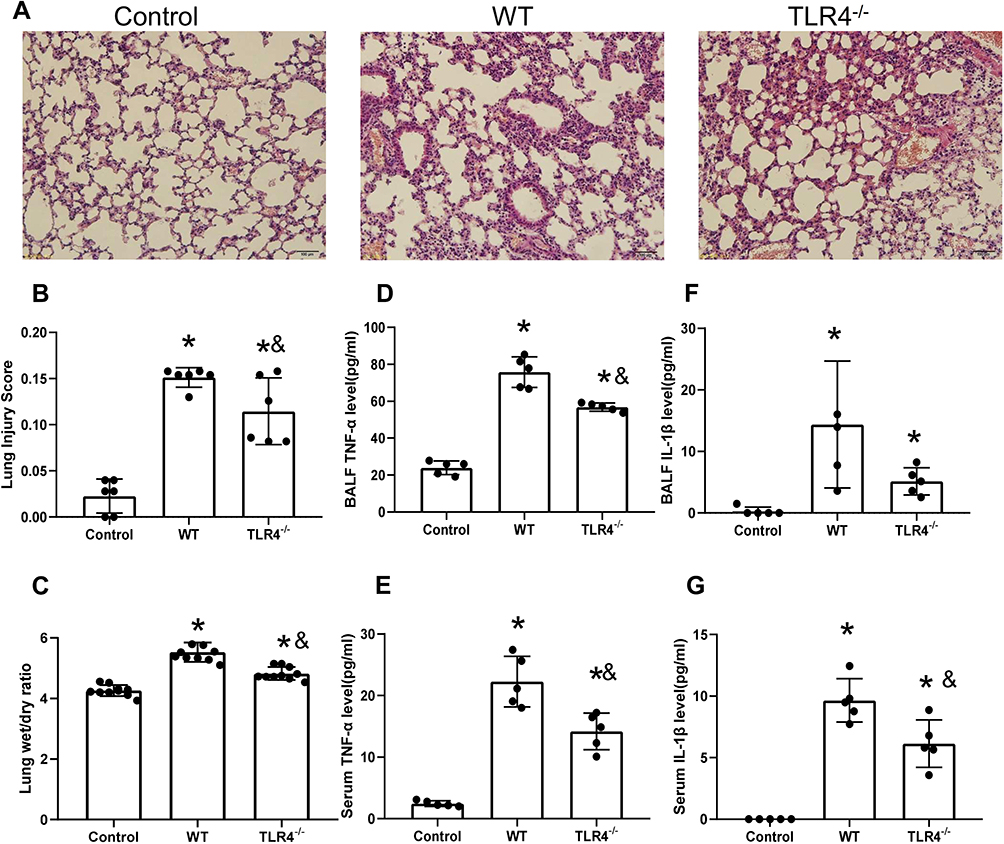 |
Figure 2 TLR4 deletion accelerates lung injury and the local inflammatory response induced by the HMGB1 instillation. (A) Pathological changes in lung tissues from TLR4 mice with different genotypes 48 h after an intratracheal instillation of HMGB1. (B) The morphological changes was quantified by using lung injury scoring system (C) Changes in the W/D weight ratio of the lung tissue 48 h after an intratracheal instillation of HMGB1 in mice with different TLR4 genotypes. (D) TNF-α levels were measured in BAL fluid from the control, WT, and TLR4-/- groups using an ELISA. (E) Serum TNF-α was measured by ELISA in the control, WT, and TLR4-/- groups. (F) IL-1β levels were measured in BAL fluid from the control, WT, and TLR4-/- groups using an ELISA. (G) Serum IL-1β levels were measured in the control, WT, and TLR4-/- groups using an ELISA, *p<0.05 between 3 groups by using Tukey’s test, &p<0.05 between WT and TLR4-/- group by using t-test. |
TLR4 Reversed CTLA-4 and FOXP3 Downregulation Caused by the HMGB1 Instillation
The induction of the inflammatory response is associated with a decreased Treg proportion. Next, CD4+CD25+ Tregs were detected in the experimental groups. CD4+CD25+ Treg cells were isolated from the spleen. Flow cytometry showed that the purity of CD4+CD25+ Treg cells sorted by immunomagnetic beads was greater than 90%. The expression of FOXP3 and CTLA-4 in the HMGB1 instillation group was significantly decreased compared with that in the control group (CTLA-4: 38.60±3.54 vs 19.65±1.46, p<0.05; FOXP3: 52.61±7.69 vs 34.91±4.92, p<0.05, Figure 3A, Table 2). However, TLR4 deletion reversed these changes and caused the upregulation of CTLA-4 and FOXP3 expression compared with the control group and HMGB1 instillation group (CTLA-A: 38.60±3.54 vs 63.09±5.72, p<0.05; FOXP3: 52.61±7.69 vs 65.16±4.93, p<0.05).
 |
Figure 3 TLR4 reversed the CTLA-4 and FOXP3 downregulation caused by the HMGB1 instillation. (A) Changes in CTLA-4 and FOXP3 expression in CD4+CD25+ Tregs from different groups of mice with acute lung injury. (B) TGF-β levels in CD4+CD25+ Tregs from different groups. (C) IL-10 levels in CD4+CD25+ Tregs from different groups. (D) TGF-β content in CD4+CD25+ Tregs from mice with different genotypes stimulated with HMGB1. (E) IL-10 content in CD4+CD25+ Tregs from mice with different genotypes stimulated with HMGB1, *p<0.05 between 3 groups by using Tukey’s test, and &p<0.05 between WT and TLR4-/- group by using t-test. |
 |
Table 2 Changes of CTLA-4 and FOXP3 Expression of CD4+CD25+Treg in Different Groups of Mice with ALI (X±s) |
TLR4 Deletion Reversed the HMGB1 Treatment-Induced Downregulation of Cytokine Release
Next, the release of TGF-β and IL-10 was detected in the different groups (Figure 3B and C). Compared with the control group, the HMGB1 instillation decreased the release of TGF-β (456.37±61.16 pg/mL vs 381.86±25.77 pg/mL, p<0.05) and IL-10 (121.55±19.02 pg/mL vs 66.78±24.37 pg/mL, p<0.05) into the culture supernatant. Notably, TLR4 deletion increased the release of TGF-β (456.37±61.16 pg/mL vs 638.41±58.18 pg/mL, p<0.05), and IL-10 (121.55±19.02 vs 136.85±14.24 pg/mL, p<0.05) compared with both the control group and the HMGB1 instillation group (p<0.05). In an in vitro system, CD4+CD25+ Tregs were isolated from WT and TLR4-/- mice and treated with HMGB1. The release of TGF-β and IL-10 into the culture supernatant was significantly decreased in the WT group (Figure 3D and E, TGF-β: 442.54±61.16 pg/mL vs 176.56±77.26 pg/mL, p<0.05; IL-10: 159.11±17.57 pg/mL vs 104.64±15.23 pg/mL, p<0.05), while TLR4 deletion increased the cytokine levels in the supernatant (TGF-β: 442.54±61.16 pg/mL vs 522.40±49.74 pg/mL, p<0.05; IL-10: 159.11±17.57 pg/mL.s. 183.27±20.50 pg/mL, p<0.05). In the next step, the proliferative effect of CD4+CD25− T cells was detected by coculture with CD4+CD25+ Tregs. CD4+CD25− T cells were activated by treatment with anti-CD3 and anti-CD28 antibodies and cocultured with CD4+CD25+ Tregs.
TLR4 Deletion Reverses HMGB1-Induced Polarization Towards Th1 Cells
After coculture with CD4+CD25+ Tregs from WT mice treated with the HMGB1 instillation, the proliferation of CD4+CD25− T cells was significantly increased, as assessed using the CCK-8 method (OD450: 0.77±0.05 vs 0.91±0.042, p<0.05, Figure 4A). The increased proliferation was inhibited by coculture with Tregs from TLR4-/- mice subjected to the same treatment (0.77±0.05 vs 0.54±0.05, p<0.05). In the supernatant of cocultures of CD4+CD25+ Treg cells and effector T cells from the control group, as well as the supernatant of cocultures of CD4+CD25+ Treg cells from wild-type mice with acute lung injury and effector T cells, the contents of IL-2 (175.42±7.11 pg/mL vs 214.76±13.35 pg/mL, p<0.05, Figure 4B) and IFN-γ (132.85±6.26 pg/mL vs 150.05±11.76 pg/mL, p<0.05, Figure 4D) were higher than those in the control group, while the contents of IL-4 (154.96±13.97 pg/mL vs 96.91±9.95 pg/mL, p<0.05, Figure 4C) were lower than those in the control group, and the ability of CD4+CD25+ Treg cells to polarize towards Th2 cells was reduced, but they polarized towards Th1 cells. In the supernatant of cocultures CD4+CD25+ Treg cells from TLR4-/- mice with acute lung injury and effector T cells, the contents of IL-2 and IFN-γ were lower than those in the control group (IL-2: 175.42±7.11 pg/mL vs 144.69±10.75 pg/mL, p<0.05; IFN-γ: 132.84±6.25 pg/mL vs 117.68±11.40 pg/mL, p<0.05), while the IL-4 content was higher than that in the control group (154.96±13.97 pg/mL vs 174.74±31.83 pg/mL, p<0.05), and the immunosuppressive effect mediated by CD4+CD25+ Treg cells caused a Th1/Th2 shift towards Th2 cell polarization.
 |
Figure 4 TLR4 deletion reverses HMGB1-induced polarization towards the Th1 phenotype. (A) The CCK-8 method was used to detect the effects of CD4+CD25+ Tregs on the proliferation of CD4+CD25− T cells from different groups. (B) Changes in IL-2 contents in cocultures of CD4+CD25+ Tregs and CD4+CD25− T cells. (C) IL-4 contents in cocultures of CD4+CD25+ Tregs and CD4+CD25− T cells. (D) IFN-γ contents in cocultures of CD4+CD25+ Tregs and CD4+CD25− T cells, *p<0.05 between 3 groups by using Tukey’s test, and &p<0.05 between WT and TLR4-/- group by using t-test. |
Discussion
ARDS/ALI is a syndrome of acute respiratory failure characterized by clinical features of hypoxemia and altered respiratory system mechanics.20 ARDS/ALI remains a common and highly morbid condition.
Although CD4+CD25+FOXP3+ Tregs are essential for mediating the resolution of lung injury,21 little is known about how HMGB1 affects Treg generation in patients with ALI. Indeed, elevated levels of HMGB1 have been observed in both plasma and lung epithelial lining fluids of patients with ALI.22,23 CD4+CD25+ Tregs expressing FOXP3 (CD4+CD25+CD127low in human Tregs) may accumulate in the injured lung.21 We showed that increased HMGB1 levels in patients with ALI induced by severe sepsis are accompanied by decreased numbers of CD4+CD25+CD127low Tregs. These data imply that circulating HMGB1 levels and the Treg frequency may be directly involved in determining disease severity in patients with ALI.
Due to the important clinical implications in patients with ALI and an inflammatory response caused by infection develop similar clinical syndromes (Table 1), the increased serum HMGB1 levels may be released from lymphocytes or infected tissues. Therefore, serum HMGB1 levels might serve as a marker to reflect the severity of tissue injury or the state of the inflammatory response.
HMGB1 is a prototypical damage-associated molecular pattern (DAMP) molecule.24 During sepsis, HMGB1 is released from tissues and promotes the systemic inflammatory response and systemic injury.25 Measurements of serum HMGB1 levels revealed that HMGB1 is associated with the severity of sepsis and is present at significantly higher levels in nonsurvivors than in survivors. The administration of HMGB1 increased mortality in a mouse model; in contrast, treatment with an HMGB1 antibody to block HMGB1 protected against lethal effects on a mouse model of LPS-induced SIRS.2 The HMGB1 antibody was used to block the effect of HMGB1. Blocking HMGB1 increased the animal survival rate, decreased lung injury, and increased the Treg proportion.18 In the present study, an intratracheal instillation of HMGB1 generated a stable and reliable mouse acute lung injury model, which provides a compact basis for further in vivo investigation.
Extracellular disulfide HMGB1 activates monocytes and macrophages to induce cytokine release and chemokine production via the TLR4 signaling pathway.26 TLR4 is one receptor for HMGB1. The activation of TLR4 by HMGB1 mediates acute lung injury in a liver ischemia reperfusion model.27 The activation of TLR4 by HMGB1 occurs via the binding between HMGB1 and MD2.28 Notably, HMGB1 and LPS do not share the same binding site on MD2. Therefore, interrupting the binding between HMGB1 and TLR4 will not influence the activation of the LPS-dependent signaling pathway.29 HMGB1-induced LTA tolerance is mediated by the TLR2 signaling pathway but not RAGE.30 Wild et al reported that HMGB1 directly enhances Treg activity via RAGE-mediated mechanisms31 and impairs the host’s ability to clear pathogens.32 In our results, deletion of TLR4 further increased the HMGB1-induced Treg elevation, which may be explained partially by the increased activation of RAGE. Although the interaction between HMGB1 and RAGE leads to the activation of NF-κB and the release of cytokines, it fails to induce cytokine release from macrophages expressing RAGE proteins without TLR4.29 Based on this finding, the HMGB1-RAGE pathway may not directly induce cytokine production. In the present study, TLR4 was deleted to confirm the effect of HMGB1 on acute lung injury. A lethal effect was observed on TLR4-deficient mice with endotoxemia in the presence of increased levels of HMGB1, which was reversed in caspase 11-deficient mice.33 The deletion of TLR4 decreased the HMGB1-induced local inflammatory response, lung edema, and tissue injury.
Sepsis is responsible for the induction of the inflammatory response but also contributes to subsequent immunosuppression.34 HMGB1 is reported to induce immune tolerance.30,35 Our results revealed that during acute lung injury, HMGB1 may affect the expression of FOXP3 and CTLA-4 in Treg cells through TLR4 at least in part, thus reducing its immunosuppressive function. Evidence suggests that TLR4 activation in the environment increases the induction of Treg generation. Iran Rashedi reported that the activation of TLR4 on mesenchymal stromal cells increased the induction of Treg cell production via notch signaling.36 Wei et al observed that a quercetin treatment decreased inflammatory injury through the induction of Treg production, which was reversed by treatment with a TLR4 antagonist.37 In chronic infection, TLR4 knockout caused a decrease in the Treg population, which was associated with increased F. nucleatum colonization in a mouse model.38 Tregs prevent tissue damage caused by immune activation through their suppressive function.39 The anti-inflammatory nature of Tregs prompted us to examine whether they might play a role in helping reverse the tissue injury caused by the inflammatory response in an infectious disease. Further evidence suggested that HGMB1-TLR4 may reduce the ability of CD4+CD25+ Treg cells to polarize towards Th2 cells at least in part, causing the cells to polarize towards Th1 cells. T-cells play a critical role in sepsis. Distinct patterns of cytokines are produced by Th1 and Th2 cells. Th1 cells produce IFN-γ and IL-2, and Th2 cells secrete IL-4, IL-5, IL-10, and IL-13. Polarization from Th1 to Th2 cells leads to immunosuppression. The suppressive effect of Tregs on the inflammatory response occurs through cytokines, eg, IL-10- and TGF-β-mediated mechanisms.40 Kapur et al reported low levels of IL-10 in patients with transfusion-related acute lung injury (TRALI) compared with patients with sepsis-induced ALI.41 TRALI is induced by sterile inflammation, and the intensity of the inflammatory response is significantly lower than that of the pathogen-induced inflammatory response. This result may explain why no significance was observed between patients in the no TRALI and TRALI groups. In animal experiments, the administration of IL-10 was protective and rescued mice from TRALI development,13 indicating that IL-10 contributes to reversing the proinflammatory response and initiating the process of tissue repair.
In conclusion, our results revealed that HMGB1 affects the expression of FOXP3 and CTLA-4 in Treg cells through TLR4, thus reducing its immunosuppressive function. During acute lung injury, HGMB1 may reduce the ability of CD4+CD25+ Treg cells to polarize towards Th2 cells, causing the cells to polarize towards Th1 cells.
Data Sharing Statement
Data are available upon reasonable request.
Ethical Approval
The human study was approved by the Ethical Review Board of Anhui Medical University in China (No: 2013005).
Consent for Publication
Consent for publication was obtained from all participants.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported in part by grants from the National Natural Science Foundation of China (No. 81871584 to MZ, No. 81870060 to RT, and No. 81971875 to FH) and Anhui Province Natural Science Foundation of China (No. KJ2017A199 to MZ).
Disclosure
The authors reported no conflicts of interest for this work.
References
1. Sweeney RM, McAuley DF. Acute respiratory distress syndrome. Lancet. 2016;388(10058):2416–2430. doi:10.1016/S0140-6736(16)00578-X
2. Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–251. doi:10.1126/science.285.5425.248
3. Yang H, Ochani M, Li J, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA. 2004;101(1):296–301. doi:10.1073/pnas.2434651100
4. Abraham E, Arcaroli J, Carmody A, et al. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165(6):2950–2954. doi:10.4049/jimmunol.165.6.2950
5. Kim JY, Park JS, Strassheim D, et al. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol. 2005;288(5):L958–65. doi:10.1152/ajplung.00359.2004
6. He M, Bianchi ME, Coleman TR, et al. Exploring the biological functional mechanism of the HMGB1/TLR4/MD-2 complex by surface plasmon resonance. Mol Med. 2018;24(1):21. doi:10.1186/s10020-018-0023-8
7. Yang H, Hreggvidsdottir HS, Palmblad K, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107(26):11942–11947. doi:10.1073/pnas.1003893107
8. Deng M, Scott MJ, Loughran P, et al. Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during sepsis. J Immunol. 2013;190(10):5152–5160. doi:10.4049/jimmunol.1300496
9. Egan CE, Sodhi CP, Good M, et al. Toll-like receptor 4-mediated lymphocyte influx induces neonatal necrotizing enterocolitis. J Clin Invest. 2016;126(2):495–508. doi:10.1172/JCI83356
10. Zhang Y, Yao Y-M, Huang L-F, et al. The potential effect and mechanism of high-mobility group box 1 protein on regulatory T cell-mediated immunosuppression. J Interferon Cytokine Res. 2011;31(2):249–257. doi:10.1089/jir.2010.0019
11. Wang LW, Chen H, Gong ZJ. High mobility group box-1 protein inhibits regulatory T cell immune activity in liver failure in patients with chronic hepatitis B. Hepatobiliary Pancreat Dis Int. 2010;9(5):499–507.
12. Li J, Wang F-P, She W-M, et al. Enhanced high-mobility group box 1 (HMGB1) modulates regulatory T cells (Treg)/T helper 17 (Th17) balance via toll-like receptor (TLR)-4-interleukin (IL)-6 pathway in patients with chronic hepatitis B. J Viral Hepat. 2014;21(2):129–140. doi:10.1111/jvh.12152
13. Kapur R, Kim M, Aslam R, et al. T regulatory cells and dendritic cells protect against transfusion-related acute lung injury via IL-10. Blood. 2017;129(18):2557–2569. doi:10.1182/blood-2016-12-758185
14. Semple JW, Rebetz J, Kapur R. Transfusion-associated circulatory overload and transfusion-related acute lung injury. Blood. 2019;133(17):1840–1853. doi:10.1182/blood-2018-10-860809
15. Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101(6):1644–1655. doi:10.1378/chest.101.6.1644
16. Bernard GR, Artigas A, Brigham KL, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149(3):818–824. doi:10.1164/ajrccm.149.3.7509706
17. Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1301–1308. doi:10.1056/NEJM200005043421801
18. Zhou M, Fang H, Du M, et al. The modulation of regulatory T cells via HMGB1/PTEN/beta-catenin axis in LPS induced acute lung injury. Front Immunol. 2019;10:1612. doi:10.3389/fimmu.2019.01612
19. Matute-Bello G, Downey G, Moore BB, et al. An official American Thoracic Society workshop report: Features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44(5):725-738. -doi:10.1165/rcmb.2009-0210ST
20. Michaeloudes C, Bhavsar P, Mumby S, et al. Role of metabolic reprogramming in pulmonary innate immunity and its impact on lung diseases. J Innate Immun. 2020;12(1):31–46. doi:10.1159/000504344
21. D’Alessio FR, Tsushima K, Aggarwal NR, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest. 2009;119(10):2898–2913. doi:10.1172/JCI36498
22. Ueno H, Matsuda T, Hashimoto S, et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med. 2004;170(12):1310–1316. doi:10.1164/rccm.200402-188OC
23. Zaher TE, Miller EJ, Morrow DMP, et al. Hyperoxia-induced signal transduction pathways in pulmonary epithelial cells. Free Radic Biol Med. 2007;42(7):897–908. doi:10.1016/j.freeradbiomed.2007.01.021
24. Deng MH, Scott MJ, Fan J, et al. Location is the key to function: HMGB1 in sepsis and trauma-induced inflammation. J Leukoc Biol. 2019;106(1):161–169. doi:10.1002/JLB.3MIR1218-497R
25. Andersson U, Tracey KJ. HMGB1 in sepsis. Scand J Infect Dis. 2003;35(9):577–584. doi:10.1080/00365540310016286
26. Yu M, Wang H, Ding A, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26(2):174–179. doi:10.1097/01.shk.0000225404.51320.82
27. Yang Z, Deng Y, Su D, et al. TLR4 as receptor for HMGB1-mediated acute lung injury after liver ischemia/reperfusion injury. Lab Invest. 2013;93(7):792–800. doi:10.1038/labinvest.2013.66
28. Yang H, Wang H, Ju Z, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. 2015;212(1):5–14. doi:10.1084/jem.20141318
29. Yang H, Wang HC, Andersson U. Targeting Inflammation Driven by HMGB1. Front Immunol. 2020;11.
30. Robert SM, Sjodin H, Fink MP, et al. Preconditioning with high mobility group box 1 (HMGB1) induces lipoteichoic acid (LTA) tolerance. J Immunother. 2010;33(7):663–671. doi:10.1097/CJI.0b013e3181dcd111
31. Wild CA, Bergmann C, Fritz G, et al. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int Immunol. 2012;24(8):485–494. doi:10.1093/intimm/dxs051
32. Patel VS, Sitapara RA, Gore A, et al. High mobility group Box–1 mediates hyperoxia-induced impairment of pseudomonas aeruginosa clearance and inflammatory lung injury in mice. Am J Respir Cell Mol Biol. 2013;48(3):280–287. doi:10.1165/rcmb.2012-0279OC
33. Hagar JA, Powell DA, Aachoui Y, et al. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341(6151):1250–1253. doi:10.1126/science.1240988
34. Van Wyngene L, Vandewalle J, Libert C. Reprogramming of basic metabolic pathways in microbial sepsis: therapeutic targets at last? EMBO Mol Med. 2018;10(8).
35. Aneja RK, Tsung A, Sjodin H, et al. Preconditioning with high mobility group box 1 (HMGB1) induces lipopolysaccharide (LPS) tolerance. J Leukoc Biol. 2008;84(5):1326–1334. doi:10.1189/jlb.0108030
36. Rashedi I, Gómez-Aristizábal A, Wang X-H, et al. TLR3 or TLR4 activation enhances mesenchymal stromal cell-mediated treg induction via notch signaling. Stem Cells. 2017;35(1):265–275. doi:10.1002/stem.2485
37. Wei CB, Tao K, Jiang R, et al. Quercetin protects mouse liver against triptolide-induced hepatic injury by restoring Th17/Treg balance through Tim-3 and TLR4-MyD88-NF-kappaB pathway. Int Immunopharmacol. 2017;53:73–82. doi:10.1016/j.intimp.2017.09.026
38. Jia YP, Wang K, Zhang Z-J, et al. TLR2/TLR4 activation induces Tregs and suppresses intestinal inflammation caused by Fusobacterium nucleatum in vivo. PLoS One. 2017;12(10):e0186179. doi:10.1371/journal.pone.0186179
39. Barbi J, Pardoll D, Pan F. Treg functional stability and its responsiveness to the microenvironment. Immunol Rev. 2014;259(1):115–139.
40. Maloy KJ, Salaun L, Cahill R, et al. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med. 2003;197(1):111–119. doi:10.1084/jem.20021345
41. Kapur R, Kim M, Rebetz J, Rondina MT, Porcelijn L, Semple JW. Low levels of interleukin-10 in patients with transfusion-related acute lung injury. Ann Transl Med. 2017;5(16):339.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.