")
Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 15
HLH Syndrome in a Community Hospital: The Challenge of an Early Diagnosis
Authors Wagner Y, Adam D, Pomeranz Engelberg G, Pomeranz A, Messinger YH
Received 14 November 2023
Accepted for publication 16 February 2024
Published 7 March 2024 Volume 2024:15 Pages 111—120
DOI https://doi.org/10.2147/PHMT.S446681
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Laurens Holmes, Jr
Video abstract presented by Wagner
Views: 47
Yuval Wagner,1,2 Dganit Adam,2,3 Galit Pomeranz Engelberg,1,2 Avishalom Pomeranz,1,2,* Yoav H Messinger4,*
1Pediatric Department, Meir Medical Center, Kfar Saba, Israel; 2Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel; 3Pediatric Intensive Care Unit, Meir Medical Center, Kfar Saba, Israel; 4Department of Hematology-Oncology, Children’s Hospitals of Minnesota, Minneapolis, MN, USA
*These authors contributed equally to this work
Correspondence: Yoav H Messinger, Department of Hematology-Oncology, Children’s Hospitals of Minnesota, 2530 Chicago Av. S, Minneapolis, MN, 55404, USA, Tel +1-612-813-5940, Fax +1-612-813-6325, Email [email protected]
Introduction: Hemophagocytic lymphohistiocytosis (HLH) is a potentially fatal hyperinflammatory cytokine storm. It can be secondary to infections, malignancies, autoimmune diseases, or the manifestation of genetic disorders, including primary immune deficiency. HLH requires a high index of suspicion and is challenging for community hospitals.
Methods: Medical records of children with HLH admitted to the Meir Medical Center in Israel between 2014 and 2017 were reviewed.
Results: Nine children met ≥ 5/8 HLH‐2004 criteria. The median age was 1.1 year, and 78% of the patients were aged < 2 years. All patients had prolonged fever, cytopenia, and elevated soluble interleukin‐2 receptor, and 89% had elevated ferritin levels. Of three children who underwent gene panel evaluation, one had heterozygote genetic variants of UNC13D and STXBP2 of unclear significance, whereas the other two had no variants. Infection was identified in 8 of 9 patients: adenovirus, HHV6, EBV, and Streptococcus Group A. Only 2 patients received HLH-2004 therapy (dexamethasone, etoposide, cyclosporin-A) and the others received dexamethasone and/or intravenous gamma globulins (IVIG), with rapid resolution of fever (median 2 days). One patient (11%) died of Pseudomonas septicemia and multiorgan failure. At a median follow-up of 7 years (range 2.6– 8.1 years), all others (8/9) are long-term survivors with no recurrent HLH, but 2 patients developed adenovirus-related bronchiolitis obliterans.
Conclusion: Children presenting with prolonged fever and abnormal blood counts should be evaluated with ferritin, triglycerides, and fibrinogen levels which indicate possible HLH. Early intervention with corticosteroids and/or IVIG may prevent deterioration, spare them from chemotherapy and provide time for more elaborate testing to identify true HLH. Unfortunately, mortality remains a significant risk for these children.
Plain Language Summary: In the emergency department, children with common infections may have a severe complication called Hemophagocytic Lymphohistiocytosis or HLH. HLH can be life threatening if not rapidly recognized. HLH is rare and challenging for doctors in community hospitals. We describe nine patients who presented to a community hospital who were later diagnosed with HLH, posing a dilemma for physicians. Most (78%) were less than 2 years, all had prolonged fever, abnormal blood counts, elevated marker of HLH called soluble interleukin‐2 receptor and 8 of 9 had elevated ferritin, which can be a marker of HLH. HLH could be genetic therefore three children had genetic studies, with one having minor abnormalities, but the contribution to HLH is unclear. Infection as cause for HLH was identified in 8 of 9 patients. Chemotherapy that is used for severe HLH was required for 2 patients and the others received steroids and/or intravenous gamma globulin with rapid improvement. One patient who received chemotherapy and had suppressed immunity died of a severe bacterial infection. Others (8 of 9) are long-term survivors with no evidence of recurrent HLH. Two patients developed a pulmonary complication from adenovirus known as bronchiolitis obliterans. We conclude that children presenting with prolonged fever and abnormal blood counts should be evaluated with ferritin and other markers of possible HLH. Early intervention may prevent deterioration, may spare them from chemotherapy, and allow further assessment of true HLH. However, the death of one (11%), demonstrates the significant risks to these children.
Keywords: hemophagocytic lymphohistiocytosis, children, community hospital, infections, early diagnosis
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is an aggressive and potentially fatal syndrome that is characterized by dysregulation and excessive activation of the immune system. It is characterized by cytotoxic T cell (CTL) dysfunction, coupled with uninhibited macrophage activity.1 Malfunction of cytotoxic T cells result in the overproduction of cytokines and, eventually, tissue damage. Familial or genetic HLH is caused by an underlying genetic defect or a complication of other genetic syndromes.1,2 Secondary HLH or syndromic HLH in children is caused by infections (most common), malignancies, and autoimmune or autoinflammatory conditions in the absence of a known genetic predisposition to HLH.1,2 Recently, a case of HLH resulting from gene therapy was reported, possibly secondary to the Adeno-associated virus (AAV) vector used in this case, adding another potential risk to gene therapy.3 HLH has an estimated prevalence of 1 in 100,000 children4 and familial HLH has a reported incidence of 1.2 in 1,000,000 children.5 Symptoms and laboratory signs of this syndrome include fever, rash, cytopenia, hepatosplenomegaly, coagulopathy, elevation in ferritin and soluble interleukin-2 receptor (sIL-2R) levels, liver dysfunction, and central nervous system (CNS) involvement. The disease can rapidly progress to acute liver failure, multiorgan failure, and death.2
For clinical studies, the Histiocyte Society developed criteria, the latest of which are those used in the HLH-2004 study; these criteria remain widely used in clinical practice.2 It is notable that HLH clinical diagnosis is complicated due to an overlap with other clinical syndromes, such as COVID-19 infection and sepsis, and can lead to overdiagnosis or severe delay in diagnosis, resulting in mortality and morbidity while waiting for the criteria. Several modifications and updates have been suggested, including those from the North American Consortium for Histiocytosis (NACHO).6 Various scores have been developed to help the diagnosis, including the MS score, which was designed to identify macrophage activation syndrome (MAS) in systemic juvenile idiopathic arthritis (sJIA)7,8 and the HScore, which was created and validated to diagnose reactive HLH in adults.9
Considering the difficulty in early recognition of HLH in children, we describe a series of patients presenting to a community hospital in Israel. In this group of children who fulfilled the 5/8 HLH criteria, only 2/9 required full HLH chemotherapy protocol.
Methods
Children presenting with clinical picture of HLH were identified from all admission to the community hospital. The study was approved by the Helsinki Committee at the Meir Medical Center, Kfar Saba, Israel; therefore, it complies with the Declaration of Helsinki. This study was conducted as a partial fulfilment of the pediatric residency requirements in Israel (YW).
Data were extracted from the medical records of the hospital, outpatient clinics, and primary community clinics. Clinical data included presenting symptoms, laboratory data, length of hospital stay, resolution of fever after treatment onset, HLH treatment, and follow-up. For this analysis, HLH diagnosis was defined according to the Histiocytic Society criteria.10 Patients who fulfilled ≥5/8 of the diagnostic criteria are considered possibly positive for HLH. The criteria were as follows: 1. fever; 2. splenomegaly; 3. cytopenia of at least two cell lines: hemoglobin < 10 g/dL, platelets < 100,000/µL, or absolute neutrophil count <1000/µL; 4. elevated triglycerides level (>265 mg/dL) and/or low fibrinogen level (<150 mg/dL); 5. elevated ferritin levels >500 ng/mL; 6. elevated sIL-2R levels (>2400 U/mL or > 2 standard deviations from the mean value); 7. hemophagocytosis in the bone marrow, spleen, lymph nodes, or liver; 8. low/absent NK function. Notably, CD107a was not performed,11 and perforin expression was available only in a minority of patients. Cytokine levels and T cell analyses, including activated T cells, were not performed.
Genetic workup was performed using sequencing specific for HLH-associated genes: PRF1, UNC13D (Munc13-4), STXBP2 (Munc18-2), STX11, RAB27A and ITK. The outcomes included subsequent hospital admissions and follow-up at the last visit to the community clinic. Viral infection data were reported qualitatively, and no viral load was available.
Statistical methods: Simple descriptive statistics are provided since the group was too small for statistical significance analysis.
Results
Nine children admitted during the years 2014–2017 at Meir Medical Center were included; their specifics are listed in Table 1 and summarized in Table 2. During this period, 103,908 children aged 0–18 years were evaluated at the pediatric emergency department, noting the rarity of this phenomenon. All patients met at least 5/8 of the HLH-2004 criteria. Most were female, all were Caucasian Jewish, median age of 1.1 year and 78% were younger than 2 years, and 3 (33%) were younger than 1 year. The HLH-2004 criteria included fever (100%), bicytopenia (100%), elevated sIL-2R (100%), ferritin > 500 ng/mL (89%), elevated triglycerides or low fibrinogen levels (66%), very low or absent NK function (33%), and splenomegaly (33%). Bone marrow evaluation of 6 children showed hemophagocytosis in 5 (83% of evaluated cases). Perforin expression evaluated in three patients was normal.
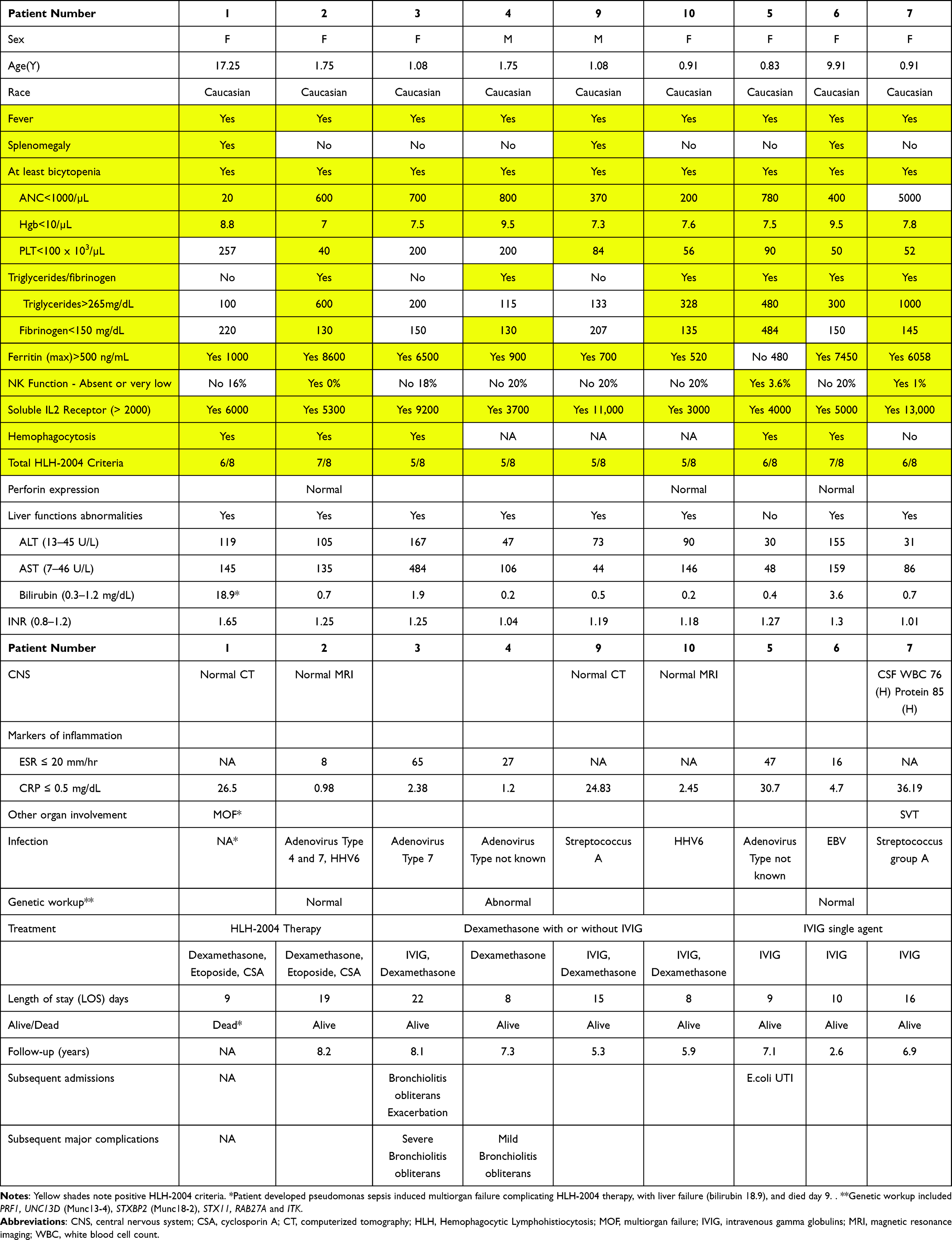 |
Table 1 Patient Data |
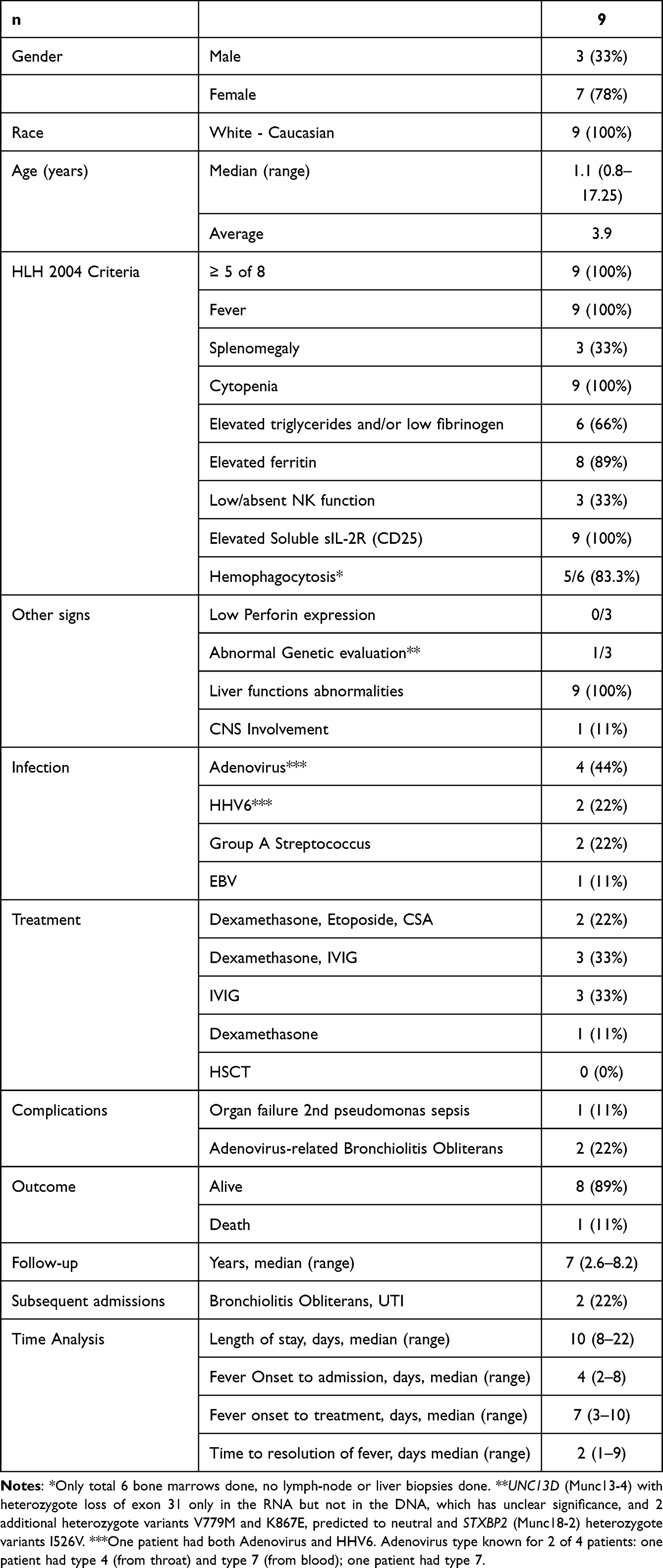 |
Table 2 Summary Data |
Mild liver dysfunction was noted in 100% of the patients. One patient developed multiorgan failure (MOF) and subsequent liver failure with bilirubin of 18.9 before death (Table 1). Most patients had minimal CNS involvement, none had seizures, cerebrospinal fluid (CSF) evaluation was performed in only one patient showing pleocytosis and elevated protein, and imaging (two CT scans and two MRI) were normal.
Infections were noted in 8 patients: adenovirus in 4 children, HHV6 in 2 (one patient had both adenovirus and HHV6), EBV in 1, and Streptococcus Group A in 2 patients.
Specific HLH-related genetic panel evaluations were performed in 3 patients for variants of PRF1, UNC13D, STXBP2, STX11, RAB27A or ITK. The decision who to test was made by the team, noting at the time that access to such testing was limited. Patient 1 died before testing was done. Patient 2 had severe HLH and was considered for HSCT thus genetic testing done and was negative. Patient 6 had genetic testing that was negative. In patient 4 gene UNC13D (which encodes Munc13-4) had heterozygote loss of exon 31 only at the RNA level but not at the DNA level, which is of unclear significance (not reported in ClinVar). The patient also had two additional heterozygous variants in UNC13D: V779M (ClinVar benign/likely benign) and K867E (ClinVar conflicting interpretations of pathogenicity). In STXBP2 (which encodes Munc18-2), the patient had the heterozygous variant I526V (ClinVar interpretation benign). No genetic testing was done in for the other 5 patients.
Treatment and Outcome
HLH-2004 therapy (dexamethasone, etoposide, and cyclosporine A [CSA]) was administered to 2 children, one of whom developed multiorgan failure due to a secondary bacterial infection (Pseudomonas) and died 9 days after admission (Table 1). The patient was 17 years old (the oldest in this group), had 6/8 HLH criteria before death, and may have deteriorated possibly due to a delay in diagnosis. The other patient responded rapidly to HLH-2004 therapy and is a long-term survivor without sequelae. Intravenous gamma globulin (IVIG) single agent was administered to three children with infections. Dexamethasone with IVIG was given to 3 resulting in a complete response and long-term survival. The child with variants of UNC13D and STXBP2 (patient 4) received a single agent, dexamethasone, which resulted in complete recovery. He did not need hematopoietic stem cell transplantation (HSCT) and had no further flares of HLH during more than seven years of follow-up. None of the patients had received intrathecal chemotherapy.
Time analysis (Table 2, Supplemental Table 1) revealed a median time from fever onset to admission of 4 days, a median time from fever onset to treatment of 7 days (89% ≥ 5 days), and a median time for resolution of fever of 2 days (range 1–9).
Access to the medical records of community clinics allowed for long-term follow-up. HSCT was not performed in any patient. Ultimately, 8/9 (89%) patients are long-term survivors at a median follow-up of seven years (range 2.6–8.2 years). Two patients had one subsequent admission each: one patient was admitted due to adenovirus-induced bronchiolitis obliterans and another for an episode of urinary tract infection. None of the patients had HLH exacerbations. To the best of our knowledge, no clinical evidence of immune deficiency is noted with long-term follow-up data. However, none had a formal immune deficiency evaluation. Two patients have ongoing bronchiolitis obliterans, which was presumed to be related to the adenovirus infection.
An additional patient (Supplemental Table 2) had Kawasaki disease, and his sIL-2R was 1990, borderline for our lab (<2000), obtained after IVIG, and thus had 3/8 HLH-2004 criteria and received only IVIG, resulting in rapid response and long-term survival. Whether this patient had MAS or HLH related to Kawasaki is not clear at this time; therefore, this patient was not included in the analysis.
Discussion
This group of patients with a clinical constellation of HLH symptoms and signs presented to a community hospital after a median of four days of fever and started treatment at a median of seven days after fever onset (89% ≥ five days). We suggest that in pediatric emergency department patients with prolonged fever, who are usually evaluated using blood counts and erythrocyte sedimentation rate (ESR), addition of ferritin, fibrinogen, and triglycerides to the evaluation can be performed in most community hospitals. This may lead to the early diagnosis of HLH, leading to further workup that can be performed at a tertiary hospital. Evaluation of the bone marrow, cytokine analysis including CXCL9, T-cell activation, CD107a degranulation study, and rapid genetic panel analysis can then be completed.2 This may allow for expedited diagnosis and early intervention, which may result in improved outcomes.
These patients demonstrate the difficulty in diagnosing and managing this disorder in a community hospital setting. Most of these patients had an infection as the most likely trigger of immune hyperactivation and an HLH picture. Infections, such as adenovirus, HHV6, EBV and Streptococcus Group A are very common in the pediatric setting. However, most patients do not require chemotherapy, corticosteroids, or IVIG and that were ultimately used here. Recognizing the deterioration from HLH remains extremely challenging for pediatricians and community hospitals.
The abundant use of the HLH-2004 criteria is problematic and may delay simple interventions such as dexamethasone or IVIG. Canna and Marsh noted the difficulty of waiting for the full HLH-2004 criteria to develop, as some patients may never develop the full five of eight criteria, especially patients with unusual presentations, such as CNS HLH or liver failure.2 On the other hand, over diagnosis of HLH using HLH-2004 criteria may also be a vexing problem especially in a community hospital with limited access to CD107a degranulation test, cytokine analysis including CXCL9 levels and rapid T-cell flow cytometry.2 One can suggest that patients 5, 6, and 7 who required only IVIG to reverse this process may not have bona fide HLH but did have full 5/8 criteria, demonstrating the limitation of the use of the HLH-2004 criteria. In this regard, the distinction between HLH and sepsis is especially problematic because they overlap clinically. Recently, a group from Cincinnati Children’s Hospital reported that activated T-cells, CD38high/HLA-DR+ (especially CD8 + cells), distinguished HLH from early sepsis.12 Unfortunately, in our community hospital (and many others) cell marker assays of this complexity are not available.
In our group of nine children, one patient died of multiorgan failure after starting HLH therapy, presumably secondary to Pseudomonas sepsis. To the best of our knowledge, death due to HLH remains an issue.
Of the three patients whose genetic evaluation was available to us, only one patient had heterozygous genetic variants of unclear significance for UNC13D which encodes Munc13-4. Heterozygote variants of UNC13D have been reported in 50/322 (15.7%) cases of familial hemophagocytic lymphohistiocytosis type 3 (FHL3).13 This patient also had a concomitant STXBP2 heterozygote variant. ClinVar citations of variants of both genes indicate that they are benign or have conflicting interpretations. Additionally, the patient had UNC13D deletion in exon 31 at the RNA level, but we could not find reports of this deletion in the literature. Whether these variants or their combinations contributed to the clinical course of the patient remains unclear. The patient had no reactivation of HLH and was not transplanted during more than seven years of follow-up.
Young patients presenting with HLH tend to have a greater genetic predisposition to HLH than older patients, require chemotherapy, and at times undergo HSCT. In contrast, most of our patients were younger than 2 years of age, required minimal therapy to recover, and are long-term survivors with no HLH recurrence. We speculate that the rapid response to minimal therapy and good outcome were most likely due to the infectious-induced HLH picture and no genetic HLH predisposition.
Adenovirus was diagnosed in four young patients (10–21 months). HLH is a rare complication of adenovirus infection, particularly adenovirus type 7 infection.14 Two of the patients (one with adenovirus type 7 and the other with an unknown type) developed subsequent bronchiolitis obliterans, which had been described after HLH and adenovirus pneumonia.15 A recent Chinese publication notes that of 15/27 children who survived HLH and adenovirus pneumonia developed bronchiolitis obliterans, suggesting that these patients should be closely monitored for this complication.16 HHV-6-induced HLH (one with co-infection with adenoviruses) were noted in two patients aged 11 and 21 months. HLH and HHV6 have been described in the context of primary immune deficiency, with pathogenic variants in IL-2R,17 XIAP deficiency18 and isolated CNS HLH.19 Both our HHV6 patients are long-term survivors with no suggested immune deficiency or CNS involvement. Two patients had Streptococcus group-A-induced HLH picture that resolved completely. We could only find a case report of fulminant Streptococcus Group B infection leading to HLH (who died within 10 hours).20 Remarkably in our group only one patient had EBV induced HLH. EBV is the most common infection associated with HLH, developing in one-third of infectious HLH cases in North America and up to three-quarters of patients in Asia, and can have poor outcomes.21 Our EBV patient recovered completely with only IVIG; thus, may not have developed the full spectrum of EBV-HLH or the deterioration was abbreviated early with IVIG.
Patients with virus-induced HLH may have abnormalities in the genes associated with the innate immune response to viral infections, especially in the IFN-1 pathway, such as STAT2, STAT1, IFNAR1, and IFNAR1.22 STAT2 deficiency was elegantly reported by López-Nevado et al,22 who suggested that these genes should be evaluated in patients with virus-induced HLH. Unfortunately, these abnormalities were not evaluated in the present study.
One additional patient with Kawasaki disease (KD) met 3/5 HLH-2004 criteria; therefore, he was not included in this series, and the patient responded to single-agent IVIG (Supplemental Table 2). Mast cell activation syndrome (MAS) clinically identical to HLH, was reported in up to 1% of KD cases, although many do not meet 5/8 criteria, and one of them died.23 Notably, our patient had cytopenia with hepatomegaly, that is uncommon in KD, and is mentioned here to increase awareness of the possible unusual course of KD.
We acknowledge the limitations of this report, especially the fact that we had no access to activated T cell numbers, CD107a degranulation tests, and cytokine data. Critically, perforin expression and CD107a degranulation tests were superior to NK cell function in screening for genetic HLH.11 Additionally, the use of NK cell function to identify HLH in this group of patients has been fraught with poor reproducibility; thus, it should not be used to screen for HLH. Additionally, three of our patients did not undergo bone marrow aspiration, which was required to rule out malignancy,2 although they are long-term survivors, suggesting that in retrospect, this test was not necessary. Genetic evaluation performed in a minority of these patients is also a notable limitation. As noted above, this report cannot exclude overdiagnosis of HLH in these patients, a problem that has been extensively discussed by the NACHO group.6 Such patients who met HLH-2004 may have been overtreated by us, like many other patients at other institutions.
Conclusions
We describe a group of patients presenting to a community hospital with infection induced HLH, most of whom required minimal therapy and are long-term survivors with no recurrence, but with 11% (1 of 9) deaths from sepsis. Considering that this is a rare phenomenon, we suggest that in children presenting to the emergency department of a community hospital with prolonged fever and abnormal blood counts, the addition of ferritin, triglycerides, and fibrinogen could expedite further workup in a tertiary center. Critically, extremely ill patients should receive dexamethasone with or without IVIG, which can provide a window of opportunity to allow rapid evaluation of sIL-2R, perforin expression, CD107a degranulation, and activated T cells, which can further support the diagnosis of HLH. Similarly, dexamethasone and/or IVIG may prevent the deterioration of HLH in some patients, sparing them from the use of chemotherapy or other treatment modalities.
Abbreviations
CNS, Central nervous system; CSF, Cerebrospinal fluid; CT, Computerized Tomography; CSA; Cyclosporine A; CTL, Cytotoxic T-cell; ESR, Erythrocyte sedimentation rate; FHL3, Familial hemophagocytic lymphohistiocytosis type 3; HLH, Hemophagocytic lymphohistiocytosis; HSCT, Hematopoietic stem-cell transplantation; IVIG, Intravenous gamma globulins; KD, Kawasaki disease; MAS, Macrophage activation syndrome; MRI, Magnetic Resonance Imaging; NACHO, North American Consortium for Histiocytosis; sIL-2R, Soluble interleukin-2 receptor; SJIA, Systemic Juvenile Idiopathic Arthritis.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article (Table 1).
Ethics Approval
The study was approved by the Helsinki Committee at the Meir Medical Center, Kfar Saba, Israel (number 0002-17-MMC). We confirm that it complies with the Declaration of Helsinki.
Consent for Publication
Owing to the retrospective nature of this study, anonymized data are presented in Table 1. The Helsinki Committee of the Meir Medical Center waived the requirement for consent for this study.
Acknowledgments
The authors acknowledge the dedicated work of the staff at the pediatric intensive care unit of the Meir Medical Center, Kfar-Saba, for their dedicated care of these patients and their families.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work did not receive any funding from any funding agency.
Disclosure
The authors declare no competing interests in this work.
References
1. Hosahalli Vasanna S, Dalal J. Traffic jam within lymphocytes: a clinician’s perspective. Front Immunol. 2022;13:1034317. doi:10.3389/fimmu.2022.1034317
2. Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. 2020;135(16):1332–1343. doi:10.1182/blood.2019000936
3. Galletta F, Cucinotta U, Marseglia L, et al. Hemophagocytic lymphohistiocytosis following gene replacement therapy in a child with type 1 spinal muscular atrophy. J Clin Pharm Ther. 2022;47(9):1478–1481. doi:10.1111/jcpt.13733
4. Niece JA, Rogers ZR, Ahmad N, Langevin AM, McClain KL. Hemophagocytic lymphohistiocytosis in Texas: observations on ethnicity and race. Pediatr Blood Cancer. 2010;54(3):424–428. doi:10.1002/pbc.22359
5. Henter JI, Elinder G, Söder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Padiatric Scand. 1991;80(4):428–435. doi:10.1111/j.1651-2227.1991.tb11878.x
6. Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66(11):e27929. doi:10.1002/pbc.27929
7. Minoia F, Bovis F, Davì S, et al. Development and initial validation of the MS score for diagnosis of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2019;78(10):1357–1362. doi:10.1136/annrheumdis-2019-215211
8. Ravelli A, Minoia F, Davì S, et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2016;68(3):566–576. doi:10.1002/art.39332
9. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613–2620. doi:10.1002/art.38690
10. Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. doi:10.1002/pbc.21039
11. Rubin TS, Zhang K, Gifford C, et al. Perforin and CD107a testing is superior to NK cell function testing for screening patients for genetic HLH. Blood. 2017;129(22):2993–2999. doi:10.1182/blood-2016-12-753830
12. Chaturvedi V, Marsh RA, Zoref-Lorenz A, et al. T-cell activation profiles distinguish hemophagocytic lymphohistiocytosis and early sepsis. Blood. 2021;137(17):2337–2346. doi:10.1182/blood.2020009499
13. Amirifar P, Ranjouri MR, Abolhassani H, et al. Clinical, immunological and genetic findings in patients with UNC13D deficiency (FHL3): a systematic review. Pediatr Allergy Immunol. 2021;32(1):186–197. doi:10.1111/pai.13323
14. Otto WR, Behrens EM, Teachey DT, et al. Human Adenovirus 7-Associated Hemophagocytic Lymphohistiocytosis-like Illness: clinical and Virological Characteristics in a Cluster of Five Pediatric Cases. Clin Infect Dis. 2021;73(7):e1532–e1538. doi:10.1093/cid/ciaa1277
15. La Fay C, Bosdure E, Baravalle-Einaudi M, Stremler-Le Bel N, Dubus JC, Mazenq J. Severe adenovirus pneumonia with hemophagocytic syndrome and respiratory failure. Arch Pediatr. 2020;27(7):383–385. doi:10.1016/j.arcped.2020.07.003
16. Zhang HY, Li CJ, Long Y, Sun DM, Wang RG, Zhang Y. Clinical features of children with severe adenovirus pneumonia and hemophagocytic syndrome: an analysis of 30 cases. Zhongguo Dang Dai Er Ke Za Zhi. 2020;22(7):744–748. doi:10.7499/j.issn.1008-8830.2003080
17. Singh P, Secord E, Pappas K, Savaşan S. An infant with severe combined immunodeficiency, osteopetrosis, chromosomally integrated herpesvirus-6 infection, and hemophagocytic syndrome: what are the links? Pediatr Blood Cancer. 2021;68(1):e28564. doi:10.1002/pbc.28564
18. Inoue K, Miura H, Hoshino A, et al. Inherited chromosomally integrated human herpesvirus-6 in a patient with XIAP deficiency. Transpl Infect Dis. 2020;22(5):e13331. doi:10.1111/tid.13331
19. Bucciol G, Willemyns N, Verhaaren B, et al. Child Neurology: familial Hemophagocytic Lymphohistiocytosis Underlying Isolated Central Nervous System Inflammation. Neurology. 2022;99(15):660–664. doi:10.1212/WNL.0000000000201124
20. Choi YB, Yi DY. Fatal case of hemophagocytic lymphohistiocytosis associated with group B streptococcus sepsis: a case report. Medicine. 2018;97(40):e12210. doi:10.1097/MD.0000000000012210
21. Marsh RA. Epstein-Barr Virus and Hemophagocytic Lymphohistiocytosis. Front Immunol. 2017;8:1902. doi:10.3389/fimmu.2017.01902
22. López-Nevado M, Sevilla J, Almendro-Vázquez P, et al. Inborn Error of STAT2-Dependent IFN-I Immunity in a Patient Presented with Hemophagocytic Lymphohistiocytosis and Multisystem Inflammatory Syndrome in Children. J Clin Immunol. 2023;43(6):1278–1288. doi:10.1007/s10875-023-01488-6
23. Wang W, Gong F, Zhu W, Fu S, Zhang Q. Macrophage activation syndrome in Kawasaki disease: more common than we thought? Semin Arthritis Rheum. 2015;44(4):405–410. doi:10.1016/j.semarthrit.2014.07.007
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.