")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Gallic acid attenuates dextran sulfate sodium-induced experimental colitis in BALB/c mice
Authors Pandurangan AK , Mohebali N, Norhaizan M, Looi CY
Received 9 April 2015
Accepted for publication 11 June 2015
Published 30 July 2015 Volume 2015:9 Pages 3923—3934
DOI https://doi.org/10.2147/DDDT.S86345
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Ashok Kumar Pandurangan,1,2 Nooshin Mohebali,2 Mohd Esa Norhaizan,1,3 Chung Yeng Looi2
1Department of Nutrition and Dietetics, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, Serdang, Selangor, Malaysia; 2Department of Pharmacology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia; 3Laboratory of Molecular Medicine, Institute of Bioscience, Universiti Putra Malaysia, Serdang, Selangor, Malaysia
Abstract: Gallic acid (GA) is a polyhydroxy phenolic compound that has been detected in various natural products, such as green tea, strawberries, grapes, bananas, and many other fruits. In inflammatory bowel disease, inflammation is promoted by oxidative stress. GA is a strong antioxidant; thus, we evaluated the cytoprotective and anti-inflammatory role of GA in a dextran sulfate sodium (DSS)-induced mouse colitis model. Experimental acute colitis was induced in male BALB/c mice by administering 2.5% DSS in the drinking water for 7 days. The disease activity index; colon weight/length ratio; histopathological analysis; mRNA expressions of IL-21 and IL-23; and protein expression of nuclear erythroid 2-related factor 2 (Nrf2) were compared between the control and experimental mice. The colonic content of malondialdehyde and the activities of superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase activity were examined as parameters of the redox state. We determined that GA significantly attenuated the disease activity index and colon shortening, and reduced the histopathological evidence of injury. GA also significantly (P<0.05) reduced the expressions of IL-21 and IL-23. Furthermore, GA activates/upregulates the expression of Nrf2 and its downstream targets, including UDP-GT and NQO1, in DSS-induced mice. The findings of this study demonstrate the protective effect of GA on experimental colitis, which is probably due to an antioxidant nature of GA.
Keywords: IL-21, NQO1, MDA, enzymic antioxidants, Nrf2
Introduction
Ulcerative colitis (UC), one of the two primary subtypes of inflammatory bowel disease (IBD), is a chronic relapsing and remitting inflammatory disorder of the gastrointestinal tract.1 Although the precise molecular basis and pathophysiology of UC remain unclear, it is generally accepted that interactions between genetic susceptibility and environmental triggers cause hyperactivation of the mucosal immune system, resulting in chronic intestinal inflammation.2 Currently, a specific causal treatment of IBD is not available, and the best treatment regimen involves regulation of the oxidant/antioxidant balance.
Dextran sulfate sodium (DSS)-induced colitis is a well-established experimental model in rodents that resembles many of the signs and symptoms of human UC, including bloody feces, diarrhea, weight loss, mucosal ulceration, and shortening of the colon. Although the mechanisms through which DSS induces colitis are unknown, one study revealed that it increases intestinal permeability and causes injury to the epithelium, suggesting that DSS causes dysfunction of the mucosal defense systems.3 In this model system, leukocytes, including neutrophils, lymphocytes, and macrophages, have been reported to infiltrate inflamed tissues.4 The production and release of reactive oxygen species by immune cells seem to play a crucial role in the physiopathology of UC.5 Increased accumulation of tissue injury produced by neutrophils and macrophages has been attributed to their ability to release reactive oxygen mediators, nitrogen metabolites, and proinflammatory cytokines, and to disrupt epithelial integrity.6–8
Numerous studies have indicated that oxidative stress is a key causative of UC. Reactive oxygen metabolites are generated as byproducts of cellular metabolism, primarily in the mitochondria. When the cellular production of these metabolites overwhelms the antioxidant capacity of cells, cellular macromolecules are damaged. This state of “oxidative stress” is thought to contribute to the pathogenesis of a number of human diseases, including UC.9,10 The end product of lipid peroxidation, malondialdehyde (MDA), is harmful and may be responsible for some of the overall effects of oxidative stress, including the release of cell contents and cell death, which cause tissue and organ damage.10
Nuclear erythroid 2-related factor 2 (Nrf2) is a basic leucine zipper redox-sensitive transcriptional factor that plays a central role in transcriptional regulation of antioxidant and/or detoxifying genes. Nrf2 is sequestered in the cytoplasm by Keap1 under normal conditions, which results in its proteosomal degradation.11,12 Under certain conditions, such as oxidative stress, Nrf2 is released from Keap1 and translocates into the nucleus where it acts in combination with other transcription factors to activate the transcription of genes containing the antioxidant response element in their promoter regions, and this results in a cytoprotective adaptive response.13,14 This adaptive response is characterized by upregulation of a battery of antioxidative enzymes and decreased sensitivity to oxidative damage and cytotoxicity.15,16
Gallic acid (GA) is a polyhydroxy phenolic compound that has been found in various natural products, such as green tea, strawberries, grapes, bananas, and many other fruits.17–19 GA exhibits a variety of biological activities, including antioxidant,20 antibacterial,21 antitumor,22 and anti-inflammatory23 activities. Ho et al24 reported that GA inhibits gastric cancer cell metastasis and invasive growth via upregulation of RhoB, downregulation of AKT/small GTPase signals, and inhibition of NF-κB activity. GA induces apoptosis via caspase-3 and mitochondrion-dependent pathways in vitro, and suppresses lung xenograft tumor growth in vivo.25 In this study, we investigated the protective effects of GA against DSS-induced colitis in a BALB/c mouse model. In addition, we examined GA’s ability to enhance enzymic antioxidants (superoxide dismutase [SOD], catalase [CAT], glutathione peroxidase [GPx], and glutathione reductase [GR]) and activate the redox sensitive transcription factor Nrf2 and its downstream targets, including NQO1 and UDP-GT.
Materials and methods
Animals
In this study, male BALB/c mice weighing 25–30 g were purchased from Sapphire Enterprises (Seri Kembangan, Malaysia). They were housed individually in plastic cages at a constant temperature (21°C±2°C) with an alternating 12-hour light/dark cycle, and animal chow and water were provided ad libitum. All animal treatments adhered strictly to institutional and international ethical guidelines of the care and use of laboratory animals. The experimental protocol was approved by the Institutional Animal Care and Use Committee, Universiti Putra Malaysia, Malaysia, for the use of animal subjects.
Experimental design
Animals were randomly divided into three experimental groups. Group 1 served as the control and received normal drinking water. In group 2 (DSS), acute colitis was induced by administering 2.5% (w/v) DSS (molecular weight: 35,000–50,000; MP Biomedicals, LLC, Santa Ana, CA, USA) through the drinking water for 7 days.26 In group 3, acute colitis was induced in the same manner as in group 2, and the animals also received GA orally at 10 mg/kg body weight for 7 days. During the experimental period, body weight, stool consistency, and gross bleeding scores were recoded to calculate disease activity index (DAI).27 The method for scoring of DAI is illustrated in Table 1.
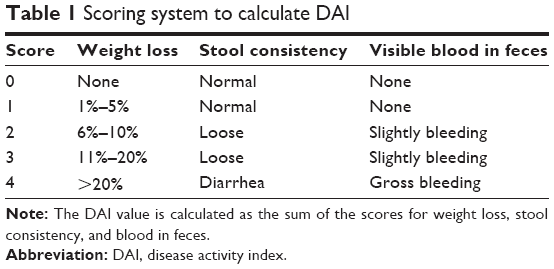 | Table 1 Scoring system to calculate DAI |
After the experimental period, each animal was sacrificed, its colon was excised, and the length and weight of the colon was measured. Then, the colon was washed with phosphate-buffered saline (PBS), a portion of it was fixed in 10% buffered formalin, and the other portion was stored in dry ice for further analyses.
Histopathology of the colon
Formalin-fixed colons were paraffin embedded and then cut into 5 μm sections on a glass slide. Sections were stained with hematoxylin and eosin for the histopathology analysis. The sections were photographed using a Nikon 80i ECLIPSE microscope (Nikon Corporation, Tokyo, Japan) at 20× magnification.
Measurement of MDA
The level of MDA in the colon was determined as an indicator of lipid peroxidation and was estimated by the method of Ohkawa et al.28 The colon tissue was homogenized in 1.15% KCl solution. The sample consisted of 0.2 mL 8.1% sodium dodecyl sulfate, 1.5 mL 20% acetic acid, 1.5 mL 0.8% thiobarbituric acid, and 0.7 mL distilled water. The samples were boiled for 1 hour at 95°C and centrifuged at 3,000× g for 10 minutes. The absorbance of the supernatant was measured spectrophotometrically at 650 nm.
Assay of enzymic antioxidants
The assay of SOD was performed following the method of Kakkar et al29 based on 50% inhibition of the formation of reduced nicotinamide adenine dinucleotide (NAD)–phenazine methosulphate–nitroblue tetrazolium (NBT) formazan at 520 nm. The amount of enzyme required for 50% inhibition of NBT reduction/minute/mg protein was considered one unit of enzyme activity. CAT activity was determined following the method of Sinha.30 The values of CAT activity are expressed as micromoles of H2O2 utilized/minute/mg protein. GR activity was assayed following the method of Carlberg and Mannervik,31 and the values are expressed as μmol of reduced NAD phosphate (NADPH) oxidized/minute/mg protein. GPx activity was assayed by the method of Rotruck et al32 and the values are expressed as μmol of reduced glutathione (GSH) utilized/minute/mg protein.
Immunohistochemical analysis
The immunohistochemical method of Pandurangan et al33 was adopted with some modifications. Paraffin-embedded tissue sections of 5 μm thickness were rehydrated in xylene and then in graded ethanol solutions. Then, the slides were incubated with HistoVT (10×, pH 7.0) (Nacalai Tesque, Tokyo, Japan) antigen retrieval solution for 20 minutes at 90°C and then cooled to room temperature. The slides were then blocked with 5% bovine serum albumin (BSA) in TBS–Tween 20 (TBST) for 2 hours. The sections were then incubated with mouse monoclonal Nrf2 antibody (Santa Cruz Biotechnology Inc., Dallas, TX, USA) overnight at 4°C for immunostaining. After washing the slides thrice with TBST, the sections were incubated with the appropriate secondary antibodies in TBST with 5% BSA for 2 hours at room temperature. The sections were then washed with TBST and incubated for 5–10 minutes with a peroxidase stain from a peroxidase stain DAB kit following the instructions provided by the manufacturer (Nacalai Tesque). Counterstaining was performed using hematoxylin (Cell Path, Mochdre, Newtown, UK), and the slides were photographed under a light microscope (Nikon ECLIPSE 80i; Nikon Corporation).
Isolation of nuclear and cytosolic fractions
Isolation of nuclear and cytosolic fractions was performed following the method of Pandurangan et al.34 A small piece of colonic tissue in PBS was washed and resuspended in 1 mL of solution A (250 mM sucrose; 1.5 mM MgCl2; 20 mM HEPES; 10 mM KCl; 1 mM ethylenediaminetetraacetic acid (EDTA), pH 8.0; 1 mM ethylene glycol tetraacetic acid; and 1× protease inhibitor cocktail; ~300 mOsm) with gently tapping and pipetting. Then, the samples were homogenized using an electric homogenizer, and the lysates were centrifuged at 720× g for 8 minutes at 4°C. The supernatant (cytosolic fraction) was transferred into a new 1.5 mL tube, and the remaining cell pellet was washed twice with 0.5 mL solution A and resuspended in 50 μL of solution B (20 mM HEPES, pH 7.9; 420 mM NaCl; 1.5 mM MgCl2; 0.2 mM EDTA, pH 8.0; 0.5 mM phenylmethanesulfonyl fluoride or phenylmethylsulfonyl fluoride (PMSF); 0.5 mM dithiothreitol; 25% glycerol; and 1× protease inhibitor cocktail; ~3,860 mOsm). The samples were incubated on ice for 1 hour and then centrifuged at 9,300× g for 10 minutes at 4°C. The resulting supernatant was the nuclear fraction and was isolated and kept at −80°C until use.
Immunoblot analysis
Immunoblot analysis was performed according to the method of Ashokkumar and Sudhandiran,35 with some modifications. Briefly, colonic tissues were removed and washed in ice cold PBS. The whole colonic tissue was cut into pieces and homogenized in five volumes of ice-cold homogenizing buffer (0.1 mM NaCl, 0.1 M Tris Cl, 0.001 M EDTA, 1 mM PMSF, 1 mg/mL aprotonin, and 0.1 mM leupeptin) and centrifuged at 3,000× g for 1 hour at 4°C. The protein contents of the supernatants were estimated using BSA as the standard. The extracts were heated in a boiling water bath for 5 minutes, and 40 μg protein samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes using a transfer apparatus (Bio-Rad Laboratories Inc., Hercules, CA, USA). The membranes were blocked overnight at 4°C with blocking reagent (20 mM Tris (pH 7.4), 125 mM NaCl, 0.2% [v/v] Tween 20, 4% [w/v] nonfat dry milk, and 0.1% [w/v] sodium azide). Then, the membranes were incubated with the Nrf2 and β-actin (Santa Cruz Biotechnology Inc.) primary antibodies diluted according to the recommendations of the supplier overnight. They were then incubated with the corresponding horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology Inc.) for 1 hour. The protein–antibody complexes were detected using the Clarity Western ECL substrate (Bio-Rad Laboratories Inc.). The quantification of the respective blots was performed using ImageJ software (NIH, Bethesda, MD, USA).
RNA isolation and quantitative polymerase chain reaction
Isolation of RNA from colonic tissue and real-time polymerase chain reaction (RT-PCR) were performed following the method described by Pandurangan et al.34 Total RNA from colonic tissue was extracted using a QIA shredder and RNeasy Kit (Qiagen NV, Venlo, the Netherlands) following the manufacturer’s instructions. Quantitative RT-PCR was performed using an Eppendorf PCR system with QuantiFast SYBR Green PCR Master Mix (Qiagen NV), primers (1 mM; Table 2), and 1 μg cDNA in a 25 mL reaction mixture. All target cDNAs and the standard β-actin cDNA were analyzed in duplicate in three independent RT-PCR assays. Thermal cycling was initiated with an activation step at 95°C for 30 seconds, followed by 40 cycles of 95°C for 5 seconds and 60°C for 30 seconds. Immediately after amplification, melt curves were generated to ensure the primer dimers and other nonspecific products had been minimized. The relative expressions of the target genes were analyzed by the ΔΔCt method.
 | Table 2 List of primers used |
Statistical analysis
The data are expressed as means and standard deviation. The data were processed using the statistical analysis software SPSS version 16.0 (SPSS Inc., Chicago, IL, USA). All P-values were two-tailed, and a P-value <0.05 was considered to indicate a statistically significant difference.
Results
Effect of GA on clinical signs
In the present study, the mice in the DSS-induced group showed body weight loss, bloody diarrhea, and severe colitis without mortality. As shown in Figure 1A, DSS-induced mice had an increased DAI score (body weight loss, stool consistency, and blood in the stool) compared to the control mice. DSS-induced mice that received oral GA treatment had obviously less weight loss and lower DAI scores than the untreated DSS-induced mice. In addition, the GA-treated DSS-induced mice showed a significant reduction in colonic weight/length ratio (Figure 1B), an index of colonic edema that is increased significantly as a consequence of the inflammatory process, compared with the untreated DSS-induced mice. Figure 1C shows the percentage of hematocrit in control and experimental animals. We observed that there was a decreased hematocrit in DSS-induced mice when compared to control mice. Administration of GA showed significantly (P<0.05) increased hematocrit in DSS-induced mice.
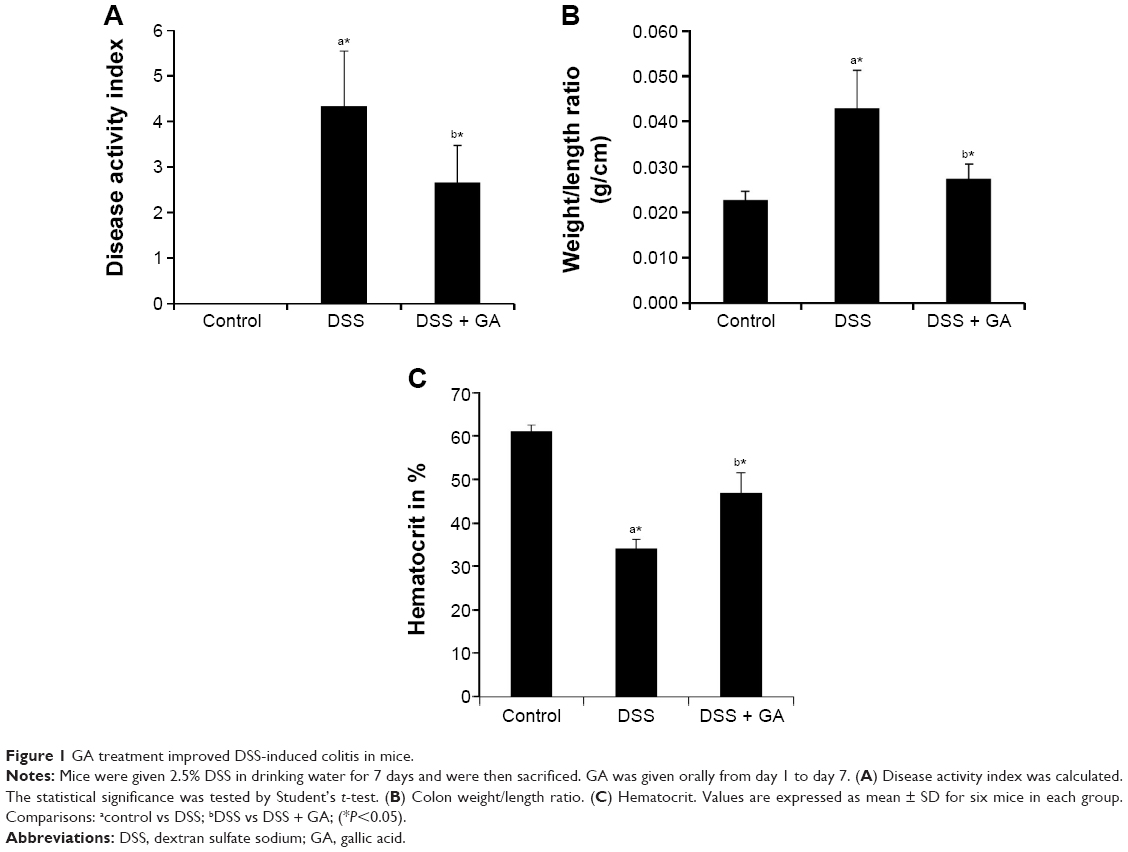 | Figure 1 GA treatment improved DSS-induced colitis in mice. |
GA improves histological signs of DSS-induced damage
Figure 2 shows the histology of the colonic tissues of the control and experimental mice. The control mice had normal colonic architecture with no damage. However, the DSS-induced mice showed mucosal ulceration, gland distortion, and leukocyte infiltration. These observations were more severe in the untreated DSS-induced mice than in the GA-treated DSS-induced mice.
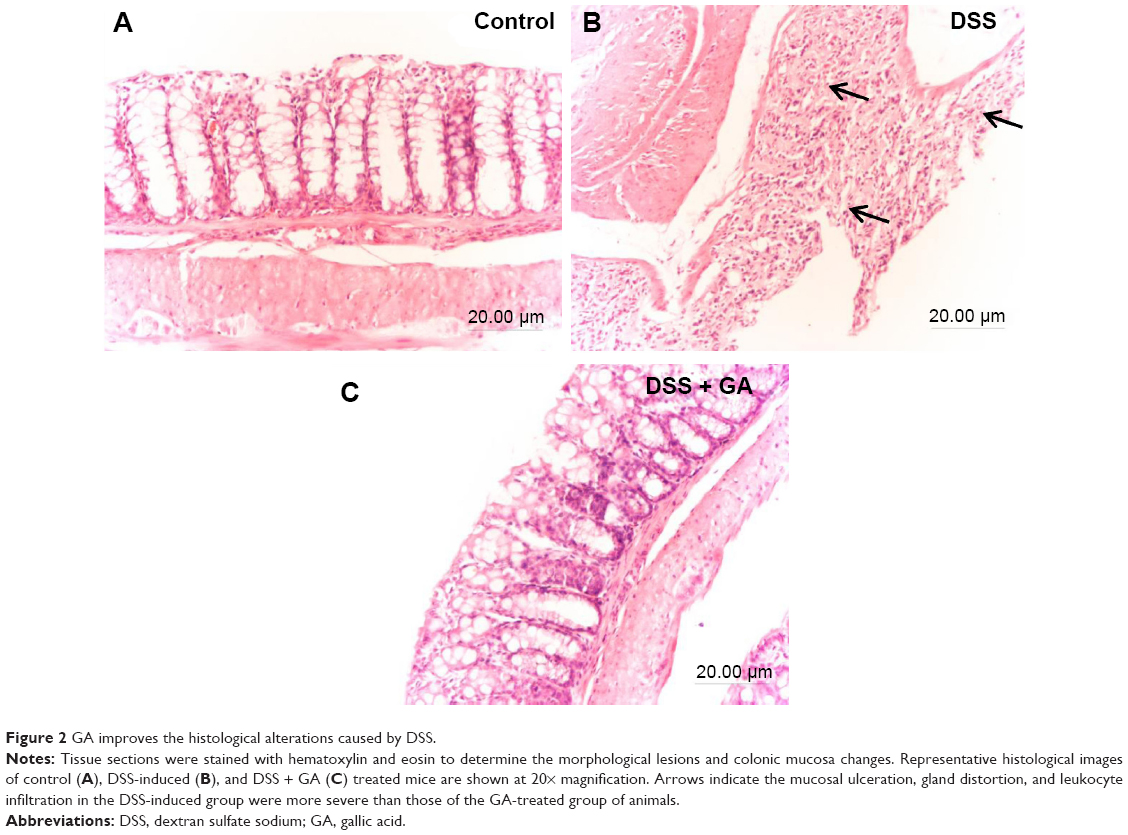 | Figure 2 GA improves the histological alterations caused by DSS. |
GA suppresses the expressions of IL-21 and IL-23
Figure 3 shows the mRNA levels of IL-21 and IL-23. The DSS-induced mice showed increased expressions of IL-21 and IL-23, and the administration of GA significantly (P<0.05) suppressed the effects of DSS on the expressions of IL-21 and IL-23.
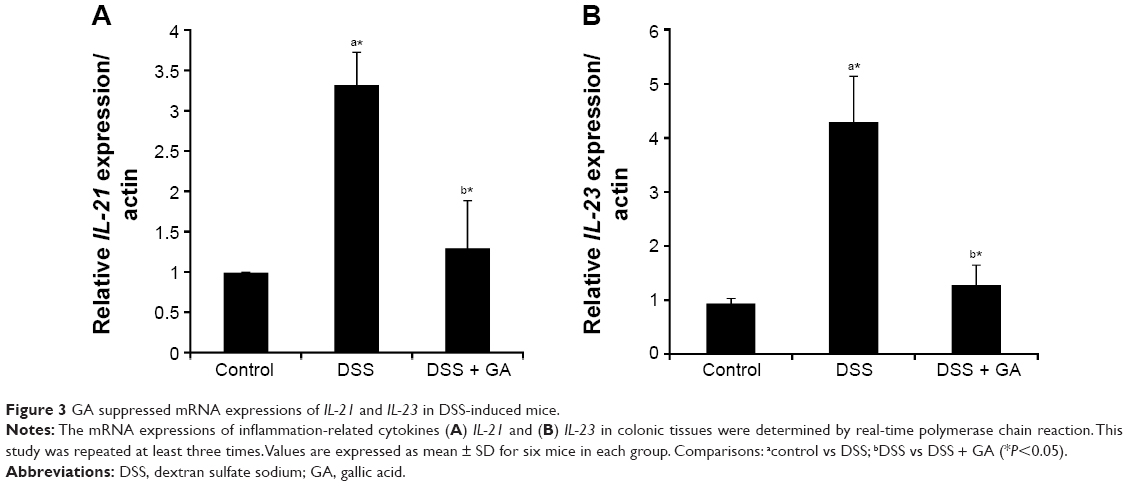 | Figure 3 GA suppressed mRNA expressions of IL-21 and IL-23 in DSS-induced mice. |
GA attenuates MDA levels in DSS-induced UC
Figure 4 shows a significant (P<0.05) increase in the level of MDA in DSS-induced mice compared with control mice. Additionally, treatment of DSS-induced mice with GA resulted in a significant reduction in the level of MDA.
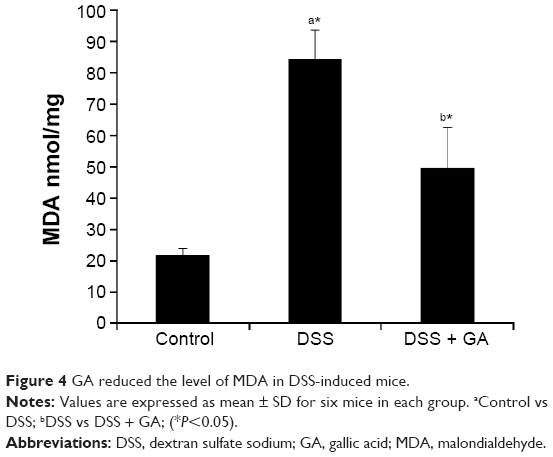 | Figure 4 GA reduced the level of MDA in DSS-induced mice. |
GA increases the activities of enzymic antioxidants
Figure 5 presents the levels of enzymic antioxidants (SOD, CAT, GPx, and GR) in the colonic mucosa of mice in the control and experimental groups. The activities of enzymic antioxidants were significantly (P<0.05) reduced in DSS-induced mice compared to the control mice (group 1). DSS-induced mice administered GA (group 3) showed significantly increased enzymic antioxidant activities compared with the untreated DSS-induced mice (group 2). We also confirmed the expressions of enzymic antioxidants by quantitative RT-PCR as shown in the Supplementary materials.
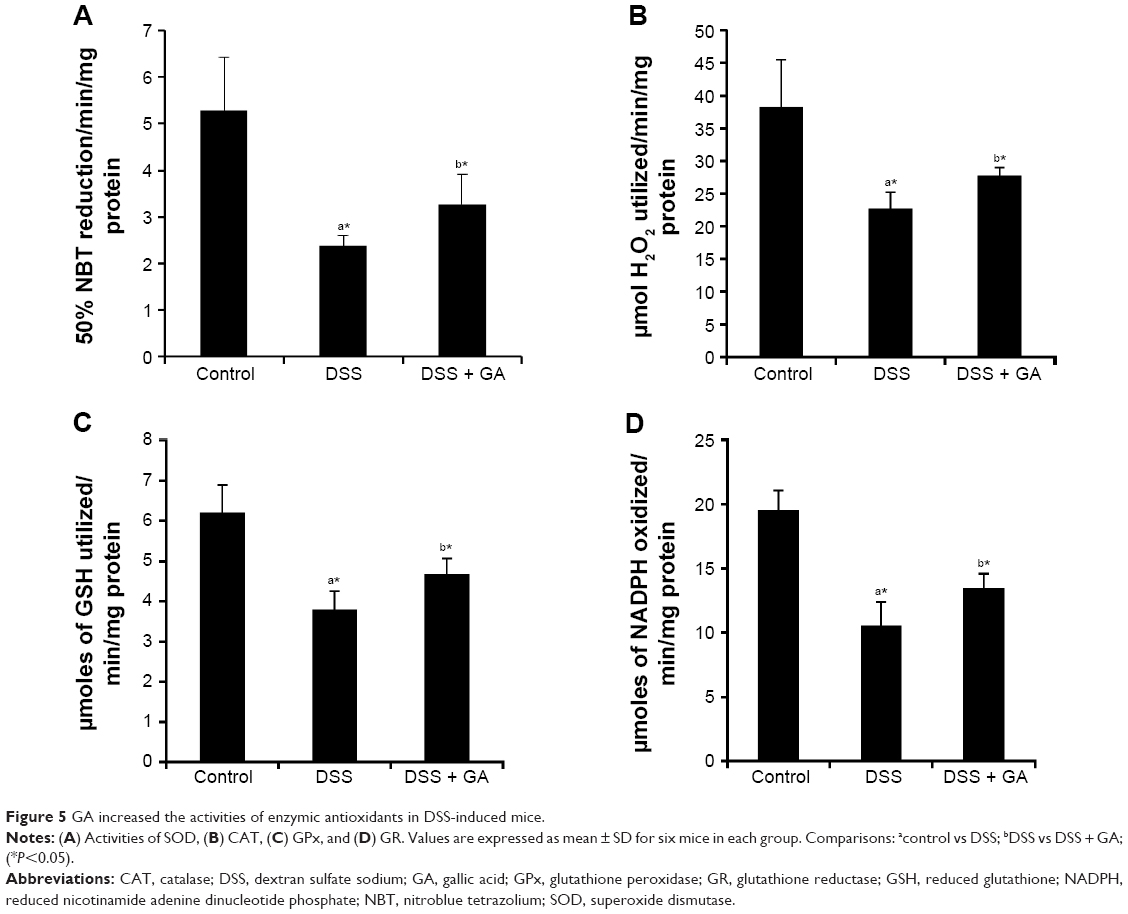 | Figure 5 GA increased the activities of enzymic antioxidants in DSS-induced mice. |
GA induces Nrf2 protein expression and nuclear translocation
We examined the protein expression and subcellular location of Nrf2 in colonic tissue during the exposure of mice to DSS. The Western blot analysis results in Figure 6A show a significant increase of Nrf2 protein expression in the GA-treated group of mice. Western blot analysis of the nuclear protein fraction (Figure 6A) and immunohistochemical analysis (Figure 6B) show Nrf2 accumulation in the nucleus of colon in the GA-treated mice. Additionally, Figure 6C and D show the mRNA expressions of NQO1 and UDP-GT, two downstream targets of Nrf2. The expressions of NQO1 or UDP-GT were not altered in the DSS-induced mice compared to the control mice. Administration of GA was shown to increase the expressions of NQO1 and UDP-GT. This clearly indicates that GA has a cytoprotective role by activating Nrf2 and its downstream targets.
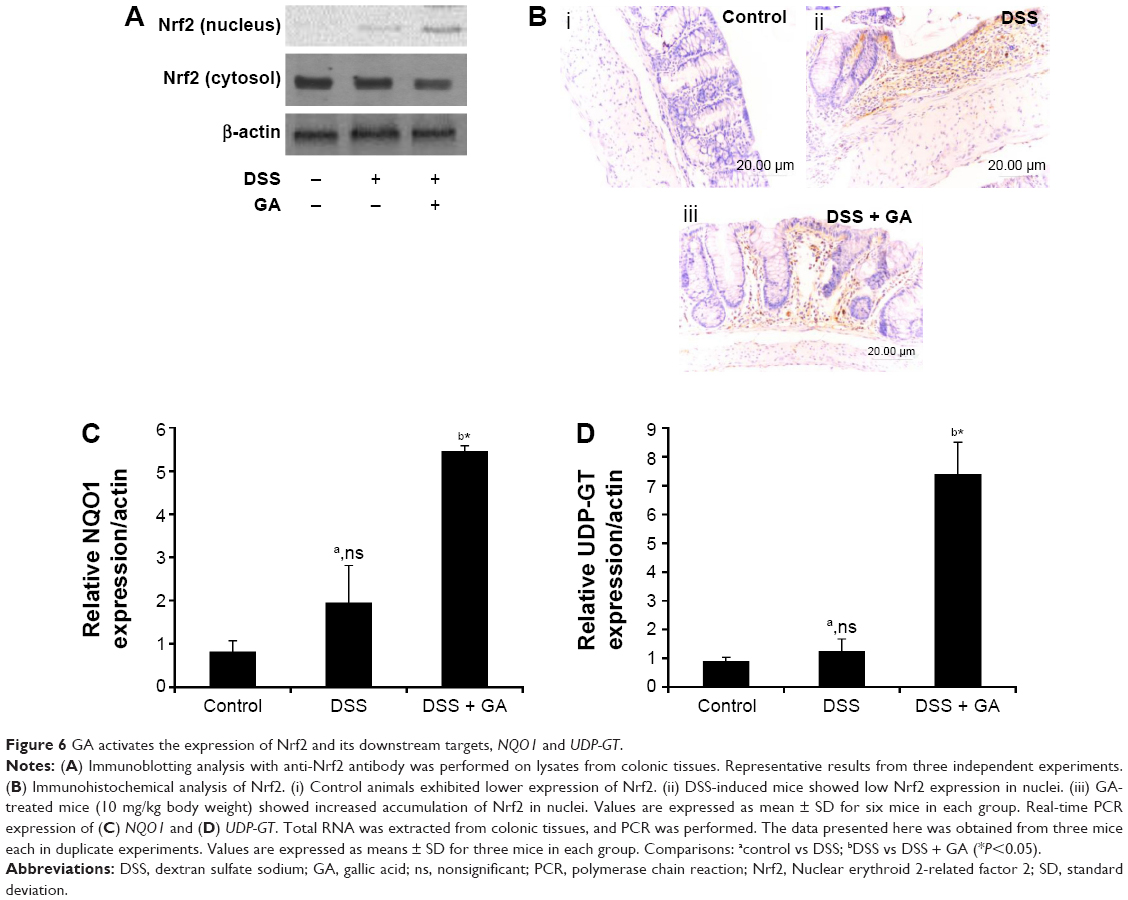 | Figure 6 GA activates the expression of Nrf2 and its downstream targets, NQO1 and UDP-GT. |
Discussion
UC is considered a chronic inflammatory disorder of the gastrointestinal tract. UC has a high prevalence worldwide and is a well-known risk factor of colitis-associated cancer.36 Generally, UC is treated as an autoimmune disease, and nonsteroidal anti-inflammatory drugs, immunosuppressants, and biological therapies targeting specific components of the immune response have been reported to be effective in controlling the symptoms of UC. However, those treatments also have potential side effects, including steroid dependence37 and serious infections.38,39 Therefore, new therapeutic options and approaches for UC need to be developed. Here, we report that the polyphenolic compound GA protects mice against DSS-induced colitis by activating the Nrf2 pathway and inhibiting IL-21 and IL-23 expression.
IL-21 is a cytokine that is produced by activated CD4+ T-cells and is also one of the major products of Th17 cells in mice.40,41 Upregulation of IL-21 was found in mice with colitis induced by DSS, in mice with colitis induced by rectal administration of 2,4,6-trinitrobenzene sulfonic acid, and in mice with colitis-associated cancer.42,43 Also, increased expression of IL-21 has been observed in the inflamed guts of IBD patients, and blocking IL-21 expression in cultures of lamina propria mononuclear cells (LPMC) isolated from the gut of CD patients reduced the production of interferon-γ and IL-17A.44,45 We noted that RT-PCR expression of IL-21 was upregulated in DSS-induced mice compared to control mice. Additionally, GA-treated DSS-induced mice showed lower IL-21 expression than untreated DSS-induced mice (Figure 3A). It has been suggested that IL-21 blockers may attenuate the ongoing inflammation present in IBD patients.41 IL-23 is a cytokine that stimulates the production of IL-17, TNF-α, and IL-6 by T-cells. Therefore, IL-23 has been proposed to play an integral role in the pathogenesis of IBD.46 The expression of IL-23 has been shown to be upregulated in both rodents and humans affected by UC.47–49 In our study, we showed an increase in the mRNA expression of IL-23 in DSS-induced mice. Additionally, administration of GA suppressed the expression of IL-23 (Figure 3B).
Previous studies have indicated that oxidative stress is one of the most important causative factors of UC.50,51 Oxygen metabolites are produced in large amounts by infiltrating leukocytes in the inflamed mucosa6 and are crucial contributors to mucosal and, eventually, submucosal tissue destruction.52 The end product of lipid peroxidation, MDA, is harmful and may be responsible for some of the overall effects of oxidative stress, including the release of cell contents and cell death, which cause tissue and organ damage.10,50,51 In our study, a markedly increased level of MDA was observed in DSS-induced mice. However, treatment with GA reduced the level of MDA in DSS-induced mice (Figure 4). This finding indicated that GA treatment of mice with colitis reduced the extent of colonic mucosal injury through its strong antioxidant effect.
Antioxidant enzymes that scavenge intermediates of oxygen reduction provide the primary defense against cytotoxic oxygen radicals.53 It is well-known that SOD, CAT, GPx, and GR play important roles as protective enzymes against lipid peroxidation in tissues. Specifically, SOD and CAT are considered primary enzymes because they are involved in the direct elimination of reactive oxygen species.54 Conversely, CAT and GPx catalyze the transformation of H2O2 to harmless byproducts to prevent cellular destruction. During H2O2 scavenging, GSH is oxidized to oxidized glutathione by the enzyme GPx.55 The reduction of oxidized glutathione to GSH is catalyzed by GR, which uses NADPH as a reducer. Numerous reports have suggested that enzymic antioxidants are depleted during the exposure of rodents to DSS.33,50,51 Consistent with the results of previous studies, we observed a depletion of enzymic antioxidants in DSS-induced mice. This study also showed that GA increased the levels of enzymic antioxidants in DSS-induced mice (Figure 5). Therefore, GA can strengthen the enzymatic defense system and reduce free radicals to subsequently alleviate inflammation.
Previous studies have confirmed that Nrf2-deficient mice are more susceptible to DSS-induced inflammation/colitis and azoxymethane-/DSS-induced colitis-associated cancer.55,57 This increased susceptibility has been shown to be associated with decreased expression of antioxidant/phase II detoxifying enzymes and concomitant increases in proinflammatory cytokines and other biomarkers.55 Trivedi and Jena58 reported that exposure to DSS results in decreased expression of Nrf2. In our study, the nuclear expression of Nrf2 was decreased in DSS-induced mice (Figure 6A and B). Polyphenols are known to increase the expression of Nrf2.16,34 Similarly, in this study, GA activated Nrf2 expression due to its polyphenolic nature. In the present study the downstream targets of Nrf2 such as UDP-GT and NQO1 were analyzed by quantitative RT-PCR. In particular, we noticed no significant change in the expressions of NQO1 and UDP-GT in DSS-induced mice when compared to control mice. But administration of GA leads to increase in the expressions of NQO1 and UDP-GT. Khor et al56,59 reported that decreased expression of the detoxifying enzymes NQO1 and UDP-GT, coupled with enhanced inflammation, could be important events that accelerate neoplastic transformation in the colonic tissues of Nrf2-deficient mice, further suggesting that these defensive enzymes are important for protecting colonic mucosa against UC and colitis associated cancer.
Conclusion
In conclusion, our data strongly suggest that GA is a polyphenol that is effective for the treatment of UC and is a new therapeutic alternative to corticosteroids, immunosuppressants, and anti-TNF-α agents. This compound exerts colonic antioxidant and anti-inflammatory activities in DSS-induced colitis through downregulation of IL-21 and IL-23 expression levels, and it also offers cytoprotection by activating enzymic antioxidants through the Nrf2 pathway.
Acknowledgment
Part of the funds used in this study were supplied by a University of Malaya Research Grant (UMRG) from the University of Malaya (RP027C-14HTM).
Disclosure
The authors report no conflicts of interest in this work.
References
Abraham C, Cho JH. Inflammatory bowel disease. New Eng J Med. 2009;361:2066–2078. | ||
Ardizzone S, Bianchi Porro G. Biologic therapy for inflammatory bowel disease. Drugs. 2005;65:2253–2286. | ||
Holma R, Salmenperä P, Virtanen I, Vapaatalo H, Korpela R. Prophylactic potential of montelukast against mild colitis induced by dextran sulphate sodium in rats. J Physiol Pharmacol. 2007;58:455–467. | ||
Tokoi S, Ohkusa T, Okayasu I, Nakamura K. Population changes in immunoglobulin-containing mononuclear cells in dextran sulfate sodium-induced colitis. J Gastroenterol. 1996;31:182–188. | ||
Damiani CR, Benetton CA, Stoffel C, et al. Oxidative stress and metabolism in animal model of colitis induced by dextran sulfate sodium. J Gastroenterol Hepatol. 2007;22:1846–1851. | ||
Kruidenier L, Verpaget HW. Review article: oxidative stress as a pathogenic factor in inflammatory bowel disease-radicals or ridiculous? Aliment Pharmacol Ther. 2002;16:1997–2015. | ||
Pavlick KP, Laroux FS, Fuseler J, et al. Role of reactive metabolites of oxygen and nitrogen in inflammatory bowel disease. Free Radic Biol Med. 2002;33:311–322. | ||
Pravda J. Radical induction theory of ulcerative colitis. World J Gastroenterol. 2005;11:2371–2384. | ||
Ran ZH, Chen C, Xiao SD. Epigallocatechin-3-gallate ameliorates rats colitis induced by acetic acid. Biomed Pharmacother. 2008;62:189–196. | ||
Amrouche-Mekkioui I, Djerdjouri B. N-acetylcysteine improves redox status, mitochondrial dysfunction, mucin-depleted crypts and epithelial hyperplasia in dextran sulfate sodium-induced oxidative colitis in mice. Eur J Pharmacol. 2012;691:209–217. | ||
Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of Keap1 in cellular protective responses. Chem Res Toxicol. 2005;18:1779–1791. | ||
Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. | ||
Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. | ||
Motohashi H, Katsuoka F, Engel JD, Yamamoto M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc Natl Acad Sci U S A. 2004;101:6379–6384. | ||
Osburn WO, Wakabayashi N, Misra V, et al. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch Biochem Biophys. 2006;454:7–15. | ||
Pandurangan AK, Ananda Sadagopan SK, Dharmalingam P, Ganapasam S. Luteolin, a bioflavonoid inhibits Azoxymethane-induced colorectal cancer through Nrf2 signaling. Toxicol Mech Methods. 2014;24(1):13–20. | ||
Ma J, Luo XD, Protiva P, et al. Bioactive novel polyphenols from the fruit of Manilkara zapota (Sapodilla). J Nat Prod. 2003;7:983–986. | ||
Singh J, Rai GK, Upadhyay AK, Kumar R, Singh KP. Antioxidant phytochemicals in tomato (Lycopersicon esculentum). Indian Journal of Agricultural Science. 2004;74:3–5. | ||
Shahrzad S, Bitsch I. Determination of some pharmacologically active phenolic acids in juices by high-performance liquid chromatography. J Chromatogr A. 1996;741(2):223–231. | ||
Subramanian V, Venkatesan B, Tumala A, Vellaichamy E. Topical application of Gallic acid suppresses the 7,12-DMBA/Croton oil induced two-step skin carcinogenesis by modulating anti-oxidants and MMP-2/MMP-9 in Swiss albino mice. Food Chem Toxicol. 2014;66:44–55. | ||
Lee DS, Je JY. Gallic acid-grafted-chitosan inhibits foodborne pathogens by a membrane damage mechanism. J Agric Food Chem. 2013;61(26):6574–6579. | ||
Daduang J, Palasap A, Daduang S, Boonsiri P, Suwannalert P, Limpaiboon T. Gallic acid enhancement of gold nanoparticle anticancer activity in cervical cancer cells. Asian Pac J Cancer Prev. 2015;16(1):169–174. | ||
Kroes BH, van den Berg AJ, Quarles van Ufford HC, van Dijk H, Labadie RP. Anti-inflammatory activity of gallic acid. Planta Med. 1992;58:499–504. | ||
Ho HH, Chang CS, Ho WC, Liao SY, Lin WL, Wang CJ. Gallic acid inhibits gastric cancer cells metastasis and invasive growth via increased expression of RhoB, downregulation of AKT/small GTPase signals and inhibition of NF-κB activity. Toxicol Appl Pharmacol. 2013;266(1):76–85. | ||
Ji BC, Hsu WH, Yang JS, et al. Gallic acid induces apoptosis via caspase-3 and mitochondrion-dependent pathways in vitro and suppresses lung xenograft tumor growth in vivo. J Agric Food Chem. 2009;57(16):7596–7604. | ||
Ito R, Kita M, Shin-Ya M, et al. Involvement of IL-17A in the pathogenesis of DSS-induced colitis in mice. Biochem Biophys Res Commun. 2008;377:12–16. | ||
Ito R, Shin-Ya M, Kishida T, et al. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin Exp Immunol. 2006;146:330–338. | ||
Ohkawa H, Ohishi N, Yagi K. Assay of lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. | ||
Kakkar P, Das B, Viswanathan PN. A modified spectrophotometric assay of superoxide dismutase. Ind J Biochem Biophys. 1984;21:130–132. | ||
Sinha KA. Colorimetric assay of catalase. Anal Biochem. 1972;47:389–394. | ||
Carlberg I, Mannervik B. Glutathione reductase. Methods Enzymol. 1985;113:484–490. | ||
Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG. Selenium: biochemical role as a component of glutathione peroxidase. Science. 1973;179:588–590. | ||
Pandurangan AK, Ismail S, Saadatdoust Z, Esa NM. Allicin Alleviates Dextran Sodium Sulfate-(DSS-) Induced Ulcerative Colitis in BALB/c Mice. Oxid Med Cell Longevity. 2015;2015:605208. | ||
Pandurangan AK, Saadatdoust Z, Esa NM, Hamzah H, Ismail A. Dietary Cocoa protects against colitis-associated cancer by activating Nrf2/Keap1 pathway. Biofactors. 2015;41(1):1–14. | ||
Ashokkumar P, Sudhandiran G. Luteolin inhibits cell proliferation during Azoxymethane-induced experimental colon carcinogenesis via Wnt/β-catenin pathway. Invest New Drugs. 2011;29(2):273–284. | ||
Pandurangan AK, Esa NM. Signal transducer and activator of transcription 3 – a promising target in colitis-associated cancer. Asian Pac J Cancer Prev. 2014;15(2):551–560. | ||
Faubion WA Jr, Loftus EV Jr, Harmsen WS, Zinsmeister AR, Sandborn WJ. The natural history of corticosteroid therapy for inflammatory bowel disease: a population-based study. Gastroenterol. 2001;121:255–260. | ||
Beaugerie L, Brousse N, Bouvier AM, et al. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet. 2009;374:1617–1625. | ||
Sultan K, Korelitz BI, Present D, Katz S, Sunday S, Shapira I. Prognosis of lymphoma in patients following treatment with 6-mercaptopurine/azathioprine for inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:1855–1858. | ||
Spolski R, Leonard WJ. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol. 2008;26:57–79. | ||
Sarra M, Pallone F, Monteleone G. Interleukin-21 in chronic inflammatory diseases. Biofactors. 2013;39(4):368–373. | ||
Jauch D, Martin M, Schiechl G, et al. Interleukin 21 controls tumor growth and tumor immunosurveillance in colitis-associated tumorigenesis in mice. Gut. 2011;60:1678–1686. | ||
Fina D, Sarra M, Caruso R, et al. Interleukin 21 contributes to the mucosal T helper cell type 1 response in coeliac disease. Gut. 2008;57:887–892. | ||
Monteleone G, Monteleone I, Fina D, et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn’s disease. Gastroenterol. 2005;128:687–694. | ||
Rovedatti L, Kudo T, Biancheri P, et al. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut. 2009;58:1629–1636. | ||
McGovern D, Powrie F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut. 2007;56(10):1333–1336. | ||
Schmidt C, Giese T, Ludwig B, et al. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19 and interleukin-27p28 in Crohn’s disease but not in ulcerative colitis. Inflamm Bowel Dis. 2005;11:16–23. | ||
Camuesco D, Rodríguez-Cabezas ME, Garrido-Mesa N, et al. The intestinal anti-inflammatory effect of dersalazine sodium is related to a down-regulation in IL-17 production in experimental models of rodent colitis. Br J Pharmacol. 2012;165:729–740. | ||
Liu Z, Yadav PK, Xu X, et al. The increased expression of IL-23 in inflammatory bowel disease promotes intraepithelial and lamina propria lymphocyte inflammatory responses and cytotoxicity. J Leukoc Biol. 2011;89:597–606. | ||
Xu L, Yang ZL, Li P, Zhou YQ. Modulating effect of Hesperidin on experimental murine colitis induced by dextran sulfate sodium. Phytomedicine. 2009;16:989–995. | ||
Lee IA, Park YJ, Yeo HK, Han MJ, Kim DH. Soyasaponin I attenuates TNBS-induced colitis in mice by inhibiting NF-κB pathway. J Agric Food Chem. 2010;58:10929–10934. | ||
Rieder F, Brenmoehl J, Leeb S, Schölmerich J, Rogler G. Wound healing and fibrosis in intestinal disease. Gut. 2007;56:130–139. | ||
Ashokkumar P, Sudhandiran G. Protective role of luteolin on the status of lipid peroxidation and antioxidant defense against azoxymethane-induced experimental colon carcinogenesis. Biomed Pharmacother. 2008;62:590–597. | ||
Halliwell B, Gutteridge JM. The antioxidants of human extracellular fluids. Arch Biochem Biophys. 1990;280(1):1–8. | ||
Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. | ||
Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. Nrf2-defcient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006;66(24):11580–11584. | ||
Osburn WO, Karim B, Dolan PM, et al. Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int J Cancer. 2007;121:1883–1891. | ||
Trivedi PP, Jena GB. Role of α-lipoic acid in dextran sulfate sodium-induced ulcerative colitis in mice: studies on inflammation, oxidative stress, DNA damage and fibrosis. Food Chem Toxicol. 2013;59:339–355. | ||
Khor TO, Huang MT, Prawan A, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res (Phila). 2008;1(3):187–191. |
Supplementary materials
Real-time polymerase chain reaction analysis of superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase
Materials and methods
The method of quantitative real-time-polymerase chain reaction was mentioned in detail in the “Materials and methods” section. The primer details are listed below in Table S1.
Results
Gallic acid upregulates the enzymic antioxidants
Figure S1 shows the real-time-polymerase chain reaction analysis of enzymic antioxidants such as SOD, CAT, GPx, and GR in control and experimental animals. We observed a significantly (P<0.05) decreased expression of enzymic antioxidants in dextran sulfate sodium-induced mice when compared to control mice. But administration of gallic acid showed significant increase in the expression of enzymic antioxidants in dextran sulfate sodium-induced mice. So, we confirm that gallic acid potentially restores the enzymic antioxidants due to its strong antioxidant potential.
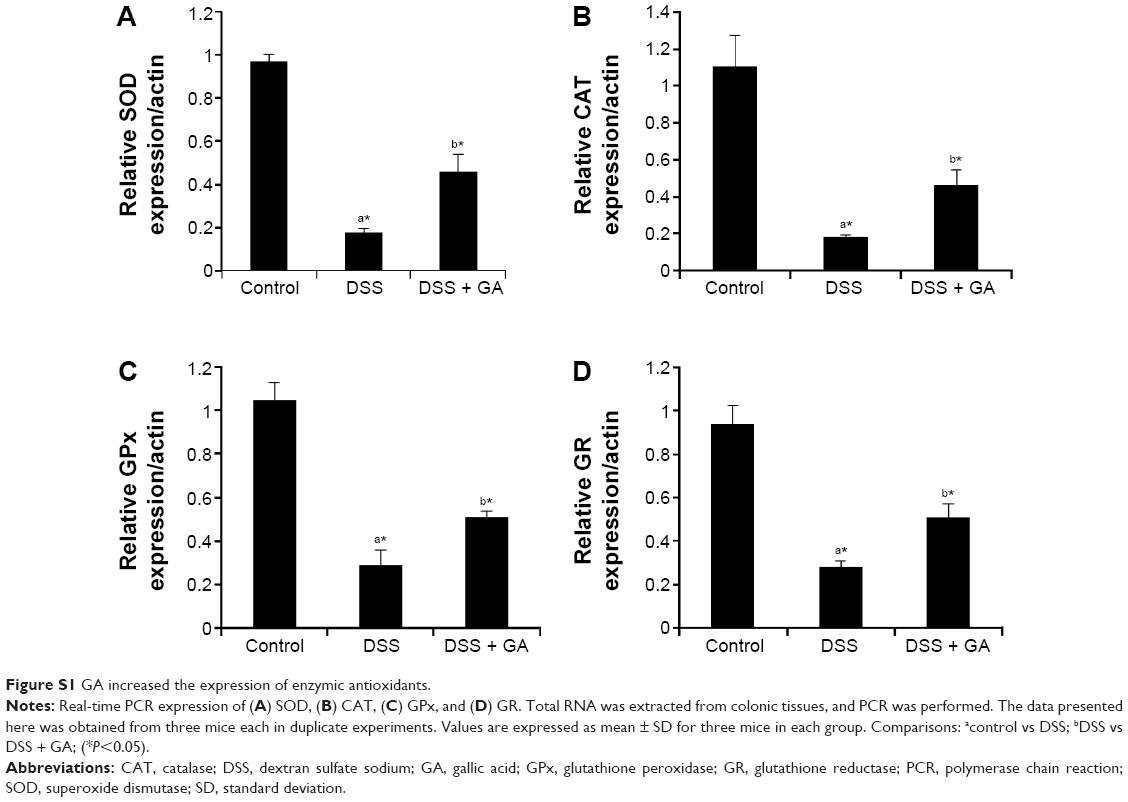 | Figure S1 GA increased the expression of enzymic antioxidants. |
 | Table S1 List of primers used |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.