")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Exploration of natural product ingredients as inhibitors of human HMG-CoA reductase through structure-based virtual screening
Authors Lin S, Huang K, Weng C, Shiuan D
Received 14 March 2015
Accepted for publication 10 April 2015
Published 26 June 2015 Volume 2015:9 Pages 3313—3324
DOI https://doi.org/10.2147/DDDT.S84641
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Shih-Hung Lin,1 Kao-Jean Huang,1,2 Ching-Feng Weng,1 David Shiuan1
1Department of Life Science and Institute of Biotechnology, National Dong Hwa University, Hualien, Taiwan, Republic of China; 2Development Center of Biotechnology, Taipei, Taiwan, Republic of China
Abstract: Cholesterol plays an important role in living cells. However, a very high level of cholesterol may lead to atherosclerosis. HMG-CoA (3-hydroxy-3-methylglutaryl coenzyme A) reductase is the key enzyme in the cholesterol biosynthesis pathway, and the statin-like drugs are inhibitors of human HMG-CoA reductase (hHMGR). The present study aimed to virtually screen for potential hHMGR inhibitors from natural product to discover hypolipidemic drug candidates with fewer side effects and lesser toxicities. We used the 3D structure 1HWK from the PDB (Protein Data Bank) database of hHMGR as the target to screen for the strongly bound compounds from the traditional Chinese medicine database. Many interesting molecules including polyphenolic compounds, polisubstituted heterocyclics, and linear lipophilic alcohols were identified and their ADMET (absorption, disrtibution, metabolism, excretion, toxicity) properties were predicted. Finally, four compounds were obtained for the in vitro validation experiments. The results indicated that curcumin and salvianolic acid C can effectively inhibit hHMGR, with IC50 (half maximal inhibitory concentration) values of 4.3 µM and 8 µM, respectively. The present study also demonstrated the feasibility of discovering new drug candidates through structure-based virtual screening.
Keywords: HMG-CoA reductase, virtual screening, curcumin, salvianolic acid C
Introduction
Cholesterol plays an important role in living cells, serving as a cell membrane structural component and as a precursor of vitamin D, bile acids, and many hormones. Approximately 70% of the total cholesterol in human body arises from endogenous biosynthesis, and the remainder is provided in the diet.1 Epidemiological studies have shown a positive relationship between total cholesterol concentrations and mortality from coronary heart disease. For people with very high cholesterol, diet alone is no longer sufficient to achieve the desired level of low-density lipoprotein (LDL), and medications that reduce cholesterol production or absorption are usually required.2
The mevalonate pathway is responsible for the endogenous synthesis of cholesterol. Human HMG-CoA reductase (hHMGR) is a transmembrane glycoprotein that is 888 amino acids long, which catalyzes the reaction that converts HMG-CoA to mevalonate. Its first 339 residues comprise the membrane anchor domain located in the endoplasmic reticulum, followed by a linker region between residues 340 and 449, and the catalytic domain, between residues 450 and 883, which resides in the cytoplasm.3 Because hHMGR is the key enzyme in the cholesterol biosynthesis pathway, it has been considered a major target for the treatment of hypercholesterolemia. Based on the criteria of inhibition of hHMGR activity, many cholesterol-lowering drugs such as statins have been developed, and their efficacies in controlling blood cholesterol levels have been well recognized.3,4
However, hHMGR also catalyzes the biosynthesis of many nonsterol isoprenoids essential for normal cell functions.3 Therefore, the widely prescribed hHMGR inhibitor drugs also cause severe adverse effects, such as distal muscle weakness, headache, and acute renal failure. Side effects, such as hepatic transaminase elevation, sensory disturbances, and depression, have also been observed on prolonged use.5 Instead of taking chemical drugs, many people today prefer natural products or alternative medicine mainly due to the beliefs that natural products are safer and cheaper.6 In addition, lovastatin, one of the currently used statin drugs, was originally found in food such as red yeast rice. Many other traditional Chinese medicines have also been demonstrated to be good alternatives to statin therapy due to their effectiveness and safety.7 Therefore, the present study aimed to identify safer and novel hHMGR inhibitors from the ingredients originally in herbs, vegetables, and fruits.8,9 We selected the 3D structure of hHMGR (1HWK from PDB [Protein Data Bank]) as the target and virtually screened the Traditional Chinese Medicine (TCM) database for tightly bound compounds, using the molecular modeling tool. The ADMET (absorption, distribution, metabolism, excretion and toxicity) properties of high-ranking compounds were further evaluated to eliminate those unfavorable compounds. Meanwhile, we have cloned, overexpressed, and purified the hHMGR to perform enzyme activity assay experiments.
Materials and methods
Bacterial strains and materials
The bacterial strains used in the present study include: E. coli strain DH5α (fhuA2 Δ(argF-lacZ)U169 phoA glnV44 Φ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17) for molecular cloning and E. coli strain BL21 Star™ (DE3): F− ompT hsdSB (rB− mB−) gal dcm rne131) for over-expression of the cloned genes. Vector pET28a and E. coli strains were obtained from Invitrogen (Waltham, MA, USA). Curcumin, atorvastatin, docosanol, and folic acid were purchased from Sigma-Aldrich (St Louis, MO, USA), salvianolic acid C (Sal C) was purchased from ApexBio (Hsinchu, Taiwan).
Molecular docking and analysis of ligand–protein interactions
The 3D structure PDB 1HWK10 of hHMGR was chosen as the molecular target and obtained from the PDB (http://www.rcsb.org/pdb). The tetrameric structure contains the catalytic domains of hHMGR (subunit A: Pro442–His861; B: Ser463–Gly860; C: Leu462–Gly860; D: Ser463–Gly860) complexed with four atorvastatin molecules at the interfaces of two adjacent monomers.10 After removing unnecessary ligands, we kept two adjacent monomers of hHMGR, fixed with the force-field CHARMM (Chemistry at HARvard Macromolecular Mechanics) equipped in DS 3.5 (http://accelrys.com/products/discovery-studio) to add up the hydrogen atoms, partial charges, and missing residues so that the structure can be used properly for molecular docking processes. The ligand-binding sites were predicted by estimating the receptor cavities or the active sites from PDB site records, using the tools of DS 3.5.11,12
The TCM database (http://tcm.cmu.edu.tw/) was used as the small molecule resource and screened using the LigandFit software of DS 3.5 (Accelrys Software Inc., San Diego, CA, USA), which provides a shape-based method for accurately docking ligands into the protein-binding sites.12–14 The ligand–protein (hHMGR) interactions were further analyzed using the view interaction tools provided by DS 4.0 visualizer (Accelrys Software Inc., San Diego, CA, USA).
ADMET prediction
The ADMET properties of the drug candidates are very critical in drug development. Therefore, we decided to estimate the ADMET properties of the virtually selected compounds before performing the costly experimental assays. The predictions were performed by using the web server DSSTox (http://www.epa.gov/ncct/dsstox/) and DS 3.5 ADMET Descriptor software.15 The DSSTox is a project of US Environmental Protection Agency, trying to build a public data foundation for improved structure-activity and predictive toxicology capabilities. The ADMET Descriptors of DS 3.5 includes models for intestinal absorption, aqueous solubility, blood–brain barrier penetration, plasma protein binding, cytochrome P450 2D6 inhibition, and hepatotoxicity. With these advanced prediction tools, those docked candidates with undesirable chemical groups can be filtered out earlier.15
Preparation and enzymatic assay of the recombinant hHMGR
The gene cloning, overexpression of the cloned genes, and protein purification were performed as described previously.16 The HMG-CoA reductase-dependent oxidation of NADPH was measured to assess the hHMGR activities.16,17 Protein concentration was determined by the Bradford method.18
MTT assay
The potential cellular toxicity of the selected compounds on HepG2 cells was assessed by the MTT method.19,20 The MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyltetrazolium bromide) assay is based on the conversion of MTT into formazan crystals by the living cells. HepG2 cells were cultured in a 96-well culture plate (1×105 cell/mL) for 24 h at 37°C in atmosphere of 5% CO2. The cultures were treated with different concentrations of the selected compounds for 24 h. The supernatants were then removed, and MTT (2.5 mg/mL) was added and incubated for an additional 4 h. The purple formazan crystals developed by the action of mitochondrial succinate dehydrogenase were extracted into DMSO. The optical density (OD570) was measured using an EnSpire Multimode enzyme linked immuno-sorbent assay Plate Reader (PerkinElmer, Waltham, MA, USA).
Results and discussion
Virtual screening for human HMG-CoA reductase inhibitors
The 3D structure of hHMGR (PDB 1HWK) was chosen as the molecular target of the present study. It is a tetramer of hHMGR proteins complexed with four atorvastatin molecules that are located at the interfaces between two adjacent monomers (Figure 1A and B). As shown in Figure 1C and D, the binding site was surrounded by the key residues Arg A590, Ser A661, Val A683, Ser A684, Asp A690, and Lys A691 from the subunit A (red); Glu B559, Cys B561, Leu B562, Ala B564, Ser B565, His B752, Lys B735, Asn B755, Leu B853, and Ala B856 from the subunit B (green) of the two adjacent hHMGR monomers.10 To facilitate the docking process, the binding sites were identified by using the tools of DS 3.5, either based on the cavities of the receptor or through the PDB site record (Figure 1C). To evaluate the feasibility of docking parameters, we selected five known statin-like molecules with known IC50 (half maximal inhibitory concentration) values against hHMGR (Figure 2) as a test kit. As shown in Table 1, they were successfully docked into the receptor-binding site with good docking scores, confirming that the –PMF (potential of mean force) scoring function is more feasible and the docking parameters were set properly. Then we went on to screen the TCM database (containing approximately 30,000 pure compounds) and found that only 4,099 received docking scores. Among them, 561 compounds exhibited –PMF scores higher than 70, comparable with the scores of the known statins.
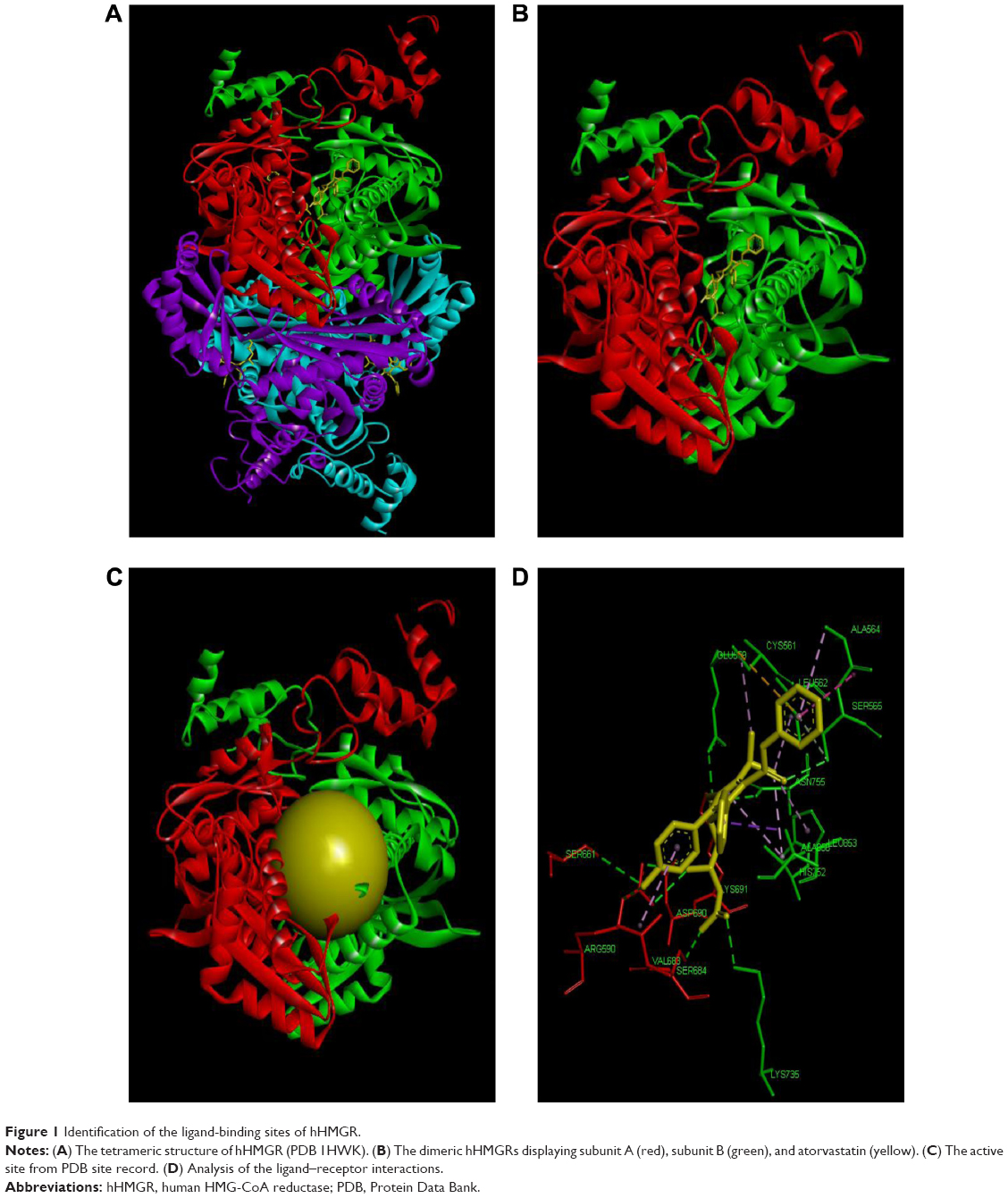 | Figure 1 Identification of the ligand-binding sites of hHMGR. |
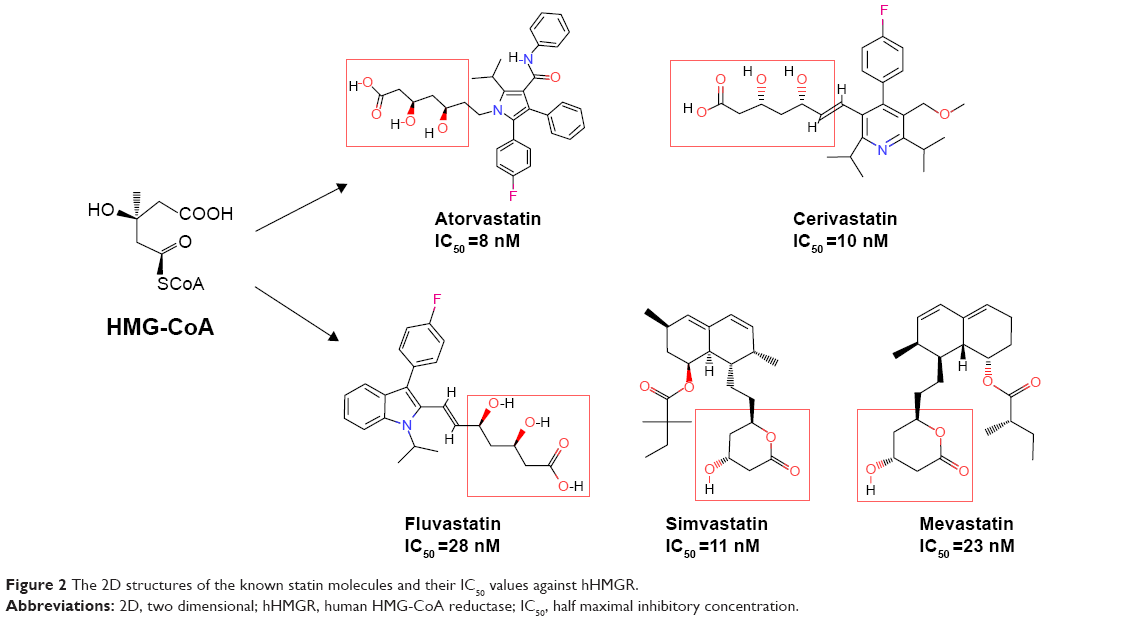 | Figure 2 The 2D structures of the known statin molecules and their IC50 values against hHMGR. |
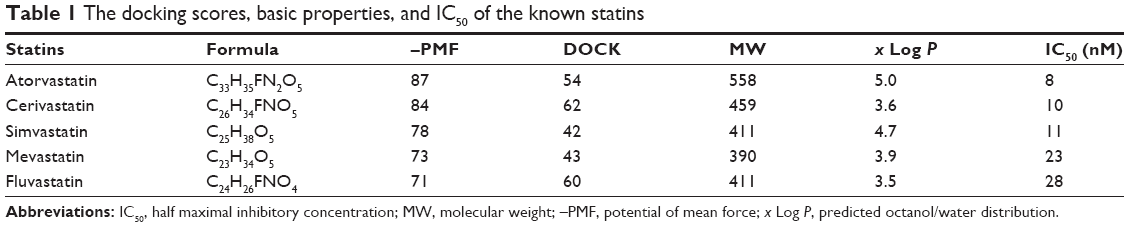 | Table 1 The docking scores, basic properties, and IC50 of the known statins |
Selection of candidate compounds after ADMET predictions
Since one of the primary factors that cause drug attrition is the poor ADMET properties, the 278 compounds with higher docking scores were evaluated in silico using the DSSTox and the ADMET prediction tools of DS 3.5. These predictions were based on certain animal and cell models and the results serve as a good reference before performing further experiments. Surprisingly, among the 278 compounds, only 51 compounds received favorable ADMET characteristics, indicating that they are both nonmutagenic and noncarcinogenic (data not shown, Table S1). Taking together the ADMET predictions and –PMF scores, ten compounds were chosen for further analysis. The ten compounds include: 1) Sal C, 2) quercetin-3-O-(6′-malonyl) glucoside, 3) curcumin, 4) ampelopsisin, 5) epigallocatechin-3-gallate, 6) Z-ligustilide-SG1a, 7) tenellin, 8) docosanol, 9) tetracosanol, and 10) folic acid. As shown in Table 2, they have similar molecular weights (MWs), but their –PMF scores span a wide range (from 70 up to 146), and their physicochemical properties are varied. The ADMET predictions of the ten selected compounds and the six statin molecules have been included in the supplementary materials. Quite different from the statins which carry a side chain similar to HMG-CoA, the ten compounds display a wide spectrum of structural features (Figure 3). Among them, compounds 1–5 possess polyphenolic moieties; compounds 1, 2, 6, and 10 are carboxylic acids with various heterocyclic branches. Policosanols, the long-chain alcohols, such as compounds 8 and 9 are also included.
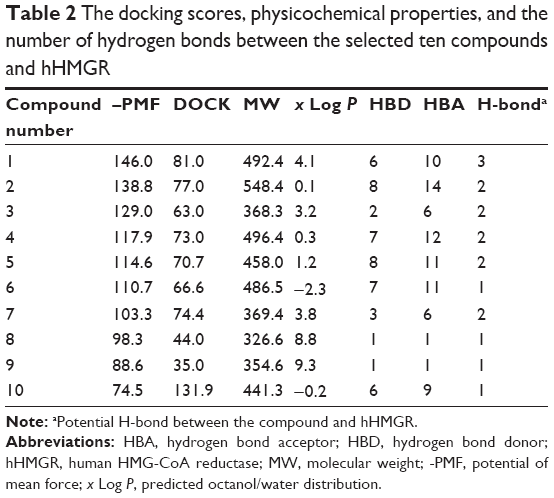 | Table 2 The docking scores, physicochemical properties, and the number of hydrogen bonds between the selected ten compounds and hHMGR |
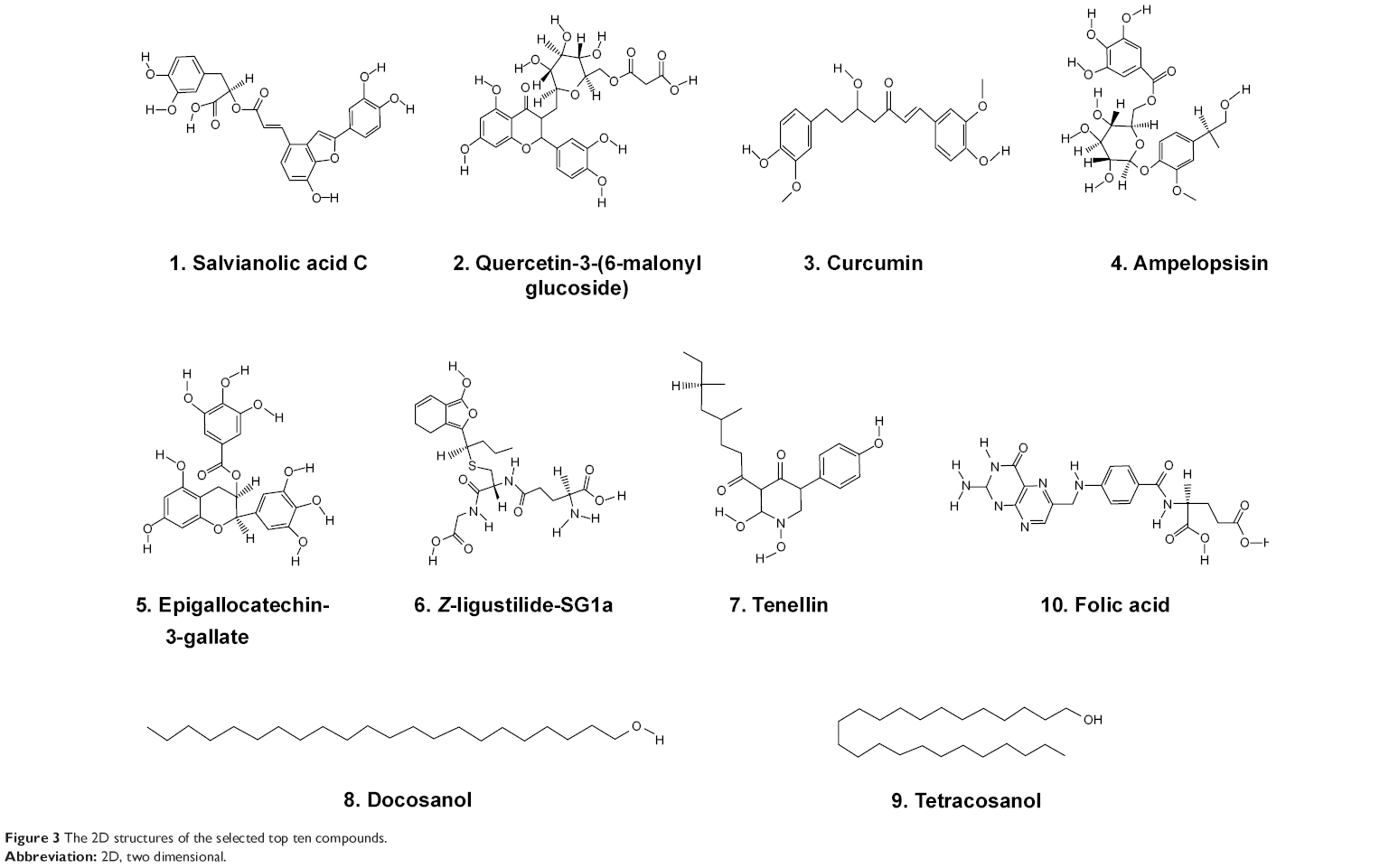 | Figure 3 The 2D structures of the selected top ten compounds. |
These ten compounds are all from very popular herbs and plants. As listed in Table 3, Sal C (compound 1) is from Salvia miltiorrhiza (also known as red sage, Dan-Shen), an extremely valued traditional Chinese medicine; Z-ligustilide-SG1a (compound 6) is from Angelica sinensis (commonly known as Dong-Quai or female ginseng), and is also a very popular traditional Chinese medicine. The rest are from very common food plants. Among them, curcumin (compound 3) is from Curcuma longa (a ginger family plant); epigallocatechin-3-gallate (compound 5) is a major polyphenol in green tea leaf and has been shown to have anti-inflammatory, anticancer, and antisteatotic effects on the liver.21 It is amazing to realize that so many ingredients with potential medicinal benefits are contained in diary food, herbs, and plants. Therefore, we can anticipate that their adverse effects could be much less and that they have very high potential to become health-improving agents.
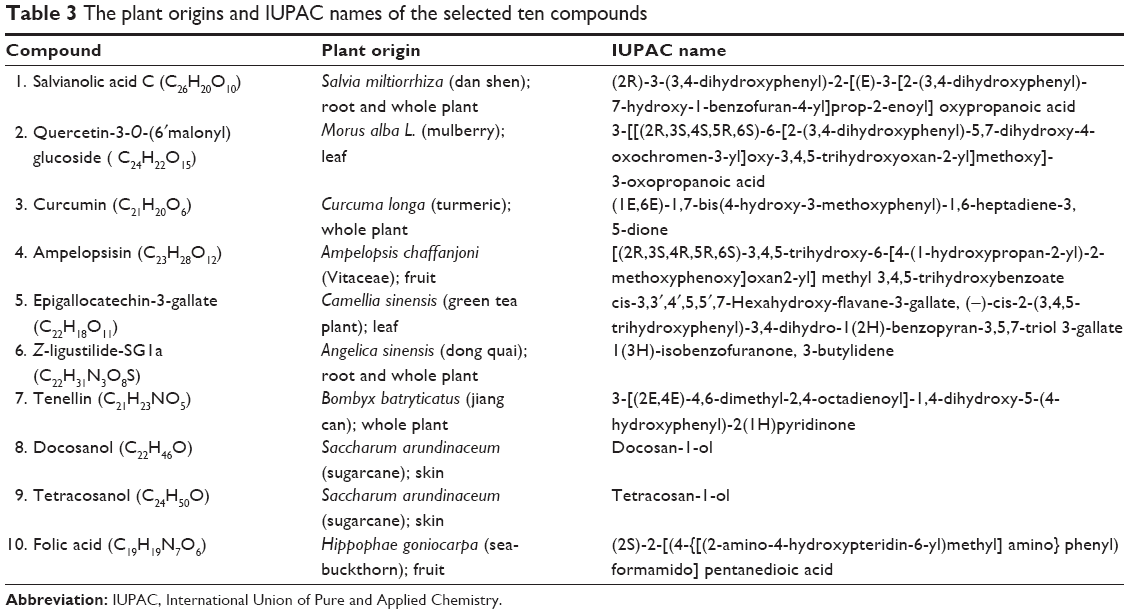 | Table 3 The plant origins and IUPAC names of the selected ten compounds |
Inhibitory effects of the selected compounds on hHMGR activities and their potential cellular toxicities
The catalytic fragment of the hHMGR protein was cloned, overexpressed, and purified as described in the “Materials and methods” section.16 The hHMGR enzymatic activities were assessed by measuring the dependent oxidation of NAPDH.17 Using the data from the Lineweaver–Burk plot, it was found that the Km value of the recombinant hHMGR was 103.4 μM, the Vmax was 10.1 μM NADPH/min/mg, under the experimental conditions. These values are comparable to the literature database on HMG-CoA-dependent oxidation of NADPH or radioisotope method.17,22,23
After evaluating the docking scores, accessibility, and known functions of the ten compounds, we were left with salvinolic acid C (compound 1), curcumin (compound 3), docosanol (compound 8), and folic acid (compound 10) for further experimental assays. As shown in Figure 4, the results indicated that Sal C, curcumin, and docosanol, can effectively inhibit hHMGR activities. However, folic acid was found to have no inhibitory capability. The IC50 of Sal C, curcumin, and docosanol upon hHMGR activities were further determined to be approximately 8 μM, 4.3 μM, and 250 μM, respectively. These values are much weaker than those of statins which have IC50 values in the nM ranges.
 | Figure 4 Inhibition on hHMGR enzyme activities by (A) salvianolic acid C, (B) curcumin, and (C) docosanol. |
The potential cytotoxic effects of Sal C, curcumin, and docosanol on HepG2 cells were then examined by the MTT assay. As shown in Figure 5, atorvastatin, such a well-known hypocholesterolemic drug, exhibited a pronounced concentration-dependent cytotoxicity effect (CC50 [50% cytotoxic concentration] near 100 μM). Curcumin retarded the HepG2 growth rate even more effectively (CC50 near 42 μM).24 On the other hand, Sal C was found to have no cytotoxic effect under the testing concentration range, and even enhanced the cell viability slightly, while docosanol had a milder cytotoxic effect.
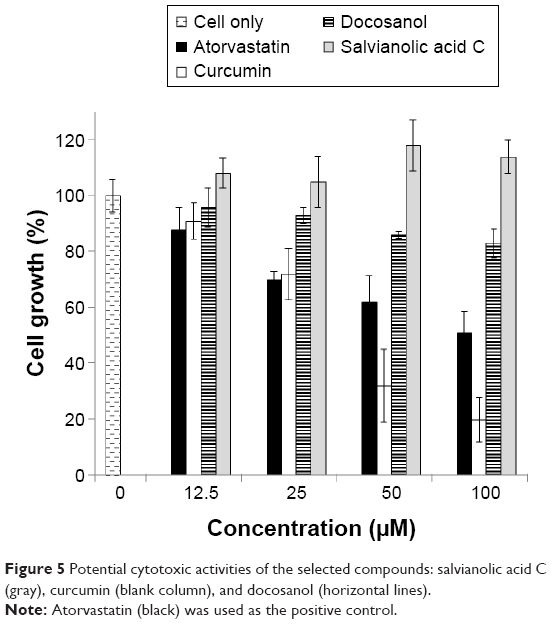 | Figure 5 Potential cytotoxic activities of the selected compounds: salvianolic acid C (gray), curcumin (blank column), and docosanol (horizontal lines). |
These four compounds not only have very different structural scaffolds, but also possess many interesting known pharmaceutical activities. The dried root of Salvia miltiorrhiza, is currently receiving worldwide attention for its potential to prevent and treat cardiovascular diseases.25 Phytochemical and pharmacological investigations have revealed that both salvianolic acids and tanshinones are responsible for the bioactive effects.26 Among them, salvianolic acid A (Sal A) and salvianolic acid B (Sal B) have been found to have potent antioxidative capabilities; however, the bioactivity of Sal C remains unknown.26 Curcumin received even more attention because of its accessibility and its many functions such as obesity prevention, clearance of free radicals, improvement of retinal function, anti-inflammatory properties, and reduction of tumor proliferation and invasion.27–30 A recent report also suggested that long-term curcumin treatment can lower the plasma and hepatic cholesterol levels and suppress early atherosclerotic lesions (comparable to the protective effects of lovastatin), but nothing was known about its action mechanism.31 Policosanols, which include a mixture of long-chain primary alcohols, have also been shown to decrease serum cholesterol in animals and in humans. However, both the suppression mechanism and the active components of policosanols are still ambiguous.32 Now, the present studies demonstrated clearly that Sal C, curcumin, and docosanol possess a new function as effective inhibitors against hHMGR. However, one should be aware that hHMGR is a highly regulated enzyme. Transcription and translation of hHMGR increase when the concentrations of products of the mevalonate pathway are low. Conversely, when sterol concentrations are high, the intracellular hHMGR concentration decreases rapidly.33,34 Therefore, the overall clinical outcomes of these potential hypocholestermeric agents demand thorough investigations.
Analysis of the ligand–receptor interactions
The detailed ligand–protein interactions of the four compounds can be further analyzed through the view interaction tools DS 4.0. Using Sal C as an example (Figure 6), it was predicted that Sal C fits well in the interface between the two subunits, quite similar to the site occupied by atorvastatin. As shown in Figure 6C and D, its interactions with hHMGR include residues from subunit A (green) and subunit B (red), potentially forming H-bond with Ser B661, Ala A751, Lys B692, and Lys A735. Others are mainly hydrophobic interactions and van der Waals interactions. Basically, the ligand–protein interaction analysis revealed that these molecules occupy similar binding sites as the normal substrate (HMG-CoA) in the catalytic domain and may form hydrogen bonds, electrostatic interactions, and hydrophobic interactions with the residues at the binding site of hHMGR (The ligand–receptor interactions of curcumin and docosanol are shown in Figure S1).
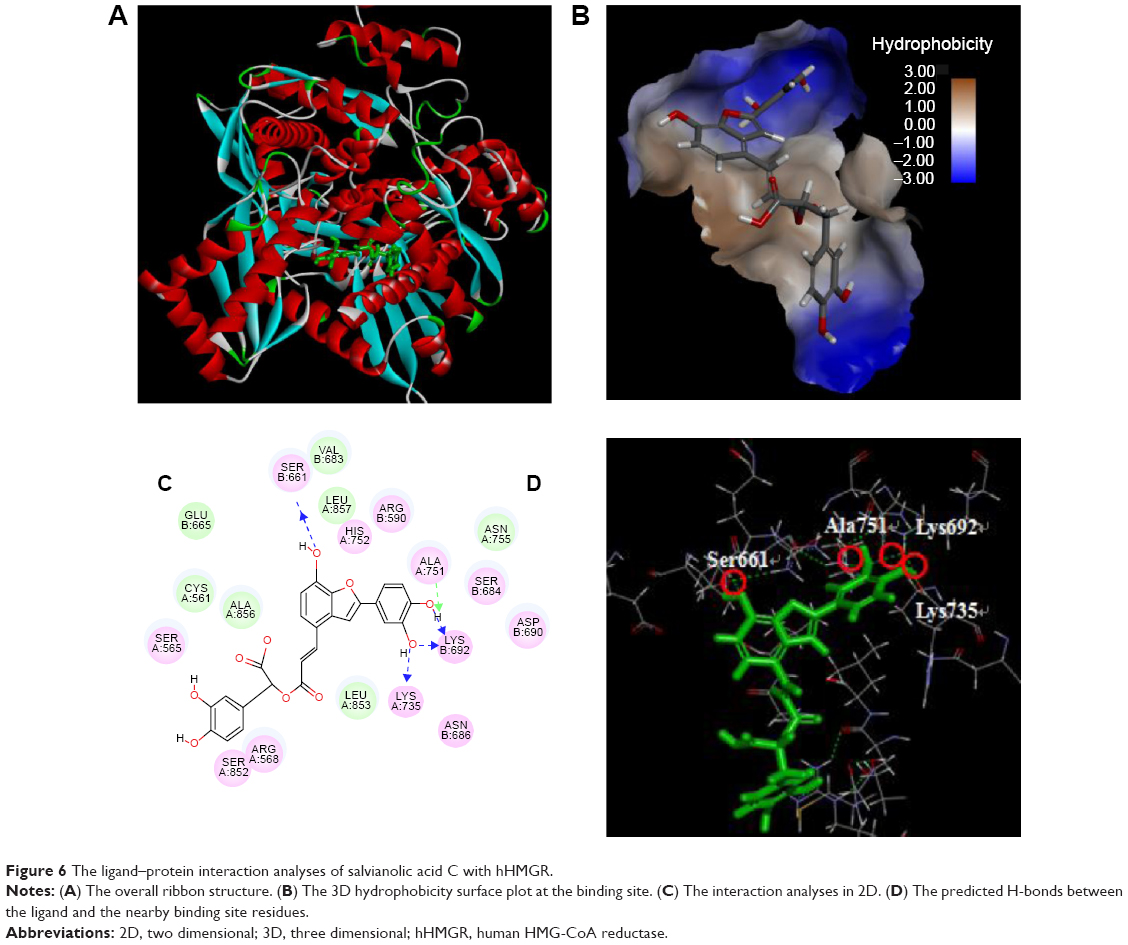 | Figure 6 The ligand–protein interaction analyses of salvianolic acid C with hHMGR. |
However, it is rather difficult to explain the fact that atorvaststin received relatively lower docking scores, but exhibited much higher inhibitory capability against hHMGR. It could be attributed partially to the limit of the docking tool, especially in the present study, and the location of the binding site at the interface between the two monomers of hHMGR. Of course, other factors such as the importance of the HMG-CoA-like moiety on atorvastatin, the natural physical properties such as hydrophobicity and solubility of these inhibitors while performing the experiments, should not be ignored.
Conclusion
Aging-associated syndromes, such as heart disease, stroke, cancer, diabetes, and arthritis, have been identified as the common and costly health problems in the 21st century.35 The current consensus is that many aging-associated diseases are preventable. Apart from changing lifestyle, exercise, and nutrition improvement, the contribution of complementary medicines such as dairy products still deserves more investigations. In the present study, we have shown that combining high-throughput virtual screening and in vitro/in vivo validation experiments can be an efficient strategy to discover new drug candidates and to confirm their action mechanisms simultaneously. Through limited trials, we have identified many novel natural candidates with very unique structural features. We have also demonstrated that Sal C, curcumin, and docosanol could be potential agents to treat hypercholesterolemia. Furthermore, convincing evidence from both in vitro and in vivo data has demonstrated that statins exert pleiotropic actions beyond their lipid-lowering effects, including cancer prevention.36 HMG-CoA reductase inhibitors were found to induce lymphoma-cell apoptosis by increasing intracellular ROS generation and p38 activation and suppressing activation of Akt and Erk pathways, through inhibition of metabolic products of the HMG-CoA reductase reaction including mevalonate, farnesyl pyrophosphate, and geranylgeranyl pyrophosphate.37 Therefore, one can speculate that the hHMGR inhibitors found in the present work may also possess similar anticancer activities. The therapeutic benefits of many dietary constituents of plant origin should be the focus of further studies, and many challenges are still ahead.38,39
Disclosure
The authors report no conflicts of interest in this work.
References
Grundy SM, Vegn GL. Plasma cholesterol responsiveness to saturated fatty acid. Am J Clin Nutr. 1988;47:822–824. | ||
Carmena R, Duriez P, Fruchart JC. Atherogenic lipoprotein particles in atherosclerosis. Circulation. 2004;109:III2–III7. | ||
Endo A. The discovery and development of HMG-CoA reductase inhibitors. J Lipid Res. 1992;33:1569–1582. | ||
Rader DJ. Regulation of reverse cholesterol transport and clinical implications. Am J Cardiol. 2003;92:42–49. | ||
Grosset KA, Grosset DG. Prescribed drugs and neurological complications. J Neurol Neurosurg Psychiatry. 2004;75:iii2–iii8. | ||
Barnes PM, Powell-Griner E, McFann K, Nahin RL. Complementary and alternative medicine use among adults: United States 2002. Adv Data. 2004;343:1–19. | ||
Xu DY, Shu J, Huang QY, et al. Evaluation of the lipid lowering ability, anti-inflammatory effects and clinical safety of intensive therapy with Zhibitai, a Chinese traditional medicine. Atherosclerosis. 2010;211:237–241. | ||
Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862–865. | ||
Daidone F, Montioli R, Paiardini A, et al. Identification by virtual screening and in vitro testing of human DOPA decarboxylase inhibitors. PLoS One. 2012;7:e31610. | ||
Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292:1160–1164. | ||
Brooks BR, Bruccoleri RE, Olafson BD, et al. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. | ||
Huang KJ, Lin SH, Lin MR, et al. Xanthone derivatives could be potential antibiotics: virtual screening for the inhibitors of enzyme I of bacterial phosphoenolpyruvate-dependent phosphotransferase system. J Antibiotics. 2013;66:453–458. | ||
Muegge I, Martin YC. A general and fast scoring function for protein-ligand interaction: a simplified potential approach. J Med Chem. 1999;42:791–804. | ||
Venkatachalam CM, Jiang X, Oldfield T, Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model. 2003;22:289–307. | ||
Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43:3867–3877. | ||
Lin KC, Chang DK, Chou MJ, Shiuan D. Peptide inhibitors of human HMG-CoA reductase as potential hypocholesterolemia agents. Biochem Biophys Res Commun. 2015;456:104–109. | ||
Pak VV, Koo M, Lee N, et al. Hypocholesterolemic soybean peptide (IAVP) inhibits HMG-CoA reductase in a competitive manner. Food Sci Biotech. 2005;14:727–731. | ||
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye-binding. Anal Biochem. 1976;72:248–254. | ||
Lin CY, Lu MC, Su JH, et al. Immunomodulatory effect of marine membrane-type diterpenoids on dendritic cells. Mar Drugs. 2013;11:1336–1350. | ||
Van Meerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol Biol. 2011;731:237–245. | ||
Tipoe GL, Leung TM, Liong EC, et al. Epigallocatechin 3-gallate (EGCG) reduces liver inflammation, oxidative stress and fibrosis in carbon tetrachloride (CCl4)-induced liver injury in mice. Toxicology. 2010;273:45–52. | ||
Pak VV, Kim SH, Koo M, Lee N, Shakhidoyatov KM, Kwon DY. Peptide design of a competitive inhibitor for HMG-CoA reductase based on statin structure. Biopolymers. 2006;84:586–594. | ||
Philipp BW, Shapiro DJ. Improved methods for the assay and activation of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J Lipid Res. 1997;20:588–593. | ||
Mendonca LM, Dos Santos GC, Antonucci GA, Dos Santos AC, Bianchi Mde L, Antunes LM. Evaluation of the cytotoxicity and genotoxicity of curcumin in PC12 cells. Mutation Res. 2009;675:29–34. | ||
Zhou L, Zuo Z, Chow MS. Dan-Shen: an overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. J Clin Pharmacol. 2005;45:1345–1359. | ||
Ho JH, Hong CY. Salvianolic acids: small compounds with multiple mechanisms for cardiovascular protection. J Biomed Sci. 2011;18:30–34. | ||
Maheshwari RK, Singh AK, Gaddipati J, Srimal RC. Multiple biological activities of curcumin: a short review. Life Sci. 2006;78:2081–2087. | ||
Mimeault M, Batra SK. Potential applications of curcumin and its novel synthetic analogs and nanotechnology-based formulations in cancer prevention and therapy. Chin Med. 2011;6:31. | ||
Johnson JJ, Mukhtar H. Curcumin for chemoprevention of colon cancer. Cancer Lett. 2007;255:170–181. | ||
Mirza M, Volz C, Karlstetter M, et al. Progressive retinal degeneration and glial activation in the CLN6 (nclf) mouse model of neuronal ceroid lipofuscinosis: a beneficial effect of DHA and curcumin supplementation. PLoS One. 2013;8:e75963. | ||
Shin SK, Ha TY, McGregor RA, Choi MS. Long-term curcumin administration protects against atherosclerosis via hepatic regulation of lipoprotein cholesterol metabolism. Mol Nutr Food Res. 2011;55:1829–1840. | ||
Singh DK, Li L, Porter TD. Policosanol inhibits cholesterol synthesis in hepatoma cells by activation of AMP-kinase. J Pharmacol Exp Ther. 2006;318:1020–1026. | ||
Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. | ||
Nakanishi M, Goldstein JL, Brown MS. Multivalent control of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mevalonate-derived product inhibits translation of mRNA and accelerates degradation of enzyme. J Biol Chem. 1988;263:8929–8937. | ||
Kung HC, Hoyert DL, Xu J, Murphy SL. Deaths: final data for 2005. Natl Vital Stat Rep. 2008;56(10):1–120. | ||
Qi XF, Zheng L, Lee KJ, et al. HMG-CoA reductase inhibitors induce apoptosis of lymphoma cells by promoting ROS generation and regulating Akt, Erk and p38 signals via suppression of mevalonate pathway. Cell Death Dis. 2013;4:e518. doi:10.1038/cddis.2013.44. | ||
Koyuturk M, Ersoz M, Altiok N. Simvastatin induces apoptosis in human breast cancer cells: p53 and estrogen receptor independent pathway requiring signalling through JNK. Cancer Lett. 2007;250:220–228. | ||
Stravic B. Antimutagens and anticarcinogens in foods. Food Chem Toxicol. 1994;32:79–90. | ||
Corson TW, Crews CM. Molecular understanding and modern application of traditional medicines: triumphs and trials. Cell. 2007;130:769–774. |
Supplementary materials
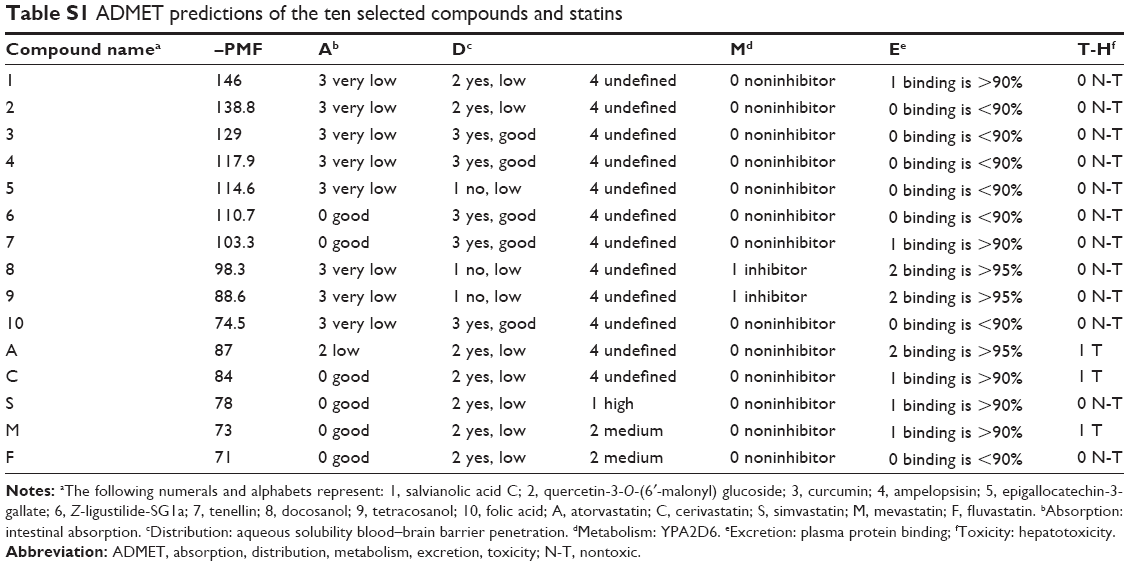 | Table S1 ADMET predictions of the ten selected compounds and statins |
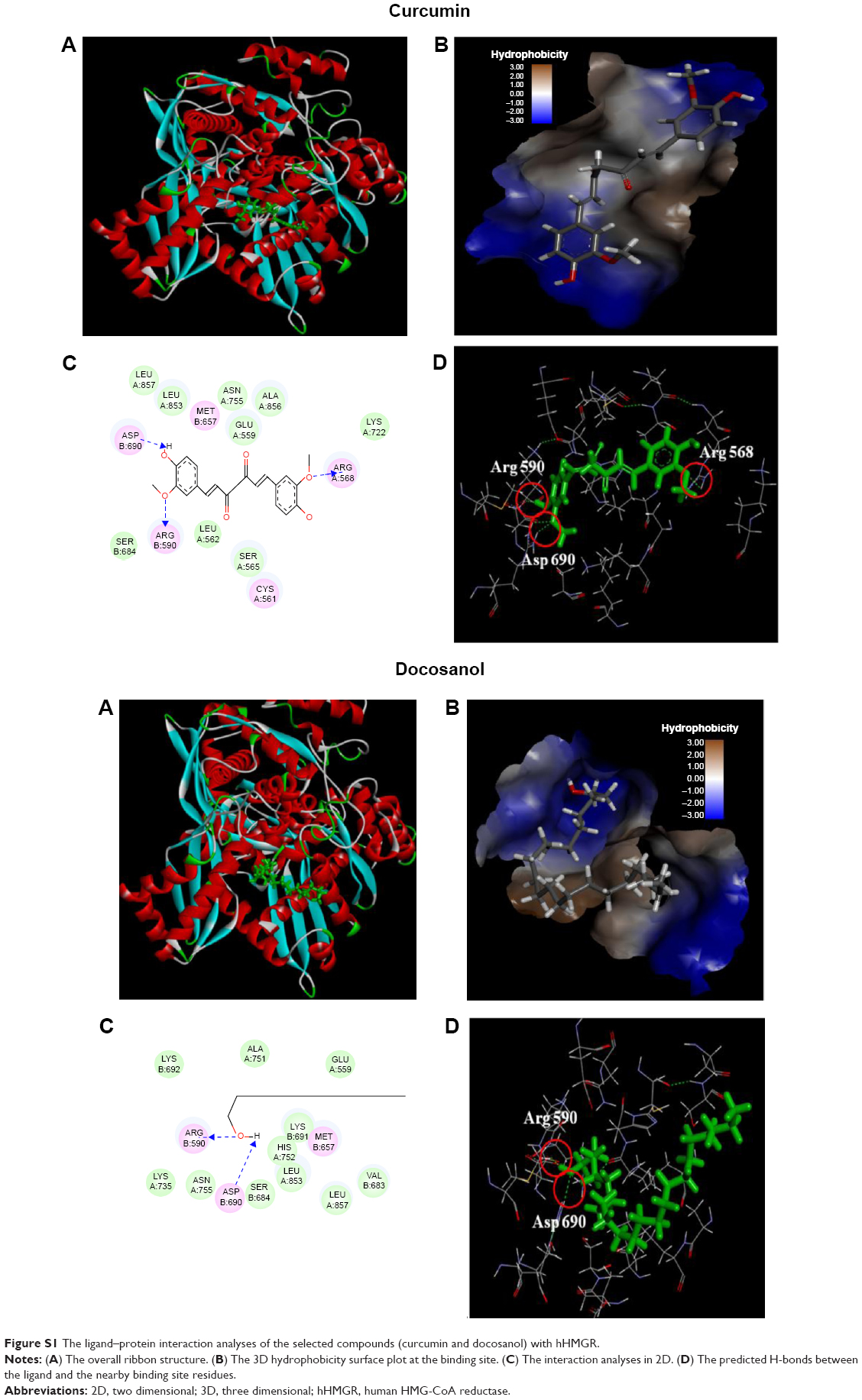 | Figure S1 The ligand–protein interaction analyses of the selected compounds (curcumin and docosanol) with hHMGR. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.