")
Back to Journals » International Journal of Nanomedicine » Volume 19
Emerging Strategies to Overcome Current CAR-T Therapy Dilemmas - Exosomes Derived from CAR-T Cells
Authors Hu D, Yang R, Wang G, Li H, Fan X, Liang G
Received 17 October 2023
Accepted for publication 27 February 2024
Published 18 March 2024 Volume 2024:19 Pages 2773—2791
DOI https://doi.org/10.2147/IJN.S445101
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Dong Hu,1 Ruyue Yang,1 Guidan Wang,2 Hao Li,1 Xulong Fan,1 Gaofeng Liang1
1School of Basic Medicine and Forensic Medicine, Henan University of Science & Technology, Luoyang, 471023, People’s Republic of China; 2School of Medical Technology and Engineering, Henan University of Science & Technology, Luoyang, 471023, People’s Republic of China
Correspondence: Gaofeng Liang, School of Basic Medicine and Forensic Medicine, Henan University of Science & Technology, Luoyang, 471023, People’s Republic of China, Email [email protected]
Abstract: Adoptive T cells immunotherapy, specifically chimeric antigen receptor T cells (CAR-T), has shown promising therapeutic efficacy in the treatment of hematologic malignancies. As extensive research on CAR-T therapies has been conducted, various challenges have emerged that significantly hampered their clinical application, including tumor recurrence, CAR-T cell exhaustion, and cytokine release syndrome (CRS). To overcome the hurdles of CAR-T therapy in clinical treatment, cell-free emerging therapies based on exosomes derived from CAR-T cells have been developed as an effective and promising alternative approach. In this review, we present CAR-T cell-based therapies for the treatment of tumors, including the features and benefits of CAR-T therapies, the limitations that exist in this field, and the measures taken to overcome them. Furthermore, we discuss the notable benefits of utilizing exosomes released from CAR-T cells in tumor treatment and anticipate potential issues in clinical trials. Lastly, drawing from previous research on exosomes from CAR-T cells and the characteristics of exosomes, we propose strategies to overcome these restrictions. Additionally, the review discusses the plight in large-scale preparation of exosome and provides potential solutions for future clinical applications.
Keywords: tumor, CAR-T cells, immune escape, exosome
Introduction
CAR-T therapy represents tremendous progress in the field of tumor treatment.1–4 After tight binding of the tumor target antigen to the single-chain variable fragment (scFv) of the specific antibody in the extracellular binding domain, the signal is transmitted through the hinge region, the transmembrane structural domain, to the intracellular structural domain and subsequently initiates activation in the T cells. Notably, the intracellular signaling domain is the initial signal for T cells activation, whereas intracellular co-stimulatory domains such as CD28/4-1BB are integral to the sustained activation and proliferation of T cells (Figure 1).5 Six CAR-T-related drugs, all of which target hematologic cancers, have been approved for commercialization by the Food and Drug Administration (FDA) to date.6 CAR-T therapy for solid tumors has been encouraged by the success of CAR-T therapy for hematological cancers. With the development of this field, many CAR therapies based on a variety of other immune cells have emerged, such as CAR-NK and CAR-M. However, despite the fact that current scientists have developed strategies and achieved remarkable remission results, there are a number of obstacles in solid tumors, such as the difficulty in infiltrating the solid tumors, T-cell exhaustion, and CRS, that have not been addressed at their root.7,8 The advent of CAR-T cell-derived exosomes offers a promising prospect for the treatment of malignancies, with the benefits of nanoscale size and cell-free nature compensating for the drawbacks of treating solid tumors and so attaining considerable remission (Figure 2).9 Most of the exosomes from CAR-T cells, which possess the properties of the parental cells, including CAR, CD3, granzyme, perforin, etc., also show promising anti-tumor effects in in vitro experiments and preclinical models and are promising to become an additional option for the treatment of tumor patients.9,10 In this review, we introduce the significant benefits of CAR-T therapy in treating tumors, discuss the limitations of the therapy and the corresponding mechanisms, and finally highlight the superior potential of exosomes from CAR-T cells in the field of tumor treatment.
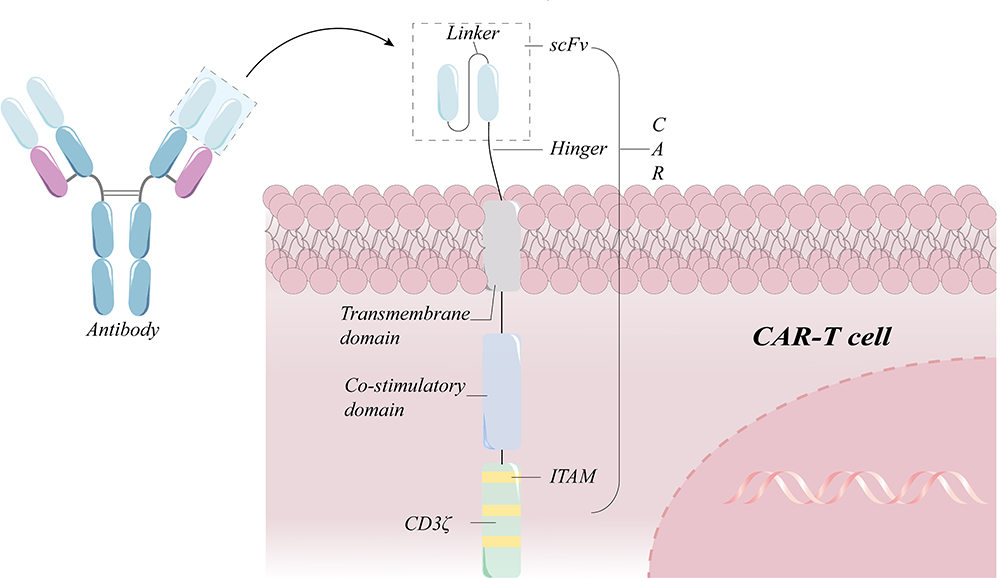 |
Figure 1 Schematic diagram of a standard second-generation CAR-T cell structure. A chimeric antigen receptor (CAR) has a heavy chain variable region (VH) and a light chain variable region (VL) in the sequence of an antibody against a target antigen. The hinge region links the transmembrane domain to the intracellular co-stimulatory domain and the CD3 signaling domain. If the scFv identifies the target antigen, it stimulates the signaling and co-stimulatory domains in order to promote sustained T cell proliferation and effector functions. |
 |
Figure 2 Biogenesis and killing mechanisms of exosomes released from CAR-T cell. The proteins on the surface of CAR-T cells enter the cell through the plasma membrane invagination and then can exchange cargo within the cell, followed by the transition of early sorters and late sorters, and finally form multivesicular bodies (MVBs), part of which enter the lysosome to be degraded and another part of which docks on the side of the plasma membrane under the action of docking proteins and is then expelled by the cell membrane, thereby releasing exosomes.11 The exosomes generated by CAR-T cells not only have CAR molecules imbedded on their surface, but they also include cytotoxic particles that can precisely target tumor cells and then be absorbed to release granzyme and trigger endogenous apoptosis. |
Limitations of CAR-T Therapy
Patients with malignant hematologic tumors have shown positive results with CAR-T therapies, and the FDA has approved six commercial CAR-T medicines, indicating the potential of this therapy in cancer treatment. However, numerous drawbacks of CAR-T therapy, particularly in the treatment of solid malignancies, have been regularly investigated.
Loss of Target Antigen
Immune escape in CAR-T-treated patients is widely reported and is currently considered a trickier dilemma in CAR-T therapies. It is reported that 50% of patients treated with CD19-CAR-T alone will experience tumor recurrence.12 Among them, the number of CAR-T cells is easily ignored but is crucial (Figure 3a). In addition, sequencing of tumor cells from relapsed patients indicated that all 12 patients had CD19 gene mutations that resulted in CD19 protein deletion.13 The expression of target antigens on tumor cells exhibited a downward trend following continuous treatment with CAR-T therapy. Tumor cells under the targeted high pressure of CAR are prone to mutating themselves, which is also a potential cause of tumor recurrence for patients in clinical trials.14 Sotillo et al showed that aberrant CD19 alternative splicing in tumor cells was the cause of tumor recurrence in patients treated with CAR-based immunotherapies that target CD19.15 Missing exon 2 and abnormalities in exons 5 and 6 are all significant for abnormal CD19 protein expression, affecting CAR’s recognition of the FMC63 (Epitopes of CD19 recognized by CAR) epitope of the CD19 protein (Figure 3b).15,16 The reduced expression of the splicing factor SRSF3 in exon 2 deletion raises the possibility that it may act as an exon retainer. These indicate that antigen evasion mechanisms include gene mutations and alternative splicing.17 Genetics plays an essential role in the stable production of proteins on the cell surface, but the role of molecular chaperones is also significant. Although research on molecular chaperones is still in its infancy, their ability to aid in the folding and translocation of proteins is a well-established fact.18,19 Breger et al found that CD81, a chaperone protein for CD19, was tightly associated with tumor recurrence by molecular analysis of CD19-negative acute lymphocytic leukemia (ALL) patients with early recurrence.20 Their findings show that the absence of accessory proteins would interfere with the normal surface expression of target antigens on tumor cells (Figure 3c).20,21 However, this is not the only mechanism of immune escape from tumor cells, as Evans et al first reported a case of a relapsed, refractory chronic lymphocytic leukemia (CLL) patient treated with CD19 protein-based CAR-T cells who later relapsed to CD19-negative plasmacytoid lymphoma (Figure 3d).22 Moreover, Jacoby et al reported that CAR-induced immunological stress on CD19 led to a variety of leukemic changes.23,24 This suggests that the targeted CAR-treated tumor cells may be able to escape from the immune cells by switching their own lineage. Further investigation revealed changes in tumor cell lineage switching markers, which showed that deletion of the Pax5 or Ebf1 transcription factors had a profound effect on B-cell gene rearrangement.25 Thus, genealogical switching is an additional mechanism that causes antigen escape in hematoma diseases, where key B-cell transcription factors or factors closely linked to them are likely to play a significant role in reversing gene reprogramming.
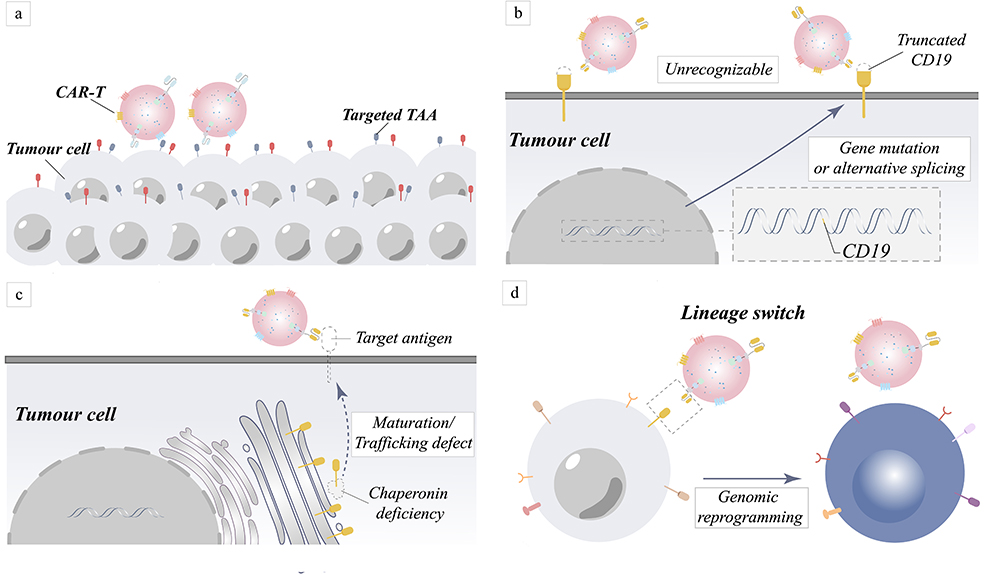 |
Figure 3 Mechanism of tumor relapse in target antigen-negative tumors caused by target antigen positivity and target antigen loss. (a) Fewer CAR-T cells may be responsible for tumor cell resistance to treatment. (b) Due to gene mutations or variable splicing, target antigens become unrecognized. (c) The absence of chaperone proteins results in improper protein maturation and membrane transport, which manifests as antigen loss. (d) Rearrangement of genes causes rearrangement of intracellular proteins, result in the loss of the target antigen. |
Decreased Antigen Identification
In addition to the loss of target antigen, the decline in the capacity of both target antigen and CAR to bind is a substantial contributor to the immune evasion of tumor cells. To evade recognition by immune cells, cancer cells also employ many approaches; under-expression of antigen-presenting MHC molecules with target antigens has become a common ploy for them.26 The discovery of the trogocytosis effect refines a further mechanism through which the recognition of tumor cells by CAR is impaired. The potential cause of the effect is still the downregulation of target antigens on the surface of tumor cells (Figure 4a).27,28 Compact and overcoming the limitations of MHC, CAR has a higher target antigen recognition threshold compared to the T cell receptor (TCR), which requires a greater amount of antigen binding to initiate recognition.29,30 Hamieh et al observed that trogocytosis action decreased the presence of recognized target antigen molecules on the cell surface of tumor cells.27 While the aforementioned are the most common mechanisms that lead to a decrease in antigen surface expression, the following scenario explains additional antigen expression downregulation pathways. Ruella et al documented a patient with Acute B Lymphoblastic Leukemia (B-ALL) whose tumor returned after nine months of treatment with CD19-targeting CAR-T cells.31 Analysis of leukemic B cells from relapsed patients who underwent CD19-targeted CAR T-cell therapy revealed that the CAR gene was accidentally introduced into the cells and that its product naturally binds to the CD19 epitope, obscuring the epitope that should be recognized by CAR (Figure 4b).32 Despite the fact that 73% of patients were in complete remission, tumor relapse was still evident and was substantially connected with a CD22 locus decrease, according to a Phase I clinical trial conducted by Fry et al.33 The results of this clinical experiment support the idea that a reduction in the variety of target antigen sites can influence the efficiency of CAR recognition and, subsequently, the effectiveness of CAR-T treatment against malignancies. However, if the number of CAR-T cells is only raised, the on-target, off-tumor (OTOT) toxicity will gradually emerge as the targeting problem develops.
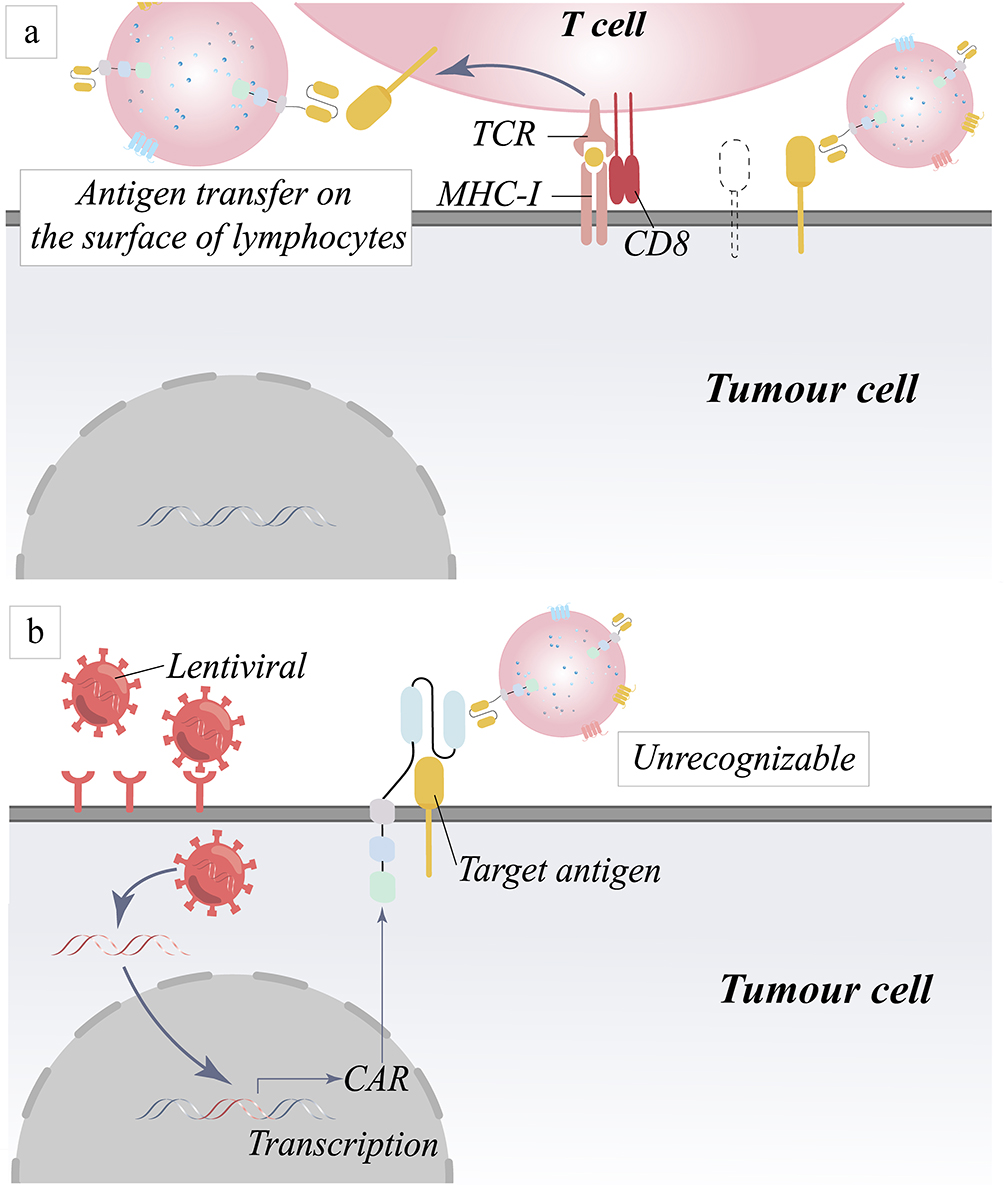 |
Figure 4 Mechanism of tumor relapse due to down-regulation of target antigen recognition. (a) Trogocytosis decreases the number of tumor antigens by transferring antigens from the surface of tumor cells to the surface of lymphocytes. (b) Masking of target antigens leads to down-regulation of recognition by CAR molecules. |
CAR-T Cell Exhaustion and Insufficient Infiltration in Solid Tumor
The initial anti-tumor potency of CAR-T treatment is astounding, but later efficacy has not been as promising. The most probable explanation is the exhaustion of CAR-T cells infiltrating solid tumors.34,35 The immunosuppressive microenvironment and poor survival environment in solid tumors are the main reasons for limiting the efficacy of CAR-T therapy; besides, the limited infiltration of CAR-T cells into the tumor also affects its efficacy.36,37 The dense stroma surface of solid tumors and the interior environment filled with weak acidity, low oxygen, and immunosuppression both affect the infiltration effect and survival of CAR-T cells, causing irreversible damage to CAR-T cells. Tumor antigens could continuously stimulate CAR-T cells and influence their development, making them susceptible to entering the exhaustion phase. Zebley et al reported the presence of exhaustion-associated epigenetic modifications found in CAR-T cells injected into ALL patients four weeks, furthermore, the results reported by Kong et al coincide with the previous studies, in which most of the exhaustion-marked T cells were also detected when tested several months after injection of a CAR-T cell product into a chronic lymphocytic leukemia (CLL) patient.38,39 TOX is reported to be a critical factor in the regulation of T-cell exhaustion (Tex), which affects the transcription factor NFAT and thus regulates Tex.34,40–42
Toxicities of the Therapy
CRS is the most common clinical complication associated with CAR-T treatment. In general, patients with CRS exhibit symptoms such as widespread fever, nausea, dyspnea, hypotension, etc., accompanied by elevated serum cytokine levels; in extreme situations, it poses a threat to the patients’ mortality. CRS is caused by the excessive proliferation and activation of T-cells, which results in the release of excessive cytokines such as IL-6 and IL-1.43,44 These cytokines activate nearby immune cells, which then release more cytokines, creating a cycle of activated inflammation and a cytokine storm in the body.45 Similarly, neurotoxins are also the most frequent toxicity in clinical trials, and generally exhibit symptoms such as aphasia, facial paralysis, and facial muscle spasms, which can be fatal in severe cases.46,47
Endeavor to Resolve the Limitations of CAR-T Therapy
Analyzing the potential reasons for the limitations of CAR-T therapy could help develop appropriate strategies to better mitigate the challenges faced by this therapy. Currently, many approaches have been reported to mitigate some of the adverse effects associated with CAR-T therapy, thus improving its therapeutic efficacy.
Designing Multi-Targeted CAR
Developing dual-targeted CARs has been reported as an effective approach to tackle the challenges associated with CAR-T therapeutic targeting.48–52 To date, five bispecific CAR constructs have been engineered and loaded onto the surface of T cells.53 Dual-targeted CARs have been designed and shown to inhibit tumor relapse in vitro and in xenograft models, while excitingly also demonstrating remarkable therapeutic efficacy in clinical trials.54,55 Similarly, a trivalent CAR-targeted product has been developed and shown to be effective in mitigating individual antigenic heterogeneity, which further boosts the recognition and killing of tumor cells and enriches the strategy of multi-targeted CARs.56
Adjusting the Affinity of CAR
Notably, adjusting the affinity of CAR is an effective approach to mitigate the decreased ability to recognize antigens. Absolutely, CARs that target several antigens need not only the selection of the appropriate target site, but also the sequential structuring of the target site based on the tumor antigen’s position. For instance, given that CD20 is expressed proximally to the tumor cell membrane, the CD20-targeting single-chain antibody could be positioned at the distal end of the CAR molecule when constructing a CAR structure for CD19 in combination with CD20.57 Furthermore, variable domain of heavy chain antibodies (VHHs), which are more adaptive to different conditions than single-chain antibodies, have attracted the interest of scientists.58 Nanobody-based CAR constructs are frequently created today and have shown encouraging results in clinical trials, with one phase I clinical trial (NCT03090659) demonstrating that a tested nanobody-based CAR-T product (LCAR-B38M) had a good safety profile and a durable response in patients.59 It is worth mentioning that ciltacabtagene autoleucel (CARVYKTI), the sixth CAR-T product approved by the FDA, is the first nanoantibody-based CAR-T cell product.60,61
Majzner et al discovered that different hinge regions could have effects that influence the response threshold of CARs and thus exhibit different levels of killing effects, suggesting that all CAR other than the scFv segment, such as hinge regions, also impact the ability of CARs to recognize tumor antigens and play an irreplaceable role.26,62 Consequently, optimizing the hinge region to promote tumor antigen recognition is a possible option. The hinge region of CARs serves to connect the extracellular structural domain to the intracellular domain, and subsequent studies suggest that it also provides the flexibility to overcome spatial constraints, hence facilitating the CAR’s ability to bind to tumor antigens.63
However, is high affinity equivalent to high antigen recognition? The analysis of CARs targeting various stages of ICAM-1 affinity by Park et al found that micromolar, lower-affinity CAR constructs exhibited greater antitumor efficacy and safety.64 However, high-affinity CARs are probably going to have unexpected negative effects. Richman et al have shown that the new CAR constructs exhibited significant anticancer efficacy and safety compared to GD2, enhancing antitumor activity against human adult neuroblastoma xenografts.65 The high affinity of CARs did detect the paracancer GD2 antigen, but significant neurotoxicity was later identified, indicating that high-affinity CARs are extremely likely to cause targeted toxicity. This suggests that there is a limit to the affinity in CARs, but it needs to be found through affinity testing experiments based on the need for different tumor antigens and fine-tuning the CAR structure with the optimal affinity to optimize recognition of tumor antigens and less binding to normal tissues to alleviate the OTOT defect.
Enhance Antigen Expression
Enhancing antigen expression is another crucial and effective approach against downregulated antigen expression. The optimal recognition of the two requires the target antigen numbers to reach a specific threshold for CAR. Otherwise, the ability to bind the two is greatly diminished.30 Hombach et al found that CD30+ hematopoietic progenitor cells (HSPC) can downregulate CD30 protein expression, thereby evading CAR detection of CD30-targeted lymphoma cells and thus avoiding attack by CAR-T cells.66 Then, artificially enhancing antigen expression is a reliable approach to countering the tumor’s own down-regulation of antigen expression to avoid being targeted by CAR. Lynn et al showed that all-trans retinoic acid (ATRA), which enhances folate receptor beta (FRβ) expression, was superior to CAR-T therapy alone in combination with FRβ-based CAR-T.67
Approaches for Artificially Modifying Tumor Cell Antigens
At present, many artificial modification approaches have been widely reported, and not only delivery vectors have become modified targets, but also tumor cells have become investigated targets. Sun et al proposed using fusion nanosystems in in-situ tumors, which use plasma membrane fusion to change the cell surface with adequate foreign antigens in an efficient and safe manner, hence offering sufficient and specific targets for CAR structure identification.68–71 More interestingly, the synergistic impact of CAR-T and the strategies for artificially modifying proteins suppressed tumor growth and prolonged survival in tumor models (Figure 5a). Notably, current research targeting antigenic modification of tumor cell surfaces is not limited to nanomodification, but also includes approaches that take advantage of the uniqueness of the tumor microenvironment to target tumor cell surfaces, such as low-pH insertion peptides (pHLIPs) depending on the weakly acidic environment of the tumor (Figure 5b).72–74
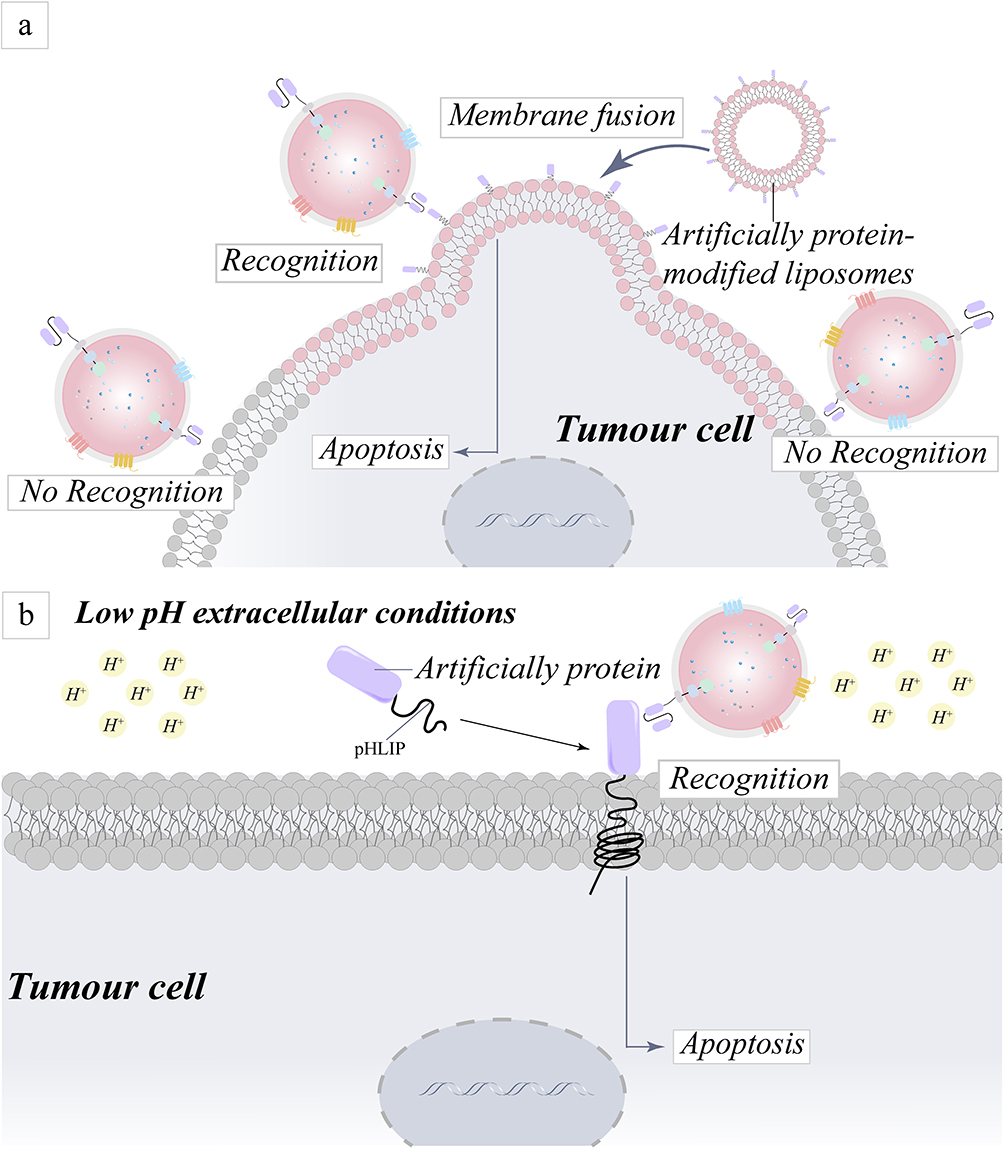 |
Figure 5 Schematic representation of the mechanism for addressing targeting challenges by artificially modifying proteins on the surface of tumor cells. (a) An approach for the precise identification and destruction of tumor cells based on the ability of liposomes to fuse cell membranes and the coating of tumor cells with artificial proteins. (b) A technique to altering tumor cell surface proteins for specific identification and death of tumor cells is reported, based on the premise that pHLIPs can be integrated into cells in an acidic environment generated by tumor cells. |
Novel Strategy Based on Exosomes Released from CAR-T Cells
CAR-T therapy, as a popular therapy for tumor treatment, is characterized by its precision, but the limitations faced with this therapy in clinical trials, such as CAR-T cells exhaustion and CRS, restricted its wide clinical application, especially in solid tumors. Recently, a novel exosome-based strategy has been developed, and holds promise for overcoming the challenges encountered in the current clinical application of CAR-T therapies.10 This strategy is currently being considered for testing in clinical trials, following its successful demonstration of significant anti-tumor effects in preclinical models.
Introduction of Exosomes
Exosomes are composed of a circular bilayer of lipid membranes as well as vesicle contents, and although previous studies have considered exosomes as mere subcellular structures produced by cellular metabolism for the sole purpose of excluding intracellular excess metabolites with no other biological function.75,76 Growing research, however, suggests that exosomes have irreplaceable functions both intracellularly and intercellularly.77
Exosome Biogenesis and Applications
The cell membrane first undergoes invagination, wrapping some of the membrane surface proteins into the cell to form the early sorting endsome. Next, the late sorting endsome is formed by exchanging cargoes such as proteins, miRNAs, and other small molecule cargoes with the intracellular Golgi apparatus. Finally, continuing to evolve intracellularly into multivesicular bodies (MVBs), some of them enter the lysosome to be broken down, and the rest enter the body fluids through exocytosis.11 Interestingly, endosomal sorting complex required for transport (ESCRT) and Vps4 proteins, which are crucial proteins involved in sorting exosomes, have also been reported.78 In any case, exosomes released from cells expressing CD47 binding to the receptor signaling regulator protein α (SIRPα) of innate immune cells such as macrophages facilitate evasion of immune clearance and thus prolong activity in vivo.76
Elimination of intracellular metabolic wastes is a common characterization of exosomes. Additionally, research has identified exosomes as an important medium of information exchange through cytoplasmic division, membrane fusion, and receptor-mediated transport, which in turn affect the metabolism and function of recipient cells.79,80 This action of exosomes links each individual cell together to form a system that synergistically regulates a number of biogenesis processes, further contributing to the internal stabilization of the organism.81 For example, exosomes secreted by NK cells contain small molecules such as granzyme B and perforin, which could boost the body’s intrinsic immunity, and similarly, exosomes secreted by tumor cells are key factors in promoting the growth and proliferation of tumor cells.82,83 Interestingly, in addition to promoting biogenesis within the body, exosomes could serve as a specific indicator for the early diagnosis of disease.84 Liquid biopsies, for example, play an essential role in cancer diagnosis and prognosis, and exosomes, which are more abundant in body fluids, are an ideal diagnostic index.85 These suggest that exosomes possess considerable potential and application, both in the maintenance of normal organismal biogenesis and in clinical applications.
In recent times, exosomes have been recognized as crucial components in cell-free therapies of regenerative medicine. Stem cell-derived exosomes have been reported to possess partial stem cell bioactivity, which can aid in bone and cartilage repair, vascular regeneration, skin repair, and even anti-skin aging and hair regeneration.86–88
The Potential of Exosomes in Tumor Therapy
Solid tumors are known to be mostly occupied by the tumor microenvironment (TME), which is an essential factor that makes solid tumors difficult to cure. In the process of remodeling TME, exosomes from tumor cells are reported to be the key to accomplishing this process. Besides, the exosomes from tumor cells could inhibit the function and growth of immune cells while also promoting the proliferation and development of cells with immunosuppressive functions, such as MDSCs, TAMs, Tregs, etc.89 This also demonstrates that exosomes from tumor cells are able to maintain promising activity and function in the TME. Therefore, strategies to reduce the release of exosomes from tumor cells have been frequently reported and have produced significant antitumor effects.90,91 Similarly, exosomes from immune cells also gain the properties of the cell of origin, making it stimulating to use exosomes released by immune cells to treat tumors with therapeutic effects such as NK cells, T cells, and CAR-T cells.10,83,92 The potential and clinical application of exosomes in cancer treatment is closely tied to their use as drug delivery systems. Firstly, exosomes are similar to liposomes, which are nanoscale materials capable of crossing various barriers, including the blood-brain barrier (BBB).93 Additionally, exosomes can enhance tumor targeting through the enhanced permeability and retention effect (EPR) of tumor cells.94 Secondly, exosomes are produced by the body’s own cells and exhibit good biocompatibility and low toxicity. This minimizes the risk of degradation and significantly improves the success rate of delivering therapeutic agents.76 Finally, exosomes can be loaded with anti-tumor chemotherapeutic drugs and easily degradable small molecule nucleic acids. The delivery methods, such as parental cell modification and engineered modification, offer diverse options to further enhance the effectiveness of tumor treatment.95
The Advantages of Exosomes from CAR-T Cells in Tumor Therapy
Exhibiting Exceptional Anti-Tumor Activity
Interestingly, the status and composition of the cargoes transported by exosomes are closely related to the cell of origin, which also transports cargoes from the mother cell, such as miRNAs, messenger RNAs, long-stranded non-coding RNAs, and proteins.75,96,97 Exosomes derived from immune cells have shown excellent results in countering tumors, such as dendritic cells (DCs), natural killer (NK) cells, and T cells, as well as CAR-T cells.92,98–101 Exosomes from CAR-T cells have been reported to have great prospects for clinical application in tumor therapy. These exosomes contain key components of CAR-T cells, including CARs that target tumor cells, granzyme, and perforin, which contribute to tumor cell killing. Therefore, the exosomes possess some of the tumor-killing capabilities of CAR-T cells. It has been observed that CAR-T cells, when stimulated by tumor cells, can release a higher quantity of exosomes.102 These exosomes exhibit a stronger ability to bind to tumors and exert an anti-tumor effect.103 Furthermore, it has been demonstrated that the stimulated exosomes possess a greater number of CARs on their surface.103 These indicate that exosomes from CAR-T cells exhibit remarkable targeting ability and killing effects on tumor cells. In addition, CAR-NK cell-derived exosomes have also been reported to exhibit CAR-NK cell-specific killing activities, demonstrating exceptional anti-tumor effects. This finding suggests that the utilization of exosomes derived from CAR-based immune cells (such as CAR-T, CAR-NK, CAR-M) is a crucial step in advancing CAR therapies towards clinical application.104
Superior Biocompatibility and Loadability
While facing solid tumors, exosomes, characterized by nanoscale dimensions, can effectively across the barrier formed by tumor-associated fibroblasts (TAFs) and dense extracellular matrix (ECM) on the surface of solid tumors.105,106 However, the tumor parenchyma is also accompanied by a hostile TME, which could be detrimental to the patient’s health. This environment is characterized by harsh conditions such as low oxygen levels, low pH, and weak acidity.107 Besides immunosuppressive checkpoint-mediated immunosuppression such as PD-L1 and CTLA-4 and suppressive soluble factors such as IL-10 and TGF-β, the tumor microenvironment (TME) also contains immunosuppressive cellular constituents such as regulatory T cells (Treg), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAM), etc.108,109 CAR-T cells facing extremely harsh conditions when entering this environment seriously affect their own situation, but it has been reported that exosomes released by CAR-T cells in this environment include RNA component of signal recognition particle 7SL1 (RN7SL1, an endogenous small RNA), which is able to activate the intrinsic immune modality in vivo through the RIG-MDA5 pathway and also limit the development of MDSCs, suggesting promising perspectives for utilizing a combination of CAR-T cells and exosomes secreted by CAR-T cells in tumor treatment.110 Clinically, researchers are struggling to overcome the initial obstacle to employing CAR-T treatment in clinical trials: cytokine release syndrome (CRS), also called cytokine storm. Despite the impressive outcomes achieved by modifying CARs and incorporating suicide genes, these additional modifications have also presented increased difficulties in the preparation process and hindered the clinical application of CAR-T cells. Nevertheless, unlike live CAR-T cells, exosomes do not respond to the surrounding environment in the same manner, allowing them to effectively avoid the occurrence of the CRS and remain unaffected by the tumor immunosuppressive microenvironment. Interestingly, exosomes themselves are excellent nanoscale drug carriers that are not only compatible and safe in vivo but have also been shown to carry additional chemotherapeutic drugs to enhance the antitumor effect.95,111 The exosomes carry therapeutic drugs and miRNAs or siRNAs, enabling them to target not only superficial tumor cells but also deep-seated tumor cells, effectively killing tumors.76,112,113 Exosomes from CAR-T cells exhibit sustained viability in the vasculature and tumor microenvironment in addition to being loaded, suggesting that the development of an engineered CAR-T exosome strategy is likely to further refine this therapy towards the goal of curing tumors. To sum up, this innovative strategy is highly efficient, precise, and practical in tumor therapy and has a broad clinical application, which likely provides patients with new option for tumor treatment.
Perspectives on CAR-T Cell-Based Exosomes
Ability to Inherit More Specific CARs
Innovative strategies to limiting tumor growth using exosomes from CAR-T cells are in development. More recently, the use of multi-targeted CAR strategies to break through the burden of tumor recurrence has been proven to be feasible, and by virtue of exosome inheritance, exosomes from multi-targeted CAR-T cells probably have multiple targets on their surface. Accordingly, modified CAR-T cell-derived exosomes are not only more effective but also safer for patient treatment. These new “CAR-T” products should also inherit greater potency in their released exosomes and combine safety and efficacy as a result of initiatives to design unique CAR to overcome the limitations of CAR-T therapies. Exosomes obtained using CAR-T cells modified from existing strategies should also inherit the corresponding properties. Furthermore, fine-tuning the variable binding region and hinge region, adjusting the fitness affinity, and other modifications of the CAR should be found on the surface of its released exosomes, which can further enhance its binding ability to tumor cells. Overall, the additional modification of the CAR strategy should be able to inherit its characteristics from the exosomes it releases, which can be utilized as a continuation of the advantages of exosomes in tumor therapy. However, the usefulness of exosomes generated by these cells must be confirmed in future investigations and preclinical models. In addition to the heritability of exosomes, their loadability cannot be ignored. Consequently, the utilization of engineered alteration of exosomes to resolve the problem at hand is a more appropriate technique at present. Similarly, chemicals such as ATRA that lead to high antigen expression on the surface of tumor cells can be loaded into exosomes formed from the discharge of CAR-T cells. Delivering gene therapy and immunotherapy to tumors by loading miRNAs or RNA interference (RNAi) into exosomes is also another way to enhance their anti-tumor effects.
Correcting Mutations and Variable Splicing
Notably, challenges in targeting may still persist, so we will utilize the properties of exosomes to look forward to strategies for mitigation. Target antigen loss-mediated immune escape has been reported to be associated with splicing abnormalities and gene mutations, which may be the greatest obstacle and difficulty in delivering exosomes from CAR-T cells for future widespread clinical applications. Alanis et al used U1 small nuclear RNA (SnRNA) to correct splicing abnormalities and also exploited an exon-specific U1SnRNA (ExSpeU1), which is capable of correcting different types of mutations leading to exon skipping and has successfully corrected splicing abnormalities in several F9 mutations, allowing complete restoration of IX factor levels.114 Loading of functional U1 hybrid plasmids into CAR-T cell-derived exosomes is achievable due to the loading nature of exosomes, which is likely to provide a new way of thinking to address future medical palliative targeting issues. The resulting engineered exosomes could overcome the challenge of target antigen loss due to splicing abnormalities, which unquestionably improves the efficacy and durability of the antitumor from the root. Also, abnormal splicing is also associated with unusual expression of splicing factors, and Sotillo et al predicted the abnormal expression of the splicing factor SRSF3 leading to loss of CD19 using Avispa’s algorithm, in addition to verifying by immunoblotting that with increasing knockdown of SRSF3 in P493-6 cells and NALM6B cells, exon 2 jumping increases significantly with the knockdown, and also showing that the correlation between the splicing factor SRSF3 and target antigens has only been demonstrated in B-ALL, and the correlation with splicing factors in different tumor cells needs to be explored in subsequent experiments.15 Using the loading properties of exosomes, it is also possible to correct variable splicing by loading small molecules of kinase or kinase inhibitors that regulate splicing factor activity into exosomes released by CAR-T cells. The underlying cause of cancer recurrence is intrinsically related to gene mutation as a result of the growing popularity of genomics and scientists’ extensive research on genes. Research that can address genetic mutations, represented by gene editing, is also in full swing. Gene editing is a genetic manipulation technique that can modify DNA sequences at the genomic level, and it is currently used in experiments to treat neurological diseases (most notably Alzheimer’s disease), which is feasible for future clinical use but has many issues that must be resolved before it can be used clinically.115,116 This method still confronts numerous problems, including the non-negligible immunogenicity of fusion enzymes and delivery barriers, which must be overcome for clinical application to be effective. Fortunately, many reports of loading CRISPR/Cas9-like gene editing technologies into exosomes to address issues of transport and immunogenicity have been published, and increasing evidences suggest that exosomes could be used as an effective delivery vector for gene editing drugs and also for targeting cancers.117–119 The “organic combination” of gene editing technology and exosomes is highly feasible, replacing the carried RNA sequence with the RNA sequence before the mutation caused by the overload of the tumor caused by CAR protein targeting, thereby completing the fundamental correction of the antigen escape caused by the mutation and permitting the fusion enzyme to serve as a “checker”. Once tumor cells mutate as a result of CAR targeting pressure, the fusion enzyme will perform a corrective function at the genetic level, forcing them to return to their pre-mutation proteins so that the exosomes released from CAR-T cells can continue to target tumor cells, thereby decreasing the likelihood of tumor recurrence and increasing the cure rate. These engineering strategies will further alleviate the limitations of exosomes in treating tumors and accelerate translational applications in clinical patients.
The Challenges of Exosomes from CAR-T Cells in Clinical Applications
Exosomes derived from CAR-T cells have shown remarkable potential in tumor therapy. They possess inherent anti-tumor activity and can serve as effective carriers for delivering chemotherapeutic drugs, thereby enhancing the inhibition of tumor growth. It is noteworthy that these exosomes do not express PD-1, indicating their resilience to the harsh tumor environment.10 Consequently, they hold great promise as active drugs for clinical application on tumor therapy. However, it is important to acknowledge that their clinical utility is currently constrained by the limitations associated with exosomes. The clinical application of exosomes has several challenges that need to be addressed. Firstly, large-scale preparation of exosomes is expensive, as it requires the use of bioreactors and costly separation methods. Additionally, the transportation and storage of exosomes pose a major challenge, as prolonged storage can lead to aggregation and reduced activity.76 Secondly, the heterogeneity of prepared exosomes can result in low purity and reduced tumor-killing activity. Therefore, there is an urgent need to develop a device for preparing uniform and purified exosomes.120 Thirdly, modifying and loading exosomes with small-molecule cargoes can enhance their affinity and lethality towards tumor cells. However, the loading efficiency of engineered exosomes is generally low, possibly due to the presence of other proteins and cargoes. Furthermore, the safety and immunogenicity of engineered exosomes need to be carefully considered.76,121 Finally, the application of exosomes from CAR-T cells in clinical trials still lacks sufficient data to justify their use, indicating that further research is required.
In summary, the following limitations are associated with exosomes from CAR-T cells: (1) high cost of isolation of exosomes; (2) storage of exosomes; (3) heterogeneity of exosomes; (4) high efficiency of engineered exosomes; (5) early stage of development in clinical trials; (6) scale-up of exosomes from CAR-T cells. While the advantages and clinical potential of CAR-T cell-derived exosomes in anti-tumor applications have been acknowledged by the public, their current clinical applications are primarily hindered by the challenges in scaling up the preparation process. In the following section, we will specifically discuss the scale-up preparation of exosomes derived from CAR-T cells.
Scale-Up Preparation of Exosomes from CAR-T Cells
Scale-Up Production of CAR-T Cells
In addition to producing exosomes at scale, mass production of CAR-T cells is also an important part of generating CAR-T cell exosomes in large quantities. Developing allogeneic CAR-T cell strategies could reduce the cost of producing CAR-T cells, which would also likely alleviate this dilemma of mass-producing CAR-T cells. Nevertheless, the infusion of allogeneic CAR-T cells into patients, while lowering costs, is associated with weaker efficacy than autologous, lower levels of cell expansion, and greater susceptibility to toxicity such as graft-versus-host reactions, all of which greatly limit the clinical utility of CAR-T therapies.122,123 Using expensive and time-consuming viral vectors is another factor hindering the development of CAR-T cells, which could be mitigated using the Suppa CAR system.124,125 The high cost of construction is commonly attributed to the specificity of CARs, the virus-based synthesis of CAR-T cells, and the availability of T cells of individual patient origin. The CAR could be updated modularly by introducing a bispecific fusion molecule between the transmembrane domain and the antigen recognition domain, which on the one hand connects the transmembrane domain to the intracellular signaling domain via a hinge, and together they can serve as a basic module (signaling module), and on the other hand, a binding domain of the fusion molecule can directly bind to the antigen recognition domain as a variable module (exchange module). Therefore, just the switching modules need to be redesigned to be able to compose the desired CAR.126 One strategy is to use a “zipper” structure for regulation, which consists of two parts: a leucine zipper of the receptor attached to a structure below the transmembrane domain of the CAR, and a homologous leucine zipper bound to the scFv fragment, making this the only way to form a complete signaling pathway. By culturing at scale the recipient T cells carrying the intracellular stimulatory domain of the leucine zipper, collecting the exosomes, purifying the homologous leucine zipper connecting the different single-chain antibodies, and identifying the two, we could obtain CAR-T cell exosomes with a complete CAR chimeric on the surface, which is more cost-effective and only requires the preparation of the antigen recognition region adapted to the cancer target to be used (Figure 6a).125 Similarly, biotin-binding immune receptors (BBIR) with novel epitope tags such as FITC and 5B9 can be utilized to generate exosomes for universal CAR-T cells (Figure 6b).127,128 Another approach to reducing the cost of CAR-T cell production could be to integrate target genes into T cells by replacing high-cost viral particles.124 Transposon-system-based preparation of CAR-T cells has been developed, which is represented by the Sleeping Beauty transposon (SB) and Piggyback transposon, having the biggest advantages over virus-based CAR-T cells in terms of being more cost-effective and safer, allowing for large-scale production and lower production costs.129–131
 |
Figure 6 Schematic diagram of the general-purpose modular CAR structure. (a) The leucine zipper joins the scFv segment to produce a full CAR structure, whereas its receptor connects to the intracellular structural domain of the CAR structure. (b) The scFv fragment is modified by biotin, and streptavidin is attached to the intracellular structural domain of the CAR molecule, forming a complete CAR once the two are combined. |
Scale-Up Production of Exosomes
Exosomes, as a natural nanocarrier, have shown extraordinary advantages and achieved remarkable results in the field of drug delivery, with 355 clinical trials on exosomes registered to date (https://clinicaltrials.gov), but unlike artificial nanomaterials, such as nanoliposomes, most exosomes are still “locked” in laboratories and are not available for public service.132,133 What contributes to this is the fact that exosomes face the dilemma of mass production, and similarly, exosomes from CAR-T cells suffer from this restriction. Typical strategies for capturing exosomes include differential centrifugation, density gradient centrifugation, ultrafiltration, and immunoassay, which could obtain exosomes with relatively impressive purity and bioactivity but are restricted to scale-up preparations. The novel strategy developed not only captures exosomes but more importantly supports large-scale preparation requirements such as tangential flow filtration (TFF), which is considered to be the most likely strategy for large-scale preparation of Current Good Manufacturing Practice (cGMP)-grade exosomes.134,135 Meanwhile, the development of artificial exosomes also provides a new strategy to realize the scale-up preparation of exosomes, which consists of three parts: nanovesicles (NVs), analog exosomes (EMs), and hybrid exosomes (HE). A top-down approach to the production of NV, which has the advantage that this strategy contains proteins, nucleic acids, and other proteins from the parental cells that largely retain the characteristics of the CAR-T cell exosome, is considered feasible.136 The most common approach to producing NVs on a top-down scale is the extrusion-based approach. Jang et al obtained NVs by sequential extrusion at 10 m, 5 m, and 1 m followed by ultracentrifugation, which not only had the same properties as exosomes but also had a 100-fold higher yield, and they demonstrated that nanovesicles can be loaded with chemotherapeutic drugs and have comparable tumoricidal effects in vivo and in vitro compared to exosomes.137 Although the NVs obtained by extrusion are closer to the desired exosomes, they remain costly to obtain using ultracentrifugation. The field of scale-up preparation of exosomes is changing radically with the development of nanomedicine. In a follow-up study, Jo et al developed a membrane filtration device for centrifugation, where cells are stretched and ruptured by centrifugal force to eventually be assembled into NVs, using this method to allow for large-scale production of NVs.138 The NVs prepared by this device showed exosome-like structures and higher productivity, saving the cost of labor even more. Other innovative methods of preparing exosomes at scale using the nitrogen-air method, acoustics, and cellular bubbles are also under development. Ingato et al discovered that the secretion of natural NVs was stimulated by adding paraformaldehyde and dithiothreitol to a cell culture membrane that had been blasted with a sulfhydryl closure solution and that the NVs produced by the “bubbles” were more homogeneous and smaller in size while retaining their normal shape.139 The stability and economic advantages of employing this technique to create NVs on a wide scale significantly highlight the viability and necessity of adopting NVs as a substitute for natural exosomes.
Conclusion
CAR-T therapy has demonstrated effectiveness and potential as a treatment for tumors. However, challenges such as tumor recurrence, CAR-T cell exhaustion, and CRS have been observed in both preclinical models and clinical trials, significantly restricting the clinical application of this therapy. An exosome-dependent cell-free-based tumor therapy could greatly alleviate these limitations. Recent studies have shown that exosomes from CAR-T cells identified as the most promising resolution to overcome the hurdle of CAR-T therapy. The loaded character of the exosomes allows further refinement of the therapeutic properties, and they can also inherit parental CARs, leading to the possibility of using CAR-T cell-derived exosomes in clinical trials. To address the challenges associated with CAR and tumor antigen targeting, researchers propose innovative approaches for exosome binding. Loading chimeric enzymes into exosomes, regulating abnormally spliced kinases, and inheriting novel CARs that could be developed in this field are all possible strategies for curing tumors. Alternatively, exosomes prepared through a top-down strategy can significantly reduce costs, making them a more economical and rational solution. Although exosomes from CAR-T cells are increasingly showing great application potential in the field of tumor treatment in the future, the issue of the large-scale, uniform, stable, and multi-component, controllable preparation of exosomes is still an urgent problem to be solved. Additionally, revealing the sorting mechanism of exosome “cargo” is a vital factor in improving the druggability of exosomes for clinical application.
Consent for Publication
All authors participated in reviewing the article and consented to publication.
Acknowledgments
This work was financially supported by the Key R&D project of Henan Province (221111310600), National Key Research and Development Program of China (2022YFE132800) and Special Foundation for Basic Research Program of Higher Education Institutions of Henan Province (22ZX005).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kong YJ, Tang L, You Y, et al. Analysis of causes for poor persistence of CAR-T cell therapy in vivo. Front Immunol. 2023;14:1.
2. Naeem M, Hazafa A, Bano N, et al. Explorations of CRISPR/Cas9 for improving the long-term efficacy of universal CAR-T cells in tumor immunotherapy. Life Sci. 2023;2023:316.
3. Baker DJ, Arany Z, Baur JA, et al. CAR T therapy beyond cancer: the evolution of a living drug. Nature. 2023;619(7971):707–715. doi:10.1038/s41586-023-06243-w
4. Ma L, Dichwalkar T, Chang JYH, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365(6449):162–+. doi:10.1126/science.aav8692
5. Byun JM. Practical issues in CAR T-cell therapy. Blood Res. 2023;58(S1):S11–S2. doi:10.5045/br.2023.2023015
6. Flugel CL, Majzner RG, Krenciute G, et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol. 2023;20(1):49–62. doi:10.1038/s41571-022-00704-3
7. Chohan KL, Siegler EL, Kenderian SS. CAR-T Cell Therapy: the Efficacy and Toxicity Balance. Curr Hematol Malignancy Rep. 2023;18(2):9–18. doi:10.1007/s11899-023-00687-7
8. Reinhard K, Rengstl B, Oehm P, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. 2020;367(6476):446–+. doi:10.1126/science.aay5967
9. Yang PX, Cao XJ, Cai HL, et al. The exosomes derived from CAR-T cell efficiently target mesothelin and reduce triple-negative breast cancer growth. Cell Immunol. 2021;2021:360.
10. Fu W, Lei C, Liu S, et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat Commun. 2019:10. doi:10.1038/s41467-018-07709-6
11. Kalluri R, Lebleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478):640–+. doi:10.1126/science.aau6977
12. Chavez JC, Bachmeier C, Kharfan-Dabaja MA. CAR T-cell therapy for B-cell lymphomas: clinical trial results of available products. Ther Adv Hematol. 2019;2019:10.
13. Orlando EJ, Han X, Tribouley C, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nature Med. 2018;10:24.
14. Walsh Z, Ross S, Fry TJ. Multi-Specific CAR Targeting to Prevent Antigen Escape. Curr Hematol Malignancy Rep. 2019;14(5):451–459. doi:10.1007/s11899-019-00537-5
15. Sotillo E, Barrett DM, Black KL, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discovery. 2015;5(12):1282–1295. doi:10.1158/2159-8290.CD-15-1020
16. Fischer J, Paret C, El Malki K, et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J Immunother. 2017;40(5):187–195. doi:10.1097/CJI.0000000000000169
17. Yang X, Wei J, Zhou J. Overcoming resistance to anti-CD19 CAR T-cell therapy in B-cell malignancies. Hematol Oncol. 2022;40(5):821–834. doi:10.1002/hon.3036
18. Reinle K, Mogk A, Bukau B. The Diverse Functions of Small Heat Shock Proteins in the Proteostasis Network. J Mol Biol. 2022;434(1):167157. doi:10.1016/j.jmb.2021.167157
19. Kim YE, Hipp MS, Bracher A, et al. Molecular Chaperone Functions in Protein Folding and Proteostasis [M]//KORNBERG R D. Annu Rev Biochem. 2013;82(1):323–355. doi:10.1146/annurev-biochem-060208-092442
20. Braig F, Brandt A, Goebeler M, et al. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood. 2017;129(1):100–104. doi:10.1182/blood-2016-05-718395
21. Huo C-D, Yang J, Gu Y-M, et al. Overcome tumor relapse in CAR T cell therapy. Clin Transl Oncol. 2022;24(10):1833–1843. doi:10.1007/s12094-022-02847-2
22. Evans AG, Rothberg PG, Burack WR, et al. Evolution to plasmablastic lymphoma evades CD19-directed chimeric antigen receptor T cells. Br J Haematol. 2015;171(2):205–209. doi:10.1111/bjh.13562
23. Gardner R, Wu D, Cherian S, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127(20):2406–2410. doi:10.1182/blood-2015-08-665547
24. Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8 + T cells. J Exp Med. 2005;202(7):907–912. doi:10.1084/jem.20050732
25. Jacoby E, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. 2016;2016:7.
26. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21(3):145–161. doi:10.1038/s41568-020-00323-z
27. Hamieh M, Dobrin A, Cabriolu A, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568(7750):112–+. doi:10.1038/s41586-019-1054-1
28. Miyake K, Karasuyama H. The Role of Trogocytosis in the Modulation of Immune Cell Functions. Cells. 2021;10(5):1255. doi:10.3390/cells10051255
29. Walker AJ, Majzner RG, Zhang L, et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol Ther. 2017;25(9):2189–2201. doi:10.1016/j.ymthe.2017.06.008
30. Watanabe K, Terakura S, Martens AC, et al. Target Antigen Density Governs the Efficacy of Anti-CD20-CD28-CD3 zeta Chimeric Antigen Receptor-Modified Effector CD8(+) T Cells. J Iimmunol. 2015;194(3):911–920. doi:10.4049/jimmunol.1402346
31. Ruella M, Barrett DM, Shestova O, et al. A cellular antidote to specifically deplete anti-CD19 chimeric antigen receptor–positive cells. Blood. 2020;135(7):505–509. doi:10.1182/blood.2019001859
32. Ruella M, Xu J, Barrett DM, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nature Med. 2018;24(10):1499–+. doi:10.1038/s41591-018-0201-9
33. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nature Med. 2018;24(1):20–+. doi:10.1038/nm.4441
34. Gumber D, Wang LD. Improving CAR-T immunotherapy: overcoming the challenges of T cell exhaustion. Ebiomedicine. 2022;2022:77.
35. Weber EW, Parker KR, Sotillo E, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. 2021;372(6537):49–+. doi:10.1126/science.aba1786
36. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4). doi:10.1038/s41408-021-00459-7
37. Allen GM, Frankel NW, Reddy NR, et al. Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science. 2022;378(6625):1186–+. doi:10.1126/science.aba1624
38. Zebley CC, Brown C, Mi T, et al. CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep. 2021;37(9):110079. doi:10.1016/j.celrep.2021.110079
39. Kong WM, Dimitri A, Wang WL, et al. BET bromodomain protein inhibition reverses chimeric antigen receptor extinction and reinvigorates exhausted T cells in chronic lymphocytic leukemia. J Clin Investig. 2021;131(16). doi:10.1172/JCI145459
40. Yin XC, He LF, Guo ZG. T-cell exhaustion in CAR-T -cell therapy and strategies to overcome it. Immunology. 2023;169(4):400–411. doi:10.1111/imm.13642
41. Kim K, Park S, Park SY, et al. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 2020;12(1). doi:10.1186/s13073-020-00722-9
42. Khan O, Giles JR, McDonald S, et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature. 2019;571(7764):211–+. doi:10.1038/s41586-019-1325-x
43. Zhang HH, Lv XF, Kong QF, et al. IL-6/IFN-γ double knockdown CAR-T cells reduce the release of multiple cytokines from PBMCs in vitro. Hum Vaccines Immunother. 2022;18(1):1–14. doi:10.1080/21645515.2021.2016005
44. Moriyama S, Fukata M, Yokoyama T, et al. Case Report: cardiac Tamponade in Association With Cytokine Release Syndrome Following CAR-T Cell Therapy. Front Cardiovasc Med. 2022;2022:9.
45. Riddell SR. Adrenaline fuels a cytokine storm during immunotherapy. Nature. 2018;564(7735):194–196. doi:10.1038/d41586-018-07581-w
46. Rubin DB, Al Jarrah A, Li K, et al. Clinical Predictors of Neurotoxicity After Chimeric Antigen Receptor T-Cell Therapy. JAMA neurol. 2020;77(12):1536–1542. doi:10.1001/jamaneurol.2020.2703
47. Lee K, Paek H, Ai L, et al. Treatment Profile of CAR-T Cell Therapy Induced Cytokine Release Syndrome and Neurotoxicity: insights from Real-World Evidence. Blood. 2022;140(Supplement 1):12750–12752. doi:10.1182/blood-2022-166716
48. Cronk RJ, Zurko J, Shah NN. Bispecific Chimeric Antigen Receptor T Cell Therapy for B Cell Malignancies and Multiple Myeloma. Cancers. 2020;12(9):2523. doi:10.3390/cancers12092523
49. Gardner RA, Annesley C, Wilson A, et al. Efficacy of SCRI-CAR19x22 T cell product in B-ALL and persistence of anti-CD22 activity. J clin oncol. 2020;38(15_suppl):3035. doi:10.1200/JCO.2020.38.15_suppl.3035
50. Schneider D, Xiong Y, Wu DR, et al. Leukemia Cell Surface Antigen Modulation Induced By Dual CD19/CD20 Chimeric Antigen Receptor (CAR)-T Cells. Biol Blood Marrow Transplant. 2017;23(3):S209–S10. doi:10.1016/j.bbmt.2016.12.418
51. Pan J, Zuo S, Deng B, et al. Sequential CD19-22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood. 2020;135(5):387–391. doi:10.1182/blood.2019003293
52. Feng YR, Liu X, Li X, et al. Novel BCMA-OR-CD38 tandem-dual chimeric antigen receptor T cells robustly control multiple myeloma. Oncoimmunology. 2021;10(1). doi:10.1080/2162402X.2021.1959102
53. Xie B, Li Z, Zhou J, et al. Current Status and Perspectives of Dual-Targeting Chimeric Antigen Receptor T-Cell Therapy for the Treatment of Hematological Malignancies. Cancers. 2022;14(13):3230. doi:10.3390/cancers14133230
54. Ruella M, Barrett DM, Kenderian SS, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Investig. 2016;126(10):3814–3826. doi:10.1172/JCI87366
55. Amrolia PJ, Wynn R, Hough R, et al. Simultaneous Targeting of CD19 and CD22: phase I Study of AUTO3, a Bicistronic Chimeric Antigen Receptor (CAR) T-Cell Therapy, in Pediatric Patients with Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia (r/r B-ALL): Amelia Study. Blood. 2018;2018:132.
56. Bielamowicz K, Fousek K, Byrd TT, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology. 2018;20(4):506–518. doi:10.1093/neuonc/nox182
57. Zah E, Lin M-Y, Silva-Benedict A, et al. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res. 2016;4(6):498–508. doi:10.1158/2326-6066.CIR-15-0231
58. Muyldermans S. Nanobodies: natural Single-Domain Antibodies [M]//KORNBERG R D. Annu Rev Biochem. 2013;82(1):775–797. doi:10.1146/annurev-biochem-063011-092449
59. Zhao WH, Liu J, Wang BY, et al. A Phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;2018:11.
60. Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398(10297):314–324. doi:10.1016/S0140-6736(21)00933-8
61. Kozani PS, Naseri A. Nanobody-based CAR-T cells for cancer immunotherapy. Biomarker Res. 2022;10(1):1.
62. Majzner RG, Rietberg SP, Sotillo E, et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discovery. 2020;10(5):702–723. doi:10.1158/2159-8290.CD-19-0945
63. Fujiwara K, Tsunei A, Kusabuka H, et al. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells. 2020;9(5):1182. doi:10.3390/cells9051182
64. Park S, Shevlin E, Vedvyas Y, et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. 2017:7. doi:10.1038/s41598-017-00035-9
65. Richman SA, Nunez-Cruz S, Moghimi B, et al. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol Res. 2018;6(1):36–46. doi:10.1158/2326-6066.CIR-17-0211
66. Hombach AA, Gorgens A, Chmielewski M, et al. Superior Therapeutic Index in Lymphoma Therapy: CD30(+) CD34(+) Hematopoietic Stem Cells Resist a Chimeric Antigen Receptor T-cell Attack. Mol Ther. 2016;24(8):1423–1434. doi:10.1038/mt.2016.82
67. Lynn RC, Poussin M, Kalota A, et al. Targeting of folate receptor beta on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood. 2015;125(22):3466–3476. doi:10.1182/blood-2014-11-612721
68. Sun ZZ, Li RT, Shen Y, et al. In situ antigen modification-based target-redirected universal chimeric antigen receptor T (TRUE CAR-T) cell therapy in solid tumors. J Hematol Oncol. 2022;15(1). doi:10.1186/s13045-022-01246-y
69. Csiszar A, Hersch N, Dieluweit S, et al. Novel Fusogenic Liposomes for Fluorescent Cell Labeling and Membrane Modification. Bioconjugate Chem. 2010;21(3):537–543. doi:10.1021/bc900470y
70. Kim B, Pang HB, Kang J, et al. Immunogene therapy with fusogenic nanoparticles modulates macrophage response to Staphylococcus aureus. Nat Commun. 2018:9. doi:10.1038/s41467-017-01881-x
71. Lee J, Kim J, Jeong M, et al. Liposome-Based Engineering of Cells To Package Hydrophobic Compounds in Membrane Vesicles for Tumor Penetration. Nano Lett. 2015;15(5):2938–2944. doi:10.1021/nl5047494
72. Svoronos AA, Engelman DM. Pharmacokinetic modeling reveals parameters that govern tumor targeting and delivery by a pH-Low Insertion Peptide (pHLIP). Proc Natl Acad Sci USA. 2021;118(1). doi:10.1073/pnas.2016605118
73. Slaybaugh G, Weerakkody D, Engelman DM, et al. Kinetics of pHLIP peptide insertion into and exit from a membrane. Proc Natl Acad Sci USA. 2020;117(22):12095–12100. doi:10.1073/pnas.1917857117
74. Liberti MV, Locasale JW. The Warburg Effect: how Does it Benefit Cancer Cells? Trends Biochem Sci. 2016;41(3):211–218. doi:10.1016/j.tibs.2015.12.001
75. Zhang HR, Lu J, Liu J, et al. Advances in the discovery of exosome inhibitors in cancer. J Enzyme Inhib Med Chem. 2020;35(1):1322–1330. doi:10.1080/14756366.2020.1754814
76. Kar R, Dhar R, Mukherjee S, et al. Exosome-Based Smart Drug Delivery Tool for Cancer Theranostics. ACS Biomater Sci Eng. 2023;9(2):577–594. doi:10.1021/acsbiomaterials.2c01329
77. Ferreira D, Moreira JN, Rodrigues LR. New advances in exosome-based targeted drug delivery systems. Crit Rev Oncol Hematol. 2022;2022:172.
78. Villarroya-Beltri C, Baixauli F, Gutiérrez-Vázquez C, et al. Sorting it out: regulation of exosome loading. Semi Cancer Biol. 2014;28:3–13. doi:10.1016/j.semcancer.2014.04.009
79. Cariello M, Squilla A, Piacente M, et al. Drug Resistance: the Role of Exosomal miRNA in the Microenvironment of Hematopoietic Tumors. Molecules. 2023;28(1):1.
80. Lau NCH, Yam JWP. From Exosome Biogenesis to Absorption: key Takeaways for Cancer Research. Cancers. 2023;15(7):1992. doi:10.3390/cancers15071992
81. Hamzah RN, Alghazali KM, Biris AS, et al. Exosome Traceability and Cell Source Dependence on Composition and Cell-Cell Cross Talk. Int J Mol Sci. 2021;22(10):5346. doi:10.3390/ijms22105346
82. Bebelman MP, Smit MJ, Pegtel DM, et al. Biogenesis and function of extracellular vesicles in cancer. Pharmacol Ther. 2018;188:1–11. doi:10.1016/j.pharmthera.2018.02.013
83. Kim HY, Min H-K, Song H-W, et al. Delivery of human natural killer cell-derived exosomes for liver cancer therapy: an in vivo study in subcutaneous and orthotopic animal models. Drug Delivery. 2022;29(1):2897–2911. doi:10.1080/10717544.2022.2118898
84. Li X, Corbett AL, Taatizadeh E, et al. Challenges and opportunities in exosome research-Perspectives from biology, engineering, and cancer therapy. APL Bioeng. 2019;3(1). doi:10.1063/1.5087122
85. Yu D, Li Y, Wang M, et al. Exosomes as a new frontier of cancer liquid biopsy. Mol Cancer. 2022;21(1). doi:10.1186/s12943-022-01509-9
86. Huang JH, Xiong JY, Yang L, et al. Cell-free exosome-laden scaffolds for tissue repair. Nanoscale. 2021;13(19):8740–8750. doi:10.1039/D1NR01314A
87. Kang Y, Xu C, Meng L, et al. Exosome-functionalized magnesium-organic framework-based scaffolds with osteogenic, angiogenic and anti-inflammatory properties for accelerated bone regeneration. Bioact Mate. 2022;18:26–41. doi:10.1016/j.bioactmat.2022.02.012
88. Ha DH, Kim H-K, Lee J, et al. Mesenchymal Stem/Stromal Cell-Derived Exosomes for Immunomodulatory Therapeutics and Skin Regeneration. Cells. 2020;9(5):1157. doi:10.3390/cells9051157
89. Xie F, Zhou X, Fang MY, et al. Extracellular Vesicles in Cancer Immune Microenvironment and Cancer Immunotherapy. Adv Sci. 2019;6(24). doi:10.1002/advs.201901779
90. Ding Y-N, Ding H-Y, Li H, et al. Photosensitive small extracellular vesicles regulate the immune microenvironment of triple negative breast cancer. Acta Biomater. 2023;167:534–550. doi:10.1016/j.actbio.2023.06.004
91. Xie LS, Li J, Wang GH, et al. Phototheranostic Metal-Phenolic Networks with Antiexosomal PD-L1 Enhanced Ferroptosis for Synergistic Immunotherapy. J Am Chem Soc. 2022;144(2):787–797. doi:10.1021/jacs.1c09753
92. Zhou QJ, Wei SY, Wang H, et al. T cell-derived exosomes in tumor immune modulation and immunotherapy. Front Immunol. 2023;2023:14.
93. Choi H, Choi K, Kim D-H, et al. Strategies for Targeted Delivery of Exosomes to the Brain: advantages and Challenges. Pharmaceutics. 2022;14(3):672. doi:10.3390/pharmaceutics14030672
94. Li SD, Hou X, Qi HZ, et al. Exosomes Provide Naturally Occurring Endogenous Nanocarriers for Effective Drug Delivery Strategies. Progress in Chemistry. 2016;28(2–3):353–362.
95. Zhang M, Shao W, Yang T, et al. Conscription of Immune Cells by Light-Activatable Silencing NK-Derived Exosome (LASNEO) for Synergetic Tumor Eradication. Adv Sci. 2022;9(22):1.
96. Beit-Yannai E, Tabak S, Stamer WD. Physical exosome: exosome interactions. J Cell Mol Med. 2018;22(3):2001–2006. doi:10.1111/jcmm.13479
97. Zheng JY, Hu XX, Zeng YY, et al. Review of the advances in lipid anchors-based biosensors for the isolation and detection of exosomes. Anal Chim Acta. 2023;2023:1263.
98. Lu JM, Wei NA, Zhu SL, et al. Exosomes Derived From Dendritic Cells Infected With Toxoplasma gondii Show Antitumoral Activity in a Mouse Model of Colorectal Cancer. Front Oncol. 2022;2022:12.
99. Di Pace AL, Tumino N, Besi F, et al. Characterization of Human NK Cell-Derived Exosomes: role of DNAM1 Receptor in Exosome-Mediated Cytotoxicity against Tumor. Cancers. 2020;12(3):661. doi:10.3390/cancers12030661
100. Haque S, Vaiselbuh SR. CD19 Chimeric Antigen Receptor-Exosome Targets CD19 Positive B-lineage Acute Lymphocytic Leukemia and Induces Cytotoxicity. Cancers. 2021;13(6):1401. doi:10.3390/cancers13061401
101. Chen Z, Yuan R, Hu SY, et al. Roles of the Exosomes Derived From Myeloid-Derived Suppressor Cells in Tumor Immunity and Cancer Progression. Front Immunol. 2022;2022:13.
102. Blanchard N, Lankar D, Faure F, et al. TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/zeta complex. J Iimmunol. 2002;168(7):3235–3241. doi:10.4049/jimmunol.168.7.3235
103. Calvo V, Izquierdo M. T Lymphocyte and CAR-T Cell-Derived Extracellular Vesicles and Their Applications in Cancer Therapy. Cells. 2022;11(5):790. doi:10.3390/cells11050790
104. Tao BL, Du RX, Zhang XM, et al. Engineering CAR-NK cell derived exosome disguised nano-bombs for enhanced HER2 positive breast cancer brain metastasis therapy. J Control Release. 2023;363:692–706. doi:10.1016/j.jconrel.2023.10.007
105. Pullan JE, Confeld MI, Osborn JK, et al. Exosomes as Drug Carriers for Cancer Therapy. Mol Pharmaceut. 2019;16(5):1789–1798. doi:10.1021/acs.molpharmaceut.9b00104
106. Shao JT, Zaro J, Shen YX. Advances in Exosome-Based Drug Delivery and Tumor Targeting: from Tissue Distribution to Intracellular Fate. Int j Nanomed. 2020;15:9355–9371. doi:10.2147/IJN.S281890
107. Jenkins S, Wesolowski R, Gatti-Mays ME. Improving Breast Cancer Responses to Immunotherapy-a Search for the Achilles Heel of the Tumor Microenvironment. Curr Oncol Rep. 2021;23(5). doi:10.1007/s11912-021-01040-y
108. Johnson A, Townsend M, O’neill K. Tumor Microenvironment Immunosuppression: a Roadblock to CAR T-Cell Advancement in Solid Tumors. Cells. 2022;11(22):1.
109. Ye ZL, Zeng DQ, Zhou R, et al. Tumor Microenvironment Evaluation for Gastrointestinal Cancer in the Era of Immunotherapy and Machine Learning. Front Immunol. 2022;2022:13.
110. Xu Q, Zhang Z, Zhao L, et al. Tropism-facilitated delivery of CRISPR/Cas9 system with chimeric antigen receptor-extracellular vesicles against B-cell malignancies. J Control Release. 2020;326:455–467. doi:10.1016/j.jconrel.2020.07.033
111. Wei HX, Chen J, Wang S, et al. A Nanodrug Consisting Of Doxorubicin And Exosome Derived From Mesenchymal Stem Cells For Osteosarcoma Treatment In Vitro. Int j Nanomed. 2019;14:8603–8610. doi:10.2147/IJN.S218988
112. Kyuno D, Zhao K, Bauer N, et al. Therapeutic Targeting Cancer-Initiating Cell Markers by Exosome miRNA: efficacy and Functional Consequences Exemplified for claudin7 and EpCAM. Transl Oncol. 2019;12(2):191–199. doi:10.1016/j.tranon.2018.08.021
113. Liu QH, Dai GR, Wu Y, et al. iRGD-modified exosomes-delivered BCL6 siRNA inhibit the progression of diffuse large B-cell lymphoma. Front Oncol. 2022;2022:12.
114. Alanis EF, Pinotti M, Dal Mas A, et al. An exon-specific U1 small nuclear RNA (snRNA) strategy to correct splicing defects. Hum Mol Genet. 2012;21(11):2389–2398. doi:10.1093/hmg/dds045
115. Dan JM, Memczak S, Belmonte JCI. Expanding the Toolbox and Targets for Gene Editing. Trends Mol Med. 2021;27(3):203–206. doi:10.1016/j.molmed.2020.12.005
116. Stepanichev MY. Using Genome Editing for Alzheimer’s Disease Therapy: from Experiment to Clinic. Neurochem J. 2021;15(4):367–375. doi:10.1134/S1819712421040139
117. Mcandrews KM, Xiao F, Chronopoulos A, et al. Exosome-mediated delivery of CRISPR/Cas9 for targeting of oncogenic Kras G12D in pancreatic cancer. Life Sci Alliance. 2021;4(9):e202000875. doi:10.26508/lsa.202000875
118. Duan L, Ouyang K, Xu X, et al. Nanoparticle Delivery of CRISPR/Cas9 for Genome Editing. Front Genetics. 2021;2021:12.
119. Ye YY, Zhang X, Xie F, et al. An engineered exosome for delivering sgRNA:Cas9 ribonucleoprotein complex and genome editing in recipient cells. Biomater Sci. 2020;8(10):2966–2976. doi:10.1039/D0BM00427H
120. Hu JW, Zhu J, Chai J, et al. Application of exosomes as nanocarriers in cancer therapy. J Mat Chem B. 2023;11(44):10595–10612. doi:10.1039/D3TB01991H
121. Sadeghi S, Tehrani FR, Tahmasebi S, et al. Exosome engineering in cell therapy and drug delivery. Inflammopharmacology. 2023;31(1):145–169. doi:10.1007/s10787-022-01115-7
122. Dinofia AM, Grupp SA. Will allogeneic CAR T cells for CD19(+) malignancies take autologous CAR T cells ‘off the shelf’? Nat Rev Clin Oncol. 2021;18(4):195–196. doi:10.1038/s41571-021-00485-1
123. Depil S, Duchateau P, Grupp SA, et al. ‘Off-The-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19(3):185–199. doi:10.1038/s41573-019-0051-2
124. Balke-Want H, Keerthi V, Cadinanos-Garai A, et al. Non-viral chimeric antigen receptor (CAR) T cells going viral. Immuno Oncology Technol. 2023;18:100375. doi:10.1016/j.iotech.2023.100375
125. Cho JH, Collins JJ, Wong WW. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell. 2018;173(6):1426. doi:10.1016/j.cell.2018.03.038
126. Liu DL, Zhao JJ, Song YP. Engineering switchable and programmable universal CARs for CAR T therapy. J Hematol Oncol. 2019;12(1). doi:10.1186/s13045-019-0763-0
127. Zhao JJ, Lin Q, Song Y, et al. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. 2018;11(1). doi:10.1186/s13045-018-0677-2
128. Lohmueller JJ, Ham JD, Kvorjak M, et al. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology. 2018;7(1):e1368604. doi:10.1080/2162402X.2017.1368604
129. Magnani CF, Tettamanti S, Alberti G, et al. Transposon-Based CAR T Cells in Acute Leukemias: where Are We Going? Cells. 2020;9(6):1337. doi:10.3390/cells9061337
130. Yagyu S, Nakazawa Y. piggyBac-transposon-mediated CAR-T cells for the treatment of hematological and solid malignancies. Int J Clin Oncol. 2023;28(6):736–747. doi:10.1007/s10147-023-02319-9
131. Lock D, Monjezi R, Brandes C, et al. Automated, scaled, transposon-based production of CAR T cells. J Immuno Ther Cancer. 2022;10(9):e005189. doi:10.1136/jitc-2022-005189
132. Kimiz-Gebologlu I, Oncel SS. Exosomes: large-scale production, isolation, drug loading efficiency, and biodistribution and uptake. J Control Release. 2022;347:533–543. doi:10.1016/j.jconrel.2022.05.027
133. Liu WZ, Ma ZJ, Kang XW. Current status and outlook of advances in exosome isolation. Anal Bioanal Chem. 2022;414(24):7123–7141. doi:10.1007/s00216-022-04253-7
134. Qu Q, Fu B, Long Y, et al. Current Strategies for Promoting the Large-scale Production of Exosomes. Curr Neuropharmacol. 2023;21(9):1964–1979. doi:10.2174/1570159X21666230216095938
135. Jalaludin I, Lubman DM, Kim J. A guide to mass spectrometric analysis of extracellular vesicle proteins for biomarker discovery. Mass Spectrom Rev. 2023;42(2):844–872. doi:10.1002/mas.21749
136. Li YJ, Wu JY, Liu J, et al. Artificial exosomes for translational nanomedicine. J Nanobiotechnol. 2021;19(1):1.
137. Jang SC, Kim OY, Yoon CM, et al. Bioinspired Exosome-Mimetic Nanovesicles for Targeted Delivery of Chemotherapeutics to Malignant Tumors. Acs Nano. 2013;7(9):7698–7710. doi:10.1021/nn402232g
138. Jo W, Kim J, Yoon J, et al. Large-scale generation of cell-derived nanovesicles. Nanoscale. 2014;6(20):12056–12064. doi:10.1039/C4NR02391A
139. Ingato D, Edson JA, Zakharian M, et al. Cancer Cell-Derived, Drug-Loaded Nanovesicles Induced by Sulfhydryl-Blocking for Effective and Safe Cancer Therapy. Acs Nano. 2018;12(9):9568–9577. doi:10.1021/acsnano.8b05377
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.