")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Eltrombopag for the treatment of aplastic anemia: current perspectives
Authors Lum SH, Grainger J
Received 27 March 2016
Accepted for publication 25 May 2016
Published 13 September 2016 Volume 2016:10 Pages 2833—2843
DOI https://doi.org/10.2147/DDDT.S95715
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Su Han Lum, John D Grainger
Department of Blood and Marrow Transplant, Royal Manchester Children’s Hospital, Manchester, UK
Abstract: Aplastic anemia (AA) is a potential life-threatening hematopoietic stem cell (HSC) disorder resulting in cytopenia. The mainstays of treatment for AA are definitive therapy to restore HSCs and supportive measures to ameliorate cytopenia-related complications. The standard definitive therapy is HSC transplantation for young and medically fit patients with suitable donors and immunosuppressive therapy (IST) with antithymocyte globulin and cyclosporine for the remaining patients. A significant proportion of patients are refractory to IST or relapse after IST. Various strategies have been explored in these patients, including second course of antithymocyte globulin, high-dose cyclophosphamide, and alemtuzumab. Eltrombopag, a thrombopoietin mimetic, has recently emerged as an encouraging and promising agent for patients with refractory AA. It has demonstrated efficacy in restoring trilineage hematopoiesis, and this positive effect continues after discontinuation of the drug. There are ongoing clinical trials exploring the role of eltrombopag as a first-line therapy in moderate to severe AA and a combination of eltrombopag with IST in severe AA.
Keywords: eltrombopag, aplastic anemia, thrombopoietin, c-Mpl receptors
Introduction
Aplastic anemia (AA) is a potential life-threatening hematopoietic stem cell (HSC) disorder with an estimated incidence of 2–3 per million per year.1,2 The incidence is triphasic, with the first peak occurring in patients at 2–5 years of age, the second peak at 20–25 years, and the third peak at >60 years of age. It is characterized by pancytopenia with a hypocellular marrow in the absence of abnormal infiltration or marrow fibrosis.3–5 Its diagnosis requires exclusion of clonal disorders, including myelodysplastic syndrome (MDS), hypocellular myeloid leukemia, and paroxysmal nocturnal hemoglobinuria. Its severity depends on the degree of peripheral cytopenias and the marrow cellularity (Table 1). Severe AA (SAA) is defined as bone marrow cellularity of <25% with two out of three of the following: neutrophils <0.5×109/L, platelet count <20×109/L, and reticulocyte count <20×109/L. The presenting features range from an incidental finding of cytopenias on a routine blood count to symptomatic cytopenias, including pallor, bleeding, and infection. AA is associated with substantial morbidity, leading to reduced quality of life and premature death. Prolonged neutropenia increases susceptibility to potentially fatal infections, while thrombocytopenia might result in catastrophic bleeding in major organs. Chronic iron overload secondary to frequent red cell transfusion inevitably leads to multisystemic dysfunctions that encompass cardiomyopathy, endocrinopathies, and liver siderosis.
 | Table 1 Overview of aplastic anemia |
The etiology of AA can be broadly classified into inherited AA and acquired AA. Inherited AA is rare, and the diagnostic clues to this are skeletal abnormalities, skin pigmentary anomalies, and marrow dysplasia. Absence of these features does not rule out inherited disorders, and genetic testing is recommended to exclude marrow failure syndrome, as it has critical impact on clinical management and donor selection for HSC transplantation (HSCT). The four important causes for inherited AA are Fanconi anemia (FA), dyskeratosis congenita, Shwachman–Diamond syndrome, and congenital amegakaryocytic thrombocytopenia (CAMT). FA is the most common of inherited AA. Although most patients with FA present in childhood, the diagnosis might be delayed until adulthood, as characteristic congenital malformations are absent in 30%–40% of patients. Hence, FA should be considered as a part of workup for patients with AA. HSCT is the only current curative treatment for inherited AA, and these patients require specific transplant considerations to prevent unexpected toxicities.
Acquired AA accounts for ~80%–90% of marrow failure. It is distinguished from constitutional marrow failure by the absence of phenotypic features and the absence of mutation in genes known to be responsible for inherited AA. The causes of acquired stem cell failure include direct stem cell destruction by drugs, chemicals, ionizing radiation, and viruses, but no identifiable cause is found in the majority of patients. The pathology underpins the marrow failure in idiopathic AA is immune dysfunction, resulting in paucity of HSCs. The most convincing evidence supporting the immune-mediated destruction in idiopathic AA is its response to immunosuppressive therapy (IST) and rebound cytopenia with discontinuation of IST. The proposed hypothesis is that there is an aberrant T-cell activation stimulated by a viral infection, a drug, or a specific environmental precipitant in a genetically susceptible host. Clonal expansion of CD8+ T-cells and Th1-cells, reduced and dysfunctional regulatory T-cells (Tregs), and increased Th2- and Th17-cells are the hallmarks of immune dysregulation. The hemostasis of hematopoiesis is interrupted by marrow-suppressing cytokines, including interferon gamma and tumor necrosis factor.6,7 The apoptotic death of HSCs in the bone marrow is mediated by increased expression of the Fas receptor and antigen, as well as possibly by cytotoxic granules.8
Current recommendation of management of patients with acquired SAA is illustrated in Figure 1.3,7,9 In general, patients with SAA need definite therapy to target the marrow failure and support care for cytopenia-associated clinical symptoms and complications. The components of the support care of AA encompass transfusion supports, antimicrobial prophylaxis, treatment for neutropenic fevers, and hematologic growth factor therapies. The definite therapy for SAA includes HSCT and IST. Both are effective and aim to abrogate the immune-mediated depletion of HSC. By stopping the immune destruction of HSC, IST allows repopulation of the marrow from the surviving, depleted HSC pool. In HSCT, administration of donor-derived HSC allows more rapid restoration of blood counts, and there is also a reduced incidence of later clonal hematologic disorders than following IST. Matched sibling donor HSCT has always been the therapy of choice for young patients with SAA.10 The event-free survival with human leukocyte antigen-matched sibling donor transplant is 75%–90%, and the risk of graft failure is 4%–14% using conditioning regimen with cyclophosphamide and antithymocyte globulin.11–14 Transplant-related morbidity, such as graft-versus-host disease and infection, has reduced significantly with better transplant care. There are few later effects since the conditioning therapy is light and growth and puberty are preserved. In addition, with better unrelated donor availability, the outcome of well-matched unrelated donor HSCT in all indications is approaching matched sibling donor.15 Some institutions and national groups are using matched unrelated donor HSCT as the preferred initial therapy rather than IST for young patients.16
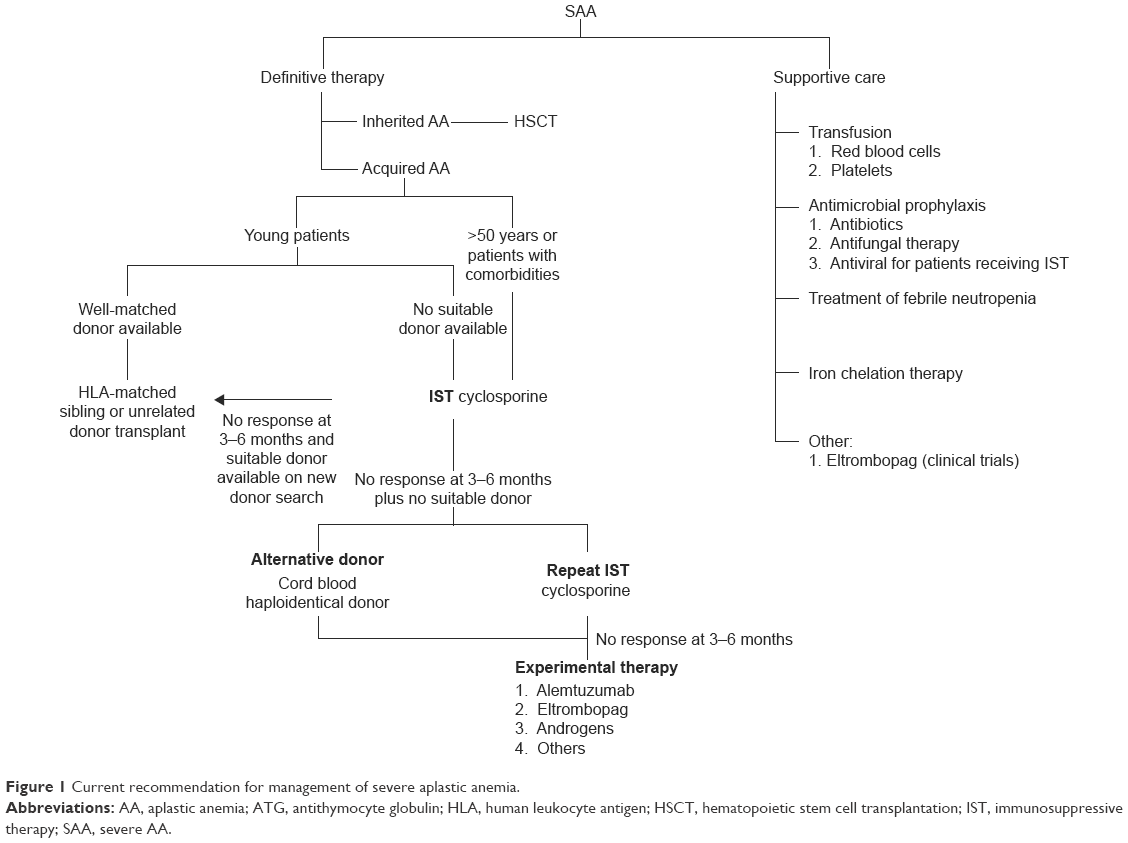 | Figure 1 Current recommendation for management of severe aplastic anemia. |
With regard to IST, the standard regimen is a combination of antithymocyte globulin and cyclosporine. The response to IST is delayed and usually seen after 3–4 months. Of those patients treated with IST, 20%–30% do not respond while 10%–30% of responders relapse.17,18 The mechanism supports the relapse, and refractory diseases might be due to an uncontrolled immune dysfunction or deficiency in residual HSC. Various strategies, including eltrombopag, alemtuzumab, and high-dose cyclophosphamide, have been proposed for these patients. Patients treated with IST are at risk of developing clonal evolution, including MDS/acute myeloid leukemia (8%), hemolytic paroxysmal nocturnal hemoglobinuria (10%), and solid tumor (11%).19–21 The article describes the principle of eltrombopag use in acquired AA and summarizes the current evidence for its use in AA.
The role of thrombopoietin in hematopoeisis
Thrombopoietin (TPO) is the most important hematopoietic growth factor that regulates the circulating platelet mass. Although the concept of this platelet regulator was first described in 1950, TPO protein was purified only in 1994.22,23 TPO is also known as megapoietin, megakaryocyte growth and developmental factor, and c-Mpl ligand. The TPO gene is located on the long arm of chromosome 6 at the position 3q27–28. It has seven exons extending along ~7,000 bp, and the first two exons are noncoding.24,25 TPO is mainly produced by liver, with small amounts being made by the kidney and bone marrow. It consists of 353 amino acids with 21-amino-acid secretory leader sequence. The mature TPO protein is a member of four-helix-bundle cytokine superfamily and consists of two domains. The amino-terminal 154 residue domain is homologous to erythropoietin and binds to the c-Mpl receptor. The main two functions of carboxyl-terminal domain of TPO are serving as an intramolecular chaperone to aid the proper folding of the polypeptide into the mature hormone and prolonging the circulatory half-life of TPO by modifying with multiple sites of both N- and O-linked carbohydrates.26
TPO is a potent endogenous cytokine that acts through the TPO receptors, known as c-Mpl receptors, which present primarily in platelets and megakaryocytes and in a small percentage of hematopoietic progenitor cells (HPC).27 The c-Mpl gene, which is located on human chromosome 1p34, was cloned in 1992.28 c-Mpl protein exists as an inactive dimer, each monomer containing two cytokine receptor homology (CHR) domains. Binding of TPO to distal cytokinecytokine receptor homology region of c-Mpl receptors stimulates multiple signal transduction pathways, including JAK/STAT and mitogen-activated protein kinase pathways. Activation of these pathways promotes megakaryocyte proliferation and maturation, as well as platelet release into circulation. TPO clearance depends on its binding to c-Mpl receptors. Increasing free TPO in thrombocytopenia state leads to stimulation of platelet production, whereas its level is low in thrombocytosis. As c-Mpl receptors are present in HPC, TPO has been shown to play an important role in HSC survival, self-renewal, and expansion.29–31 Mutations in the c-Mpl gene have been reported in association with familial AA, and patients with c-Mpl-associated CAMT are at high risk of developing AA.32–34 Deficiency of c-Mpl receptors in mouse model demonstrated reduced numbers of HPC.35,36 This evidence supports the role of TPO beyond its primary function as a platelet regulator.
With the discovery of TPO, a number of TPO receptor agonists or TPO mimetics have been developed (Table 2). The two, first generation of TPO mimetics, recombinant human TPO (rHuTPO) and PEGylated rHuTPO (PEG-rHuMGDF),37 were introduced in 1997. rHuTPO shares the same amino acid structure as endogenous TPO but a slightly lower molecular weight. rHuTPO was administered intravenously with a half-life of 30–40 hours. PEG-rHuMGDF was composed of the amino-terminal 163 amino acids of endogenous TPO coupled with polyethylene glycol moiety. It had a half-life of 25–35 hours after an intravenous injection.38 Although both have demonstrated positive platelet increment, clinical experience with the first-generation TPOs was disappointing because of the development of endogenous antibodies against endogenous TPO, resulting in secondary thrombocytopenia. Developments of PEG-rHuMGDF and rHuTPO were stopped in 1998 and 2002, respectively.39
 | Table 2 Key milestones in eltrombopag |
The experience from the first generation of TPO mimetics has formed an important learning platform for the new generation of thrombopoietic growth factors. The second-generation molecules, including small molecular peptides and nonpeptide agents, were first introduced in 1997. Romiplostim is an injectable peptide TPO mimetic that activates the TPO receptors just like native TPO. It is composed of 14-amino-acid peptides attached to two disulfide-bonded human immunoglobulin G1 kappa heavy chain constant regions by glycine bridges. As the peptide has no sequence homologous with endogenous TPO, production of antibody against romiplostim would not cross-react with endogenous TPO.37 It has been used in patients with immune thrombocytopenia (ITP), MDS, chemotherapy-induced thrombocytopenia, and hepatitis C-related thrombocytopenia.40–46 Nonpeptide TPO receptor agonists have been derived from small molecule screening techniques that uncovered a number of chemical compounds that interact with c-Mpl receptors. Eltrombopag, which has been derived from hydrazinonaphthalene, has a number of advantages compared to peptide TPO mimetics, including oral preparation and a different mechanism of action that confers additive effect. Eltrombopag is the only orally available nonpeptide TPO receptor agonist approved by the US Food and Drug Administration for the treatment of adults with ITP, patients with hepatitis C-related thrombocytopenia who are being treated with interferons, and patients with AA who are refractory to IST.47
Pharmacology of eltrombopag
Eltrombopag is a member of the biarylhydrazone class of compound with a chemical structure of C25H22N4O4. In clinical studies, it has been used as eltrombopag olamine, which is the bis-monoethanolamine salt form of eltrombopag.28 It is rapidly absorbed following oral administration and achieves peak plasma concentrations at a median time of 2.5 hours (Table 3). The biological half-life is ~21–32 hours, and it is mainly excreted in feces and urine. It is highly bound (>99%) to plasma proteins, mainly albumin, and extensively metabolized by monooxygenation, glucuronidation, hydrazine cleavage, and secondary oxidation and conjugation. In vitro, it is an inhibitor of CYP2C8 and CYP2C9, several glucuronosyltransferases isoenzymes, breast cancer resistance protein, and organic anion transporting polypeptide (OATP0 1B1).28,48
 | Table 3 Drug property of eltrombopag |
In vitro studies have demonstrated that the activity of eltrombopag is dependent on the expression of TPO receptors.49 It binds to the transmembrane domain of the TPO receptor, leading to signal transduction through various pathways, including Janus kinase/signal transducer and activator of transcription and mitogen-activated protein kinase. Activation of TPO receptor results in proliferation and differentiation of megakaryocytes and increased platelet production. As eltrombopag binds at transmembrane domain, it does not compete with endogenous TPO binding at the extracellular TPO receptor domain (Figure 2).49 Hence, eltrombopag provides an additive effect with endogenous TPO in promoting megakaryocyte maturation and platelet production.
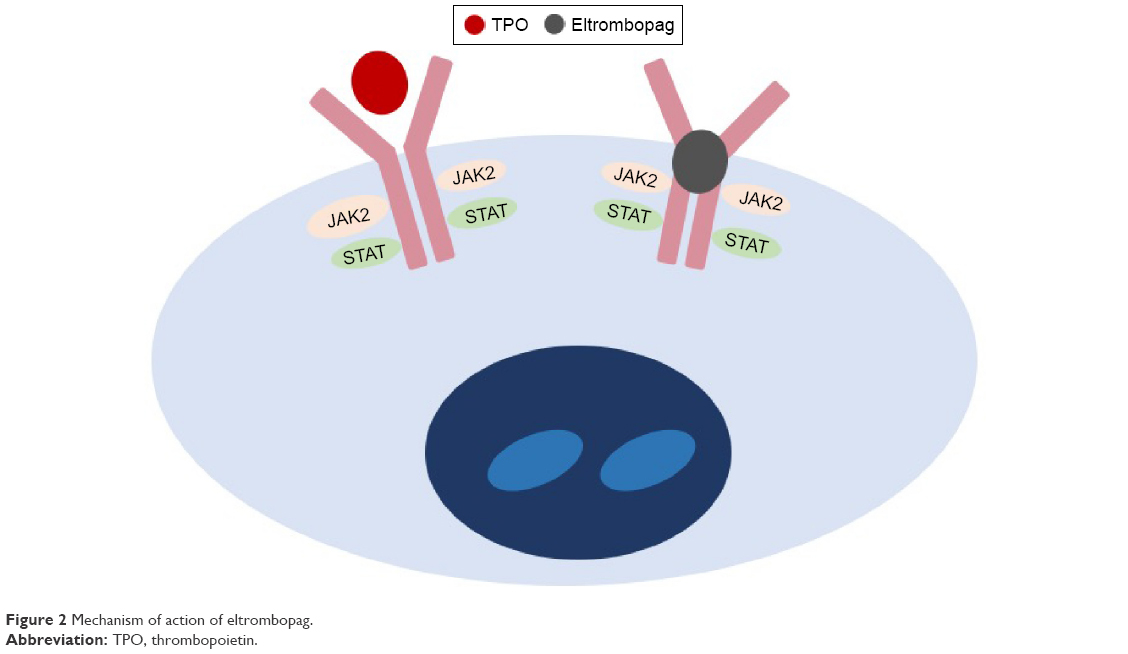 | Figure 2 Mechanism of action of eltrombopag. |
Studies using eltrombopag in chronic ITP have demonstrated that prolonged administration of eltrombopag is safe and well tolerated.50–53 Similar to other TPO mimetics, the potential toxicities of eltrombopag include thrombocytosis, thrombosis, reversible bone marrow fibrosis, rebound thrombocytopenia, cataract formation, and reversible hepatic dysfunction. The proposed mechanisms for thrombosis of TPO mimetics are rapid elevation of platelet count and increased rate of microparticle formation in patients with ITP.54 Eltrombopag might be associated with a risk of portal vein thrombosis in patients with advanced liver disease. Of 142 patients with chronic liver disease treated with eltrombopag, seven patients developed portal vein thrombosis compared to three patients in the control group (n=147) (odds ratio 3.04, 95% CI 0.62–14.82).55 Similar association was not seen in 74 patients with hepatitis C treated with eltrombopag.56 The use of eltrombopag in ITP did not increase the rate of thrombosis compared to placebo.57
With regard to marrow fibrosis, mouse models demonstrated higher fibrosis after TOP gene transfection and reversible fibrosis after discontinuation of TPO.58,59 In a recent paper by Brynes et al, of 232 marrow biopsy specimens taken from 117 patients with chronic ITP treated with eltrombopag in the EXTEND study, two patients were found to have moderate to marked reticulin fibrosis. After discontinuing the treatment, the repeat biopsy was normal in one patient and unchanged in another patient. None of the 117 patients developed MDS and acute myeloid leukemia.60 Similar findings of reversible marrow fibrosis were reported by Kuter et al on 271 patients with chronic ITP treated with romiplostim. Although there is no long-term prospective bone marrow evaluation in patients treated with eltrombopag, the reported studies of eltrombopag in AA and post-HSCT thrombocytopenia did not show evidence of marrow fibrosis in their cohorts.61,62
The experience on the use of eltrombopag in children is limited, and most of the studies are on adults with chronic ITP. PETIT, the first international trial on eltrombopag for the treatment of children with persistent and chronic ITP, identified the dose to be used in Phase III studies.63 PETIT2 was done to study the efficacy of eltrombopag in children with chronic ITP aged 6–17 years. In these studies, there were no unexpected safely concerns, and few children discontinued treatment because of adverse effects.51
Eltrombopag in AA
The thrombocytopenia in AA is caused by inadequate HSC numbers and function, which lead to impaired megakaryopoesis and reduced mature platelet production. The consequences of thrombocytopenia in these patients range from silent hemorrhage, such as minor cerebral bleeds, to troublesome mucocutaneous hemorrhage and fatal major organ hemorrhage. The mainstay of therapy for thrombocytopenia in these patients is prophylactic and therapeutic platelet transfusion while waiting for definitive therapy. Antibody-mediated platelet refractoriness is the momentous sequela from multiple donor exposure, although its incidence has reduced from 50% to 12% with the use of leukodepleted blood products.64 The use of hematopoietic growth factor (HGF) in correcting bone marrow failure syndrome has been explored since 1960. The early clinical trials using erythropoietic growth factors, G-CSF, GM-CSF, stem cell factor, interleukin (IL)-1, IL-3, and IL-6 have failed to show beneficial effects. The lack of positive therapeutic effects of these growth factors might be explained by: 1) normal mRNA expression and secretion of these HGFs and cytokines in long-term marrow cultures from AA stromal cells, 2) marked elevation of serum HGF in patients with AA, and 3) activity of these cytokines on more committed myeloid progenitors.65 In addition, significant toxicities were reported with most agents, except G-CSF.65 As TPO receptors, c-Mpl receptors present on more primitive HSCs, which supports the ability of TPO to act on less differentiated progenitor cells and the theoretical concept of treating AA.
As discussed earlier, knockout mice that are deficient in TPO and c-Mpl receptors have reduced numbers of multiple hematopoietic lineage progenitor cells.35,36 Similarly, patients with CAMT are associated with c-Mpl mutation and develop AA at a median age of 3.7 years.66 These findings have reaffirmed the role of TPO and its receptors in multilineage hematopoeisis.
Although the clinical trials of eltrombopag are limited, they have demonstrated the efficacy of the drug in increasing platelet count in patients with chronic ITP and thrombocytopenia associated with hepatitis C infection. The first and only published clinical trial of eltrombopag in AA was performed by Olnes et al in 2012 (Figure 3). In this Phase II study, 25 patients with refractory AA were treated with eltrombopag using a dose escalation schedule starting at 50 mg and increasing every 2 weeks by 25 mg, if the platelet count remained less than 20×109/L to a maximum dose of 150 mg. The primary endpoint was hematological response at 3–4 months. The median platelet count was 9×109/L (range 5–15×109/L), and the median TPO level in these patients was 2,767 pg/mL (range 1,615–4,618 pg/mL). It was observed that 44% of patients (11/25) demonstrated at least a response at one lineage. Of these eleven patients, four patients had bilineage response and one patient had trilineage response. In addition, 36% (9/25) had a median platelet count increment of 44×109/L, 24% (6/25) had a median hemoglobin increase of 44 g/L, and 63% (9/25) had a median increased neutrophil count of 1.35×109/L. Seven of the eleven responders continued treatment for a median of 16 months, and six achieved a trilineage response.67
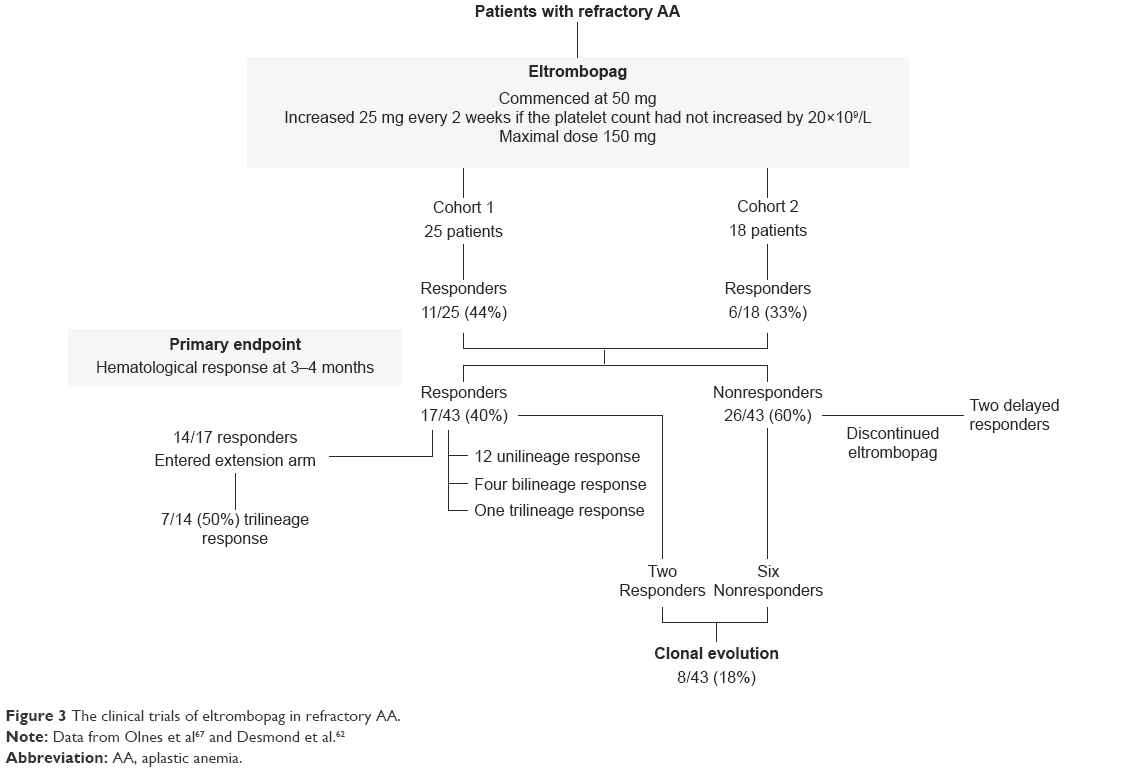 | Figure 3 The clinical trials of eltrombopag in refractory AA. |
In the recent update on this study by Desmond et al,62 a second cohort, which consisted of an additional 18 patients, were added to the original cohort. The median age of these 43 patients was 44 years (range 17–77 years). These patients were heavily treated with a median of two courses of IST and transfusion dependent for red blood cells and platelets. Of these 43 patients, 40% (17/43) showed hematologic response at 3–4 months (Figure 3). Of the 14 responders who continued the treatment for a median of 16 months, seven (50%) patients achieved trilineage response. Of the five patients with near-normal blood counts who had discontinued eltrombopag at a median of 20.5 months of therapy (range 9–37 months), all continued to have stable counts for a median of 13 months (range 1–15 months). Of the 26 nonresponders, two patients demonstrated hematological improvement after discontinuing eltrombopag. A total of eight patients, two responders, and six nonresponders developed clonal evolution on eltrombopag, including five with chromosome 7 abnormalities. None of the studied patients developed acute myeloid leukemia. All the patients with chromosome 7 changes were successfully transplanted. The major toxicities were irreversible deranged hepatic transaminases.
In contrast to low TPO level in ITP, the TPO level in these patients is much higher than the normal values of 7–99 pg/mL; this study demonstrated unequivocal efficacy of eltrombopag in trilineage hematopoeisis in these patients.68 All patients had bone marrow biopsies done at study entry and every 3–6 months. Eltrombopag has demonstrated an increase in marrow cellularity in responders, and none had evidence of myelofibrosis. The marrow cellularity seen in patients treated with eltrombopag is different from that observed in patients treated with standard IST, where residual hypocellularity is frequently seen.69
Discussion
The small molecular nature of eltrombopag, and lack of competitive binding with endogenous TPO, may maximize the potential for eltrombopag to enter the stem cell niche leading to direct expansion of the few HSCs that remain in SAA. In addition, the use of TPO-receptor agonist in patients with ITP has been postulated to increase Treg activity by increasing total circulating transforming growth factor-β1 levels.70 In patients with SAA, a similar increase in Treg activity with eltrombopag may provide an immunomodulatory role in the treatment of SAA.
The stimulation of HSC driven by eltrombopag may lead to genomic instability and clonal evolution to MDS or acute myeloid leukaemia. The development of monosomy 7 in five patients is a concern. Monosomy 7 is a high-risk cytogenetic clone associated with an evolution to acute myeloid leukemia. It is probable that these clones exist at low levels, beyond conventional cytogenetics, with a diagnosis of SAA, and the use of targeted DNA sequencing to detect such clones may be a future direction to exclude such patients from eltrombopag.69 Eltrombopag is, however, tolerated well by the majority of patients with no toxicity and absence of clonal evolution.
In view of encouraging and propitious results from Olnes et al, eltrombopag has been shown to have the potential to be effective as initial therapy. On the back of these results, both the European Medicines Agency and the US Food and Drug Administration have granted expedited approval for the use of eltrombopag in adults with acquired SAA who did not respond to IST and cannot receive HSCT. There are two further ongoing nonrandomized pilot Phase II studies evaluating its use as a first-line therapy in patients with SAA and moderate AA with significant thrombocytopenia or anemia (NCT01703169 and NCT013228587). Another two clinical trials have been started to study its safety and effectiveness by adding eltrombopag to IST in SAA (NCT02099747 and NCT01623167). One of the studies, NCT01623167, has included children with AA aged 2 years and above. These clinical trials are aimed to address unresolved doubts, such as selection of candidates, duration of therapy, safety of prolonged therapy, and risk of clonal evolution. Additional studies may also be appropriate for constitutional AA where the risk and long-term toxicities of HSCT are much higher.
Conclusion
Eltrombopag has increasingly convincing efficacy in restoring trilineage hematopoiesis. There is no doubt that it is going to change the strategy for managing AA in the future. It can be potentially used as a frontline therapy in moderate to severe AA alone or in combination with IST, as well as a salvage therapy for patients who are refractory to IST or relapse after IST. Its potential to be used as monotherapy in AA could be explained by its action on c-Mpl receptors, which present on more primitive progenitor HSCs. Moreover, the convincing results from Olnes et al and Desmond et al have highlighted its role in patients with refractory AA despite having a median of two courses of IST.62,67 The use of eltrombopag monotherapy could reduce IST treatment-related complications, such as immunosuppression, hypertension, and renal dysfunction. Results from ongoing trials will provide us robust data on its use in AA with an optimal strategy.
Disclosure
John Grainger has received honoraria, travel support, and speaker fees from Novartis, GSK, and Amgen. Su Han Lum reports no conflicts of interest in this work.
References
Montané E, Ibáñez L, Vidal X, et al; Catalan Group for Study of Agranulocytosis and Aplastic Anemia. Epidemiology of aplastic anemia: a prospective multicenter study. Haematologica. 2008;93(4):518–523. | ||
Young NS, Kaufman DW. The epidemiology of acquired aplastic anemia. Haematologica. 2008;93(4):489–492. | ||
Killick SB, Bown N, Cavenagh J, et al; British Society for Standards in Haematology. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172(2):187–207. | ||
Marsh JC, Ball SE, Cavenagh J, et al; British Committee for Standards in Haematology. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147(1):43–70. | ||
Marsh JC, Ball SE, Darbyshire P, et al; British Committee for Standards in Haematology. Guidelines for the diagnosis and management of acquired aplastic anaemia. Br J Haematol. 2003;123(5):782–801. | ||
Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108(8):2509–2519. | ||
Young NS. Current concepts in the pathophysiology and treatment of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2013;2013:76–81. | ||
Xu JL, Nagasaka T, Nakashima N. Involvement of cytotoxic granules in the apoptosis of aplastic anaemia. Br J Haematol. 2003;120(5):850–852. | ||
Davies JK, Guinan EC. An update on the management of severe idiopathic aplastic anaemia in children. Br J Haematol. 2007;136(4):549–564. | ||
Doney K, Leisenring W, Storb R, Appelbaum FR. Primary treatment of acquired aplastic anemia: outcomes with bone marrow transplantation and immunosuppressive therapy. Seattle bone marrow transplant team. Ann Intern Med. 1997;126(2):107–115. | ||
Myers KC, Davies SM. Hematopoietic stem cell transplantation for bone marrow failure syndromes in children. Biol Blood Marrow Transplant. 2009;15(3):279–292. | ||
Champlin RE, Perez WS, Passweg JR, et al. Bone marrow transplantation for severe aplastic anemia: a randomized controlled study of conditioning regimens. Blood. 2007;109(10):4582–4585. | ||
Kahl C, Leisenring W, Deeg HJ, et al. Cyclophosphamide and antithymocyte globulin as a conditioning regimen for allogeneic marrow transplantation in patients with aplastic anaemia: a long-term follow-up. Br J Haematol. 2005;130(5):747–751. | ||
Locatelli F, Bruno B, Zecca M, et al. Cyclosporin A and short-term methotrexate versus cyclosporin A as graft versus host disease prophylaxis in patients with severe aplastic anemia given allogeneic bone marrow transplantation from an HLA-identical sibling: results of a GITMO/EBMT randomized trial. Blood. 2000;96(5):1690–1697. | ||
Passweg JR, Socié G, Hinterberger W, et al. Bone marrow transplantation for severe aplastic anemia: has outcome improved? Blood. 1997;90(2):858–864. | ||
Dufour C, Veys P, Carraro E, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on behalf of the UK Paediatric BMT working party, paediatric diseases working party and severe aplastic anaemia working party of EBMT. Br J Haematol. 2015;171(4):585–594. | ||
Schrezenmeier H, Marin P, Raghavachar A, et al. Relapse of aplastic anaemia after immunosuppressive treatment: a report from the European bone marrow transplantation group SAA working party. Br J Haematol. 1993;85(2):371–377. | ||
Bacigalupo A, Bruno B, Saracco P, et al. Antilymphocyte globulin, cyclosporine, prednisolone, and granulocyte colony-stimulating factor for severe aplastic anemia: an update of the GITMO/EBMT study on 100 patients. European group for blood and marrow transplantation (EBMT) working party on severe aplastic anemia and the Gruppo Italiano Trapianti di Midolio Osseo (GITMO). Blood. 2000;95(6):1931–1934. | ||
Tichelli A, Socié G, Henry-Amar M, et al. Effectiveness of immunosuppressive therapy in older patients with aplastic anemia. European group for blood and marrow transplantation severe aplastic anaemia working party. Ann Intern Med. 1999;130(3):193–201. | ||
Socié G, Henry-Amar M, Bacigalupo A, et al. Malignant tumors occurring after treatment of aplastic anemia. European bone marrow transplantation-severe aplastic anaemia working party. N Engl J Med. 1993;329(16):1152–1157. | ||
Frickhofen N, Heimpel H, Kaltwasser JP, Schrezenmeier H; German Aplastic Anemia Study Group. Antithymocyte globulin with or without cyclosporin A: 11-year follow-up of a randomized trial comparing treatments of aplastic anemia. Blood. 2003;101(4):1236–1242. | ||
Kuter DJ, Beeler DL, Rosenberg RD. The purification of megapoietin: a physiological regulator of megakaryocyte growth and platelet production. Proc Natl Acad Sci U S A. 1994;91(23):11104–11108. | ||
Kato T, Ogami K, Shimada Y, et al. Purification and characterization of thrombopoietin. J Biochem. 1995;118(1):229–236. | ||
Foster DC, Sprecher CA, Grant FJ, et al. Human thrombopoietin: gene structure, cDNA sequence, expression, and chromosomal localization. Proc Natl Acad Sci U S A. 1994;91(26):13023–13027. | ||
Gurney AL, Kuang WJ, Xie MH, Malloy BE, Eaton DL, de Sauvage FJ. Genomic structure, chromosomal localization, and conserved alternative splice forms of thrombopoietin. Blood. 1995;85(4):981–988. | ||
Hitchcock IS, Kaushansky K. Thrombopoietin from beginning to end. Br J Haematol. 2014;165(2):259–268. | ||
Zeigler FC, de Sauvage F, Widmer HR, et al. In vitro megakaryocytopoietic and thrombopoietic activity of c-Mpl ligand (TPO) on purified murine hematopoietic stem cells. Blood. 1994;84(12):4045–4052. | ||
Stasi R. Eltrombopag: the discovery of a second generation thrombopoietin-receptor agonist. Expert Opin Drug Discov. 2009;4(1):85–93. | ||
Qian H, Buza-Vidas N, Hyland CD, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell. 2007;1(6):671–684. | ||
Yoshihara H, Arai F, Hosokawa K, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007;1(6):685–697. | ||
Miyazaki H. Physiologic role of TPO in thrombopoiesis. Stem Cells. 1996;14(suppl 1):133–138. | ||
Walne AJ, Dokal A, Plagnol V, et al. Exome sequencing identifies MPL as a causative gene in familial aplastic anemia. Haematologica. 2012;97(4):524–528. | ||
Geddis AE. Congenital amegakaryocytic thrombocytopenia. Pediatr Blood Cancer. 2011;57(2):199–203. | ||
Ballmaier M, Germeshausen M. Congenital amegakaryocytic thrombocytopenia: clinical presentation, diagnosis, and treatment. Semin Thromb Hemost. 2011;37(6):673–681. | ||
Kimura S, Roberts AW, Metcalf D, Alexander WS. Hematopoietic stem cell deficiencies in mice lacking c-Mpl, the receptor for thrombopoietin. Proc Natl Acad Sci U S A. 1998;95(3):1195–1200. | ||
Alexander WS, Roberts AW, Nicola NA, Li R, Metcalf D. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood. 1996;87(6):2162–2170. | ||
Kuter DJ. New thrombopoietic growth factors. Blood. 2007;109(11):4607–4616. | ||
Kuter DJ, Begley CG. Recombinant human thrombopoietin: basic biology and evaluation of clinical studies. Blood. 2002;100(10):3457–3469. | ||
Li J, Yang C, Xia Y, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood. 2001;98(12):3241–3248. | ||
Sekeres MA, Kantarjian H, Fenaux P, et al. Subcutaneous or intravenous administration of romiplostim in thrombocytopenic patients with lower risk myelodysplastic syndromes. Cancer. 2011;117(5):992–1000. | ||
Kantarjian H, Fenaux P, Sekeres MA, et al. Safety and efficacy of romiplostim in patients with lower-risk myelodysplastic syndrome and thrombocytopenia. J Clin Oncol. 2010;28(3):437–444. | ||
Kantarjian HM, Giles FJ, Greenberg PL, et al. Phase 2 study of romiplostim in patients with low- or intermediate-risk myelodysplastic syndrome receiving azacitidine therapy. Blood. 2010;116(17):3163–3170. | ||
Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med. 2006;355(16):1672–1681. | ||
Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet. 2008;371(9610):395–403. | ||
Kuter DJ, Rummel M, Boccia R, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med. 2010;363(20):1889–1899. | ||
Kuter DJ, Bussel JB, Newland A, et al. Long-term treatment with romiplostim in patients with chronic immune thrombocytopenia: safety and efficacy. Br J Haematol. 2013;161(3):411–423. | ||
Kuter DJ. New thrombopoietic growth factors. Clin Lymphoma Myeloma. 2009;9(suppl 3):S347–S356. | ||
Deng Y, Madatian A, Wire MB, et al. Metabolism and disposition of eltrombopag, an oral, nonpeptide thrombopoietin receptor agonist, in healthy human subjects. Drug Metab Dispos. 2011;39(9):1734–1746. | ||
Erickson-Miller CL, Delorme E, Tian SS, et al. Preclinical activity of eltrombopag (SB-497115), an oral, nonpeptide thrombopoietin receptor agonist. Stem Cells. 2009;27(2):424–430. | ||
Milosevic I, Slade E, Drysdale H; COMPare Project Team. Eltrombopag for chronic immune thrombocytopenia. Lancet. 2016;387(10016):336. | ||
Grainger JD, Locatelli F, Chotsampancharoen T, et al. Eltrombopag for children with chronic immune thrombocytopenia (PETIT2): a randomised, multicentre, placebo-controlled trial. Lancet. 2015;386(10004):1649–1658. | ||
González-López TJ, Alvarez-Román MT, Pascual C, et al. Eltrombopag safety and efficacy for primary chronic immune thrombocytopenia in clinical practice. Eur J Haematol. 2015. Epub 2015 Dec 28. | ||
Saleh MN, Bussel JB, Cheng G, et al; EXTEND Study Group. Safety and efficacy of eltrombopag for treatment of chronic immune thrombocytopenia: results of the long-term, open-label EXTEND study. Blood. 2013;121(3):537–545. | ||
Fontana V, Jy W, Ahn ER, et al. Increased procoagulant cell-derived microparticles (C-MP) in splenectomized patients with ITP. Thromb Res. 2008;122(5):599–603. | ||
Afdhal NH, Giannini EG, Tayyab G, et al; ELEVATE Study Group. Eltrombopag before procedures in patients with cirrhosis and thrombocytopenia. N Engl J Med. 2012;367(8):716–724. | ||
McHutchison JG, Dusheiko G, Shiffman ML, et al; TPL102357 Study Group. Eltrombopag for thrombocytopenia in patients with cirrhosis associated with hepatitis C. N Engl J Med. 2007;357(22):2227–2236. | ||
Ulich TR, del Castillo J, Senaldi G, et al. Systemic hematologic effects of PEG-rHuMGDF-induced megakaryocyte hyperplasia in mice. Blood. 1996;87(12):5006–5015. | ||
Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood. 2009;114(18):3748–3756. | ||
Villeval JL, Cohen-Solal K, Tulliez M, et al. High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice. Blood. 1997;90(11):4369–4383. | ||
Brynes RK, Orazi A, Theodore D, et al. Evaluation of bone marrow reticulin in patients with chronic immune thrombocytopenia treated with eltrombopag: data from the EXTEND study. Am J Hematol. 2015;90(7):598–601. | ||
Tanaka T, Inamoto Y, Yamashita T, et al. Eltrombopag for treatment of thrombocytopenia after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2016;22(5):919–924. | ||
Desmond R, Townsley DM, Dumitriu B, et al. Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. Blood. 2014;123(12):1818–1825. | ||
Bussel JB, de Miguel PG, Despotovic JM, et al. Eltrombopag for the treatment of children with persistent and chronic immune thrombocytopenia (PETIT): a randomised, multicentre, placebo-controlled study. Lancet Haematol. 2015;2(8):e315–e325. | ||
Killick SB, Win N, Marsh JC, et al. Pilot study of HLA alloimmunization after transfusion with pre-storage leucodepleted blood products in aplastic anaemia. Br J Haematol. 1997;97(3):677–684. | ||
Marsh JC, Ganser A, Stadler M. Hematopoietic growth factors in the treatment of acquired bone marrow failure states. Semin Hematol. 2007;44(3):138–147. | ||
King S, Germeshausen M, Strauss G, Welte K, Ballmaier M. Congenital amegakaryocytic thrombocytopenia: a retrospective clinical analysis of 20 patients. Br J Haematol. 2005;131(5):636–644. | ||
Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367(1):11–19. | ||
Makar RS, Zhukov OS, Sahud MA, Kuter DJ. Thrombopoietin levels in patients with disorders of platelet production: diagnostic potential and utility in predicting response to TPO receptor agonists. Am J Hematol. 2013;88(12):1041–1044. | ||
Marsh JC, Mufti GJ. Eltrombopag: a stem cell cookie? Blood. 2014;123(12):1774–1775. | ||
Bao W, Bussel JB, Heck S, et al. Improved regulatory T-cell activity in patients with chronic immune thrombocytopenia treated with thrombopoietic agents. Blood. 2010;116(22):4639–4645. | ||
Jenkins JM, Williams D, Deng Y, et al. Phase 1 clinical study of eltrombopag, an oral, nonpeptide thrombopoietin receptor agonist. Blood. 2007;109(11):4739–4741. | ||
Bussel JB, Cheng G, Saleh MN, et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med. 2007;357(22):2237–2247. | ||
Cheng G, Saleh M, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomized phase 3 study. Lancet. 2011;377(9763):393–402. | ||
Platzbecker U, Wong RS, Verma A, et al. Safety and tolerability of eltrombopag versus placebo for treatment of thrombocytopenia in patients with advanced myelodysplastic syndromes or acute myeloid leukaemia: a multicentre, randomised, placebo-controlled, double-blind, phase 1/2 trial. Lancet Haematol. 2015;2(10):e417–e426. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.