")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Effect and mechanism of fluoxetine on electrophysiology in vivo in a rat model of postmyocardial infarction depression
Authors Liang JJ , Yuan X, Shi S, wang F, chen Y, qu C, chen J, Hu D, Yang B
Received 15 October 2014
Accepted for publication 20 November 2014
Published 10 February 2015 Volume 2015:9 Pages 763—772
DOI https://doi.org/10.2147/DDDT.S75863
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Jinjun Liang,1,2 Xiaoran Yuan,1,2 Shaobo Shi,1,2 Fang Wang,1,2 Yingying Chen,1,2 Chuan Qu,1,2 Jingjing Chen,1,2 Dan Hu,1–3 Yang Bo1,2
1Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan, People’s Republic of China; 2Cardiovascular Research Institute, Wuhan University, Wuhan, People’s Republic of China; 3Masonic Medical Research Laboratory, Utica, NY, USA
Background: Major depression is diagnosed in 18% of patients following myocardial infarction (MI), and the antidepressant fluoxetine is shown to effectively decrease depressive symptoms and improve coronary heart disease prognosis. We observed the effect of fluoxetine on cardiac electrophysiology in vivo in a rat model of post-MI depression and the potential mechanism.
Methods and results: Eighty adult male Sprague Dawley rats (200–250 g) were randomly assigned to five groups: normal control (control group), MI (MI group), depression (depression group), post-MI depression (model group), and post-MI depression treated with intragastric administration of 10 mg/kg fluoxetine (fluoxetine group). MI was induced by left anterior descending coronary artery ligation. Depression was developed by 4-week chronic mild stress (CMS). Behavior measurement was done before and during the experiment. Electrophysiology study in vivo and Western blot analysis were carried on after 4 weeks of CMS. After 4 weeks of CMS, depression-like behaviors were observed in the MI, depression, and model groups, and chronic fluoxetine administration could significantly improve those behaviors (P<0.05 vs model group). Fluoxetine significantly increased the ventricular fibrillation threshold compared with the model group (20.20±9.32 V vs 14.67±1.85 V, P<0.05). Expression of Kv4.2 was significantly reduced by 29%±12%, 24%±6%, and 41%±15%, respectively, in the MI group, CMS group, and model group, which could be improved by fluoxetine (30%±9%). But fluoxetine showed no improvement on the MI-induced loss of Cx43.
Conclusion: The susceptibility to ventricular arrhythmias was increased in depression and post-MI depression rats, and fluoxetine may reduce the incidence of ventricular arrhythmia in post-MI depression rats and thus improve the prognosis. This may be related in part to the upregulation of Kv4.2 by fluoxetine.
Keywords: depression, myocardial infarction, electrophysiology, arrhythmias, pharmacology
Introduction
Major depression following myocardial infarction (MI) is a common disorder, affecting approximately 18% of all MI patients. It is a major predictor for disability and poor quality of life in the year post-MI and an independent risk factor of sudden cardiac death. Post-MI depressive symptoms have been shown to be associated with increased hospital admission, particularly cardiac admission, and with a significantly elevated risk of mortality.1,2 One mechanism proposed to explain the effects of depression on mortality is the increased vulnerability to ventricular arrhythmias.3 Depressive symptoms may be a predictive factor of ventricular arrhythmias in patients with coronary heart disease (CHD).3 Increased susceptibility to ventricular arrhythmias is observed in a rat model of experimental depression,4 but the underlying mechanism is still unknown. Thus, it is essential to investigate its potential mechanism and find an effective way to treat it.
Selective serotonin reuptake inhibitors (SSRIs) are widely used as medication for post-MI depression and are reported to effectively decrease depressive symptoms and improve CHD prognosis.5,6 The Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial shows a clinically meaningful antidepressant effect of citalopram, a first-line SSRI antidepressant, which can significantly reduce depressive symptoms and improve CHD prognosis.7
Although previous research has provided clinical evidence of prolonged PR, QRS, and QT intervals on standard electrocardiogram (ECG) for fluoxetine and citalopram,8–10 the underlying molecular mechanism still remains unidentified. Kv4 channels produce rapidly activating and inactivating currents such as Ito, which mediates the early repolarization phase of the cardiac action potential. Kv4.2 is predominant in rat heart, and connexin 43 (Cx43) is the predominant protein forming gap junctions in the mammalian ventricular myocardium and is essential for normal conduction of action potential and maintaining a normal electrical coupling in the heart Thus, these two molecules are both important in the genesis of arrhythmia (for details, see “Discussion”). In the present study, we focus on Kv4.2 and Cx43 to investigate the effect of fluoxetine on cardiac electrophysiology and the potential mechanisms in a rat model of post-MI depression.
Materials and methods
Animals
The rats were supplied by the animal experiment center of Wuhan University, Wuhan, People’s Republic of China. Eighty adult male Sprague Dawley rats (200–250 g) were used for the experimental procedures. Rats were allowed 1 week to acclimate to the surroundings before any experimentation began. Animals were housed in individual plastic cages with bedding. Food and tap water were available ad libitum for the duration of the experiments unless otherwise noted. The temperature in the rat colony was maintained at 22°C±2°C. The light cycle was held at 12:12 hours, with lights on at 6 am unless otherwise noted.11
Experimental protocol
A rat model of post-MI depression was developed by left anterior descending (LAD) coronary artery ligation and then 4 weeks of chronic mild stress (CMS) as previously described.12 After the first week, all animals were assigned to five groups according to behavior scores, respectively receiving the following treatments: normal control rats treated with intragastric administration of saline (control group, n=10), MI rats with intragastric administration of saline (MI group, n=20), depression rats treated with 4 weeks of CMS and intragastric administration of saline (depression group, n=10), MI rats with CMS and intragastric administration of saline (model group, n=20), and post-MI depression rats treated with intragastric administration of 10 mg/kg fluoxetine daily for 4 weeks (fluoxetine group, n=20).13 The experimental protocol are shown in Figure 1.
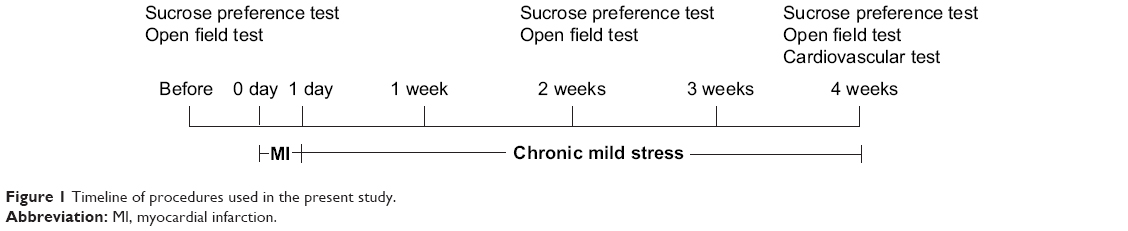 | Figure 1 Timeline of procedures used in the present study. |
Acute infarction model
After the rats were anesthetized with 1% sodium pentobarbital (40 mg/kg intraperitoneal injection), a thoracotomy was performed. The pericardium was incised, and the anterior wall of the left ventricle was exposed. Left ventricular MI was induced by LAD coronary artery ligation.14 The LAD was ligated with a silk suture approximately midway between the left atrium and the apex of the heart. A successful MI model was confirmed by ST segment elevation >0.2 mV in leads I and II and aVL. After the operation, all rats received penicillin (200,000 IU) by intramuscular injection twice a day for 1 week.14,15
Depression model
CMS was used to induce depression in rats.11,16,17 The CMS procedure was a variation of methods described previously and was designed to maximize the unpredictable nature of the stressors. The rats were exposed to the following stressors in random order for 28 days: continuous lighting, tail clipping, 45° cage tilt along the vertical axis, soiled cage (300 mL of water spilled in the bedding), restraint in a small cage (equipped with breathing holes), food deprivation, water deprivation, and tail clipping for 1 minute. During the CMS period, all the rats were treated with intragastric administration of saline or fluoxetine daily, respectively, based on group.
Behavior measurements: sucrose preference test
A sucrose preference test was employed to operationally define anhedonia. A sucrose preference test consisted of first removing the food and water from each rat’s cage for a period of 20 hours. Water and 1% sucrose were then placed on the cages in preweighed glass bottles, and the animals were allowed to consume the fluids for a period of 1 hour. The bottles were then removed and weighed. Two baseline preference tests were performed, separated by at least 5 days, and the results were averaged. Preference tests were conducted weekly throughout the CMS period.11,16
Behavior measurements: open field test
According to previous methods, the apparatus consisted of a square box with dimensions 120×90×35 cm. Rats were placed into the center of the open box under a dark light and allowed to explore the arena for 10 minutes between 9 am and 10 am. A video-computerized tracking system (Ethovision3.0, Noldus Information Technology, Wageningen, the Netherlands) was used to record the distance traveled as a measure of locomotor activity.18
Electrophysiology recording
After 4 weeks of CMS, eight rats in each group underwent rethoracotomy to expose the heart. Bipolar electrode was penetrated into the peri-infarct zone or the corresponding zone in the normal control group. The peri-infarct zone was defined as the zone with no more than 3 mm width located between the infarct zone and the noninfarct zone. The boundary was visually identified by a different color.18,19 All signals were recorded with a polygraph (LEAD2000B, Jinjiang Ltd, Chengdu, People’s Republic of China) and were filtered between 50 Hz and 300 Hz.
Electrophysiology study: monophasic action potential
The band pass filter was set at 50–300 Hz for standard cardiac electrogram recording and at open 300 Hz for recording of monophasic action potential (MAP). The MAP duration (MAPD) was determined as the interval between the onset of the MAP trace and the 50% repolarization time (MAPD50) and 90% repolarization time (MAPD90).20
Electrophysiology study: ventricular effective refractory period
The ventricular effective refractory period (VERP), defined as the longest interval at which a single premature impulse consistently failed to propagate, was determined during right ventricular pacing at a fixed 300 ms drive cycle length (S1). Premature stimuli (S2) were delivered via the left ventricular bipolar gold electrodes after every eight paced beat, at increasing S1–S2 intervals, starting at 30 ms. ERP was measured to the nearest 2 ms immediately before the measurement of ventricular fibrillation threshold (VFT).21
Electrophysiology study: VFT measurement
Electric stimulation was supplied with a cycle length of 20 ms at the peri-infarct zones. Each stimulation was given for 5 seconds. The interval between each episode of stimulation was 2 seconds. The initial pacing voltage was 12 V and progressively increased by a step of 1 V. The lowest voltage inducing sustained VF (>2 seconds, sustaining till next episode) was defined as the VFT.20,21
Protein preparation and immunoblotting analysis
Left ventricular samples were quickly removed and immediately frozen in liquid nitrogen and stored at −80°C. We used RIPA lysis buffer (P0013, Beyotime, Shanghai, People’s Republic of China) with 1 mM phenylmethylsulfonyl fluoride and homogenized with a tissue homogenizer (L7-65 Ultracentrifuge, Beckman Coulter, Inc., Brea, CA, USA). Homogenates were centrifuged at 14,000 g for 5 minutes. Supernatants remained. For immunoblotting analysis, proteins separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis with 10% polyacrylamide gels were transferred electrophoretically to polyvinylidene difluoride sheets (EMD Millipore, Billerica, MA, USA). After 2 hours of blocking in the 5% skimmed milk solution, the membrane was incubated overnight with anti-Cx43 antibody (Abcam, Hong Kong, diluted 1:10,000) and anti-Kv4.2 antibody (Abcam, diluted 1:1,000), respectively. Blots were then incubated for 1 hour with horseradish peroxidase-conjugated goat antimouse immunoglobulin G developed with an electrochemiluminescence reagent (Beyotime) and exposed to a medical X-ray film. Equal protein content in all the samples was confirmed by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression.15 For immunoblotting analysis, the optical density value of each panel was analyzed by ImageJ. The relative intensity of each sample was normalized to the value of GAPDH in the same lane. The relative intensity of target protein in diseased model groups was normalized to those in the control group, which was considered as 1 (100%).
Immunohistochemistry
Transmural blocks of left ventricular myocardium from selected hearts were immersed in a fixative containing 4% paraformaldehyde and embedded in paraffin. Sections were deparaffinized and boiled in a microwave oven for 10 minutes to enhance specific immunostaining. After blockage with goat serum for 15 minutes, the sections were then incubated overnight with anti-Cx43 antibody diluted 1:100 and then incubated for 2 hours in fluorescein isothiocyanate-conjugated rabbit antimouse immunoglobulin G diluted 1:100.15 Specimens were examined under a microscope (Olympus BX51, Tokyo, Japan). The positive area was analyzed using a computer image analysis system (Image-Pro Plus 6.0, Media Cybernetics, Rockville, MD, USA).
Statistical analysis
All data were expressed as mean ± standard deviation. Comparisons among groups were performed with analysis of variance by SPSS Version 17.0, and least-significant difference was used for post hoc multiple comparisons. Differences were considered significant at P<0.05.
Results
Experimental model
The study was completed in 64 surviving animals (ten in the control group, 16 in the MI group, ten in the depression group, 13 in the model group, and 15 in the fluoxetine group). The surviving rate in the fluoxetine group (75.0%, 15/20) was higher than that in the model group (65.0%, 13/20), but given the fact that rats died for a variety of reasons, including surgical manipulations, it is unwarranted to conclude that fluoxetine increased the surviving rate.
Sucrose preference test
Figure 2 displays the fluid intake during the sucrose preference test used to define anhedonia in five experimental groups after the experiment. There were no differences in sucrose intake between groups at baseline (P>0.05). However, after 4 weeks of the experiment, the preference for sucrose was significantly reduced in the MI group, depression group, and model group, and chronic fluoxetine administration could significantly improve this change in model rats (P=0.008 vs model group).
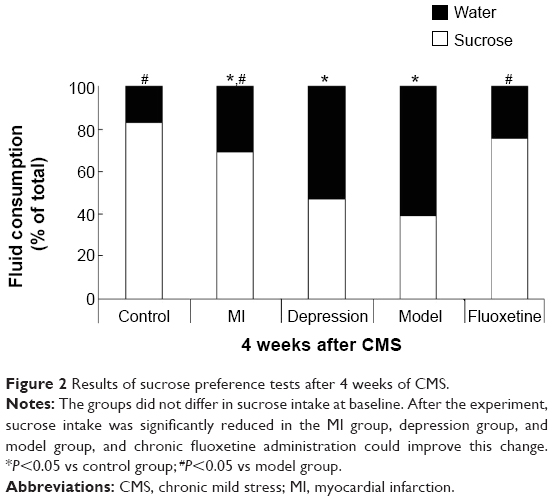 | Figure 2 Results of sucrose preference tests after 4 weeks of CMS. |
Open field test
Figure 3A displays the distance traveled, and Figure 3B the numbers of rearing in 10 minutes during the experiment. After 4 weeks of the experiment, the distance traveled and the numbers of rearing were markedly decreased in the depression group and model group compared with the control, which was reversed by fluoxetine administration (P<0.05 vs model group).
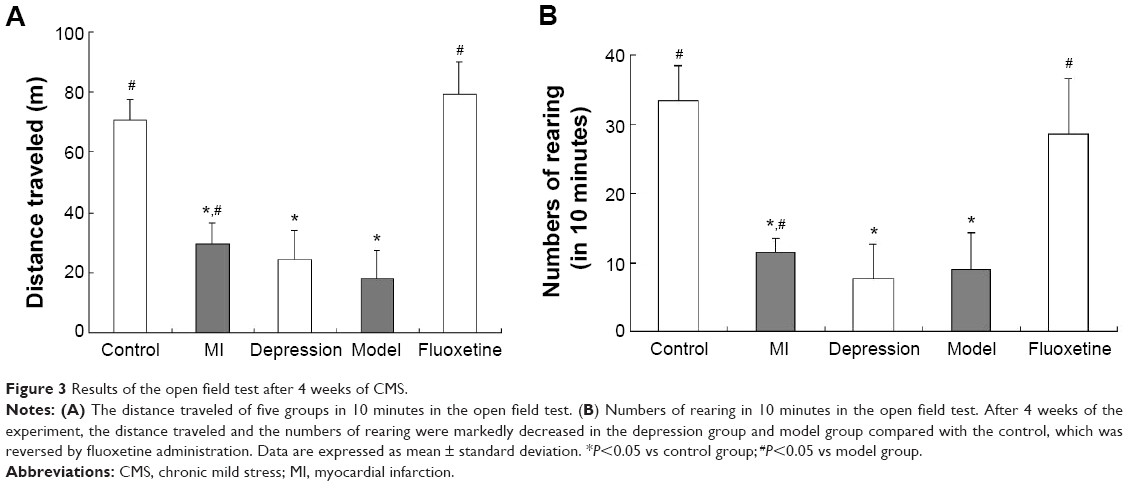 | Figure 3 Results of the open field test after 4 weeks of CMS. |
Electrophysiology study in vivo
Table 1 displays the electrophysiological properties after the experiment. Compared with the control, MAPD90 and VERP were shortened, and VFT was lower in the MI group, CMS group, and model group. Fluoxetine prolonged MAPD90 and VERP and induced a rise of VFT compared with the model. Figure 4 shows representative original AP traces during the electrophysiology study.
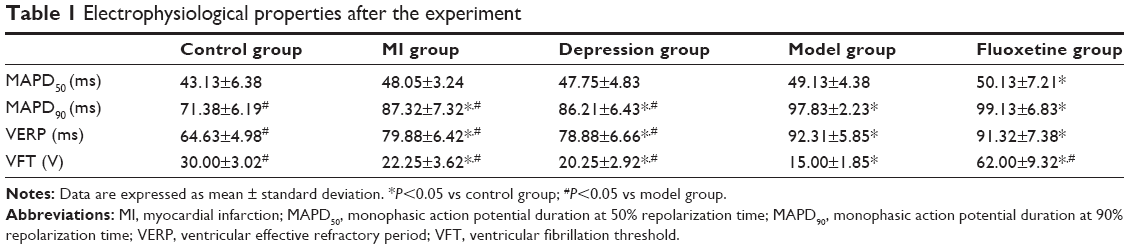 | Table 1 Electrophysiological properties after the experiment |
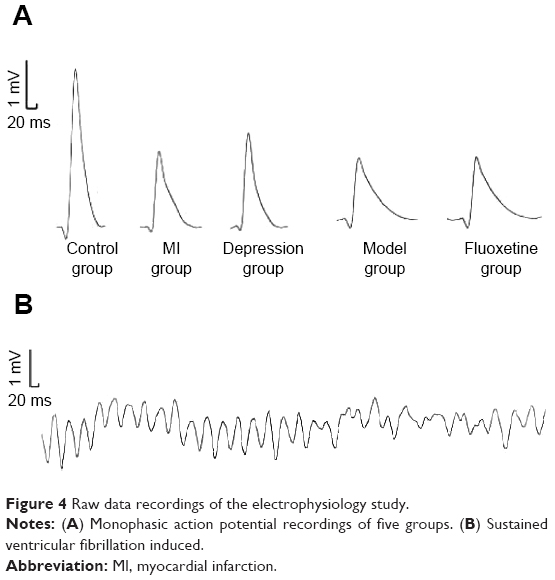 | Figure 4 Raw data recordings of the electrophysiology study. |
Effect of fluoxetine on the expression of Kv4.2 and Cx43
Figure 5 displays the results of immunoblotting. Kv4.2 was detected at a band nearly 74 kDa. Expression of Kv4.2 in myocardial cells was significantly reduced by 29%±12%, 24%±6%, and 41%±15%, respectively, in the MI group, depression group, and model group. It could be improved by fluoxetine by 11%±9% compared with the model. Two predominant forms of Cx43 were detected: phosphorylated form (P-Cx43, 43 kDa) and nonphosphorylated form (NP-Cx43, 41 kDa). MI induced a loss of phosphorylated Cx43. The expression of phosphorylated Cx43 was significantly reduced by 61%±14% and 67%±20% in the MI group and model group, and the fluoxetine did not show a marked improvement. The expression of Cx43 in the depression group was reduced slightly by 17%±9% but was not statistically significant. Immunochemistry analysis showed the same tendency (Figure 6). Expression of Kv4.2 in the model group was the lowest, and fluoxetine could increase it. However, fluoxetine showed no effect on the MI-induced reduction of Cx43.
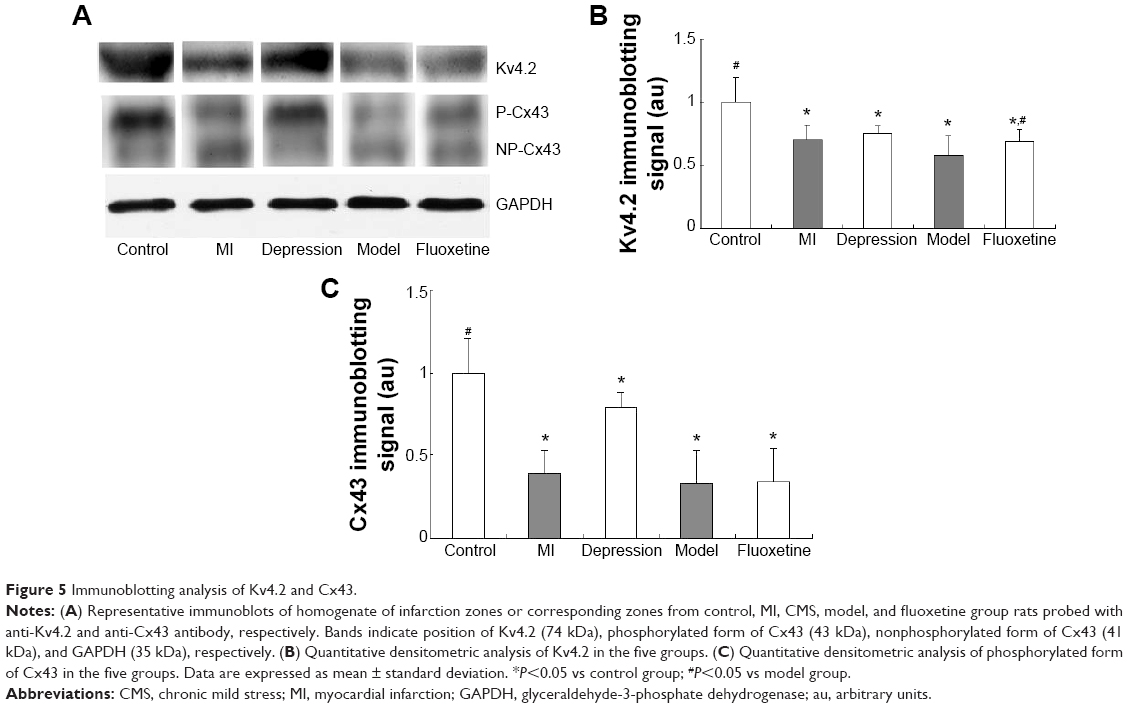 | Figure 5 Immunoblotting analysis of Kv4.2 and Cx43. |
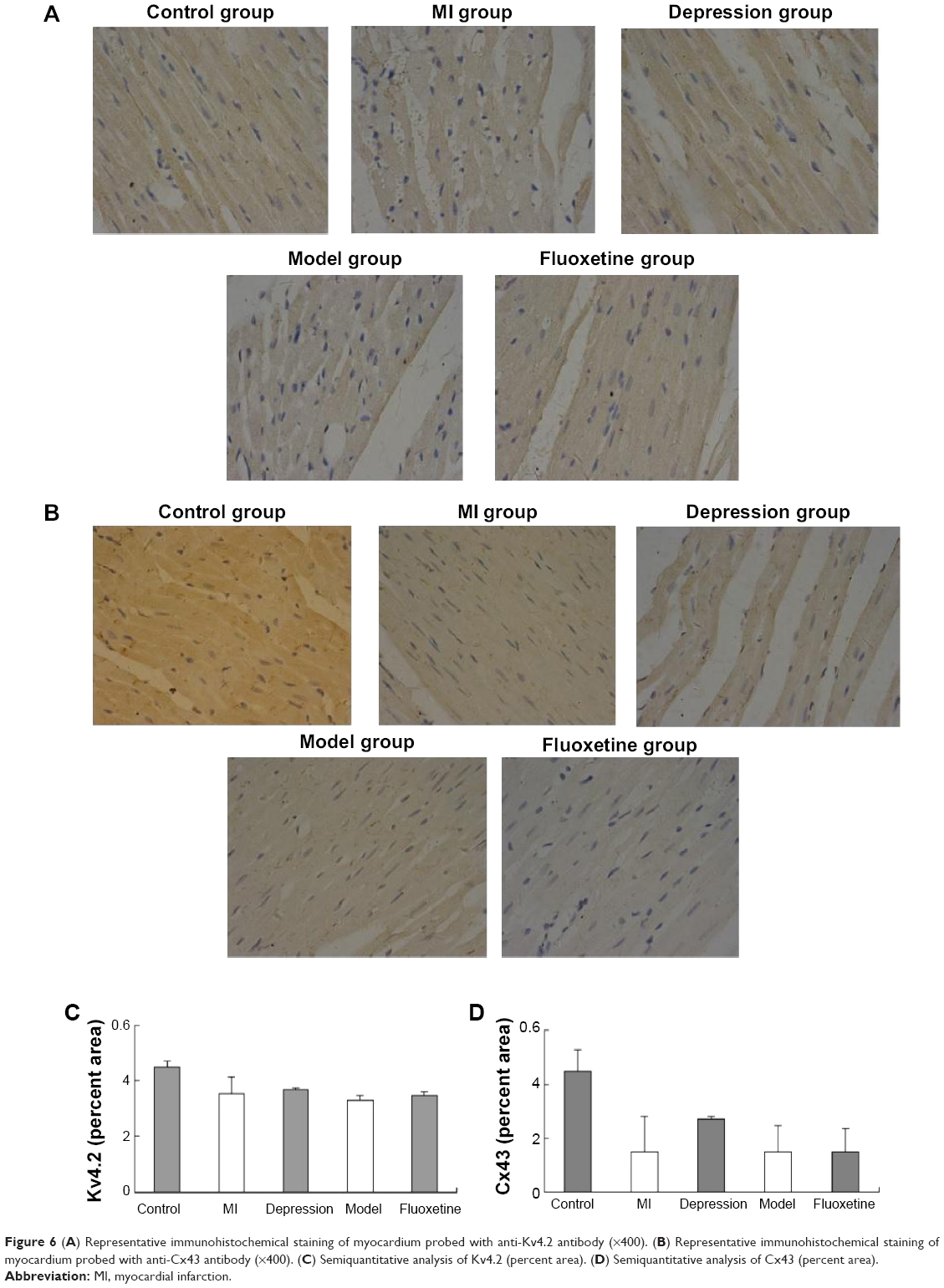 | Figure 6 (A) Representative immunohistochemical staining of myocardium probed with anti-Kv4.2 antibody (×400). (B) Representative immunohistochemical staining of myocardium probed with anti-Cx43 antibody (×400). (C) Semiquantitative analysis of Kv4.2 (percent area). (D)Semiquantitative analysis of Cx43 (percent area). |
Discussion
In MI patients, the prevalence of depressive disorder is 15%–30%, about five times that of the general population. Depression is an independent risk factor of CHD. A meta-analysis shows that post-MI depression is associated with a 2.4-fold increased risk of cardiac mortality and a two-fold increased risk of new cardiovascular events.22 SSRIs have been proven to safely and effectively decrease depressive symptoms and improve CHD prognosis.
Our group recently demonstrated in the same model that sympathetic hyperactivation and exacerbated myocardial remodeling may be a plausible mechanism linking depression to an adverse prognosis after MI.23 In the present study, we investigated the effect of fluoxetine on cardiac electrophysiology in a rat model of post-MI depression and the potential mechanism.
Experimental model
Animal models are helpful to our understanding of human health to the extent to which they faithfully mirror the specific aspect of the disease under study. As mentioned previously, an experimental rat model of post-MI depression was developed by LAD coronary artery ligation and following 28 days of CMS.
The model was then tested by ECG, sucrose preference test, and open field test and proven successful. After LAD coronary artery ligation, the surface ECG showed an ST segment elevation, suggesting MI. Current diagnostic criteria for major depression suggest that patients experience anhedonia and reduced physical activity. Animals exposed to 4 weeks of CMS exhibit a similar phenomenon to humans. Thus, CMS is widely used to develop a depression model.11,12 Similar to previous investigations,11,12 sucrose intake and activity were significantly reduced in rats exposed to 4 weeks of CMS in the present investigation.
Effect of fluoxetine on behavior changes in post-MI rats
Depression is a multifaceted psychological disorder characterized by behavioral, neuroendocrine, and physiological alterations. Depressed patients often experience anhedonia (a reduced responsiveness to pleasurable stimuli), difficulty sleeping, appetite changes, and fatigue. A similar phenomenon was observed in rats exposed to CMS in the present investigation.14
The sucrose preference test and the open field test are commonly used to observe behavior changes in rat models. The sucrose deficit in the CMS group appeared to be specific to anhedonia rather than a generalized attenuation of fluid ingestion, as both water intake and body weight were unaffected by the CMS procedure.4,11 Reduced sucrose intake preference relative to baseline values and the sucrose intake of the control group indicate anhedonia resulting from CMS. The open field test was used to evaluate anxiety-like behavior by measuring the spontaneous exploratory behavior of animals in aversive environments.13
Similar to the reduction in sucrose intake, the exploratory activity was reduced in the MI group, depression group, and model group in our study. This indicated an increase in depression-like behavior after MI in model rats. Chronic treatment with fluoxetine could significantly improve the depression-like symptoms in post-MI depression rats. This was consistent with previous clinical studies.
Effect of fluoxetine on cardiac electrophysiology in post-MI rats
MAP demonstrates how the local electroactivity of myocardial cell mass in extracellular recordings and its waveforms could reflect the morphology and phases of the transmembrane action potentials, especially the phases of depolarization. In recent years, MAP has been widely applied in studies on the mechanism of arrhythmic occurrence, the evaluation of antiarrhythmic drugs, and other fields.
In the present study, we found that both depression and MI could significantly prolong the MAPD90 and ERP and lower VFT in the peri-infarct zone. This prolongation caused by depression may be explained by the following aspects:
- Disturbances in autonomic regulation and elevated sympathetic activity lead to increased resting heart rate, impaired baroreflex sensitivity, and reduced heart rate variability. Sympathetic hyperactivity increases the density of local catecholamine in myocardium, thus activating L-type calcium channel, and finally influences cardiac repolarization.3,24,25
- Hypothalamic–pituitary–adrenal axis hyperactivity and elevated cortisol levels influence the function and expression of L-type calcium channel, sodium–calcium exchanger, and transient outward potassium channel (Ito).4,5
- Increased production of cytokines such as interleukin (IL)-1, IL-2, and IL-6 and interferon gamma affects the expression and function of ion channels.26,27
All of these three aspects of changes, which can be caused by MI as well, ultimately influence cardiac repolarization and lead to the prolongation of MAPD90 and ERP.
In the model group, MAPD90 and ERP in the peri-infarct zone were prolonged compared with the MI group, suggesting the potential cumulative effect of depression and MI on cardiac electrophysiology. This may be one of the reasons why the risks of mortality and cardiovascular events are higher in CHD patients with depression than in those without. Fluoxetine did not significantly reverse the prolongation of MAPD90 or ERP but induced a rise of VFT in model rats. Thus, fluoxetine may reduce the incidence of ventricular arrhythmia in post-MI depression rats.
Effect of fluoxetine on the expression of Kv4.2 and Cx43
In this study, we reported that Kv4.2 expression was significantly reduced in myocardial cells in model rats, which could be reversed by fluoxetine. The expression of Cx43 was reduced in any MI group, and the fluoxetine did not show marked improvement.
Kv4 channels produce rapidly activating and inactivating currents such as Ito, which mediate the early repolarization phase of the cardiac action potential. And Kv4.2 is predominant in rat heart. Kv4.2 channels, along with Kv channel-interacting protein 2, may represent the molecular correlate for Ito current gradient across the RV myocardium, and may be a cause of an atypical anterior J wave pattern associated with sudden cardiac death.28 Reduction in expression of Kv4.2 and Ito current density will affect the repolarization of myocardial cells and hence increase vulnerability to ventricular arrhythmia, as indicated by decreased heart rate variability and increased sensitivity to ventricular fibrillation.29,30 Thus, we suppose that the downregulation of Kv4.2 could potentially contribute to the cardiac electrophysiology change of depression patients, and fluoxetine may prevent the reduction, which consequently decreases the prevalence of ventricular arrhythmia in post-MI depression patients.
Cx43, the predominant protein forming gap junctions in the mammalian ventricular myocardium, is essential for normal conduction of action potential and to maintain a normal electrical coupling in the heart.31 It has been known that the expression and degradation, the distribution, and the density of Cx43 play an important role in modulating the magnitude of cell-to-cell electrical coupling in the heart. Acute ischemia induced by MI can result in marked reduction and dephosphorylation of Cx43, which may lead to delayed myocardial electrical activity and increased anisotropic conduction, prompting ventricular arrhythmias.32,33 In our study, model rats showed decreased expression of Cx43 but were not improved by fluoxetine. Thus, Cx43 may not be the target protein of fluoxetine.
Study limitations
There are several limitations in our study. First, because the electrophysiological and hemodynamic parameters were evaluated under an open-chest anesthetized state in our study, each parameter may be influenced by anesthesia. Second, although we measured the MADP, ERP, and VFT of heart in vivo, we did not carry on 24-hour Holter ambulatory ECG monitoring to observe the difference in prevalence of ventricular arrhythmias among different groups. Third, we did not simultaneously measure the MAPD and ERP of the normal peri-infarct zone and infarct area, because in the in vivo study, these zones were usually located at the lateral and posterior walls, and it is very difficult to record the MAP without altering the position and status of the heart.
Conclusion
MI rats exhibited an increase in depression-like behavior, and fluoxetine could significantly decrease the depression-like behavior in post-MI depression rats. The susceptibility to ventricular arrhythmias was increased in depression and post-MI depression rats, and fluoxetine may reduce the incidence of ventricular arrhythmia in post-MI depression rats and thus improve the prognosis. This was consistent with previous clinical studies. The decrease of ventricular arrhythmia induced by fluoxetine may be related in part to the upregulation of Kv4.2. Further molecular mechanism investigation is needed in the future.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81200139), the Natural Science Foundation of Hubei (2011CDC157, 2014CFB793, 2014CFA061), and the National Key Basic Research Development Program of China (The “973” Program, Grant Number: 2012CB518604). Statement of authorship: the authors take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
Disclosure
All authors have reported that they have no relationships to disclose, and there are no potential conflicts of interest.
References
Frasuresmith N, Lesperance F, Talajic M. Depression and 18-month prognosis after myocardial-infarction. Circulation. 1995;91:999–1005. | ||
Myers V, Gerber Y, Benyamini Y, Goldbourt U, Drory Y, Acute ISGF. Post-myocardial infarction depression: Increased hospital admissions and reduced adoption of secondary prevention measures: a longitudinal study. J Psychosom Res. 2012;72:5–10. | ||
Brown ADH, Barton DA, Lambert GW. Cardiovascular abnormalities in patients with major depressive disorder autonomic mechanisms and implications for treatment. CNS Drugs. 2009;23:583–602. | ||
Jokinen J, Nordström P. HPA axis hyperactivity and cardiovascular mortality in mood disorder inpatients. J Affect Disord. 2009;116(1–2):88–92. | ||
Shively CA, Musselman DL, Willard SL. Stress, depression, and coronary artery disease: modeling comorbidity in female primates. Neurosci Biobehav Rev. 2009;33:133–144. | ||
Pizzi C, Rutjes AWS, Costa GM, Fontana F, Mezzetti A, Manzoli L. Meta-analysis of selective serotonin reuptake inhibitors in patients with depression and coronary heart disease. Am J Cardiol. 2011;107:972–979. | ||
Lesperance F, Frasure-Smith N, Koszycki D, et al. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA. 2007;297:367–379. | ||
Pacher P, Ungvari Z, Nanasi PP, Furst S, Kecskemeti V. Speculations on difference between tricyclic and selective serotonin reuptake inhibitor antidepressants on their cardiac effects. Is there any? Curr Med Chem. 1999;6:469–480. | ||
Pacher P, Kecskemeti V. [Cardiovascular effects of selective serotonin reuptake inhibitor antidepressants]. Orvosi Hetilap. 2004;145:425–431. Hungarian. | ||
Park KS, Kong ID, Park KC, Lee JW. Fluoxetine inhibits l-type Ca2+ and transient outward K+ currents in rat ventricular myocytes. Yonsei Med J. 1999;40:144–151. | ||
Grippo AJ, Santos CM, Johnson RF, et al. Increased susceptibility to ventricular arrhythmias in a rodent model of experimental depression. Am J Physiol-Heart C. 2004;286:H619–H626. | ||
Wann BP, Bah TM, Boucher M. Vulnerability for apoptosis in the limbic system after myocardial infarction in rate: a possible model for human postinfarct major depression. J Neurosci. 2007;32(1):11–16. | ||
Lapmanee S, Charoenphandhu J, Charoenphandhu N. Beneficial effects of fluoxetine, reboxetine, venlafaxine, and voluntary running exercise in stressed male rats with anxiety- and depression-like behaviors. Behav Brain Res. 2013;250:316–325. | ||
Dai WD, Hale SL, Martin BJ, et al. Allogeneic mesenchymal stem cell transplantation in postinfarcted rat myocardium: short- and long-term effects. Circulation. 2005;112:214–223. | ||
Ando M, Katare RG, Kakinuma Y, et al. Efferent vagal nerve stimulation protects heart against ischemia-induced arrhythmias by preserving connexin 43 protein. Circulation. 2005;112:164–170. | ||
Grippo AJ, Beltz TG, Johnson AK. Behavioral and cardiovascular changes in the chronic mild stress model of depression. Physiol Behav. 2003;78:703–710. | ||
Grippo AJ, Moffitt JA, Johnson AK. Cardiovascular alterations and autonomic imbalance in an experimental model of depression. Am J Physiol Regul Integr Comp Physiol.2002;282:R1333–R1341. | ||
Keers R, Pedroso I, Breen G, et al. Reduced anxiety and depression-like behaviours in the circadian period mutant mouse afterhours. Plos One. 2012;7:e38263. | ||
Wakisaka Y, Niwano S, Niwano H, et al. Structural and electrical ventricular remodeling in rat acute myocarditis and subsequent heart failure. Cardiovasc Res. 2004;63:689–699. | ||
Wen HZ, Jiang H, Lu ZB, et al. Carvedilol ameliorates the decreases in connexin 43 and ventricular fibrillation threshold in rats with myocardial infarction. Tohoku J Exp Med. 2009;218:121–127. | ||
Anastasiou-Nana MI, Tsagalou EP, et al. Effects of transient myocardial ischemia on the ventricular defibrillation threshold. Pace. 2005;28:97–101. | ||
van Melle JP, de Jonge P, Spijkerman TA, et al. Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta-analysis. Psychosom Med. 2004;66:814–822. | ||
Shi S, Liang J, Liu T, et al. Depression increases sympathetic activity and exacerbates myocardial remodeling after myocardial infarction: evidence from an animal experiment. PLoS One. 2014;9(7):e101734. | ||
Servant D, Logier R, Mouster Y, Goudemand M. Heart rate variability. Applications in psychiatry. Encephale. 2009;35:423–428. | ||
Lache B, Meyer T, Herrmann-Lingen C. Social support predicts hemodynamic recovery from mental stress in patients with implanted defibrillators. J Psychosom Res. 2007;63:515–523. | ||
Leonard B, Maes M. Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci Biobehav Rev. 2012;36:764–785. | ||
Su JA, Chou SY, Tsai CS, Hung TH. Cytokine changes in the pathophysiology of poststroke depression. Gen Hosp Psychiat. 2012;34:35–39. | ||
Perrin MJ, Adler A, Green S, et al. Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J wave syndrome associated with sudden cardiac death. Circulation. Epub 2014 Sep 11. | ||
Ren CY, Wang FZ, Li G, et al. Nerve sprouting suppresses myocardial I(to) and I(K1) channels and increases severity to ventricular fibrillation in rat. Auton Neurosci. 2008;144:22–29. | ||
Kim LA, Furst J, Butler MH, Xu SH, Grigorieff N, Goldstein SAN. I-to channels are octomeric complexes with four subunits of each kv4.2 and K+ channel-interacting protein 2. J Biol Chem. 2004;279:5549–5554. | ||
Boengler K, Schulz R, Heusch G. Connexin 43 signalling and cardioprotection. Heart. 2006;92:1724–1727. | ||
Miura T, Miki T, Yano T. Role of the gap junction in ischemic preconditioning in the heart. Am J Physiol-Heart C. 2010;298:H1115–H1125. | ||
Saffitz JE, Laing JG, Yamada KA. Connexin expression and turnover: implications for cardiac excitability. Circ Res. 2000;86:723–728. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.