")
Back to Journals » Journal of Inflammation Research » Volume 14
Dysregulation of the Retromer Complex in Brain Endothelial Cells Results in Accumulation of Phosphorylated Tau
Authors Filippone A, Smith T, Pratico D
Received 30 September 2021
Accepted for publication 16 December 2021
Published 29 December 2021 Volume 2021:14 Pages 7455—7465
DOI https://doi.org/10.2147/JIR.S342096
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Alessia Filippone, Tiffany Smith, Domenico Pratico
Alzheimer’s Center at Temple, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, 19140, USA
Correspondence: Domenico Pratico
Alzheimer’s Center at Temple, Lewis Katz School of Medicine, Temple University, 3500 North Broad Street, MERB, suite 1160, Philadelphia, PA, 19140, USA
Tel +1 215-707-9380
Fax +1 215-707-2746
Email [email protected]
Introduction: Transport through endothelial cells of the blood–brain barrier (BBB) involves a complex group of structures of the endo-lysosome system such as early and late endosomes, and the retromer complex system. Studies show that neuronal dysregulation of the vacuolar protein sorting 35 (VPS35), the main component of the retromer complex recognition core, results in altered protein trafficking and degradation and is involved in neurodegeneration. Since the functional role of VPS35 in endothelial cells has not been fully investigated, in the present study we aimed at characterizing the effect of its downregulation on these pathways.
Methods: Genetic silencing of VPS35 in human brain endothelial cells; measurement of retromer complex system proteins, autophagy and ubiquitin-proteasome systems.
Results: VPS35-downregulated endothelial cells had increased expression of LC3B2/1 and more ubiquitinated products, markers of autophagy flux and impaired proteasome activity, respectively. Additionally, compared with controls VPS35 downregulation resulted in significant accumulation of tau protein and its phosphorylated isoforms.
Discussion: Our findings demonstrate that in brain endothelial cells retromer complex dysfunction by influencing endosome-lysosome degradation pathways results in altered proteostasis. Restoration of the retromer complex system function should be considered a novel therapeutic approach to rescue endothelial protein transport.
Keywords: retromer complex, brain endothelial cells, endosomal trafficking, tau protein, autophagy, ubiquitin-proteasome, Alzheimer’s disease
Introduction
The blood–brain barrier (BBB) represents a dynamic interface between systemic circulation and the brain and possesses unique properties regulating the micro-environment of the central nervous system (CNS).1 The differentiated BBB is composed of a complex cellular system of highly specialized endothelial cells (ECs) organized as monolayer, glial cells such as astrocytes and microglia, and pericytes.2 The BBB is recognized as a dynamic system that provides a safe environment for neuronal functionality via regulation of various mechanisms including nutrients supply, removal and protection from unwanted substances.3 Within the BBB, highly specialized ECs lining the brain microvasculature are recognized as major players in regulating endocytosis processes at the apical and basolateral membranes by producing its own early endosomes and other vesicle components.4,5
Thus, at the time when the BBB homeostasis fails, changes in vesicular trafficking and transport across brain endothelial cells, extravasation of pro-inflammatory mediators or metals exposure and accumulation in the brain could contribute to the development of many CNS pathologies. Abundant data in the literature have provided evidence for BBB disruption in several neurodegenerative diseases such as multiple sclerosis,6 Alzheimer’s disease (AD) and related tauopathies.7 Recent works showed that in polarized cells, such as brain endothelial cells, the retromer complex system, which is composed by the vacuolar protein sorting-associated proteins (VPSs), VPS26–VPS29–VPS35, could play an important role in the transcytosis process of cargo across the endothelium of the BBB. Interestingly, Wang et al recently reported that impairment of retromer-mediated endosomal trafficking can also promote mitochondrial and lysosomal dysfunction which ultimately result in disruption of the BBB integrity.8 However, together with BBB disruption, dysfunction of the retromer complex system is another important feature of the pathophysiology of AD and Down syndrome.9,10 Normally the system oversees vesicular transport from endosomes to trans Golgi-network (TGN), and involves not only endosomes but also lysosomes, proteasomes, and other degradative organelles in the trafficking of unwanted proteins.11 Despite the recognized importance of the retromer complex as a master regulator of protein sorting and trafficking in diseases of the CNS, and the wealth of knowledge in neurons little is known on the role that it plays in brain endothelial cells. Our study aimed to investigate the effect that retromer complex dysfunction, via downregulation of its main recognition core component VPS35, has on degradative pathway(s) and cellular proteostasis, two systems important for a normal BBB function. To reach this goal we implemented the human brain endothelial cell line hCMEC/D3 which represents an established in vitro model for studying endo-lysosomal structure and function.12,13
Materials and Methods
Cells
The immortalized hCMEC/D3 (Millipore/Sigma, catalog number SCC066) cell line was cultured in EBM-2 basal medium (catalog #00190860, Lonza, Walkersville, MD, USA) supplemented with 5% fetal bovine serum, 2% growth factors (GFs) (catalog #0000981202, Lonza, Walkersville, MD, USA) following the manufacturer’s instructions and maintained at 37°C with 5% CO2 exposure. For knockdown experiments, cells were cultured to 70% confluence in six-well plates and then transfected. Briefly, a mixture of opti-MEM (Thermo Fisher Scientific, catalog # 11058–021) with 15 nM of human control siRNA (Thermo Fisher Scientific, catalog #4392420) was prepared and incubated at room temperature (RT) for 5 minutes. Another mixture using opti-MEM with 15 nM of human VPS35 siRNA (Thermo Fisher Scientific, catalog # 4390843) was prepared and incubated for 5 minutes. Both solutions were mixed and incubated for 15 minutes, by using Lipofectamine RNAiMAX transfection reagent (Invitrogen, catalog # 13778–150) according to the manufacturer’s instructions. After 6 hrs incubation, fresh complete medium was replaced up to 48 hrs. In some experiments, cells were incubated with Bafilomycin A1 (BafA1) (Sigma-Aldrich, catalog # B1793-10; final concentration of 100 mM), or Bortezomib (BTZ) (Thermo Fisher Scientific, catalog # J60378; final concentration 100 μM) for 4 hrs.
Western Blot Analysis
Cell lysates were used for Western blot analyses as previously described.14 The following antibodies were used: LC3B (1:500, Cell Signaling Technology, catalog # CST-27755), SQSTM1/p62 (or p62) (1:500, Abcam, ab 56,416), ATG9 (1:300, Abcam, ab 108,338), ATG7 (1:300, Cell Signaling Technology, CST 8558S), ATG5 (1:300, Cell Signaling Technology, CST 2630S), VPS35 (1:250, ab 157,220, Abcam), VPS26 (1:250, Abcam, ab 23,892), VPS29 (1:250, Abcam, ab 10,160), ubiquitin (1:500, Cell Signaling Technology, CST 3933S), CI-MPR (1:250, Abcam, ab 124,767), Sorl-1 (1:250, Cell Signaling Technology, CST 79322S), Beclin-1 (Cell Signaling Technology, CST 3738S), HT-7 (1:500, Thermo Fisher Scientific, catalog # MN1000) AT270 (1:500, Thermo Fisher Scientific, catalog # MN10500), PHF13 (1:500, Cell Signaling Technology, catalog # CST 9632), AT8 (1:500, Thermo Fisher Scientific, catalog # MN1020), MC-1 (gift of Dr. Peter Davis), GSK3-α/β (1:100, Cell Signaling Technology, catalog # CST 5676S), p-GSK3-α/β (p-GSK-3 α/β) (1:100, Cell Signaling Technology, catalog # CST 9331S), protein phosphatase 2A (PP2A) (1:100, Invitrogen, catalog # PAS 17510), cyclin-dependent kinase 5 (Cdk-5) (1:100, Santa Cruz Biotechnology, catalog # sc249) and p25 (1:100, Abcam, catalog # 35271). Signals were developed with Odyssey Infrared Imaging Systems (LI-COR Bioscience). GAPDH (1:500, Cell Signaling Technology, catalog # CST 2118S) was always used as internal loading control.
Immunofluorescence Analysis
Cells were cultured on glass coverslips, and treated with control siRNA or VPS35 siRNA, as previously described.15 After fixing, cells were incubated overnight with LC3B (1:250), and p62 (1:250) primary antibodies. After rinsing with PBS, cells were incubated with the Alexa Fluor 488/568-conjugated secondary antibodies (1:300) (Abcam, anti-goat ab 150,133, anti-mouse ab 175,700, anti-rabbit ab 175,693) for 30 min, 37°C. The nucleus was stained with diamidino-2-phenylindole (DAPI solution) (1:1000, Thermo Fisher Scientific, catalog # 62248). Coverslips were mounted using ProLong™ Glass Antifade Mountant (Thermo Fisher Scientific, catalog # P36980) and images were taken by using ECLIPSE Ti2 fluorescent microscope (Nikon) with 100× objective (scale bar 10 μm).
LysoTrackerRed Staining
Cells were cultured on glass coverslips, and treated with control siRNA or VPS35 siRNA, as described above. After rinsing with PBS, cells were incubated with 75 nM LysoTracker Red DND-99 (Thermo Fisher Scientific, catalog # L7528) in 10% Donkey serum for 90 min at RT followed by washing with fresh PBS. Next, cells were fixed as described above. Images were taken by using ECLIPSE Ti2 fluorescent microscope (Nikon) with 100× objective (scale bar 10 μm).
Statistical Analysis
All the data are expressed as mean ± standard error of the mean. Comparisons between two groups were made using an unpaired two-tailed t-test. Comparisons between more than two groups were made using a one-way ANOVA with Bonferroni's multiple comparisons test. The p-values for each comparison are listed in each figure legend with p < 0.05 considered statistically significant. All statistical tests were performed using GraphPad Prism 5.0 (GraphPad Software).
Results
Effect of VPS35 Downregulation on the Other Retromer Complex System Components in Brain Endothelial Cells
In neuronal cells depletion of one component of the retromer recognition core leads to reduction of the other subunits.15,16 By using Western blot analysis, we investigated the effect that downregulation of VPS35 had also on the expression levels of VPS26, and VPS29 in hCMEC/D3 cells. As shown in Figure 1, when we downregulated VPS35 in these cells using VPS35 siRNA, we found that not only the expression levels of this retromer subunit but also VPS26 were significantly decreased compared to control cells. However, although we observed a reduction of VPS29 expression levels, compared with controls the difference did not reach statistical significance (Figure 1A and B). By contrast, downregulation of VPS35 did not influence the steady state levels of retromer cargo receptor and cargo protein such as the cation-independent mannose 6-phosphate receptor (CI-MPR), and sortilin-related receptor 1 (Sorl-1), respectively (Figure 1A and B).
 |
Figure 1 Downregulation of VPS35 influence on retromer complex system proteins in brain endothelial cells. The hCMEC/D3 cells were transfected with VPS35 or Ctr siRNA for 48 hours, then cell lysates were harvested for biochemistry. (A) Representative Western blot analysis for VPS35, VPS26, VPS29, CI-MPR, and Sorl-1 in cells lysates transfected with 15 nM VPS35 siRNA, Ctr siRNA, or lipofectamine (Lipof) alone. (B) Densitometric analysis of the immunoreactivity to the antibodies shown in (A) (***p < 0.001; **p < 0.01). Results are mean ± SEM (N = 2 per group, three individual experiments). |
Retromer Complex Defects Influences Degradation Pathways in Brain Endothelial Cells
Next, we assessed whether in brain endothelial cells retromer downregulation had an influence on degradation processes driven by autophagy and the ubiquitin proteasome system (UPS) as recently described in neuronal cells.17 Compared with controls, downregulation of VPS35 resulted in an accumulation of LC3B2 and an increase in the LC3B2/1 ratio, a known marker of autophagy flux activation.18 This change was associated with an increase in the transmembrane autophagy-related protein 9 (ATG9) levels, and a significant decrease in p62 expression (Figure 2A and B). By contrast, cells with VPS35 downregulation did not show any significant changes in the levels of AGT5, AGT7 and Beclin-1 protein compared with controls (Figure 2A and B).
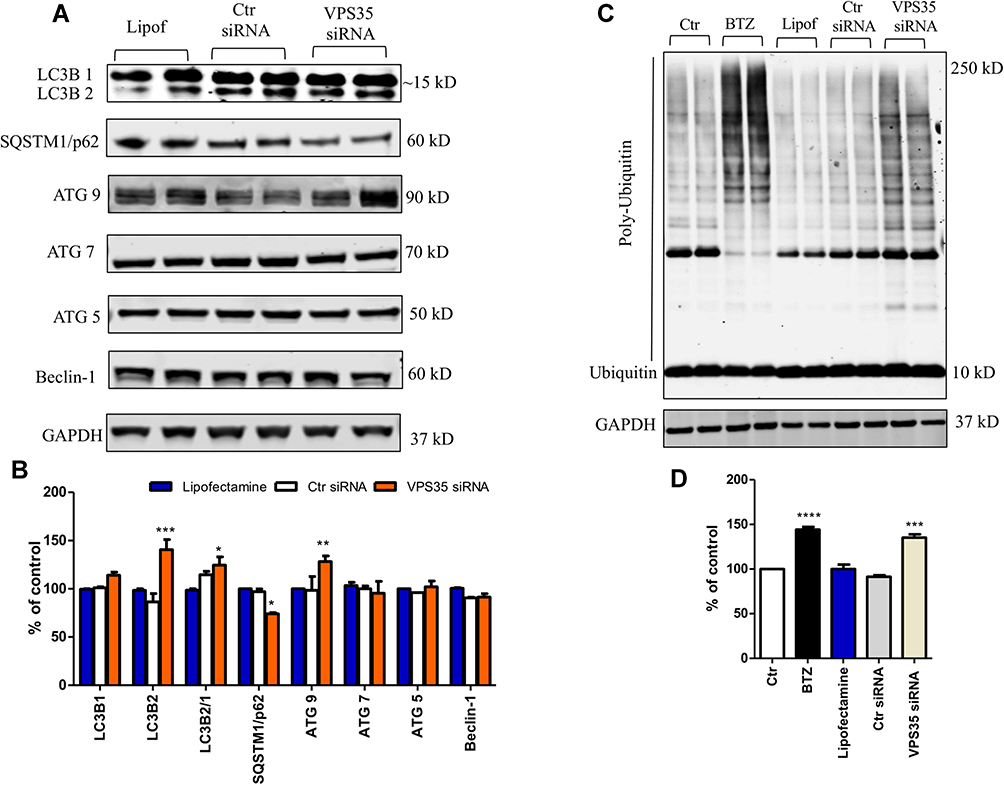 |
Figure 2 VPS35 silencing alters autophagy and promotes accumulation of poly-ubiquitinylated proteins in brain endothelial cells. The hCMEC/D3 cells were transfected with VPS35 or Ctr siRNA for 48 hrs, then cell lysates were harvested for biochemistry. (A) Representative Western blot analysis for LC3B, SQSTM1/p62, ATG9, ATG7, ATG5, and Beclin-1. (B) Densitometric analysis of the immunoreactivity to the antibodies shown in (A) (***p < 0.001 vs Lipofectamine; **p < 0.01 vs Lipofectamine; *p< 0.05 vs Lipofectamine). Results are mean ± SEM. (C) Representative Western blot analysis for ubiquitin, in cells lysates transfected as previously described. (D) Densitometric analysis of the immunoreactivity to the antibody shown in (C) (***p < 0.001 vs Lipofectamine), (****p < 0.0001 vs Lipofectamine). Results are mean ± SEM (N = 2 per group, three individual experiments). |
Besides autophagy, the UPS is another important degradative pathway which relies on the integrity of endosomal trafficking and sorting system through the retromer complex.19 Under our experimental conditions, first we incubated the cells with BTZ, a specific proteasome inhibitor,20 as a positive control, to make sure that indeed the cells produce polyubiquitinated signals. Next, we found that compared with control cells VPS35 downregulation in brain endothelial cells resulted in a significant increase in poly-ubiquitinylated proteins levels (Figure 2C and D).
Autophagy and Lysosome are Affected by VPS35 Silencing in Brain Endothelial Cells
To further support our biochemistry measures showing alteration of the autophagy and UPS degradation pathway systems, next endothelial cells were immuno-reacted with LC3B and p62 antibodies, and the LysoTracker red dye. Confirming the Western blot analysis results, compared with control cells we observed an increase in LC3B immunoreactivity in brain endothelial cells receiving VPS35 siRNA suggesting an activation of the autophagy flux following VPS35 downregulation (Figure 3A and B). Additionally, these changes were associated with a decrease in p62 immunoreactivity (Figure 3G and H).
 |
Figure 3 Immunoreactivity for LC3B, SQSTM1/p62 antibodies following VPS35 silencing in brain endothelial cells. Representative microscopy images of control cells (A, A1 inset) and VPS35 silenced cells (B, B1 inset) stained for LC3B (TXred), VPS35 (Cy5 channel pseudo-colored purple), nuclear stain DAPI (blue); (Scale bar = 10 µm). Representative microscopy images of control (C, C1 inset) and VPS35 silenced cells (D, D1 inset) stained for LC3B (TXred), VPS35 (Cy5 channel pseudo-colored purple), nuclear stain DAPI (blue) also treated with BafA1; (Scale bar = 10 µm). (E) LC3B fluorescence intensity nucleus area (***p < 0.001 vs Ctr siRNA cells). (F) LC3B fluorescence intensity nucleus area in BafA1 treated cells (***p < 0.001 vs Ctr siRNA cells). Representative microscopy images of control (G, G1 inset) and VPS35 silenced cells (H, H1 inset) stained for SQSTM1/p62 (TXred), VPS35 (FITC: green) nuclear stain DAPI (blue); (Scale bar = 10 µm). Representative microscopy images of control (I, I1 inset) and VPS35 silenced cells (J, J1 inset) stained for SQSTM1/p62 (TXred), VPS35 (FITC: green), nuclear stain DAPI (blue) also treated with BafA1; (Scale bar = 10 µm). (K) SQSTM1/p62 fluorescence intensity nucleus area (L) SQSTM1/p62 fluorescence intensity nucleus area in BafA1 treated cells. (***p < 0.001 vs Ctr siRNA cells) (N = 3 individual experiments). |
Next, we assessed the effect of VPS35 downregulation on acidic vesicles accumulation in the same cells. Since the less acidic vesicles are located in the projections of the cells and the most acidic ones with higher fluorescent intensity are closer to the nucleus in all groups of brain endothelial cells,21 treatment with BafA1, a specific V-ATPase pump inhibitor22 was used to prove the exclusive signal of LC3B and p62 antibodies for acidophilic components around the nuclear region. As shown in Figure 3, we detected fluorescent organelles positivity in most of the cells in the presence of BafA1 following VPS35 silencing compared to control cells (Figure 3C, D, I and J) confirming that proton pumps inhibition was responsible for the creation of higher pH inside the vesicles. Under these experimental conditions, we also observed that LysoTracker red dye highlighted an accumulation of autophagy-associated lysosomal activity products in the VPS35 down regulated brain endothelial cells compared to control cells (Figure 4A and B respectively, see % area of Lysotracker Red panel Figure 4C).
 |
Figure 4 Lysosomal staining following VPS35 silencing in brain endothelial cells. Representative microscope images for control cells (A and A1 inset) and VPS35 siRNA-treated cells (B and B1 inset) labeled for VPS35 antibody (Cy5 channel pseudo-colored purple) and LysoTracker (LT) live cells dye (TXred channel, red). The distribution of acidic vesicles was visualized using fluorescence microscopy (Scale bar: 10 µm). Percentage area of LT (C) (**p < 0.01 vs Ctr siRNA). Results are mean ± SEM (N=3 individual experiments). |
Accumulation of Tau Following VPS35 Silencing in Brain Endothelial Cells
The balance between formation and degradation of different proteins, also known as proteostasis, within the cell is an important function responsible for cellular health and its alteration is a common feature of neurodegeneration. Among the different proteins recent attention has been focused on the accumulation of pathological tau protein, which has been shown to spread throughout the brain in a transgenic mouse model of AD-related tauopathy which expresses human mutant tau (rTg4510 mice). Interestingly, a recent study reported on the key role that the endolysosome system plays not only in tau accumulation, but also in tau diffusion and intercellular spreading.23 For this reason, next we assessed changes in tau levels in cerebral endothelial cells following dysregulation of the retromer complex system. As shown in Figure 5, compared with controls, VPS35-downregulated cells had a significant accumulation of total tau levels as shown by the immunoreactivity to the HT-7 antibody. In addition, we found that downregulation of VPS35 resulted in higher amount of tau protein phosphorylated at the residues Threonine 181 (tau pThr181) and Serine 396 (tau pSer396) as identified by AT270 and PHF13 antibodies, respectively (Figure 5A and B). Interestingly, using the antibody MC-1, which is directed against tau having pathological conformational changes, compared with controls we also observed a significant increase in its immunoreactivity when VPS35 was downregulated. By contrast, despite a trend toward an increase no statistically significant changes were observed for the immunoreactivity to the antibody AT8, which recognizes tau isoform phosphorylated at both Serine 202 and Threonine 205 (Ser201/Thr205), when VPS35-silenced cells were compared with controls (Figure 5A and B). Next, looking for mechanisms potentially involved in the observed changes in tau phosphorylation, we examined some of the kinases and phosphatases that are considered major players of these tau modifications. As depicted in Figure 5, we found that compared with controls, endothelial cells with downregulation of VPS35 did not manifest any changes in the expression levels of Cdk5, p25, total or phosphorylated GSK-3α, GSK-3β, and protein phosphatase 2A (PP2A) (Figure 5C and D).
 |
Figure 5 VPS35 affects pathological tau accumulation in brain endothelial cells. Brain endothelial cells were transfected with VPS35 or Ctr siRNA for 48 hrs, then cell lysates were harvested for biochemistry analysis. (A) Representative Western blot analysis for total tau (HT-7), phosphorylated tau at residues S202/T205 (AT8), T181 (AT270), and S396 (PHF13), and pathological tau (MC-1). (B) Densitometric analysis of the immunoreactivity to the antibodies shown in (A). (***p < 0.001 vs Lipofectamine; **p < 0.01 vs Lipofectamine; *p < 0.05 vs Lipofectamine). (C) Representative Western blot analysis for Cdk5, p25, GSK-3α/β, p-GSK-3α/β and PP2A. (D) Densitometric analysis of the immunoreactivity to the antibodies shown in (C). Results are mean ± SEM (N = 2 per group, three individual experiments). |
Discussion
Abundant literature has clearly shown that the retromer complex system normally ensures the degradation of autophagic cargo via the maintenance of endo-lysosome system efficiency, and that its failure results in the accumulation of autophagosomes and unwanted proteins, such as Aβ, tau and α-synuclein, secondary to a decreased ability for degrading them.24 However, the majority of the published works on this particular topic have been so far focused mainly on one cell type: neurons and neuronal cell lines.25–27
In the present study, we provide the first demonstration that in human brain endothelial cells dysfunction of the retromer complex system alters two degradation pathways by influencing autophagy flux, and the UPS which ultimately result in the accumulation of pathological forms of tau protein. We have used hBEC cell line since it contains all the necessary machinery to investigate the endosomal and lysosomal system and it is a well-accepted and established in vitro model for investigating BBB vesicular transport.28 In particular, we were interested in assessing how the retromer complex system dysregulation would influence degradation pathways and protein homeostasis in the context of a BBB-like microenvironment. This is very important since within the BBB endothelial cells lining the CNS microvasculature possess unique transcytosis properties that govern selective molecular trafficking between the blood and the brain parenchyma.29
First, we confirmed the known concept that stability of the retromer complex is strictly dependent on the precise stoichiometric formation of the trimeric recognition core and that depletion or reduction of one subunit within the VPS35-VPS26-VPS29 complex leads to reductions in some or all the other subunits. Thus, like for neurons in brain endothelial cells VPS35 downregulation resulted in reduction of VPS26 and VPS29 which would support an overall dysfunction of the whole complex sorting system. By contrast, no significant changes were observed for some of the main retromer cargo receptors such as CI-MPR or Sorl-1. These findings are in line with other reports in neurons showing that dysfunction of the retromer complex system does not directly and necessarily influence the steady state levels of its cargo receptors.30,31
In recent years emerging evidence has indicated a possible biological link between the retromer complex with both autophagy and UPS systems also in neuronal cells. Interestingly, these cellular pathways which are actively involved in the clearance of abnormally folded proteins (ie, Aβ and tau) have been shown to be altered in various neurodegenerative diseases like AD and related tauopathies.32,33 In AD, it is known that BBB breakdown is present and occurs as consequence of different insults to neuronal cells including neuroinflammation and leukocytes infiltrations, which are exacerbated by subsequent autophagy impairments and ultimately contribute to the pathogenesis of the disease progression.34 However, while there is abundant literature on this topic for neurons, no data are available for endothelial cells. To assess whether VPS35 gene silencing and subsequent retromer dysfunction would also influences autophagy in brain endothelial cells, we measured some of the main components of this system. Initially, we looked at the microtubule-associated protein 1 light chain 3 (LC3) which is known to be converted from the soluble form (LC3B 1) to the autophagosome-associated form (LC3B 2) and to play an important role in the formation of the autophagosomes. Under our experimental conditions of VPS35 downregulation in brain endothelial cells, we found that the LC3B 2/1 ratio was significantly increased whereas p62 levels, which inversely correlates with autophagic activation, were significantly reduced.35
These findings suggest that retromer complex system dysfunction contributes to the activation of autophagy and this effect is at least partially mediated by changes in some of ATG proteins such as ATG9, which was indeed upregulated. According to our results, cells possessed more acidic organelles after VPS35 silencing than control cells, a phenomenon that could be explained by the increased immunopositivity of autophagic structures for LC3B but a lesser content of them for p62 after VPS35 downregulation and BafA1 exposures. Moreover, downregulation of VPS35 had an effect also on the UPS system since the number of lysosomes and acidic organelles was greatly increased compared to control cells as highlighted by the lysosome-specific dye used. While we cannot exclude that the change in autophagy and not the retromer dysfunction per se may have also influenced the UPS machinery, it is important to stress that the alterations we observed for both systems in brain endothelial cells could have significant impact on brain health. Our findings can also offer a basis for understanding the final step of the ubiquitin-proteasome pathway that could be influenced by alterations of the retromer complex system.35 Thus, by demonstrating that VPS35 silencing increases expression levels of poly-ubiquitinylated proteins in brain endothelial cells we begin to characterize the relationship between the retromer complex and the ubiquitin-proteasome system at the BBB level in terms of clearance of unwanted proteins. This fact is in line with recent studies showing the retromer complex vital role in the clearance of aggregates of unfolded tau protein in the brain that could form intracellular inclusions under pathological conditions termed “tauopathies”.36 To this end, we were interested in studying whether in brain endothelial cells the perturbed endosomal and degradative pathways resulting from VPS35 silencing would influence total tau and any of its various phosphorylated isoforms which have been implicated in AD pathogenesis.37 Our analysis of tau protein species in these cells showed a pattern of progressive accumulation of them upon VPS35 silencing. Similar to the neuronal accumulation of pathologic tau aggregates secondary to retromer complex dysfunction,14 we found that also brain endothelial cells with genetically induced retromer complex deficiency presented a significant increase in total tau, together with an elevation of 2 specific phosphorylated isoforms at residues Threonine 181 and Serine 396. Interestingly, these post-translational modifications of tau protein were independent from any involvement of several kinases and a phosphatase which have been considered important for them.38 Further supporting an alteration of tau proteostasis secondary to retromer complex dysfunction in endothelial cells are the results showing a significant increase in the immunoreactivity to the specific antibody MC-1, which is known to target specifically pathological conformational changes of tau protein, one of the earliest detectable events in the brain of AD patients.39 Besides these aspects on tau neurobiology, the mechanisms we described herein could have also potential implications for the development of cerebral amyloid angiopathy, a clinical condition often associated with AD, which is characterized by the accumulation of Aβ within the wall of small blood vessels.40
In summary, the current study supports the novel idea that in cerebral endothelial cells the retromer dysfunction is directly responsible for the accumulation of pathological tau secondary to a deficit in its clearance rather than changes in its post-translational modifications. Whether the endosomal sorting and trafficking network system failure via the silencing of VPS35 is also implicated in tau spreading across endothelial cells remains to be investigated.
Conclusions
Here, we present the first experimental evidence that in brain endothelial cells dysfunction of the retromer complex system secondary to VPS35 downregulation influences two major degradation pathways which ultimately results in accumulation of pathological tau protein. Considering the central role that brain endothelial cells play in the maintenance of a healthy BBB, our data have potential implications not only for brain health but also for neurodegenerative diseases such as AD and related tauopathies where an alteration of BBB function and permeability has been widely described as an early event in the pathogenesis of these diseases.
Abbreviations
BBB, blood–brain barrier; VPS, vacuolar protein sorting; CNS, central nervous system; TGN, trans Golgi-network; AD, Alzheimer’s disease; DAPI, diamidino-2-phenylindole; CI-MPR, mannose 6-phosphate receptor; Sorl-1, sortilin-related receptor 1; SQSTM1 or p62, sequestrosome-1; tau pThr181, tau protein phosphorylated at the residues threonine 181; tau pSer396, tau protein phosphorylated at serine 396; Ser201/Thr205, tau protein phosphorylated serine 202 and threonine 205; BafA1, bafilomycinA1.
Acknowledgments
Domenico Praticò is the Scott Richards North Star Charitable Foundation chair for Alzheimer’s research.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The work described in the article was in part supported by grants from the National Institute of Health (AG055707, and AG056689).
Disclosure
All authors declare no conflicts of interest.
References
1. Abbott NJ. Blood–brain barrier structure and function and the challenges for CNS drug delivery. J Inherit Metab Dis. 2013;36:437–449. doi:10.1007/s10545-013-9608-0
2. Engelhardt B. Development of the blood-brain barrier. Cell Tissue Res. 2003;314:119–129. doi:10.1007/s00441-003-0751-z
3. Abbott NJ, Patabendige AAK, Dolman DEM, et al. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37(1):13–25. doi:10.1016/j.nbd.2009.07.030
4. Gonzalez-Carter D, Liu X, Tockary TA, et al. Targeting nanoparticles to the brain by exploiting the blood–brain barrier impermeability to selectively label the brain endothelium. Proc Natl Acad Sci USA. 2020;117(32):19141–19150. doi:10.1073/pnas.2002016117
5. Haqqani AS, Delaney CE, Brunette E, et al. Endosomal trafficking regulates receptor-mediated transcytosis of antibodies across the blood brain barrier. J Cereb Blood Flow Metab. 2018;38:727–740. doi:10.1177/0271678X17740031
6. Bell L, Koeniger T, Tacke S, et al. Characterization of blood–brain barrier integrity in a B-cell-dependent mouse model of multiple sclerosis. Histochem Cell Biol. 2019;151(6):489–499. doi:10.1007/s00418-019-01768-6
7. Wu S, Liu H, Zhao H, et al. Environmental lead exposure aggravates the progression of Alzheimer’s disease in mice by targeting on blood brain barrier. Toxicol Lett. 2020;319:138–147. doi:10.1016/j.toxlet.2019.11.009
8. Wang H, Qi W, Zou C, et al. NEK1-mediated retromer trafficking promotes blood–brain barrier integrity by regulating glucose metabolism and RIPK1 activation. Nat Commun. 2021;12:4826. doi:10.1038/s41467-021-25157-7
9. Filippone A, Praticò D. Endosome dysregulation in down syndrome: a potential contributor to Alzheimer disease pathology. Ann Neurol. 2021;90(1):4–14. doi:10.1002/ana.26042
10. Vagnozzi A, Praticò D. Endosomal sorting and trafficking, the retromer complex and neurodegeneration. Mol Psychiatry. 2019;24:857–868. doi:10.1038/s41380-018-0221-3
11. Tang FL, Zhao L, Zhao Y, et al. Coupling of terminal differentiation deficit with neurodegenerative pathology in Vps-35-deficient pyramidal neurons. Cell Death Differ. 2020;27:2099–2116. doi:10.1038/s41418-019-0487-2
12. Toth AE, Nielsen SSE, Tomaka W, et al. The endo-lysosomal system of Bend.3 and hCMEC/D3 brain endothelial cells. Fluids Barriers CNS. 2019;16(1):14. doi:10.1186/s12987-019-0134-9
13. Weksler B, Romero IA, Couraud PO. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS. 2013;10(1):16. doi:10.1186/2045-8118-10-16
14. Li JG, Chiu J, Praticò D. Full recovery of the Alzheimer’s disease phenotype by gain of function of vacuolar protein sorting 35. Mol Psychiatry. 2020;25(10):2630–2640. doi:10.1038/s41380-019-0364-x
15. Vagnozzi AN, Li JG, Chiu J, et al. VPS35 regulates tau phosphorylation and neuropathology in tauopathy. Mol Psychiatry. 2019. doi:10.1038/s41380-019-0453-x
16. Ekyune K, Lee Y, Lee H, et al. Implication of mouse Vps26b–Vps29–Vps35 retromer complex in sortilin trafficking. Biochem Biophys Res Commun. 2010. doi:10.1016/j.bbrc.2010.10.121
17. Filippone A, Li JG, Pratico’ D. VPS35 downregulation alters degradation pathways in neuronal cells. J Alzheimers Dis. 2021;84(3):1079–1089. doi:10.3233/JAD-210701
18. Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci. 2017;18(9):1865. doi:10.3390/ijms18091865
19. Fuse A, Furuya N, Kakuta S, et al. VPS29-VPS35 intermediate of retromer is stable and may be involved in the retromer complex assembly process. FEBS Lett. 2015;589:1430–1436. doi:10.1016/j.febslet.2015.04.040
20. Pekol T, Daniels JS, Labutti J, et al. Human metabolism of the proteasome inhibitor bortezomib: identification of circulating metabolites. Drug Metab Dispos. 2005;33(6):771–777. doi:10.1124/dmd.104.002956
21. Colacurcio DJ, Nixon RA. Disorders of lysosomal acidification. The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev. 2016;32:75–88. doi:10.1016/j.arr.2016.05.004
22. Yamamoto A, Tagawa Y, Yoshimori T, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi:10.1247/csf.23.33
23. Polanco JC, Hand GR, Briner A, et al. Exosomes induce endolysosomal permeabilization as a gateway by which exosomal tau seeds escape into the cytosol. Acta Neuropathol. 2021;141(2):235–256. doi:10.1007/s00401-020-02254-3
24. Maruzs T, Lőrincz P, Szatmári Z, et al. Retromer ensures the degradation of autophagic cargo by maintaining lysosome function in drosophila. Traffic. 2015;16(10):1088–1107. doi:10.1111/tra.12309
25. Lőrincz P, Lakatos Z, Maruzs T, et al. Atg6/UVRAG/Vps34-containing lipid kinase complex is required for receptor downregulation through endolysosomal degradation and epithelial polarity during drosophila wing development. BioMed Res Int. 2014;2014:851349. doi:10.1155/2014/851349
26. Falk MM, Fong JT, Kells RM, et al. Degradation of endocytosed gap junctions by autophagosomal and endo-/lysosomal pathways: a perspective. J Membr Biol. 2012;245(8):465–476. doi:10.1007/s00232-012-9464-0
27. Zhao YG, Codogno P, Zhang H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat Rev Mol Cell Biol. 2021;1–18. doi:10.1038/s41580-021-00392-4
28. Weksler BB, Subileau EA, Perriere N, et al. Blood–brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–1874. doi:10.1096/fj.04-3458fje
29. Predescu SA, Predescu DN, Malik AB. Molecular determinants of endothelial transcytosis and their role in endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2007;293:L823–L842. pmid: 17644753. doi:10.1152/ajplung.00436.2006
30. Sager KL, Wuu J, Herskowitz JH, et al. Neuronal LR11 expression does not differentiate between clinically defined Alzheimer’s disease and control brains. PLoS One. 2012;7(8):e40527. doi:10.1371/journal.pone.0040527
31. Li Y, Rowland C, Catanese J, et al. SORL1 variants and risk of late-onset Alzheimer’s disease. Neurobiol Dis. 2008;29(2):293–296. doi:10.1016/j.nbd.2007.09.001
32. Chung KM, Hernández N, Sproul AA, Yu WH. Alzheimer’s disease and the autophagic-lysosomal system. Neurosci Lett. 2019;697:49–58. doi:10.1016/j.neulet.2018.05.017
33. Hartz A, Zhong Y, Wolf A, et al. Aβ40 reduces P-glycoprotein at the blood–brain barrier through the ubiquitin–proteasome pathway. J Neurosci. 2016;36(6):1930–1941. doi:10.1523/JNEUROSCI.0350-15.2016
34. Jucker M, Walker L. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501(7465):45–51. doi:10.1038/nature12481
35. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2021;17(1):1–382.
36. Carosi JM, Hein L, Hurk M, et al. Retromer regulates the lysosomal clearance of MAPT/tau. Autophagy. 2020. doi:10.1080/15548627.2020.1821545
37. Chen X, Kordich J, Williams E, et al. Parkinson’s disease-linked D620N VPS35 knockin mice manifest tau neuropathology and dopaminergic neurodegeneration. Proc Natl Acad Sci USA. 2019;116(12):5765–5774. doi:10.1073/pnas.1814909116
38. Gao L, Xiao H, Ai L, et al. Vps35 deficiency impairs cdk5/p35 degradation and promotes the hyperphosphorylation of tau protein in retinal ganglion cells. Invest Ophthalmol Vis Sci. 2020;61(1):1. doi:10.1167/iovs.61.1.1
39. Vitale F, Giliberto L, Ruiz S, et al. Anti-tau conformational scFv MC1 antibody efficiently reduces pathological tau species in adult JNPL3 mice. Acta Neuropathol Commun. 2018;6(1):82. doi:10.1186/s40478-018-0585-2
40. Inoue Y, Ando Y, Misumi Y, Ueda M. Current management and therapeutic strategies for cerebral amyloid angiopathy. Int J Mol Sci. 2021;22(8):3869. doi:10.3390/ijms22083869
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.