")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Disposition and metabolism of [14C]-levomilnacipran, a serotonin and norepinephrine reuptake inhibitor, in humans, monkeys, and rats
Authors Brunner V, Maynadier B, Chen L, Roques L, Hude I, Séguier S, Barthe L, Hermann P
Received 14 January 2015
Accepted for publication 19 March 2015
Published 23 June 2015 Volume 2015:9 Pages 3199—3215
DOI https://doi.org/10.2147/DDDT.S80886
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Shu-Feng Zhou
Valérie Brunner,1 Bernadette Maynadier,2 Laishun Chen,3 Louise Roques,2 Isabelle Hude,2 Sébastien Séguier,2 Laurence Barthe,1 Philippe Hermann1
1Pierre Fabre Médicament, Centre de R&D, Toulouse, 2Centre Experimental PreClinque, Campans, France; 3Forest Research Institute Inc., an affiliate of Actavis Inc., Jersey City, NJ, USA
Abstract: Levomilnacipran is approved in the US for the treatment of major depressive disorder in adults. We characterized the metabolic profile of levomilnacipran in humans, monkeys, and rats after oral administration of [14C]-levomilnacipran. In vitro binding of levomilnacipran to human plasma proteins was also studied. Unchanged levomilnacipran was the major circulating compound after dosing in all species. Within 12 hours of dosing in humans, levomilnacipran accounted for 52.9% of total plasma radioactivity; the circulating metabolites N-desethyl levomilnacipran N-carbamoyl glucuronide, N-desethyl levomilnacipran, and levomilnacipran N-carbamoyl glucuronide accounted for 11.3%, 7.5%, and 5.6%, respectively. Similar results were seen in monkeys. N-Desethyl levomilnacipran and p-hydroxy levomilnacipran were the main circulating metabolites in rats. Mass balance results indicated that renal excretion was the major route of elimination with 58.4%, 35.5%, and 40.2% of total radioactivity being excreted as unchanged levomilnacipran in humans, monkeys, and rats, respectively. N-Desethyl levomilnacipran was detected in human, monkey, and rat urine (18.2%, 12.4%, and 7.9% of administered dose, respectively). Human and monkey urine contained measurable quantities of levomilnacipran glucuronide (3.8% and 4.1% of administered dose, respectively) and N-desethyl levomilnacipran glucuronide (3.2% and 2.3% of administered dose, respectively); these metabolites were not detected in rat urine. The metabolites p-hydroxy levomilnacipran and p-hydroxy levomilnacipran glucuronide were detected in human urine (≤1.2% of administered dose), and p-hydroxy levomilnacipran glucuronide was found in rat urine (4% of administered dose). None of the metabolites were pharmacologically active. Levomilnacipran was widely distributed with low plasma protein binding (22%).
Keywords: FETZIMA, metabolites, mass balance, excretion, tissue distribution
Introduction
Major depressive disorder is a disabling mental illness that affects a significant number of individuals worldwide. Results of the World Health Organization World Mental Health survey, which analyzed data from 18 countries using the Composite International Diagnostic Interview1 for the Diagnostic and Statistical Manual of Mental Disorders, fourth edition,2 estimated that the average lifetime and 12-month prevalence of major depressive disorder was 14.6% and 5.5% in ten high-income countries and 11.1% and 5.9% in eight low- to middle-income countries, respectively.3 Current therapies used to treat major depressive disorder include selective serotonin reuptake inhibitors, dopamine–norepinephrine reuptake inhibitors, and serotonin–norepinephrine reuptake inhibitors (SNRIs), among others.4
Levomilnacipran is an SNRI, and its antidepressant action is thought to be related to potentiation of norepinephrine and serotonin in the central nervous system through inhibition of reuptake at serotonin and norepinephrine transporters.5 Levomilnacipran has a greater potency in vitro for norepinephrine reuptake inhibition than for serotonin reuptake inhibition without directly affecting the uptake of dopamine or other neurotransmitters in vitro.6 An extended-release formulation of levomilnacipran (FETZIMA®) received US Food and Drug Administration (FDA) approval in July 2013 for the treatment of major depressive disorder in adults, with a recommended dose range of 40–120 mg once daily.5 FDA approval was based on the results of three phase III studies (ClinicalTrials.gov identifiers: NCT01377194, NCT01034462, NCT00969709).7–9 Results from additional published trials provide further information on the efficacy and safety of levomilnacipran.10–12
This report presents the disposition and metabolic profile of [14C]-levomilnacipran in humans, monkeys, and rats, and describes the in vitro binding of [14C]-levomilnacipran to human plasma proteins.
Materials and methods
Materials
The specific activity of [14C]-levomilnacipran that was used in both the human and animal studies was 564.25 MBq/mmol (15.25 mCi/mmol), 1.99 MBq/mg (53.9 μCi/mg). For the human study, the stock solution of [14C]-levomilnacipran (0.74 MBq/mL [14C] and 12 mg/mL levomilnacipran) was prepared at PRA International Pharmacy within 48 hours before oral administration and was stored in a refrigerator (+2°C/+8°C) until dosing time. Isolated human plasma proteins, human serum albumin (HSA), alpha-1-acid-glycoprotein (AAG), and gamma globulins (GGs), for use in the binding study were obtained from Sigma-Aldrich (Saint-Quentin Fallavier, France).
For the animal studies, levomilnacipran and [14C]-levomilnacipran oral dosing solutions (0.222 MBq/mL [14C] and 6 mg/mL levomilnacipran [study 1: 09-0027], and 2.47 MBq/mL [14C] and 10 mg/mL levomilnacipran [study 2: 11-0267] for monkeys; and 0.37 MBq/mL and 10 mg/mL levomilnacipran [study 3: 11-0426] for rats) were made up on the dosing day in ultrapure water and stored at 5°C±3°C until dosing. Levomilnacipran was obtained from Plantes & Industrie (Gaillac, France), and [14C]-levomilnacipran was obtained from Selcia (Ongar, UK). A total of six monkeys (age: 6 years; weight: 4.2–5.5 kg) and 60 rats (weight: 245–286 g) were used in the pharmacokinetic (PK) and mass balance studies; Macaca fascicularis monkeys were obtained from Bioprim (Baziège, France), and Sprague Dawley rats were obtained from Charles River Laboratories (L’arbresle, France). Sixteen albino and eight hooded rats (weight: 220–318 g) were used in the tissue distribution study; Sprague Dawley (albino) and Lister hooded (pigmented) rats were obtained from Harlan UK Ltd.
Mass balance studies
Human study
In a single-center, open-label, single, oral radiolabeled dose study, healthy young men received a 5 mL solution containing 60 mg of [14C]-levomilnacipran with 100 μCi (3.7 MBq) of radioactivity in the morning on Day 1 within approximately 30 minutes after the start of breakfast. The dose of 60 mg was selected because this dose had shown acceptable tolerability/safety in previous studies and was within the investigated therapeutic range of doses (40–120 mg/day) for use in major depressive disorder (FETZIMA US prescribing information). The dose of radioactivity (100 μCi/3.7 MBq) was chosen after review of human dosimetry calculations (Health Protection Agency-RPA DA 88, 2009), and the associated radiation exposure falls within International Commission on Radiological Protection (ICRP-1992) Guidelines for Category IIa studies (effective dose range: 0.1–1 mSv in adults).
Healthy male volunteers who were 18–45 years of age, had a body mass index of 18.5–28 kg/m2, and had laboratory test results within normal ranges and normal clinical examinations (including vital sign and electrocardiograph measurements and physical examinations) at screening were eligible to participate in the single-dose study. All study participants completed an informed consent form at prescreening for the study. The study was approved by the Independent Ethics Committee of the Foundation “Evaluation of Ethics in Biomedical Research” (Stichting Beoordeling Ethiek Biomedisch Onderzoek), Assen, the Netherlands, and was performed in accordance with the Good Clinical Practice (CPMP/ICH/135-95) for trials on medicinal products and the principles stated in the Declaration of Helsinki (1964 and its subsequent amendments), and with the European Directive 95/46/CE.
Nine men were enrolled in the study; PK analysis and mass balance were performed on data from four men who did not vomit after oral administration of study drug. Calculations of blood-to-plasma ratio of [14C]-radioactivity were performed on data from all nine men. Blood, urine, and feces samples for mass balance and PK analyses were collected from Day 1 to Day 6. Blood samples were collected by venipuncture or drawn from a catheter in the forearm into polyethylene tubes containing lithium heparinate as anticoagulant. Samples for radioactivity measurement and for levomilnacipran and N-desethyl levomilnacipran assay were collected at the following times: pre-dose and then 1 hour, 2 hours, 2.5 hours, 3 hours, 3.5 hours, 4 hours, 5 hours, 6 hours, 8 hours, 10 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, and 120 hours post-dose. Samples for metabolite profile analysis were collected at the following times: pre-dose and then 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, and 12 hours post-dose. Urine for radioactivity measurement and for metabolite profile analysis was collected before study drug administration and at 0- to 4-hour, 4- to 8-hour, 8- to 12-hour, 12- to 24-hour, 24- to 48-hour, 48- to 72-hour, 72- to 96-hour, and 96- to 120-hour intervals following drug administration. Feces for radioactivity measurement was collected before study drug administration and at 0- to 24-hour, 24- to 48-hour, 48- to 72-hour, 72- to 96-hour, and 96- to 120-hour intervals following drug administration.
After collection, whole-blood and plasma samples for radioactivity measurement and for levomilnacipran and N-desethyl levomilnacipran assay were kept frozen at −20°C until analysis. Plasma, urine, and feces samples for metabolite profile analysis were kept frozen at −80°C until analysis.
Animal studies
Four studies were performed in animals: studies 1 (09-0027) and 4 (09-0025) in monkeys and rats, respectively, which were designed to investigate the PKs and determine the excretion mass balance and metabolic profile of levomilnacipran; and studies 2 (11-0267) and 3 (11-0426) in monkeys and rats, respectively, which were designed to quantify and characterize the glucuronide metabolites of levomilnacipran and N-desethyl levomilnacipran. Animals were weighed on the day of dose administration (30 mg/kg for monkeys and 50 mg/kg for rats), and the appropriate volume of dosing solution was administered by gavage. Blood samples were taken from the saphenous vein in monkeys and from the abdominal aorta under isoflurane anesthesia in rats. All blood samples were collected into lithium heparin and were centrifuged to separate plasma from blood cells. Plasma, urine (including cage wash), and feces samples were stored at −80°C±10°C until analysis.
In the monkey studies, blood-sampling times (three male monkeys per study) were as follows: pre-dose and at 1 hour, 2 hours, 4 hours, 6 hours, 10 hours, 24 hours, 48 hours, 72 hours, and 120 hours after dosing in study 1; and pre-dose and at 1 hour, 2 hours, 4 hours, 6 hours, and 10 hours after dosing in study 2. Urine and feces were collected at the following intervals: 24-hour period before dosing, and at 0- to 10-hour and 10- to 24-hour (0–24 hours for feces), 24- to 48-hour, 48- to 72-hour, 72- to 96-hour, and 96- to 120-hour intervals after dosing in study 1; urine was collected at the following intervals: 24-hour period before dosing and at 0- to 8-hour and 8- to 24-hour intervals after dosing in study 2.
In the rat studies, blood-sampling times (three rats per sampling time) were as follows: pre-dose (control animals) and at 0.25 hour, 0.5 hour, 1 hour, 2 hours, 4 hours, 6 hours, 10 hours, 24 hours, 32 hours, 48 hours, and 72 hours after dosing in study 4; and pre-dose (control animals) and at 1 hour, 2 hours, 4 hours, 6 hours, and 10 hours after dosing in study 3. Urine and feces from three male and three female rats were collected at the following intervals: 24-hour period before dosing and at 0–8 hours and 8–24 hours, 24–48 hours, and 48–72 hours after dosing in study 4. Urine from three male rats was collected at the following intervals: 24-hour period before dosing and at 0–8 hours and 8–24 hours after dosing in study 3.
Radioactivity analysis of whole-blood, plasma, urine, and feces samples
Radioactivity in whole-blood, plasma, urine, and feces samples was measured by liquid scintillation counting using a Packard Tri-carb™ 3100 TR liquid scintillation analyzer (Perkin Elmer LAS (UK) Ltd, Downers Grove, IL, USA) equipped with low-level and normal counting modes.
Specimens were prepared for analysis as follows: aliquots (100 μL) of whole-blood samples were weighed, placed onto a Combusto-Pad™ (Perkin Elmer LAS (UK) Ltd), dried under an extraction hood for a minimum of 12 hours, and then burnt in a Packard Sample Oxidizer model 307 (Perkin Elmer LAS (UK) Ltd, USA). The combustion recovery was determined for each batch of samples using five standards spiked with 14C Spec-Chec (Perkin Elmer LAS (UK) Ltd). The recovery value was used for the determination of radioactivity in whole-blood and in the feces homogenate. Combustion gases were trapped in a glass vial with 6 mL of Carbo-sorb® (Perkin Elmer LAS (UK) Ltd), and then, 11 mL of Permafluor® (Perkin Elmer LAS (UK) Ltd) was added. The aliquots were mixed vigorously and placed in the dark (at least 12 hours at room temperature) before liquid scintillation counting. Combustion of feces samples (200–400 μL) homogenized in ethanol was performed as described for whole-blood samples.
Following centrifugation (2,500× g for 5 minutes) of plasma and urine samples, aliquots of the samples (100–200 μL) were weighed in a plastic vial, 5 mL of Ultima Gold (PerkinElmer LAS (UK) Ltd) was added, and the vials were mixed vigorously before liquid scintillation counting.
Drug concentration measurements and metabolite profile analysis
The quantification of levomilnacipran and its metabolites in plasma and urine was performed by the bioanalysis unit of Institut de Recherche Pierre Fabre (Castres, France). Concentrations of levomilnacipran and its metabolites were determined using liquid chromatography–tandem mass spectrometry (LC–MS/MS) with electrospray ionization. Processing of plasma and urine samples was performed by liquid–liquid extraction. Specimens were prepared for analysis as follows: aliquots of plasma (200 μL) were placed in a conical polypropylene tube, and 10 μL of internal standard solutions (levomilnacipran-D10 and N-desethyl levomilnacipran-D5 solutions at 0.5 ng/μL) was added. After mixing for few seconds, 3 mL of ethyl acetate was added, and the sample was shaken for 10 minutes by means of an oscillating shaker. Then, the sample was centrifuged at 2500× g for 5 minutes at 4°C. The organic phase was transferred to another conical polypropylene tube, and 0.1 mL of 0.5% formic acid solution was added. The sample was centrifuged at 2500× g for 5 minutes at 4°C. After centrifugation, the organic phase was discarded, and the excess of ethyl acetate was evaporated by a slight nitrogen stream for 2 minutes. The extract was then transferred to an injection vial for analysis. The analytical system used was the Acquity™ sample and binary solvent manager combined with Quattro Premier XE mass spectrometer and Waters MassLynx® software (version 4.1). Reverse-phase chromatography was applied before electrospray ionization in positive mode and tandem mass spectrometry detection.
Chromatography was performed on a Synergi Polar-RP 100A column (50 mm ×2.0 mm, 2.5 μm, Phenomenex) and a Synergi Polar-RP security guard cartridge (4 mm ×2.0 mm, 4 μm, Phenomenex) at room temperature. The mobile phase consisted of a mixture of ultrapure water with formic acid 0.2% and acetonitrile 54/46 (v/v), used in isocratic conditions at 0.2 mL/min. For human plasma and urine, the calibration curve ranged from 1 ng/mL to 250 ng/mL and from 1 ng/mL to 5,000 ng/mL, respectively; for monkey plasma and urine, the calibration curve ranged from 1 ng/mL to 500 ng/mL; and for rat plasma and urine, the calibration curve ranged from 1 ng/mL to 500 ng/mL and from 50 ng/mL to 24,750 ng/mL, respectively. To determine the conjugated metabolite concentrations of levomilnacipran and N-desethyl levomilnacipran, and p-hydroxy levomilnacipran, concentrations of these compounds were measured before and after deconjugation (incubation with beta-glucuronidase/sulfate for 15 hours at 37°C).
Structural identification of levomilnacipran metabolites
The metabolite profiling of plasma and urine was performed by the bioanalysis unit of Institut de Recherche Pierre Fabre. Total radioactivity in feces never reached 5%; therefore, metabolite profiling of feces was not performed. The chemical structures of levomilnacipran metabolites were characterized using liquid chromatography coupled with multistage accurate mass spectrometry (LC–MSn) and online radioactivity detection. Mass spectrometry data, fragmentation schemes, and retention times of the compounds in chromatograms were used to identify levomilnacipran and its metabolites. Metabolites were identified by comparing fragmentation schemes of pure authentic standards, when available, with those of the metabolites in complex matrices, and by comparing the retention times of the authentic standards with those of the metabolites in the chromatography mobile phase.
Plasma protein binding
Human blood was obtained from three healthy donors (two men and one woman) from Biopredic International (Rennes, France). Blood samples were collected in lithium heparin tubes and stored at approximately +4°C until use. Equilibrium dialysis was used to measure the binding of [14C]-levomilnacipran to human plasma and isolated human plasma proteins. Time to equilibrium was determined by incubating [14C]-levomilnacipran at a concentration of 100 ng/mL in plasma against Sörensen buffer at 37°C for 5 hours; binding was measured at various time intervals (0.5 hour, 1 hour, 2 hours, 3 hours, 4 hours, and 5 hours). Nonspecific binding to the dialysis membrane and walls of the dialysis cell was also determined. Protein binding of [14C]-levomilnacipran was assessed at the concentrations of 10 ng/mL, 50 ng/mL, 100 ng/mL, 250 ng/mL, 500 ng/mL, and 1,000 ng/mL at 37°C for 30 minutes. The concentration of each isolated human plasma protein (HSA [40 g/L], AAG [0.74 g/L], and GGs [18.5 g/L]) was adjusted as close as possible to that found in pooled plasma. Binding of [14C]-levomilnacipran to plasma and isolated human plasma proteins was measured by dialyzing 200 μL of plasma or protein solution containing [14C]-levomilnacipran against 200 μL of Sörensen buffer. At the end of dialysis, 50 μL samples were collected and analyzed by liquid scintillation counting using a liquid scintillation spectrometer (PerkinElmer Tri Carb2800 Tr) with automatic quench transformation from counts per minute (cpm) to disintegrations per minute (dpm) by an external standardization technique. Plasma samples and protein solutions were mixed with 3 mL of Picofluor 40, and liquid scintillation counting was carried out with 2% precision for a maximum duration of 5 minutes. The sensitivity limit of the radioactivity measurement was 15 dpm; therefore, the limit of quantification was set at 30 dpm. Appropriate dilutions were performed in order to measure radioactivity within the range of 30–9.9×105 dpm. Assay results expressed in dpm per milliliter were converted to megabecquerel per milliliter (microcurie per milliliter), and then, drug concentrations were calculated from specific activity.
Tissue distribution
The tissue distribution of compound-related radioactivity following oral administration of [14C]-levomilnacipran was performed at the Aptuit testing facility (Riccarton, Edinburgh, UK). Male and female albino rats and male pigmented rats were used in the tissue distribution study. Each rat received a single oral administration of [14C]-levomilnacipran (5 mg/kg). One rat was sacrificed by carbon dioxide narcosis at each of the following time points: 0.5 hour, 2 hours, 8 hours, 24 hours, 72 hours, 168 hours, and 336 hours after dosing. Immediately after sacrifice, the rats were frozen rapidly by immersion in a mixture of hexane and solid CO2. When frozen, carcasses were set left lateral side uppermost, in a block of sodium carboxymethyl cellulose (approximately 2% [w/v] in water) by further immersion in a hexane and solid CO2 mixture. Each block was mounted in a CM3600 cryomicrotome (Leica Instruments GmbH, Milton Keynes, UK) maintained at approximately −20°C. After initial trimming of the block, sagittal whole-body sections (40 μm) were obtained at various levels through the carcass. Calibration and quality control standard blocks were sectioned in an identical manner to the animal blocks. A block of frozen sodium carboxymethyl cellulose containing standards of known radioactivity was prepared on a separate occasion. For the calibration line, a series of paper straws were fixed in the block, and liver homogenate containing increasing amounts of a 14C-radiolabeled compound (3–19,000 nCi/g) was dispensed into the straws and frozen. This range was chosen to cover the extent of likely tissue concentration values. Quality control samples were prepared separately with values of 44.3 nCi/g (bottom range), 7,999 nCi/g (middle range), and 17,675 nCi/g (top range). Calibration line and quality control standard values were obtained by combusting known weights of radioactive solutions used in preparation of the standards. Samples were combusted in a Packard Model 307 automatic sample oxidizer (PerkinElmer LAS (UK) Ltd) before liquid scintillation counting. Quantitative whole-body phosphor imaging was carried out using a Fuji model FLA5000 phosphor imager system (Raytek Scientific Ltd, Sheffield, UK). Aida™ software (v3.27) was used to calibrate individual scans and to quantify levels of radioactivity. Freeze-dried sections were placed against a phosphor screen (Raytek Scientific Ltd) for an exposure period of 18 hours. Following exposure, the phosphor screen was scanned by a laser beam, and the latent image was captured and stored in an electronic data file. Tissue radioactivity concentrations within individual sections were quantified and annotated, and representative images of the selected sections at each time point were produced. Terminal blood samples were taken from all animals immediately before sacrifice and analyzed for radioactivity.
Data analysis
PKs and mass balance
Plasma PK parameters (maximum plasma drug concentration [Cmax], time of maximum plasma drug concentration [Tmax], area under the plasma concentration-versus-time curve from time 0 to the last observed plasma concentration [AUClast] and from time 0 to infinity [AUCinf], and terminal elimination half-life [T1/2]) of levomilnacipran and N-desethyl levomilnacipran were derived from a non-compartmental analysis using WinNonlin® (version 5.2) for human data or Kinetica™ (version 4.2, InnaPhase Corporation) for animal data. Descriptive statistics of percentage of the dose excreted in urine and feces were reported. The mass balance (corresponding to the percentage of [14C]-levomilnacipran excreted in urine and feces) was calculated after measuring the radioactivity recovered in urine and feces samples.
The amount of the excreted dose in a sample was calculated using the following equation:
|
|
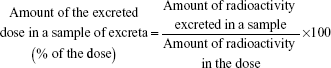
Determination of the binding of [14C]-levomilnacipran to plasma or to plasma proteins
The percentage of [14C]-levomilnacipran binding to plasma or to plasma proteins after equilibrium dialysis was determined according to the following formula:
|
|
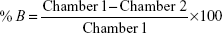
|
|
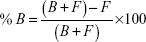
|
|
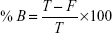
where Chamber 1 is the concentration in the first chamber of the dialysis cell, Chamber 2 is the concentration in the second chamber of the dialysis cell, F is the free ligand concentration, B is the bound ligand concentration, and T = (B + F) = (Chamber 1) is the total ligand concentration.
Results
Plasma concentration and PKs of levomilnacipran and N-desethyl levomilnacipran
As shown in Figure 1, the mean concentration-versus-time profiles of total radioactivity, levomilnacipran, and N-desethyl levomilnacipran in humans were similar after oral administration of [14C]-levomilnacipran. Median time to maximum plasma concentration (Tmax) was 2.75 hours for levomilnacipran and 5.5 hours for N-desethyl levomilnacipran (Table 1). The Tmax for total radioactivity was also 2.75 hours. The average peak plasma concentration was 179 ng/mL and 296 ng Eq/mL for levomilnacipran and total radioactivity, respectively. The average extent of exposure, as evidenced by AUCinf, was 2,089 h·ng/mL for levomilnacipran compared with 4,614 h·ng Eq/mL for total radioactivity. Regardless of the sampling time, the mean blood-to-plasma ratio was close to 1 (individual values ranged from 0.741 to 1.110).
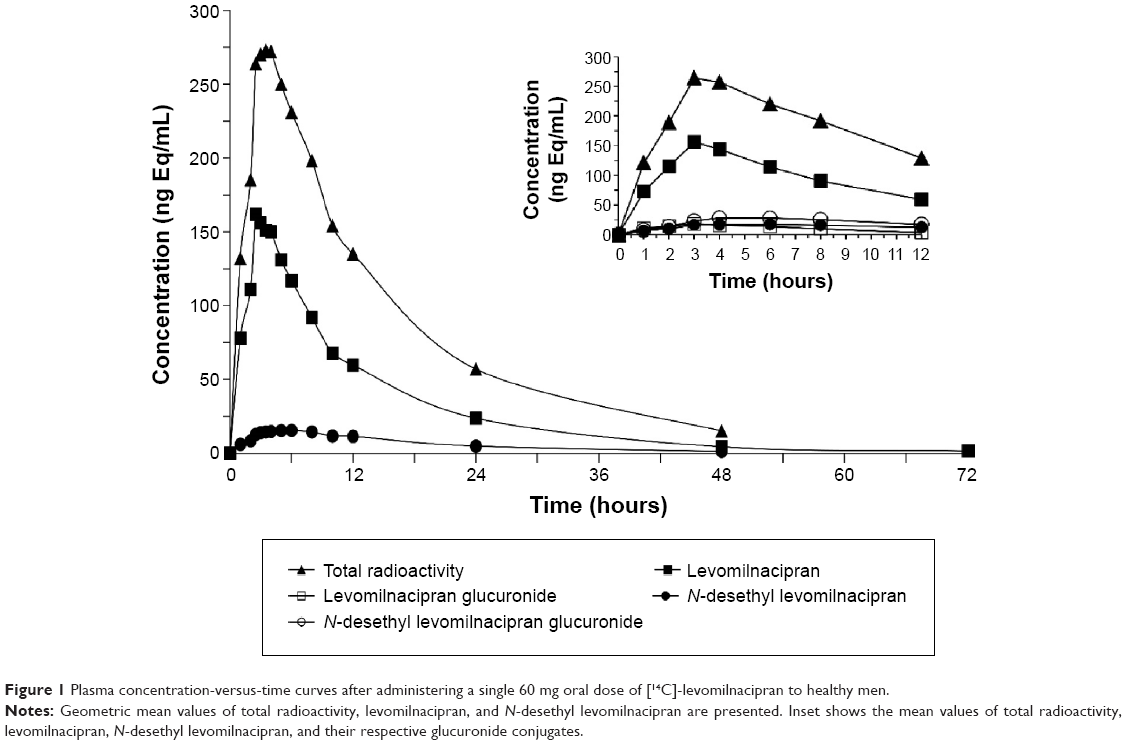 | Figure 1 Plasma concentration-versus-time curves after administering a single 60 mg oral dose of [14C]-levomilnacipran to healthy men. |
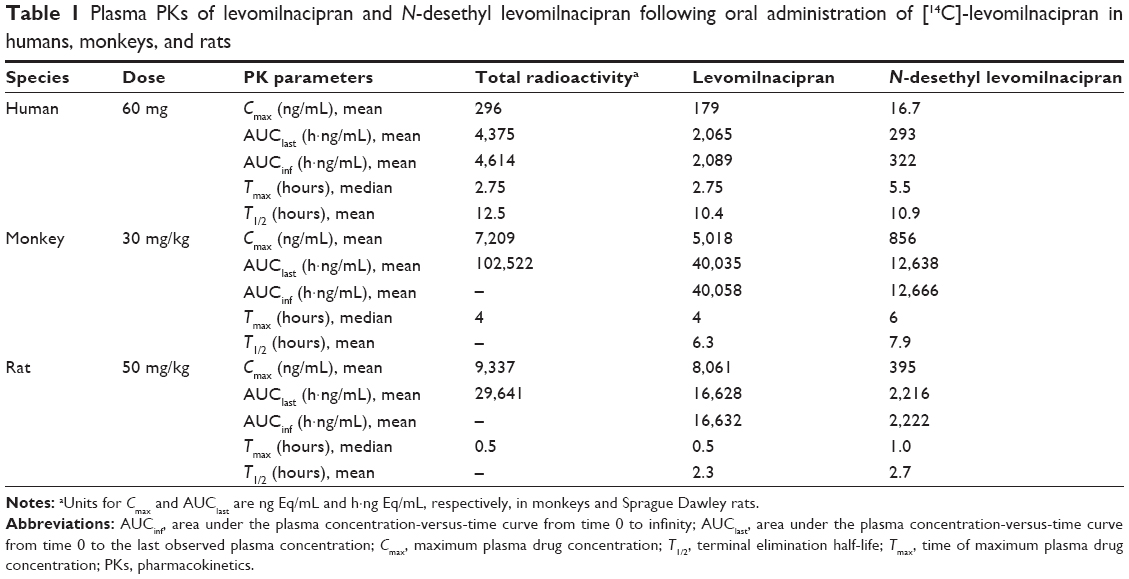 | Table 1 Plasma PKs of levomilnacipran and N-desethyl levomilnacipran following oral administration of [14C]-levomilnacipran in humans, monkeys, and rats |
After oral administration of [14C]-levomilnacipran at a dose of 30 mg/kg in male monkeys, the mean maximal concentrations of levomilnacipran and N-desethyl levomilnacipran were observed at 4 hours and 6 hours, respectively (Figure 2). Levomilnacipran was the major circulating compound in plasma up to 10 hours after dosing. The average peak plasma concentration was 5,018 ng/mL for levomilnacipran and 7,209 ng Eq/mL for total radioactivity. The average extent of exposure was 40,058 h·ng/mL for levomilnacipran compared with 102,522 h·ng Eq/mL for total radioactivity, and the average half-lives were 6.3 hours and 7.9 hours for levomilnacipran and N-desethyl levomilnacipran, respectively. The mean blood-to-plasma ratio in male monkeys was 1.032 and 0.809 at 2 hours and 10 hours after administration of [14C]-levomilnacipran, respectively. Mean plasma concentrations, PK parameters, and blood-to-plasma ratios were similar in female monkeys.
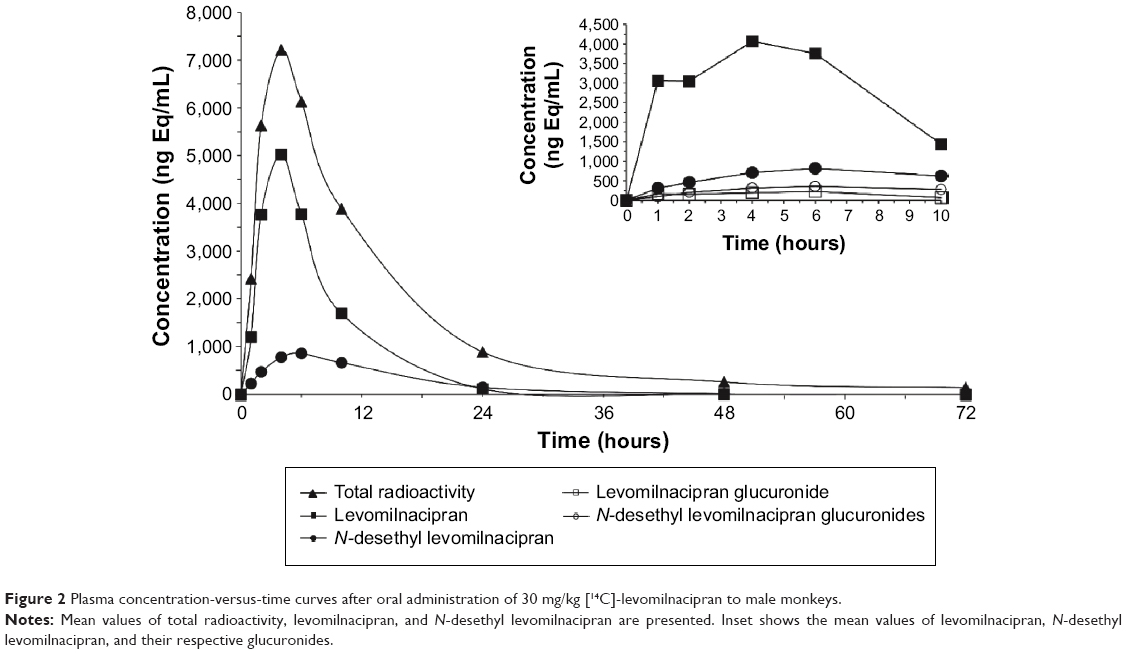 | Figure 2 Plasma concentration-versus-time curves after oral administration of 30 mg/kg [14C]-levomilnacipran to male monkeys. |
Absorption of [14C]-levomilnacipran was rapid after oral dosing of 50 mg/kg to male rats (Figure 3). Levomilnacipran was the major circulating compound in plasma up to 6 hours after dosing, and its maximum concentrations were comparable between male and female rats. Tmax was 0.5 hour for levomilnacipran and 1 hour for N-desethyl levomilnacipran. The average peak plasma concentration was 8,061 ng/mL for levomilnacipran and 9,337 ng Eq/mL for total radioactivity. Elimination from plasma was rapid. The average extent of exposure and average half-life of levomilnacipran were 16,632 h·ng/mL and 2.3 hours, respectively; and the average extent of exposure and average half-life of N-desethyl levomilnacipran was 2,222 h·ng/mL and 2.7 hours, respectively. Up to 24 hours after single oral administration of [14C]-levomilnacipran of 50 mg/kg to male rats, the blood/plasma radioactivity concentration ratio was approximately 1 (0.80–1.34).
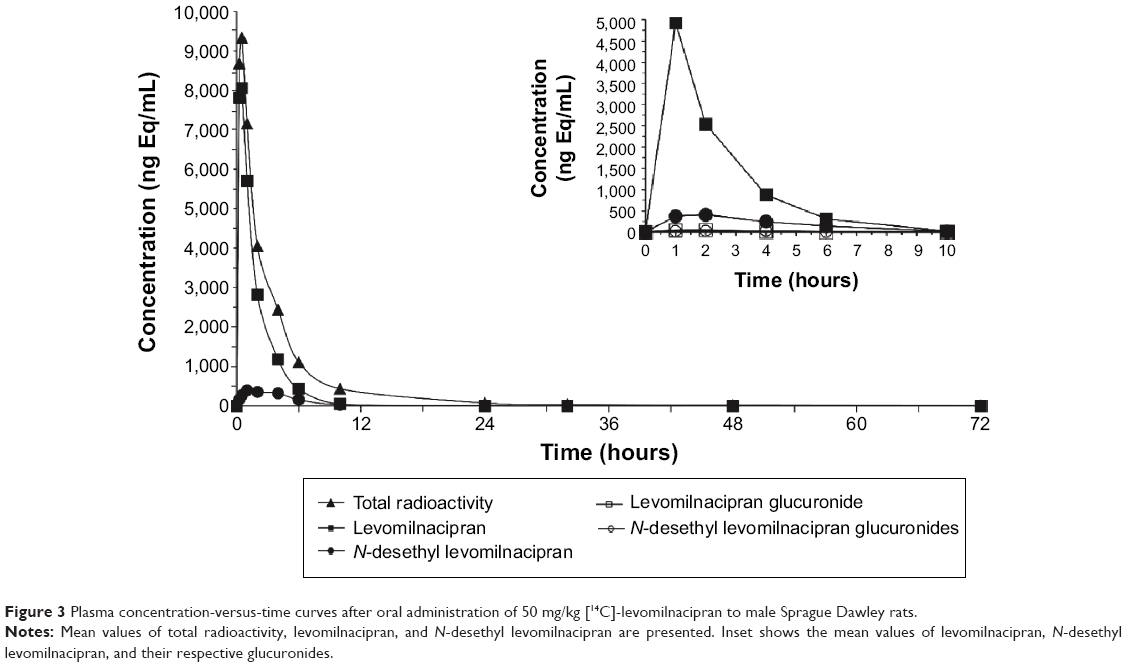 | Figure 3 Plasma concentration-versus-time curves after oral administration of 50 mg/kg [14C]-levomilnacipran to male Sprague Dawley rats. |
Mass balance
Cumulated values of total radioactivity recovered for humans, monkeys, and rats are provided in Table 2. In humans, individual cumulative recoveries in urine and feces ranged from 87.6% to 102%. An average of 93.6% and 3.8% of the administered dose was excreted in the urine and feces, respectively. The rate of excretion was rapid with about 90% of the radioactivity in the administered dose being recovered in the 0- to 48-hour urine collection.
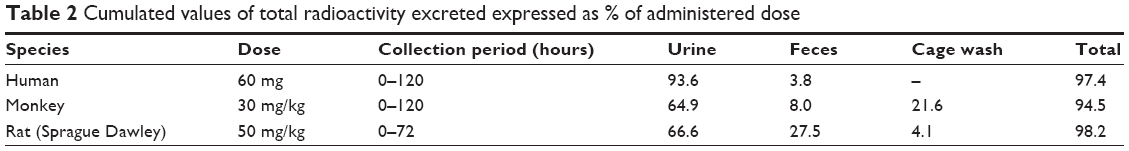 | Table 2 Cumulated values of total radioactivity excreted expressed as % of administered dose |
In monkeys, urine, feces, and cage wash samples were collected up to 120 hours after oral administration of a 30 mg/kg dose of [14C]-levomilnacipran. Mean total recovery of the radioactive dose was 94.6%, and most of the radioactivity excretion occurred during the first 48 hours. The major route of excretion was the urine, with 86.5% of the radioactive dose recovered within 120 hours in urine and cage washes. Fecal elimination accounted for about 8% of the administered dose.
In rats, the major route of excretion was the urine; 70.7% of the radioactive dose was recovered in the urine and cage washes and 27.5% in the feces within 72 hours after oral dose administration.
Structural identification of levomilnacipran metabolites
Levomilnacipran metabolites were identified in monkey plasma and urine, and human urine. Representative radiochromatograms are shown in Figures 4 and 5 (for monkey plasma and urine and human urine, respectively). Metabolite profiles of monkey and human urine were qualitatively similar, as unchanged levomilnacipran was the principal component. A second notable radiolabeled peak present in urine was the N-desethyl levomilnacipran metabolite. Levomilnacipran and N-desethyl levomilnacipran were identified by their respective retention times, mass spectra (Table 3), and comparison with corresponding authentic standards. Three additional radiolabeled chromatographic peaks were observed at lower levels in monkey plasma and urine and in human urine. Further analysis of monkey urine indicated that these additional peaks corresponded to the phase II glucuronides of levomilnacipran and the phase I metabolite of N-desethyl levomilnacipran. In the 0- to 8-hour monkey urine sample, two different carbamoyl glucuronides of N-desethyl levomilnacipran were identified, with corresponding retention times of 12.5 minutes and 13.8 minutes. Both metabolites had the same molecular ion (MH+ m/z 439 or sodium adduct m/z 461) corresponding to the addition of carbamic acid to N-desethyl levomilnacipran and subsequent conjugation with glucuronic acid. Table 3 lists the product ions of the two N-desethyl levomilnacipran conjugates, and Figures 6 and 7 illustrate the proposed fragmentation pathways of the sodium adduct of each conjugate. The indicative fragments revealed that the conjugate with a retention time of 12.5 minutes is the N-desethyl levomilnacipran carbamoyl glucuronide obtained by glucuronidation of the amine moiety, whereas the conjugate with a retention time of 13.7 minutes is the N-desethyl levomilnacipran carbamoyl glucuronide obtained by glucuronidation of the amide moiety. The presence of both carbamoyl glucuronides was confirmed in monkey plasma and human urine.
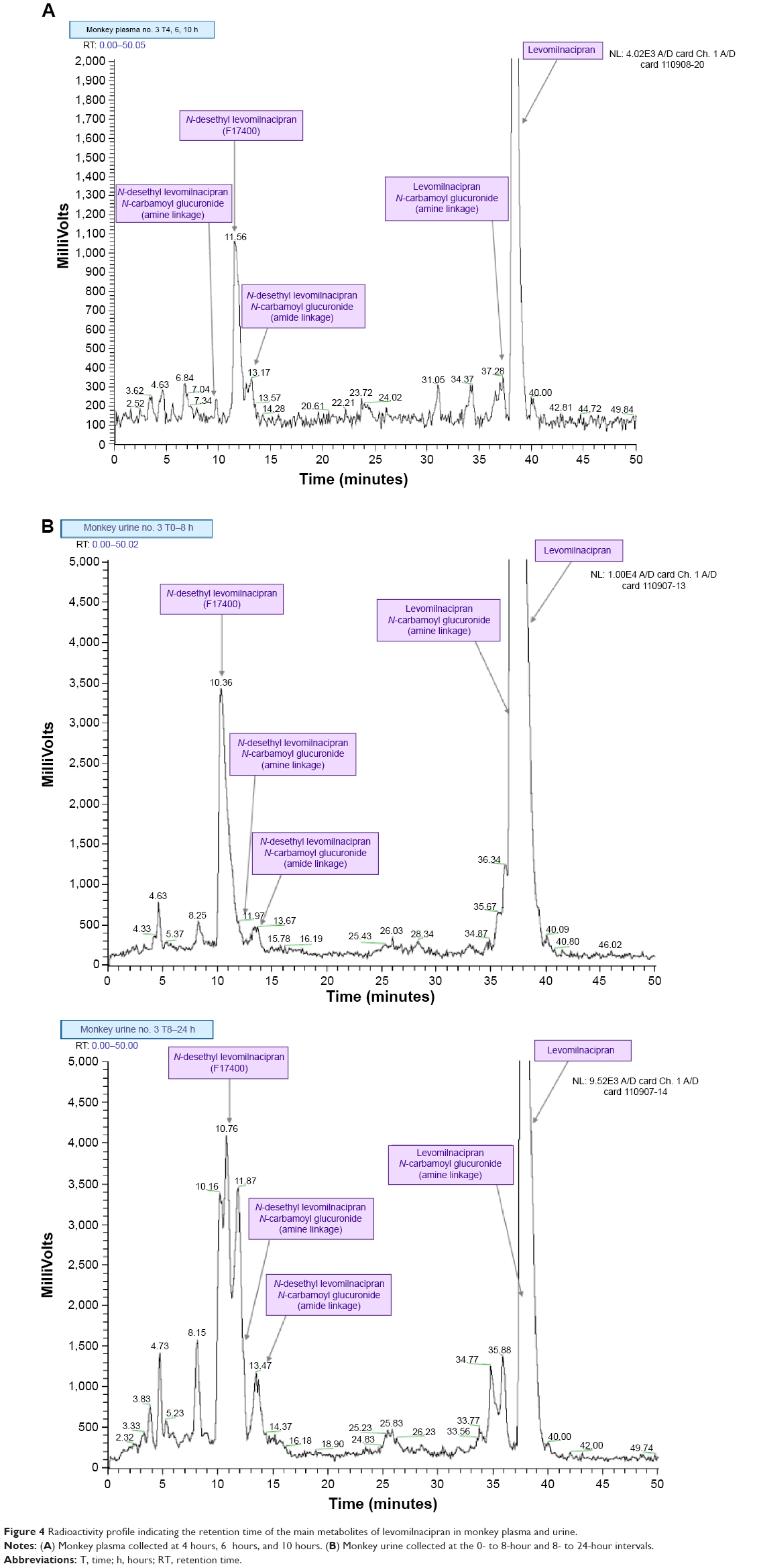 | Figure 4 Radioactivity profile indicating the retention time of the main metabolites of levomilnacipran in monkey plasma and urine. |
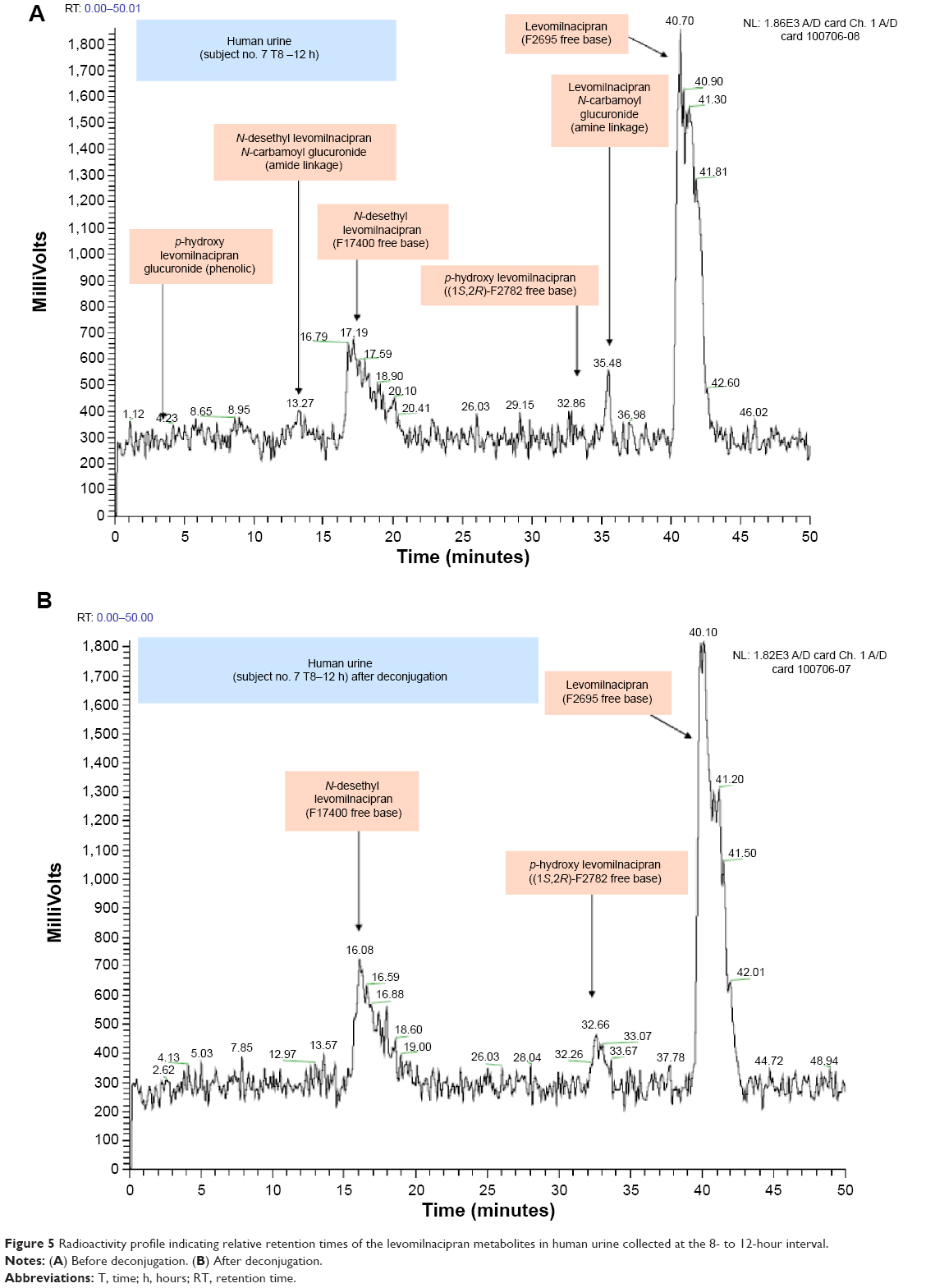 | Figure 5 Radioactivity profile indicating relative retention times of the levomilnacipran metabolites in human urine collected at the 8- to 12-hour interval. |
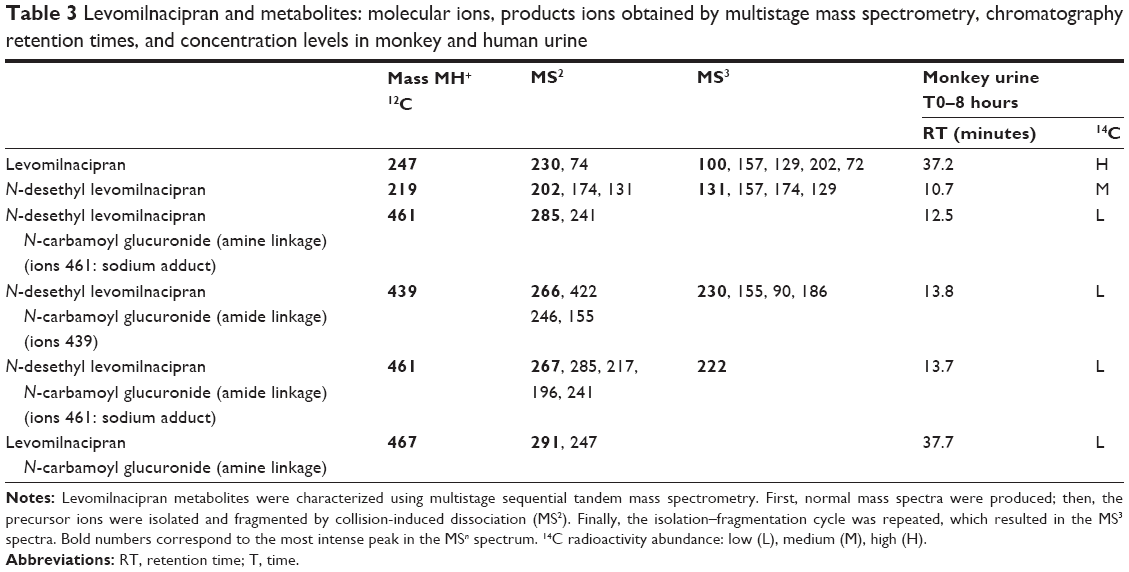 | Table 3 Levomilnacipran and metabolites: molecular ions, products ions obtained by multistage mass spectrometry, chromatography retention times, and concentration levels in monkey and human urine |
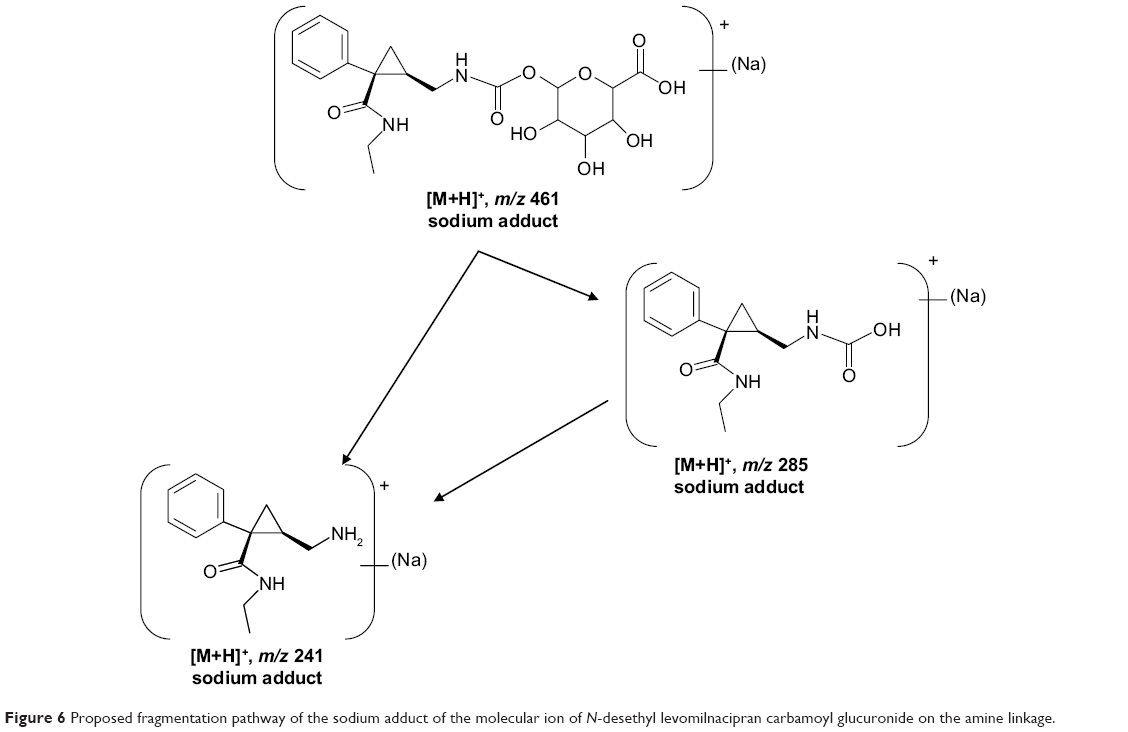 | Figure 6 Proposed fragmentation pathway of the sodium adduct of the molecular ion of N-desethyl levomilnacipran carbamoyl glucuronide on the amine linkage. |
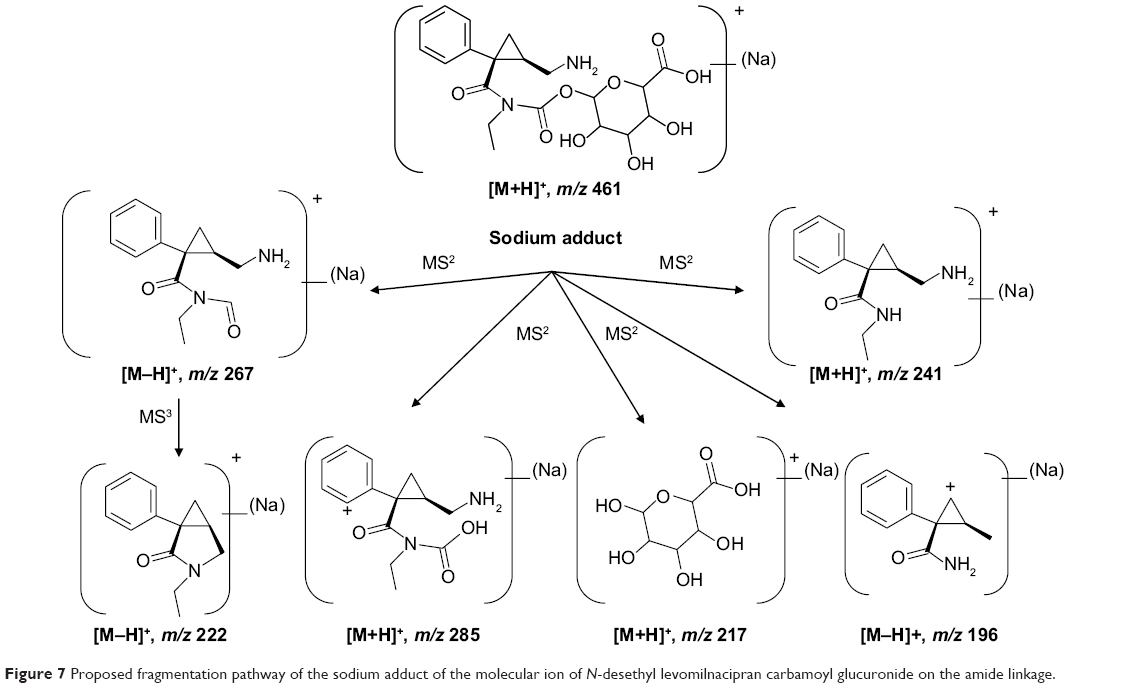 | Figure 7 Proposed fragmentation pathway of the sodium adduct of the molecular ion of N-desethyl levomilnacipran carbamoyl glucuronide on the amide linkage. |
Plasma metabolite profile
In humans, levomilnacipran was the major circulating compound post-dose (Figure 1, inset). The major circulating metabolites were the N-carbamoyl glucuronide conjugate of N-desethyl levomilnacipran and N-desethyl levomilnacipran; their plasma exposure accounted for 21.8% and 14.4% of the plasma exposure of the parent drug (levomilnacipran), respectively.
In monkeys, levomilnacipran was the major circulating compound after dosing (Figure 2, inset). The major circulating metabolite was N-desethyl levomilnacipran, which accounted for 27.5% of the plasma exposure of the parent drug. In plasma, three conjugates were observed after oral administration of 30 mg/kg to monkeys; one was formed from levomilnacipran and two from N-desethyl levomilnacipran. Levomilnacipran N-carbamoyl glucuronide and N-desethyl levomilnacipran accounted for 5.3% and 9.3% of the parent drug, respectively.
Levomilnacipran was also the major circulating compound post-dose in male rats; it represented about 60% of the circulating total radioactivity (Figure 3, inset). The two major circulating metabolites were N-desethyl levomilnacipran and p-hydroxy levomilnacipran glucuronide, which accounted for 14.9% and 12.4% of the plasma levomilnacipran exposure, respectively. In plasma, on the basis of their respective AUC, levomilnacipran N-carbamoyl glucuronide and N-desethyl levomilnacipran N-carbamoyl glucuronide accounted for 1.4% and 1.3% of the parent drug, respectively.
Urine metabolite profile
Mean urinary excretion values (expressed as a percentage of the dose administered) for humans, monkeys, and rats are provided in Table 4. After dosing in humans, the following compounds (expressed as % of the dose) were excreted in the urine: levomilnacipran (58.4%), N-desethyl levomilnacipran (18.2%), levomilnacipran glucuronide (3.8%), N-desethyl levomilnacipran glucuronide (3.2%), p-hydroxy levomilnacipran glucuronide (1.2%), and p-hydroxy levomilnacipran (0.9%). Other minor metabolites were present in human urine, but because of their low levels, they were not identified. The proposed metabolic pathway of levomilnacipran in human urine is illustrated in Figure 8.
 | Table 4 Mean urinary excretion values of levomilnacipran and its metabolites in humans, monkeys, and rats |
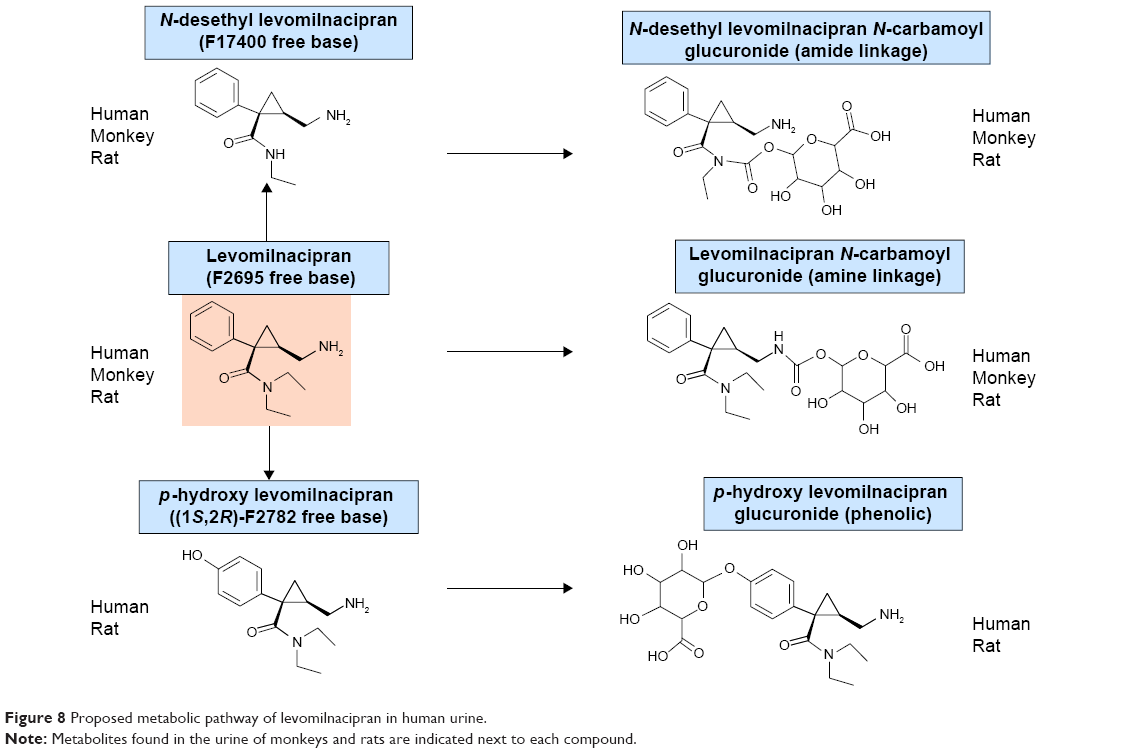 | Figure 8 Proposed metabolic pathway of levomilnacipran in human urine. |
In monkeys, excretion of levomilnacipran and N-desethyl levomilnacipran accounted for 35.5% and 12.4% of the administered dose excreted, respectively. Excretion of levomilnacipran N-carbamoyl glucuronide and the N-desethyl levomilnacipran N-carbamoyl glucuronides accounted for 4.1% and 2.3% of the administered dose, respectively.
In rats, excretion of levomilnacipran, N-desethyl levomilnacipran, p-hydroxy levomilnacipran, levomilnacipran N-carbamoyl glucuronide, and N-desethyl levomilnacipran N-carbamoyl glucuronide accounted for 40.2%, 7.9%, 4.0%, 1.25%, and 0.58% of the administered dose, respectively.
Plasma protein binding
The binding of [14C]-levomilnacipran to human blood cells, plasma proteins, and isolated plasma proteins (HSA, AAG, GGs) was determined using equilibrium dialysis. Binding of [14C]-levomilnacipran to human blood cells and to plasma was low and non-saturable. When concentrations of [14C]-levomilnacipran were increased from 0.025±0.001 μM to 1.901±0.186 μM, the binding of [14C]-levomilnacipran to plasma remained constant and was 21.7%±3.42%. Binding of [14C]-levomilnacipran to HSA and AAG was also low (12.8% and 6.3%, respectively) when evaluated at concentrations of 1–1,000 ng/mL, which corresponds to plasma concentrations expected when levomilnacipran is administered at therapeutic doses to humans. Binding to GGs was not detected under these conditions.
Tissue distribution
Tissue distribution of levomilnacipran and/or its metabolites was studied by quantitative whole-body phosphor imaging following oral administration of 5 mg/kg [14C]-levomilnacipran/kg to albino male and female and pigmented male rats. Following oral administration, the distribution of radioactivity was rapid and extensive, and tissue concentrations of radioactivity in all three groups of rats were comparable. Radioactivity was observed in almost all anatomical and biological structures. The tissue/blood ratio of radioactivity concentration in the majority of tissues was greater than 1. The highest radioactivity concentrations were generally observed in the organs and tissues associated with absorption, metabolism, and elimination (gastrointestinal tract, liver, and kidneys). In addition, high radioactivity concentration was observed in numerous glands (testes, thyroid, pituitary gland, pancreas, thymus, submaxillary gland, preputial gland, and adrenal gland). Radioactivity was also observed in cerebral tissues, and the thalamus had the highest brain concentration of radioactivity. At 2 hours post-dose, measurable levels of radioactivity were observed in all tissues with the exception of the bone, brain, eye lens, and spinal cord; highest levels of radioactivity were associated with the liver, kidney medulla, kidney cortex, pituitary gland, submaxillary salivary gland, pancreas, preputial gland, and lung (12,518 ng Eq/g, 2,952 ng Eq/g, 2,083 ng Eq/g, 1,815 ng Eq/g, 1,761 ng Eq/g, 1,592 ng Eq/g, 1,501 ng Eq/g, and 1,437 ng Eq/g, respectively).
Following the oral administration to pigmented rats, a relatively stable and high concentration of radioactivity was observed in the choroid layer of the eyes for 24 hours post-dose, but the radioactivity was reduced to below the level of detection at 336 hours post-dose. The radioactivity of tissues declined rapidly and was not detectable in the majority of tissues at 72 hours post-dose. The elimination of radioactivity was complete for all tissues at 336 hours post-dose.
Elimination of radioactivity from tissues was generally rapid with decreased tissue levels observed at 24 hours post-dose. At 72 hours post-dose, levels of radioactivity in tissues had fallen substantially, with only low concentrations of radioactivity quantifiable in a few tissues. By 168 hours, elimination was almost complete, with low levels of radioactivity only measurable in the liver and kidney cortex and in the fur and choroid layer of the eye of the pigmented rats; elimination was complete by 336 hours. Binding to the melanin of pigmented tissues was evident with levels in the eye choroid layer and pigmented fur still present at 168 hours post-dose. Whole-body phosphor images from a male albino rat after oral administration of 5 mg/kg [14C]-levomilnacipran are provided in Figure 9. Five sections from a single rat are provided to show the distribution of [14C]-levomilnacipran across the various tissues in the rat at 2 hours and 24 hours.
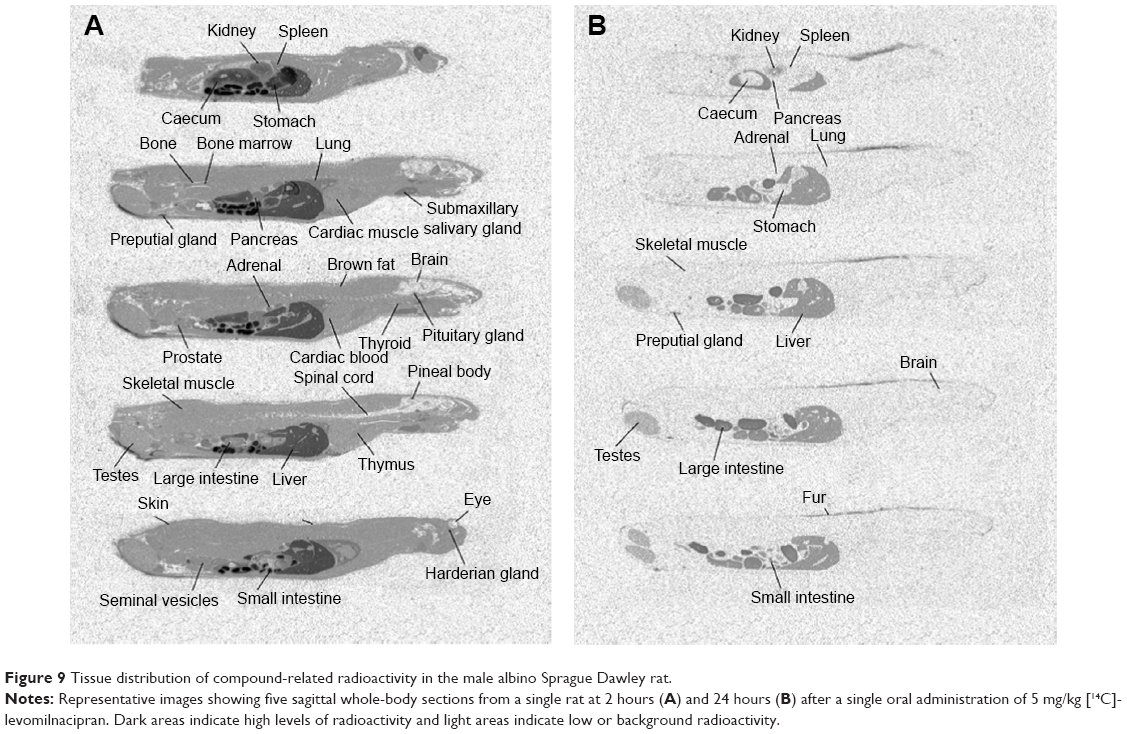 | Figure 9 Tissue distribution of compound-related radioactivity in the male albino Sprague Dawley rat. |
Discussion
To investigate the metabolic profile of levomilnacipran in humans and standard toxicology species, a single oral dose of [14C]-levomilnacipran was administered to humans, monkeys, and rats. We also evaluated the in vitro binding of [14C]-levomilnacipran to plasma proteins (HSA, AAG, and GGs). In humans, average peak plasma concentrations of levomilnacipran and total radioactivity indicated that levomilnacipran was contributing 61.0% of the total plasma radioactivity at peak plasma levels. The average extent of systemic exposure in terms of area under the curve revealed that the parent drug was contributing 45.1% of the total radioactivity exposure, suggesting that levomilnacipran is a major circulating product after oral administration. On average, N-desethyl levomilnacipran contributed 7.1% of the total radioactivity plasma exposure. The observed half-lives of the total radioactivity are comparable with the average half-lives of levomilnacipran and N-desethyl levomilnacipran, indicating the absence of long-half-life circulating metabolites. Measurable quantities of levomilnacipran glucuronide and N-desethyl levomilnacipran glucuronide were detected in human and monkey plasma, but no glucuronide metabolites were detected in rat plasma. Mean blood-to-plasma ratio of total radioactivity was close to 1 regardless of sampling time, suggesting that the radioactivity was equally distributed between the plasma and cellular component of blood. Protein binding to plasma proteins was 22% and not saturable in the concentration range of 1–1,000 ng/mL, meaning that the likelihood of drug–drug interactions due to competitive binding to plasma proteins is low.
Mass balance results indicated that levomilnacipran was predominantly excreted in the urine. Studies in monkeys and rats support the findings obtained in the human study, namely, that levomilnacipran was the major circulating compound post-dose, and levomilnacipran was mostly excreted unchanged. Differences in the excretion of relatively minor metabolites were observed across species. Human and monkey urine contained measurable quantities of levomilnacipran glucuronide and N-desethyl levomilnacipran glucuronides, but these metabolites were present in lower amounts in rat urine. The metabolites of levomilnacipran are not pharmacologically active.5 Because renal excretion is the predominant elimination pathway for levomilnacipran, dosing adjustment is recommended for patients with moderate (creatinine clearance of 30–59 mL/min) or severe (creatinine clearance of 15–29 mL/min) renal impairment; the maintenance dose for individuals with moderate renal impairment should not exceed 80 mg once daily, and for those with severe renal impairment, the maintenance dose should not exceed 40 mg once daily.5 Levomilnacipran is not recommended for patients with end-stage renal disease.5
Levomilnacipran is the active enantiomer of the racemic drug milnacipran.13,14 The metabolism and excretion of milnacipran in humans have been reported previously.13 As already described for the racemic drug, milnacipran,15,16 the primary route of metabolism of levomilnacipran is N-desethylation. Direct conjugation of levomilnacipran is a minor route of metabolism. Also an important, but apparently less pronounced, pathway is the subsequent conjugation of N-desethyl levomilnacipran to give rise to two different carbamoyl glucuronide derivatives on either the primary amine or the amide moiety. The latter is predominant compared with the amine linkage pathway.
There are similarities and differences in the PK properties of levomilnacipran and other SNRIs. In humans, the extended-release formulation of levomilnacipran has a half-life (T1/2=12 hours),5 which is longer than the half-life of milnacipran (T1/2=8 hours) and venlafaxine (T1/2=5 hours), and similar to that of desvenlafaxine (T1/2=11 hours) and duloxetine (T1/2=12 hours).17 Levomilnacipran is minimally bound to plasma proteins (22%), which is comparable to that of milnacipran (13%), venlafaxine (27%), and desvenlafaxine (30%), while plasma protein binding of duloxetine is greater than 90%.17 Like milnacipran and desvenlafaxine, levomilnacipran is not extensively metabolized; urinary excretion as unchanged drug accounts for 58%, 50%–60%, and 45% of levomilnacipran, milnacipran, and desvenlafaxine, respectively. Urinary excretion of venlafaxine and duloxetine as the parent drug is 5% and less than 1%, respectively.17 In contrast, duloxetine and venlafaxine are extensively metabolized by oxidation in the liver, where duloxetine is metabolized primarily by the cytochrome P450 (CYP) isozymes, CYP2D6 and CYP1A2, and venlafaxine is metabolized primarily by CYP2D6.17
Although not extensively metabolized, levomilnacipran does undergo desethylation to form N-desethyl levomilnacipran and hydroxylation to form p-hydroxy levomilnacipran. Both oxidative products are further metabolized by conjugation with glucuronide. Desethylation is catalyzed primarily by CYP3A4.5 Dose adjustment is recommended when levomilnacipran is coadministered with strong inhibitors of CYP3A4, such as ketoconazole, because an in vivo study has shown that a clinically meaningful increase in levomilnacipran exposure occurred when levomilnacipran was coadministered with ketoconazole.5,18 Further, in vivo studies showed no clinically meaningful changes in levomilnacipran exposure when levomilnacipran was coadministered with the CYP3A4 inducer, carbamazepine.5,18 Therefore, no dose adjustment of levomilnacipran is needed when administered with a CYP3A4 inducer. Other than CYP3A4 drug interactions, levomilnacipran is predicted to have a low potential to be involved in clinically significant PK drug interactions.5 Because hepatic metabolism of levomilnacipran is low, dose adjustment for individuals with hepatic impairment is not recommended.5,19
Results obtained in the rat distribution study indicated that tissues associated with absorption, biotransformation, and elimination had the highest maximum concentrations of radioactivity. Levomilnacipran and/or its metabolites crossed the blood–brain barrier, and the thalamus had the highest concentration of radioactivity in the brain. There was no accumulation of radioactivity in various glands (testes, thyroid, pituitary gland, pancreas, thymus, submaxillary gland, preputial gland, and adrenal gland). The uptake of radioactivity by the choroid layer of the eyes in pigmented rats suggests that levomilnacipran and/or its metabolites have an affinity for melanin-containing tissues. Elimination of radioactivity from tissues was generally rapid, with decreased tissue levels observed at 24 hours post-dose. These findings are consistent with those obtained in the mass balance and metabolite profile studies.
Conclusion
Levomilnacipran was the major circulating compound after dosing, and renal excretion of levomilnacipran was the major route of its elimination in humans, monkeys, and rats. The major circulating metabolite observed in all species was N-desethyl levomilnacipran, which was not pharmacologically active. Additionally, in humans and monkeys, N-desethyl levomilnacipran glucuronide and levomilnacipran glucuronide are circulating compounds, which are also inactive metabolites. Levomilnacipran was widely distributed, and plasma protein binding was low, which reduces the likelihood of drug–drug interactions. Knowledge of the PKs, metabolism, and plasma protein binding of levomilnacipran may help physicians prevent adverse effects from drug–drug interactions when using levomilnacipran to treat adults with major depressive disorder.
Acknowledgments
The authors thank Joann Hettasch, PhD, of Arbor Communications, Inc., Ann Arbor, MI, USA, for medical writing and editorial assistance.
The study was supported by funding from Forest Research Institute, Inc., an affiliate of Actavis Inc., Jersey City, NJ, USA, and Pierre Fabre Médicament, Boulogne, France. Medical writing and editorial assistance were funded by Pierre Fabre Médicament, Boulogne, France. Forest Research Institute, Inc., and Pierre Fabre Médicament were involved in developing the study design, data collection, data analysis and interpretation, and the decision to publish these results.
Disclosure
V Brunner, B Maynadier, L Roques, I Hude, S Séguier, L Barthe, and P Hermann are employees of Pierre Fabre Médicament, Boulogne, France, and may own shares of Pierre Fabre Médicament. L Chen is an employee of Forest Research Institute, Inc., an affiliate of Actavis Inc., Jersey City, NJ, USA, and may own shares of Actavis plc. The authors report no other conflicts of interest in this work.
References
Kessler RC, Ustün TB. The World Mental Health (WMH) survey initiative version of the World Health Organization (WHO) Composite International Diagnostic Interview (CIDI). Int J Methods Psychiatr Res. 2004;13:93–121. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association; 2000. [text rev]. | ||
Bromet E, Andrade LH, Hwang I, et al. Cross-national epidemiology of DSM-IV major depressive episode. BMC Med. 2011;9:90. | ||
Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice Guideline for the Treatment of Patients With Major Depressive Disorder. 3rd ed. Available from: http://psychiatryonline.org/guidelines. Accessed December 11, 2014. | ||
FETZIMA™ (levomilnacipran) extended-release capsules [US prescribing information]. St Louis, MO: Forest Pharmaceuticals, Inc.; 2014. | ||
Auclair AL, Martel JC, Assié MB, et al. Levomilnacipran (F2695), a norepinephrine-preferring SNRI: profile in vitro and in models of depression and anxiety. Neuropharmacology. 2013;70:338–347. | ||
Bakish D, Bose A, Gommoll C, et al. Levomilnacipran ER 40 mg and 80 mg in patients with major depressive disorder: a phase III, randomized, double-blind, fixed-dose, placebo-controlled study. J Psychiatry Neurosci. 2014;39:404–409. | ||
Sambunaris A, Bose A, Gommoll CP, Chen C, Greenberg WM, Sheehan DV. A phase III, double-blind, placebo-controlled, flexible-dose study of levomilnacipran extended-release in patients with major depressive disorder. J Clin Psychopharmacol. 2014;34:47–56. | ||
Asnis GM, Bose A, Gommoll CP, Chen C, Greenberg WM. Efficacy and safety of levomilnacipran sustained release 40 mg, 80 mg, or 120 mg in major depressive disorder: a phase 3, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74:242–248. | ||
Gommoll C, Greenberg WM, Chen C. A randomized double-blind, placebo-controlled, study of flexible doses of levomilnacipran ER (40–120 mg/day) in patients with major depressive disorder. J Drug Assessment. 2014;3:10–19. | ||
Montgomery SA, Mansuy L, Ruth A, Bose A, Li H, Li D. Efficacy and safety of levomilnacipran sustained release in moderate to severe major depressive disorder: a randomized, double-blind, placebo-controlled, proof-of-concept study. J Clin Psychiatry. 2013;74:363–369. | ||
Mago R, Forero G, Greenberg WM, Gommoll C, Chen C. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33:761–771. | ||
Deprez D, Chassard D, Baille P, Mignot A, Ung HL, Puozzo C. Which bioequivalence study for a racemic drug? Application to milnacipran. Eur J Drug Metab Pharmacokinet. 1998;23:166–171. | ||
Alliot J, Gravel E, Pillon F, Buisson DA, Nicolas M, Doris E. Enantioselective synthesis of levomilnacipran. Chem Commun (Camb). 2012;48:8111–8113. | ||
Li F, Chin C, Wangsa J, Ho J. Excretion and metabolism of milnacipran in humans after oral administration of milnacipran hydrochloride. Drug Metab Dispos. 2012;40:1723–1735. | ||
Puozzo C, Albin H, Vinçon G, Deprez D, Raymond JM, Amouretti M. Pharmacokinetics of milnacipran in liver impairment. Eur J Drug Metab Pharmacokinet. 1998;23:273–279. | ||
Shelton RC. Serotonin norepinephrine reuptake inhibitors: similarities and differences. Prim Psychiatry. 2009;16(5):25–35. | ||
Chen L, Boinpally R, Gad N, Periclou A, Ghahramani P, Greenberg WM. Drug-drug interactions of levomilnacipran sustained release capsule with ketoconazole, carbamazepine, or alprazolam in healthy subjects. New Research Poster NR9-16, American Psychiatric Association 166th Annual Meeting, 18–22 May 2013; San Francisco, CA. | ||
Chen L, Boinpally R, Greenberg WM, Wangsa J, Periclou A, Ghahramani P. Effect of hepatic impairment on the pharmacokinetics of levomilnacipran following a single oral dose of a levomilnacipran extended-release capsule in human participants. Clin Drug Investig. 2014;34:351–359. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.