")
Back to Journals » Journal of Experimental Pharmacology » Volume 15
Discovery of Fungus-Derived Nornidulin as a Novel TMEM16A Inhibitor: A Potential Therapy to Inhibit Mucus Secretion in Asthma
Authors Pongkorpsakol P, Yimnual C , Satianrapapong W, Worakajit N, Kaewin S , Saetang P, Rukachaisirikul V, Muanprasat C
Received 25 June 2023
Accepted for publication 18 October 2023
Published 15 November 2023 Volume 2023:15 Pages 449—466
DOI https://doi.org/10.2147/JEP.S427594
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Pawin Pongkorpsakol,1,* Chantapol Yimnual,2,* Wilasinee Satianrapapong,3 Nichakorn Worakajit,4 Suchada Kaewin,2 Praphatsorn Saetang,5 Vatcharin Rukachaisirikul,5 Chatchai Muanprasat2
1Princess Srisavangavadhana College of Medicine, Chulabhorn Royal Academy, Bangkok, Thailand; 2Chakri Naruebodindra Medical Institute, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Samut Prakan, Thailand; 3Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; 4Program in Translational Medicine, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; 5Division of Physical Science and Center of Excellence for Innovation in Chemistry, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
*These authors contributed equally to this work
Correspondence: Chatchai Muanprasat, Chakri Naruebodindra Medical Institute, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Samut Prakan, Thailand, Tel +66-85-2949658, Email [email protected]
Introduction: Inhibition of Ca2+-activated transmembrane protein 16A (TMEM16A) Cl− channels has been proposed to alleviate mucus secretion in asthma. In this study, we identified a novel class of TMEM16A inhibitors from natural sources in airway epithelial Calu-3 cells and determine anti-asthmatic efficacy of the most potent candidate in a mouse model of asthma.
Methods: For electrophysiological analyses, IL-4-primed Calu-3 cell monolayers were mounted in Ussing chamber and treated with various fungus-derived depsidones prior to the addition of UTP, ionomycin, thapsigargin, or Eact to stimulate TMEM16A Cl− current. Ca2+-induced mucus secretion in Calu-3 cell monolayers was assessed by determining MUC5AC protein remaining in the cells using immunofluorescence staining. OVA-induced female BALB/c mice was used as an animal model of asthma. After the course of induction, cellular and mucus components in bronchoalveolar lavage were analyzed. Lungs were fixed and undergone with H&E and PAS staining for the evaluation of airway inflammation and mucus production, respectively.
Results: The screening of fungus-derived depsidones revealed that nornidulin completely abolished the UTP-activated TMEM16A current in Calu-3 cell monolayers with the IC50 and a maximal effect being at ~0.8 μM and 10 μM, respectively. Neither cell viability nor barrier function was affected by nornidulin. Mechanistically, nornidulin (10 μM) suppressed Cl− currents induced by ionomycin (a Ca2+-specific ionophore), thapsigargin (an inhibitor of the endoplasmic reticulum Ca2+ ATPase), and Eact (a putative TMEM16A activator) without interfering with intracellular Ca2+ ([Ca2+]i) levels. These results suggest that nornidulin exerts its effect without changing [Ca2+]i, possibly through direct effect on TMEM16A. Interestingly, nornidulin (at 10 μM) reduced Ca2+-dependent mucus release in the Calu-3 cell monolayers. In addition, nornidulin (20 mg/kg) inhibited bronchoalveolar mucus secretion without impeding airway inflammation in ovalbumin-induced asthmatic mice.
Discussion and Conclusion: Our study revealed that nornidulin is a novel TMEM16A inhibitor that suppresses mucus secretion without compromising immunologic activity. Further development of nornidulin may provide a new remedy for asthma or other diseases associated with allergic mucus hypersecretion without causing opportunistic infections.
Keywords: TMEM16a, nornidulin, asthma, mucus secretion, airway epithelium
Introduction
Asthma is a heterogeneous chronic airway inflammatory disease with reversible airway obstruction that affects more than 260 million people and was considered a leading cause of disability globally in 2019.1,2 According to the CDC, around 25 million people perceive to have asthma with approximately 15% being children under 18 years old and the highest prevalence being among late teenagers and young adults in the United States.3 Several studies have identified pollution, certain viral and microbial particles, and obesity as risk factors for asthma that interact with genetic predisposition.4 Once exposed to these allergens, antigen-presenting cells capture and present these molecules to naïve T cells causing T helper 2 (Th2) cells accompanied by IgE production and binding to the surface of mast cells.5,6 Apart from this process, growing evidence has shed light on the role of basophils and NKT cells in inducing Th2 polarization and subsequent secretion of cytokines including interleukin-4 (IL-4) to increase the production of inflammatory mediators in mast cells.7,8 At the second time of allergen exposure, mast cells degranulate their contents causing airway inflammation, mucus secretion, and smooth muscle contraction, eventually leading to airway obstruction.9
Transmembrane protein 16A (TMEM16A), or anoctamin 1, is a component of the Ca2+-activated Cl− channel (CaCC) endogenously found in several cell types, including epithelial cells and smooth muscles in the cardiovascular, gastrointestinal, and respiratory systems.10–14 Several studies have suggested that TMEM16A contributes to the pathogenesis of numerous diseases including hypertension, cancer, and asthma.15–17 In head and neck squamous cell carcinoma, TMEM16a is overexpressed and is positively associated with cancer progression and poor prognosis.18 Although TMEM16A expression and function are limited in normal airway epithelial cells, their expression is considerably increased in the asthmatic epithelia.19,20 Importantly, stimulated airway mucus secretion, smooth muscle contraction, and asthmatic airway resistance are abrogated by the pharmacological or genetic silencing of TMEM16A.17,19,21–23 In addition, a putative TMEM16A activator, Eact or TMEM16A potentiator, Brevenal, augmented airway constriction of the tracheal ring and promoted mucus release associated with breathlessness in ovalbumin (OVA)-induced asthmatic mice.24,25 These data indicated that TMEM16A is required for excessive mucus secretion and airway constriction in asthma.
Expression of TMEM16A transcript and protein is upregulated by IL-4,11 the key pathogenic Th2 cytokine in asthma.26 Indeed, purinergic ligands, such as ATP and UTP, have been reported as endogenous activators of TMEM16A-dependent apical Cl− secretion.27 Notably, purinergic activation leads to an increase in intracellular Ca2+ ([Ca2+]i).27 Purinergic ligands are increased in patients with asthma and contribute to tracheal mucus secretion.28–30 In response to [Ca2+]i, TMEM16A mediates transepithelial Cl− secretion in the respiratory tract.31 Although Ca2+ is a key activator of the TMEM16A-dependent Cl− channel, Ca2+ also triggers calmodulin-dependent protein kinase II (CaMKII) TMEM16A inactivation.32,33 Therefore, CaMKII has been proposed to be a negative regulator of TMEM16A. A study on mouse airway smooth muscle suggested that TMEM16A allows Cl− efflux, induces membrane depolarization, and activates voltage-dependent Ca2+ channels, leading to airway smooth muscle contraction with a relatively unrevealed mechanism of TMEM16A-mediated mucus release.19,23 Notably, mucus hypersecretion represents an important pathogenesis of fatal asthma, convincing us to discover inhibitors of TMEM16A to alleviate both airway mucus hypersecretion and airway constriction simultaneously.34
Natural products derived from plants and microorganisms are promising sources of starting chemical scaffolds for drug discovery. Several natural-product-derived pharmacophores have been approved by the US Food and Drug Administration (FDA).35 It is well accepted that fungal species produce highly diverse bioactive metabolites including depsidones.36–38 However, the biological activity of fungal depsidones against TMEM16A has not yet been explored. Therefore, this study aimed to identify TMEM16A inhibitors from a collection of fungus-derived depsidones using electrophysiological techniques in airway epithelial (Calu-3) cells, and to investigate the basic pharmacological properties (ie, potency, cytotoxicity, and mechanism of action) of the most potent metabolites, as well as their potential application in attenuating mucus hypersecretion in Calu-3 cell monolayers and animal models of asthma.
Materials and Methods
Animal Study Approval and Ethical Statement
All animal experiments and protocols in this study were approved by the Institutional Animal Care and Use Committee of the Faculty of Science, Mahidol University (protocol no. MUSC62-028-492). Animal studies were performed in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (USA).
Mouse Model of OVA-Induced Allergic Asthma
BALB/c mice (8-week-old female, 18–22 g) were randomly divided into four groups (n = 5): control, OVA-treated, OVA plus nornidulin (20 mg/kg), and OVA plus dexamethasone (5 mg/kg; positive control; catalog number: D2915, Sigma Aldrich Co., Saint Louis, MO, USA) groups. A mouse model of OVA-induced allergic asthma was established as previously described,39 with certain modifications as shown in Figure 1. Briefly, mice were sensitized twice by intraperitoneal injection of OVA (50 µg)/Al(OH)3 (2 mg) mixed in PBS on days 0 and 14. On days 14 and 25–27, mice were challenged with intranasal instillation of PBS as a control or OVA (50 µg), with or without an intraperitoneal injection of nornidulin or dexamethasone. Bronchoalveolar lavage fluid (BALF) was collected for mucin secretion and inflammatory cell analysis. In addition, airway tissues were collected to evaluate the inflammatory reactions.
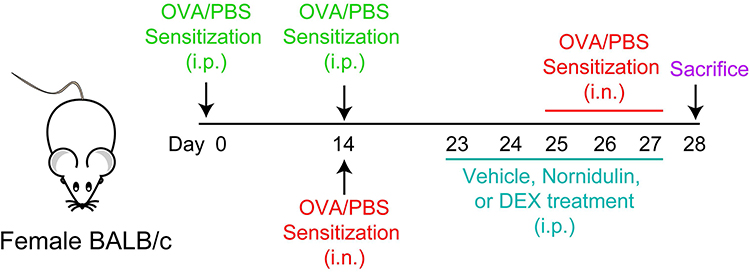 |
Figure 1 Protocol for mouse model of asthma and compound administration. Abbreviations: OVA, ovalbumin; PBS, phosphate buffer saline; DEX, dexamethasone; i.p., intraperitoneal injection; i.n., intranasal instillation. |
Chemicals
All fungus-derived depsidones used in this study, including the active compound nornidulin (N9), were prepared from the soil-derived fungus Aspergillus unguis PSU-RSPG204 as previously described.40 UTP (catalog number: U6875), N-((4-Methoxy)-2-naphthyl)-5-nitroanthranilic acid (MONNA; a specific TMEM16A inhibitor; catalog number: SML0902), 1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan, thiazolyl blue formazan (MTT formazan; catalog number: M2003), 4 kDa fluorescein isothiocyanate-dextran (FITC-dextran; catalog number: 46944), forskolin (catalog number: F6886), amphotericin B (catalog number: A4888), CFTRinh-172 (catalog number: 219670), ionomycin (catalog number: I9657), thapsigargin (catalog number: T9033), 3,4,5-trimethoxy-N-(2-methoxyethyl)-N-(4-phenyl-2-thiazolyl)-benzamide (Eact, a putative activator of TMEM16A; catalog number: SML1157), and ovalbumin (OVA; catalog number: A5503) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Human interleukin-4 (IL-4) recombinant (catalog number: A42602) was obtained from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Other reagents were purchased from Merck Millipore (Billerica, MA, USA) and Cell Signaling Technology (Danvers, MA, USA).
Cell Culture
Airway epithelial cell line, Calu-3 cells, was obtained from American Type Culture Collection (Manassas, VA, USA) and nourished in Eagle’s Minimum Essential Medium supplemented with 10% fetal bovine serum (FBS) (catalog number: F7524) (Sigma-Aldrich Co., St. Louis, MO, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin (catalog number: 15140122) (Thermo Fisher Scientific Inc., Waltham, MA, USA) at 37 °C in a humidified incubator under an atmosphere of 95% O2/5% CO2. For Ussing chamber experiments, Calu-3 cells were seeded onto Snapwell inserts (catalog number: 3801) (Costar, Cambridge, Massachusetts, USA) at a density of 5×105 cells/well and grown in a humidified incubator with culture medium renewal every day for two weeks.
Electrophysiological Analyses
For all analyses of TMEM16A-mediated Cl− currents, Ussing chamber experiments were performed as previously described.41,42 Briefly, fully differentiated Calu-3 cell monolayers with transepithelial electrical resistance (TER) > 500 Ω.cm2 as measured by EVOM2 volt-ohm meter (World Precision Instruments, Sarasota, Florida, USA) were primed with IL-4 (10 ng/mL) for 24 h. Then, IL-4-primed differentiated Calu-3 cell monolayers were mounted in Ussing chambers. To measure apical Cl− currents, the basolateral and apical hemi-chambers were filled with high Cl− and low Cl− buffers, respectively, to generate a basolateral-to-apical Cl− gradient. High Cl− basolateral solution (pH 7.3) contained NaCl (130 mM), KCl (2.7 mM), KH2PO4 (1.5 mM), CaCl2 (1 mM), MgCl2 (0.5 mM), Na-HEPES (10 mM) and glucose (10 mM). In the low Cl− apical solution, 65 mM NaCl was replaced with 65 mM sodium gluconate and the concentration of CaCl2 was adjusted to 2 mM. The basolateral membrane was permeabilized with amphotericin B (250 mg/mL) before the application of agonists/test compounds. Permeabilized Calu-3 cell monolayers were pretreated with fungus-derived metabolites from the library and incubated for 30 min prior to the stimulation of the Cl− current with UTP, ionomycin, thapsigargin, or Eact. The specific TMEM16A inhibitor MONNA was used to validate the contribution of TMEM16A. The TMEM16A inhibitory percentage of specific compounds was calculated based on their ability to reduce peak currents induced by TMEM16A activators compared to vehicle-treated inserts.
Evaluation of Cytotoxic Effect and Airway Epithelial Barrier Integrity
Calu-3 cells were seeded into 96-well plates (2 x 104 cells/well) and cultured overnight. Calu-3 cells were treated for 24 h with the vehicle (DMSO; control) or nornidulin (0.1–20 µM). MTT assay was performed as previously described.43 Formazan was quantified by measuring absorbance at 590 nm. The percentage cell viability after exposure to nornidulin was evaluated by comparing the absorbance of the nornidulin-treated group to that of the vehicle-treated control.
To demonstrate the effect of nornidulin on airway epithelial barrier function, real-time monitoring of TER was performed using an EVOM2 volt-ohm meter (World Precision Instruments, Inc., Sarasota, FL, USA) as previously described.44 The TER of Calu-3 cell monolayers was measured before and after treatment with DMSO, nornidulin (0.1–20 µM), or EGTA (5 mM; catalog number: E3889, Sigma-Aldrich Co., Saint Louis, MO, USA). The percentage change in TER was calculated by matching the TER at each specific time point to the initial TER.
The monolayers of Calu-3 cell barrier integrity were evaluated based on the rates of permeability of fluorescein isothiocyanate (FITC)-labeled dextran (molecular weight of ∼4 kDa), as previously described.42 Briefly, Calu-3 cells were plated onto Transwell membrane inserts (Costar, Cambridge, Massachusetts, USA) at a density of 5×105 cells/well and grown for 14 days, when TER was > 500 Ω.cm2. Calu-3 cell monolayers were treated for 24 h with DMSO (control), nornidulin (0.1–20 µM), or EGTA (5 mM). FITC-dextran (1 mg/mL) was added to the apical chamber of the Transwell inserts to assess the flux of FITC-dextran (4 kDa) across the Calu-3 cell monolayers. At 1-h post-incubation, the culture media were sampled from the basolateral chamber to measure the concentrations of FITC-dextran using a Wallac Victor2 microplate reader (Perkin Elmer, Waltham, Massachusetts, USA).
Intracellular Calcium Measurement
A calcium-sensitive fluorescent indicator, indo-1 (catalog number: I1223, Life Technologies, Carlsbad, CA, USA) was used for cytosolic calcium ([Ca2+]i) measurement. Calu-3 cells at a density of 5×105 cells were collected, washed with phosphate buffer saline (PBS), and incubated with indo-1 tracer at 37 °C in the dark. An hour later, the indo-1 indicator was completely removed by washing the cells twice with fresh calcium buffer containing KCl (5.33 mM), KH2PO4 (0.441 mM), NaHCO3 (4.17 mM), NaCl (137.93 mM), Na2HPO4 (0.338 mM), d-glucose (5.56 mM), CaCl2 (1 mM), and 1% FBS. Calu-3 cells were pre-incubated with nornidulin (10 µM) for 30 min before the addition of DMSO or ionomycin. Fluorescence intensity (excitation/emission wavelengths of 338/405 and 338/490 nm for Ca2+-bound and Ca2+-free indo-1, respectively) was measured using an FP-6200 spectrofluorometer (JASCO, Essex, UK).
Western Blot
Calu-3 cells were plated onto 60-mm Petri dishes at a density of 1×106 cells/dish (Corning Life Sciences, Tewksbury, MA, USA). Calu-3 cells were treated with DMSO, nornidulin (10 µM), or ionomycin (5 µM). At 1-h post-treatment, Calu-3 cells were lysed and collected using a lysis buffer. The cell lysates were centrifuged to obtain the proteins isolated from the supernatant. Proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (0.45 μm pore size). Membrane-containing proteins from Calu-3 cell lysates were blocked with 5% non-fat milk (Bio-Rad, Hercules, CA, USA) at room temperature for an hour. The membrane was probed with rabbit anti-β-actin polyclonal antibody and rabbit anti-phospho-CaMKII (Thr286) (D21E4) monoclonal antibody (Cell Signaling Technology, Boston, MA, USA) for an hour, washed with Tris-buffered saline containing Tween-20 (TBST), and incubated for an hour with HRP-conjugated goat anti-rabbit IgG antibody. Luminata™ Forte Western HRP Substrate (Merck Millipore, Billerica, MA, USA) was used to measure the protein expression using an Omega Lum G Imaging System (Aplegen, San Francisco, CA, USA). The expression of phospho-CaMKII was analyzed using the ImageJ software (NIH, Bethesda, Maryland, USA).
Immunofluorescence Staining for Mucin Secretion Assay
Calu-3 cells were seeded onto a transwell permeable support as previously described.45 After 7 days at the air-liquid interface, fully confluent Calu-3 cells were primed with IL-4 (10 ng/mL) for 24 h to induce TMEM16A upregulation. IL-4-primed Calu-3 cells were then treated with ionomycin (10 µM) for 10 min to stimulate Ca2+-dependent mucin secretion, with or without pretreatment with nornidulin (10 µM) for 30 min. The cells were fixed with 4% paraformaldehyde and permeabilized with 0.05% Triton X before overnight incubation with mouse anti-MUC5AC monoclonal antibody (45M1) (catalog number: MA5-12178, Thermo Fisher Scientific Inc., Waltham, MA, USA). The fixed cells were washed five times with PBS and incubated for an hour with Alexa Fluor 488-conjugated goat anti-rabbit IgG, followed by DNA staining with Hoechst 33342 for 15 min to determine cell boundaries. Fluorescently labeled was visualized using an inverted confocal laser-scanning microscope, FV1000 (Olympus, Tokyo, Japan). In this assay, the depletion of mucin storage inside cells indicated mucin secretion.
The Enzyme-Linked Immunosorbent Assay (ELISA) for Measurement of MUC5AC
Mucin secretion was estimated from the levels of MUC5AC, a pathogenic mucin protein, in BALF. The Mouse MUC5AC ELISA Kit (catalog number: CSB-E11040m, CUSABIO, Houston, TX, USA), a sandwich-type ELISA kit, was used. The MUC5AC levels were measured according to the manufacturer’s instructions.
Histological Analyses of Mouse Airway Tissues
Airway tissues were dissected from OVA-challenged mice with or without nornidulin. All isolated mouse airway tissues were immediately fixed for 24 h with 10% formaldehyde, followed by tissue dehydrated using ethanol (molecular grade), and embedded in paraffin. Mouse airway tissues fixed in paraffin were cut into 5 µm-thick sections and stained with hematoxylin and eosin (H&E) or periodic-acid Schiff (PAS) reagents. The histopathological morphology of tissue slices was observed under a light microscope. Eosinophils were counted in H&E-stained sections under high-power field (400x magnification) at average for each lung as described previously with some modifications.46
Statistical Analyses
For all experimental data presented in this study, the specific numbers of in vitro experiments and mice in each group are indicated in the figure legends. Data are presented as mean ± S.E.M., and all data are presented as at least three to seven independent experiments. The differences between the control and treatment groups were examined using one-way ANOVA or two-way ANOVA followed by Bonferroni’s post hoc test, where appropriate, with a P value of < 0.05, which was considered statistically significant. The statistical analysis was performed using GraphPad Prism software version 5 (GraphPad Software, La Jolla, CA, USA)
Results
Validation of in vitro Model for Assessment of TMEM16A Activity and Screening of Fungal Metabolites
As TMEM16A is not functionally expressed in normal airway epithelial cells,19,20 IL-4, one of the key pathogenic cytokines in asthma, was used to upregulate TMEM16A and establish an in vitro model of the asthmatic airway.11,26 The airway epithelial-like Calu-3 cell line, which endogenously expresses TMEM16a, was used as the in vitro model.45,47 In this experiment, airway epithelial-like Calu-3 cell monolayers were pretreated with IL-4 (10 ng/mL) for 24 h prior to electrophysiological analysis. We found that the peak of the UTP-induced Cl− current in IL-4-primed Calu-3 monolayers was higher than that in the IL-4-untreated group (Figure 2A and B). Treatment with MONNA (10 µM), a TMEM16A inhibitor, significantly suppressed the UTP-induced Cl− current, after which forskolin stimulated the CFTR-mediated Cl− current (Figure 2A and B). It was noted that MONNA reduced the UTP-induced Cl− current in the IL-4-primed Calu-3 cells to a level lower than that in the IL-4-untreated Calu-3 cells, indicating that MONNA inhibited the UTP-induced Cl− current contributed by basal/endogenous TMEM16A expression in Calu-3 cells. In addition, CFTRinh-172 (5 µM), a specific CFTR inhibitor, reduced the UTP-induced Cl− current by approximately 30% (data not shown). These results indicate that the Cl− current upon purinergic stimulation in IL-4-primed Calu-3 monolayers was mainly mediated by TMEM16A, with less contribution from CFTR. Next, we used this model to screen 11 depsidone derivatives from the soil-derived fungus, A. unguis PSU-RSPG204. To identify compounds with submicromolar potency, we selected depsidone derivatives with more than 50% inhibition of UTP-induced Cl− current at 1 µM for subsequent studies. N9 (nornidulin) and N15 (2-chlorounguinol) fulfilled the aforementioned criteria (Figure 2C and D).
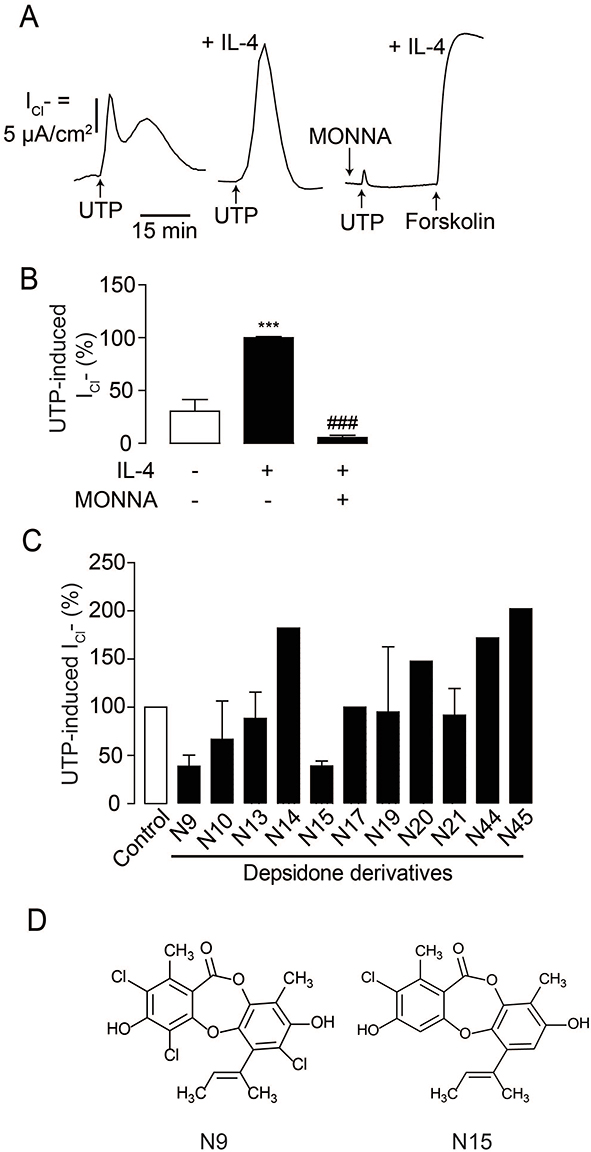 |
Figure 2 Screening of depsidone derivatives on UTP-induced Cl− secretion in IL-4-primed Calu-3 cell monolayers. (A) Validation of an in vitro model of TMEM16A-dependent Ca2+-activated Cl− channel. Apical Cl− current analyses were performed in Calu-3 cell monolayers. IL-4 was used to upregulate TMEM16A and UTP-induced Cl− current was increased in IL-4-primed Calu-3 cell monolayers compared to IL-4-untreated group. MONNA (10 µM) was shown to inhibit UTP-induced Cl− current. A representative apical Cl− current tracing is shown. (B) Graphs indicating analyses and comparisons of UTP-induced apical Cl− currents in IL-4-free and IL-4-treated groups with or without MONNA. Results were expressed as % of TMEM16-dependent Cl− current peak ± S.E.M. (n = 4–6). ***p < 0.001 compared with the control group; ###p < 0.001 compared with the IL-4-treated group (one-way ANOVA). (C) Screening of depsidone derivatives (1 µM) isolated from a soil-derived fungus Aspergillus unguis PSU-RSPG204 on TMEM16-dependent Cl− current. In the screening step, we performed experiment in singlicate, especially inactive compounds. (D) Chemical structures of active depsidone derivatives N9 (nornidulin) and N15 (2-chlorounguinol). |
Effect of Bioactive Depsidones on TMEM16A-Mediated Apical Cl− Transport in Airway Epithelial Cell Monolayers
To investigate the effects of the depsidone derivatives N9 (nornidulin) and N15 (2-chlorounguinol) on TMEM16A Cl− channel activity, apical Cl− current analyses were performed in IL-4-primed Calu-3 cell monolayers. In this experiment, the basolateral membrane of Calu-3 cell monolayers was permeabilized with amphotericin B, and a Cl− gradient was established using asymmetrical Cl− buffers (high [Cl−] in the basolateral solution and low [Cl−] in the apical solution). Concentration-dependence of nornidulin and 2-chlorounguinol on TMEM16A inhibition was evaluated. As shown in Figure 3A–C, TMEM16A-mediated apical Cl− current induced by UTP (100 µM) in Calu-3 cell monolayers was inhibited by nornidulin in a concentration-dependent manner, with IC50 of ~0.8 µM and almost complete inhibition being observed at 10 µM. Conversely, TMEM6A-mediated apical Cl− currents were incompletely inhibited by 2-chlorounguinol (Figure 3D and E). Notably, at 10 µM, 2-chlorounguinol reduced TMEM16A-mediated apical Cl− currents by only 60% of the UTP-induced Cl− current peak (Figure 3D and E). Therefore, N9 (nornidulin), but not N15 (2-chlorounguinol), was used to evaluate its pharmacological properties and potential utility in asthma therapy in subsequent experiments.
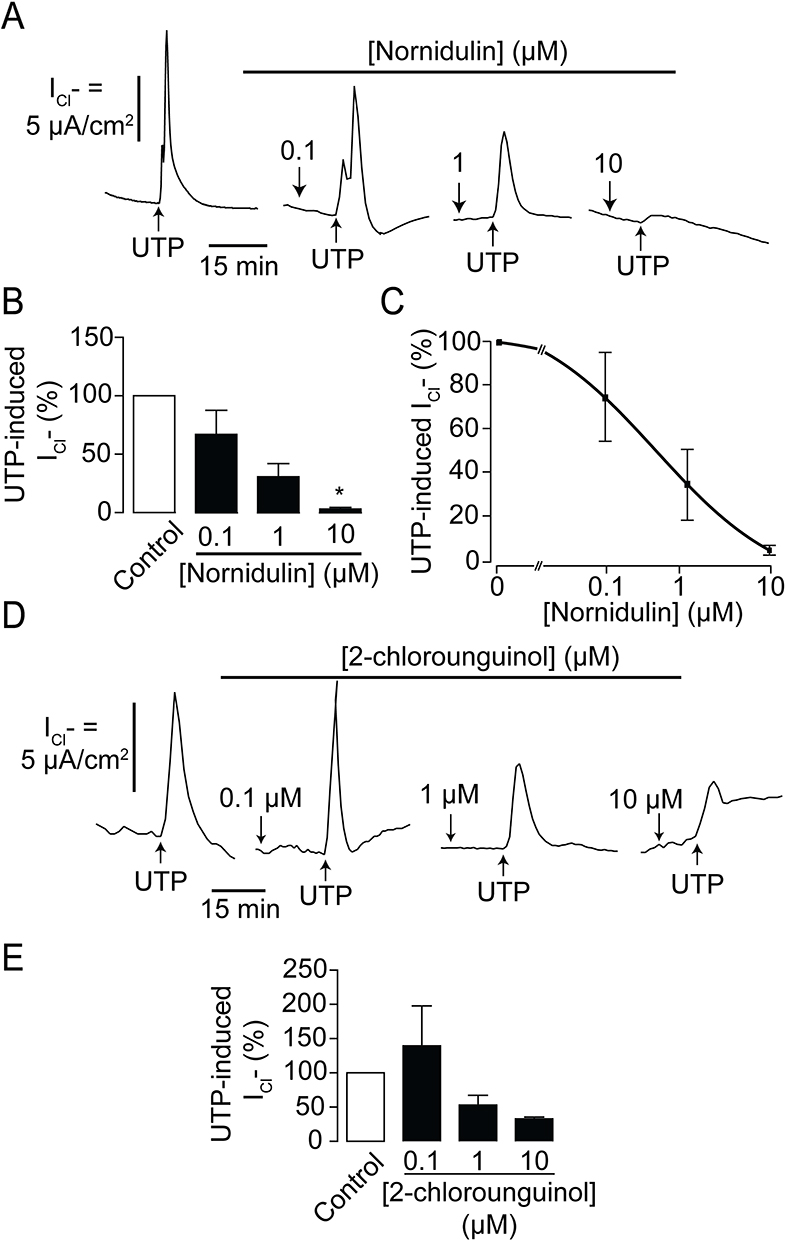 |
Figure 3 Effect of depsidone derivatives on TMEM16A activity. (A) Dose-inhibition studies of N9 (nornidulin) on UTP-induced Cl− secretion across IL-treated Calu-3 cell monolayers. A representative apical Cl− current tracing is shown. (B) Analyses and comparisons of apical Cl− current in IL-4-treated Calu-3 cell monolayers with or without N9 (nornidulin) at various concentrations (0.1 µM, 1 µM, and 10 µM). (C) IC50 determination. Data are analyzed by Hill’s equation and expressed as means of % UTP-activated apical Cl− current ± S.E.M. According to the Hill coefficient, IC50 was approximately 0.8 µM. (D) Dose-inhibition studies of N15 (2-chlorounguinol) on UTP-induced Cl− secretion across IL-treated Calu-3 cell monolayers. Representative apical Cl− current tracings are shown. (E) Analyses and comparisons of apical Cl− current in IL-4-treated Calu-3 cell monolayers with or without N15 (2-chlorounguinol) at various concentrations (0.1 µM, 1 µM, and 10 µM). The summary of data is shown. Results were expressed as % of TMEM16-dependent Cl− current peak ± S.E.M. (n = 4–6). *, p < 0.05 compared with the reference group (one-way ANOVA). |
Evaluation of Cytotoxic Potential of Nornidulin in Calu-3 Cells
To evaluate the potential toxicity of nornidulin in airway epithelial cells, its effects on Calu-3 cell viability and barrier function were tested. MTT assay was used to determine the viability of Calu-3 cells. As illustrated in Figure 4A, 24-h exposure to nornidulin at concentrations ranging from 0.1 µM to 20 µM had no significant effect on Calu-3 cell viability. TER measurements were performed to further investigate the effect of nornidulin on the integrity of airway epithelial barrier function. Nornidulin at concentrations of 0.1 µM to 10 µM did not change TER of Calu-3 cell monolayers (Figure 4B). However, 20 µM nornidulin significantly reduced the TER by approximately 50% relative to that of the control (P = <0.0001). As a positive control, treatment with ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA; 5 mM), a Ca2+ chelator known to disrupt the integrity of epithelial tight junctions, decreased the TER value by > 95% (P = <0.0001). Fluorescein isothiocyanate (FITC)-dextran (molecular weight of 4.4 kDa), a fluorescence-tagged paracellular probe, was used to evaluate tight junction integrity across Calu-3 cell monolayers. At 2 h post-treatment, nornidulin at concentrations of 0.1 µM to 10 µM had no remarkable effect on flux of FITC-dextran over Calu-3 cell monolayers compared with control (Figure 4C). Notably, 20 µM nornidulin slightly increased FITC-dextran flux (P = <0.0020), with EGTA as a positive control, causing a significant increase in FITC-dextran flux across Calu-3 cell monolayers (P = <0.0001) (Figure 4C).
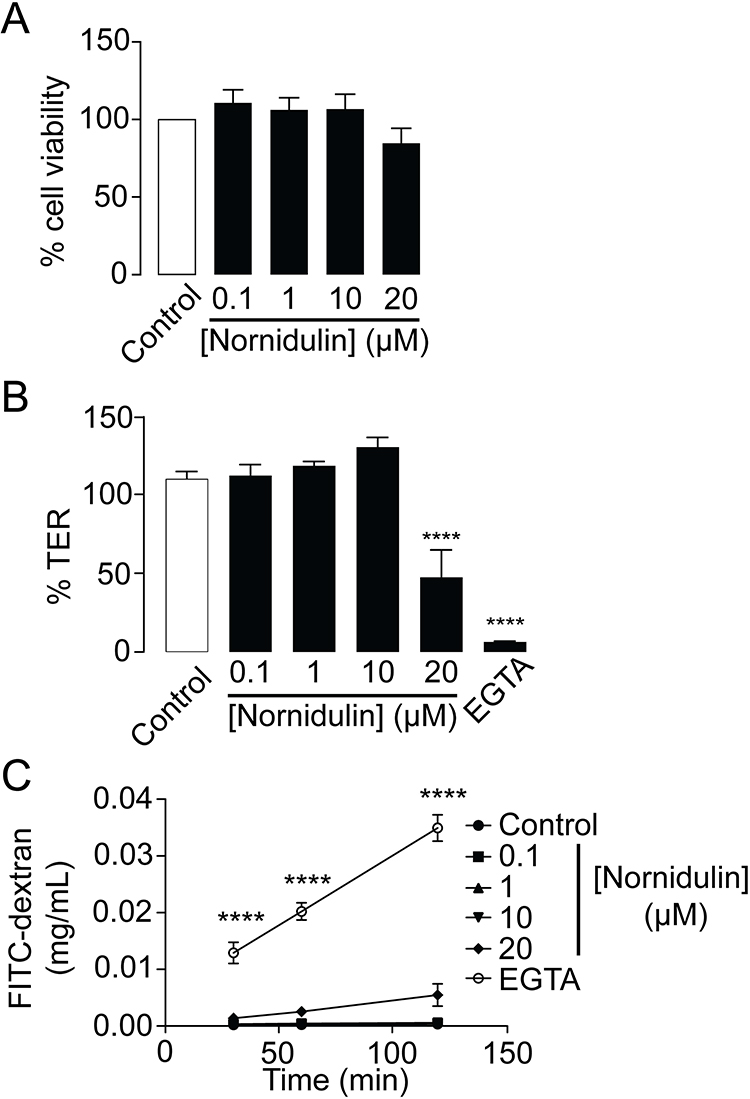 |
Figure 4 Evaluation of cytotoxic potential of nornidulin in Calu-3 cell monolayers. (A) Effect of nornidulin on Calu-3 cell viability. Calu-3 cells grown in 96-well plates were incubated for 24 h with nornidulin at the indicated concentrations (0.1–20 µM). MTT assay was performed to determine the viability of Calu-3 cell. (B) Effect of nornidulin on transepithelial electrical resistance (TER) of Calu-3 cell monolayers. Calu-3 cells grown on permeable support for 14 days were treated for 24 h with nornidulin. At 24-hour post-incubation with nornidulin at the indicated concentrations (0.1–20 µM), TER was measured by the epithelial volt/ohms meter. EGTA was used as a positive control for experiment. (C) Effect of nornidulin on paracellular flux across monolayers of Calu-3 cell. FITC-dextran (molecular weight ∼4.4 kDa) was used as a paracellular fluorescent probe. Fully differentiated Calu-3 cell monolayers grown on permeable support were exposed for 2 h to nornidulin at the indicated concentrations (0.1–20 µM). FITC-dextran was added into the apical chamber of Calu-3 cell monolayers and incubated for an hour. Culture media from basolateral chamber of permeable support were sampled to measure the rate of paracellular flux of FITC-dextran. EGTA was used to dissociate tight junctions as a positive control for this experiment. Data are expressed as means of % control ± S.E.M. (n = 5). ****p < 0.0001 compared with the control group. One-way ANOVA was used for.Figure 3A and B, while two-way ANOVA was for Figure 3C. |
Mechanisms of Inhibition of TMEM16A-Mediated Cl− Secretion by Nornidulin in Calu-3 Cells
To investigate whether nornidulin inhibited TMEM16A-mediated Cl− secretion by interfering with Ca2+ signaling, electrophysiological analyses and [Ca2+]i measurements were performed. To determine whether nornidulin could inhibit TMEM16A independent of signaling transduction to [Ca2+]i elevation, TMEM16A-mediated Cl− secretion was induced by either ionomycin (a Ca2+-specific ionophore; 5 µM) or thapsigargin (an inhibitor of the endoplasmic reticulum Ca2+ ATPase; 5 µM). Nornidulin (10 µM) completely abolished ionomycin- and thapsigargin-induced Cl− secretion in Calu-3 cell monolayers (Figure 5A–D) with overall P-values of 0.0128 and 0.0002, respectively. In addition, ionomycin with or without nornidulin significantly increased [Ca2+]i compared to control with overall P = 0.0005 and P = 0.0004, respectively. Indeed, nornidulin (10 µM) did not significantly suppress ionomycin-induced [Ca2+]i elevation (P = 0.8843) (Figure 5E and F). Notably, the effect of nornidulin on TMEM16A was much greater than that on [Ca2+]i elevation (Figure 5B and F). These results indicate that restraining TMEM16A function by nornidulin is not through events upstream of [Ca2+]i elevation, favoring mechanisms downstream of [Ca2+]i elevation.
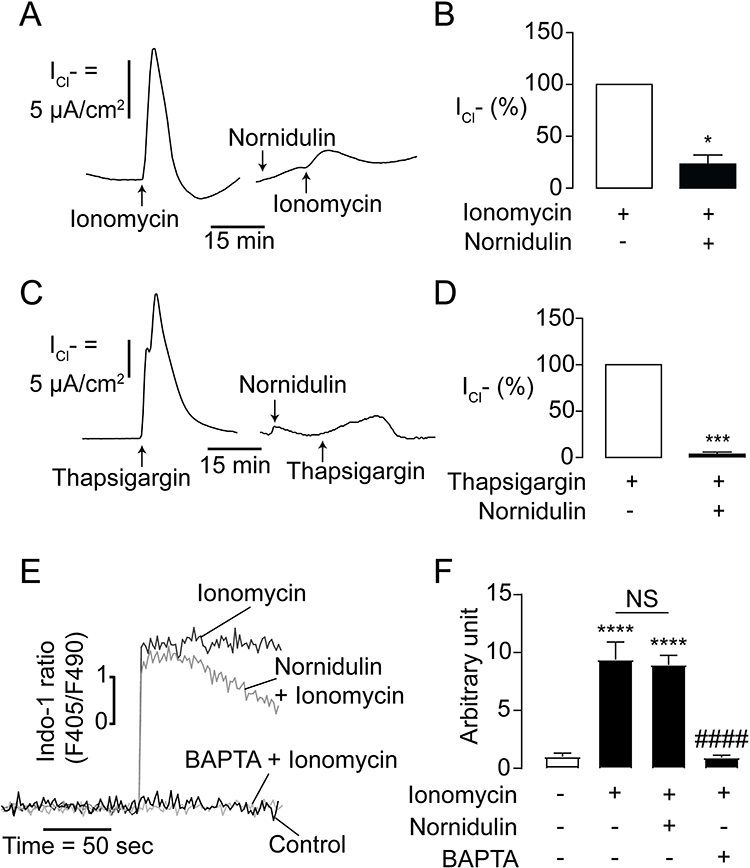 |
Figure 5 Mechanism of TMEM16A inhibition by nornidulin in IL-4-primed Calu-3 cell monolayers. (A) Effect of nornidulin on ionomycin-induced Cl- secretion in IL-4-treated Calu-3 cell monolayers. A representative ionomycin-induced, TMEM16A-dependent apical Cl− current tracing is shown. (B) Graphs indicating analyses and comparisons of apical Cl− current in IL-4-treated Calu-3 cell monolayers induced by ionomycin treatment with or without nornidulin (10 µM). The summary and statistical analyses of data are shown. (C) Effect of nornidulin on thapsigargin-induced Cl− secretion in IL-4-treated Calu-3 cell monolayers. A representative thapsigargin-induced, TMEM16A-dependent apical Cl− current tracing is shown. (D) Graphs indicating analyses and comparisons of apical Cl− current in IL-4-treated Calu-3 cell monolayers induced by thapsigargin treatment with or without nornidulin (10 µM). The summary and statistical analyses of data are shown. (E) Effect of nornidulin on [Ca2+]i increase induced by ionomycin. A representative tracing of indo-1-sensitive fluorescent intensities is shown. (F) Analyses of intracellular indo-1-sensitive fluorescent intensities of control show the Calu-3 cells exposed to ionomycin with or without nornidulin pretreatment (10 µM). Results were expressed as % of control ± S.E.M. (n = 4–6). *p < 0.05; ***p < 0.001; ****p < 0.0001 compared with the control group; ####p < 0.0001 compared with the ionomycin-treated group (one-way ANOVA). |
As CaMKII is known to be a negative regulator of TMEM16A following an increase in [Ca2+]i, we examined the effect of nornidulin on CaMKII phosphorylation, which is the active state of CaMKII, using Western blotting. We found that nornidulin (10 µM) did not affect CaMKII phosphorylation compared to the control (Figure 6A and B). Ionomycin was used as a positive control to enhance Ca2+-dependent CaMKII phosphorylation. Moreover, we tested whether nornidulin inhibited TMEM16A activity using the putative TMEM16A activator Eact (10 µM). Interestingly, nornidulin (10 µM) significantly reduced the Cl− current peak induced by Eact (Figure 6C and D). Previous studies have shown that Eact augments [Ca2+]i which is required for TMEM16A activation.48,49 We also found that Eact elevated [Ca2+]i, which was virtually unaffected by nornidulin, when BAPTA (a Ca2+ chelator) was used as a positive control (Figure 6E and F). These results indicate that TMEM16A inhibition by nornidulin in Calu-3 cells does not result from indirect mechanisms, including CaMKII phosphorylation or alterations in calcium signaling.
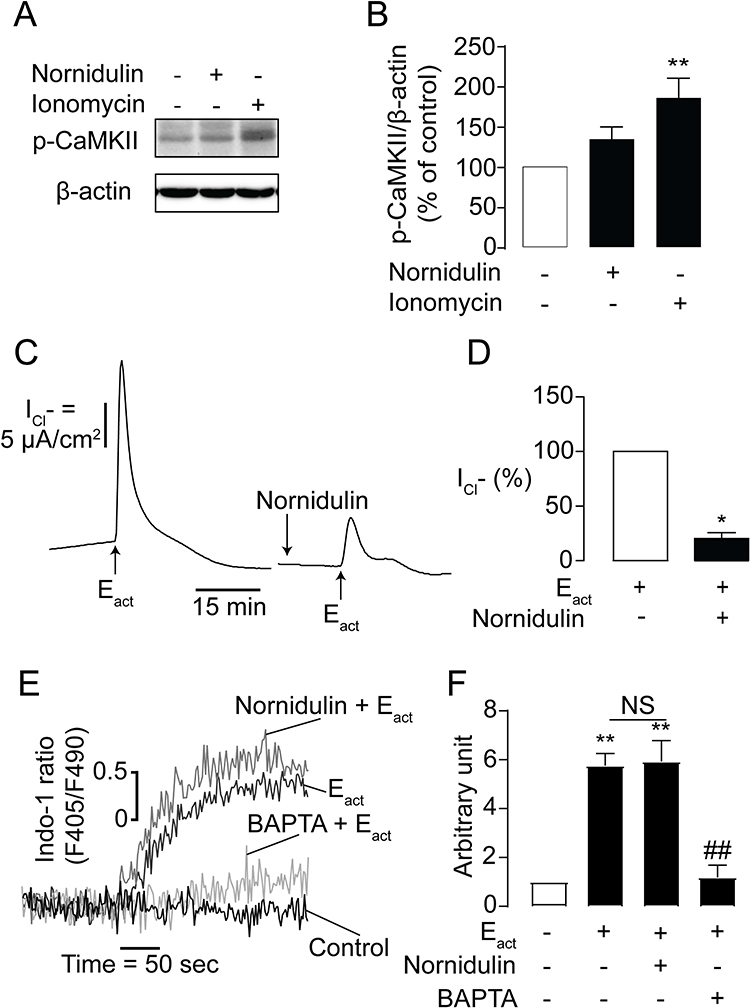 |
Figure 6 Effects of nornidulin on CaMKII phosphorylation and Eact-induced TMEM16A-dependent Cl− secretion. (A) Representative Western blot bands of p-CaMKII and β-actin. (B) Band intensity evaluation of CaMKII phosphorylation in Calu-3 cells treated with or without nornidulin. Results were analyzed from p-CaMKII/β-actin ratio and expressed as % of control ± S.E.M. (C) Effect of nornidulin on Eact-stimulated TMEM16A Cl− channel. A representative tracing of Eact-induced TMEM16A-dependent Cl− secretion is shown. (D) Graphs indicating analyses and comparisons of Eact-induced TMEM16A-dependent apical Cl− current in IL-4-treated Calu-3 cell monolayers with or without nornidulin (10 µM). (E–F) Effect of nornidulin on Eact-induced [Ca2+]i elevation in Calu-3 cells. The summary and statistical analyses of data are shown. Results were expressed as % of control ± S.E.M. (n = 3–6). *p < 0.05; **p < 0.01 compared with the control group; ##p < 0.01 compared with Eact (one-way ANOVA). |
Effect of Nornidulin on Ionomycin-Induced Mucus Release in Calu-3 Cells
Hypersecretion of mucus is a hallmark of asthmatic airway.34 Increased TMEM16A expression and activity plays an important role in mediating mucus hypersecretion in asthma.19,22,50 To examine whether nornidulin inhibits ionomycin-induced mucin secretion in inflamed Calu-3 cell monolayers, co-staining with intracellular MUC5AC and Hoechst was performed. Calu-3 cell monolayers were incubated for 24 h with IL-4 to mimic asthmatic inflammatory conditions and to induce TMEM16A upregulation. IL-4-primed Calu-3 cell monolayers were treated with ionomycin (10 µM) with or without nornidulin (10 µM). As shown in Figure 7, ionomycin significantly reduced intracellular MUC5AC levels, indicating Ca2+-dependent mucin secretion. Interestingly, nornidulin (10 µM) abrogated the ionomycin-induced intracellular MUC5AC depletion (58.3054±7.1338 vs 104.3716±15.2886; p=0.0092; overall p=0.0038). Similarly, MONNA (10 µM) suppressed the ionomycin-induced decrease in intracellular MUC5AC levels (p=0.0308). These data suggest that nornidulin attenuates mucus secretion in an in vitro asthma model.
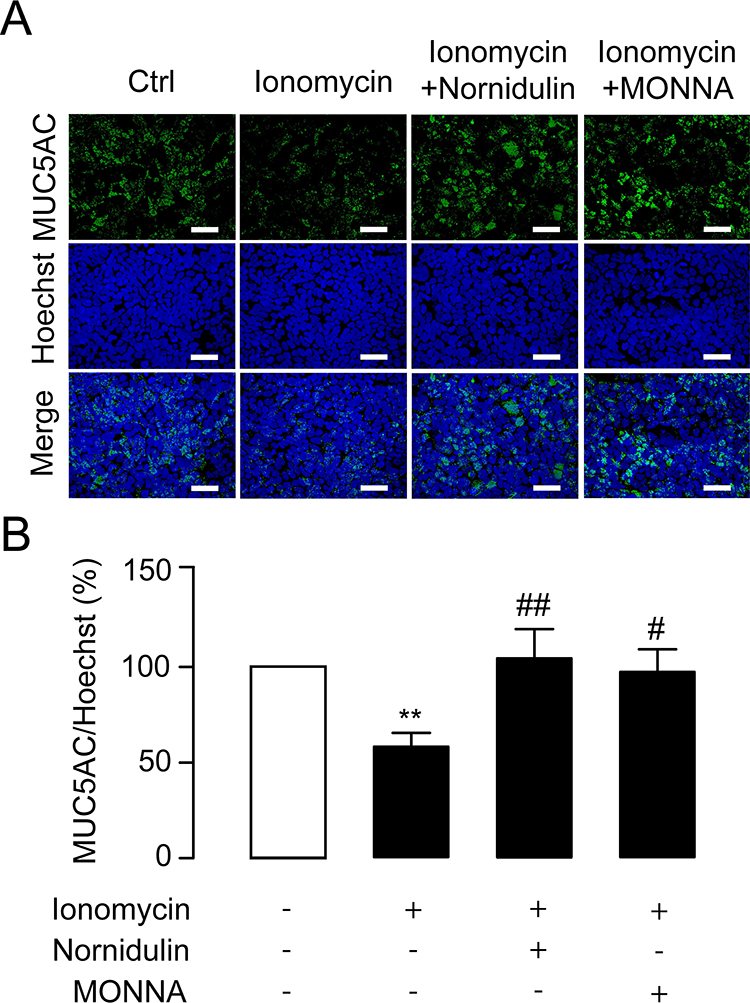 |
Figure 7 Effect of nornidulin on Ca2+-dependent mucin secretion in Calu-3 cells. (A) Immunofluorescence staining of MUC5AC. Hoechst staining was used to determine the cell area. MUC5AC, a marker of pathogenic mucin in asthma, was labeled to evaluate depletion of intracellular mucin store that indicates mucin secretion. Ionomycin treatment was performed to promote Ca2+-dependent mucin secretion. Nornidulin (10 µM) and a TMEM16A inhibitor MONNA were able to suppress ionomycin-induced depletion of intracellular mucin store, suggesting that nornidulin was capable of inhibiting mucin secretion. The scale bars of 50 µm were shown. (B) Analyses of intracellular mucin levels. The summary and statistical analyses of data are shown. Results were expressed as % of control ± S.E.M. (n = 4–6). **p < 0.01 compared with control group (one-way ANOVA). #p < 0.05; ##p < 0.01 compared with ionomycin-treated group (one-way ANOVA). |
Attenuation of Mucin Hypersecretion by Nornidulin in an Experimental Asthmatic Mouse Model
To prove a proof of concept that nornidulin is effective in the treatment of mucus hypersecretion in asthma, the effect of nornidulin was evaluated in a mouse asthma model. Female BALB/c mice were primed and challenged with OVA with or without intraperitoneal administration of nornidulin (Figure 1). Because pharmacokinetic profiles of nornidulin have not been previously investigated, we adapted the treatment protocol of nornidulin from a previous study that showed the systemic effects of a compound structurally related to nornidulin.51 As shown in Figure 8A, OVA significantly increased the spleen weight (p=0.0263; overall p<0.0001), indicating the induction of systemic inflammation. Dexamethasone (5 mg/kg; p<0.0001), an immunosuppressive drug but not nornidulin (p=0.9648), reduced OVA-induced spleen weight gain. In addition, H&E staining revealed that nornidulin had no effect on OVA-induced deterioration of lung pathology and eosinophil infiltration in the mouse airway epithelial tissues as shown in Figure 8B–D (inflammatory score 3 vs 2.6±0.2449; p= 0.573; overall p<0.0001 and Eosinophil count 36.3±4.1309 vs 31.5±6.6427; p=0.8891; overall p=0.0003). Interestingly, PAS staining showed that nornidulin significantly reduced OVA-induced mucus production as displayed in Figure 8B and E (27.3081±3.1098 vs 17.5601±1.9405; p=0.0422; overall p<0.0001). Dexamethasone suppressed OVA-induced lung inflammation (p=0.0056) and eosinophil infiltration (p=0.0263) and mucus production (p=0.0015) in the mouse airway epithelial tissues (Figure 8B–F). To investigate the effect of nornidulin on OVA-induced mucin secretion, the levels of MUC5AC, a pathogenic mucin protein, in bronchoalveolar lavage fluid (BALF) were measured using ELISA kits. Interestingly, nornidulin significantly mitigated MUC5AC levels (1.9809±0.2267 vs 0.8337±0.1654; p=0.0011; overall p=0.0020) without affecting the total number of immune cells (p=0.1348; overall p=0.0034) in the BALF, whereas dexamethasone significantly suppressed both parameters (Figure 8F and G; MUC5AC p=0.0396; total cells p=0.0202).
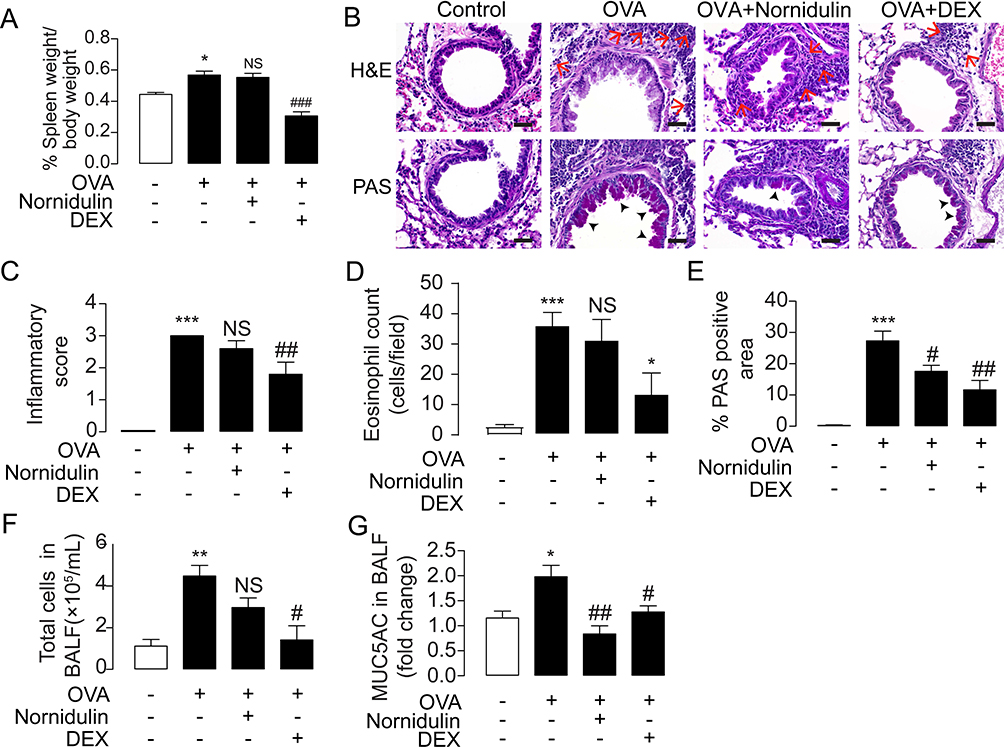 |
Figure 8 Effect of nornidulin in the treatment of inflammation-associated mucus hypersecretion in OVA-challenged mouse model of asthma. (A) Timeline and detailed protocol of OVA-challenged mouse model of asthma establishment. (B) Effect of nornidulin and dexamethasone (DEX) on percent spleen weight. (C) H&E and PAS staining of airway tissues from OVA-challenged mice treated with or without nornidulin. Dexamethasone (DEX) was used as a control of this experiment. The scale bars of 40 µm were shown with the red arrows and the black arrow heads indicating clusters of eosinophils and mucus-producing cells, respectively. (D) Inflammatory score and (E) % PAS-positive area of airway tissues from OVA-challenged mice treated with or without nornidulin. Dexamethasone (DEX) was used as a control of these experiment. (F) Effect of nornidulin on in vivo mucus hypersecretion in OVA-challenged mice. Mucus hypersecretion was evaluated by the levels of MUC5AC, a pathogenic mucin, collected from bronchoalveolar lavage fluid (BALF). Both nornidulin and dexamethasone (DEX) significantly reduced mucus hypersecretion. (G) Effect of nornidulin on total immune cell number. BALF was collected and immune cells were observed and counted under microscope. Dexamethasone (DEX), but not nornidulin, significantly reduced total cells in BALF. Results were analyzed from 5–7 independent experiments and shown as a means of control ± S.E.M. *p < 0.05; **p < 0.01; ***p < 0.001 compared with control group (one-way ANOVA). #p < 0.05; ##p < 0.01; ###p < 0.001; NS, non-statistical difference compared with OVA-challenged groups without any treatment (one-way ANOVA). |
Discussion
In this study, we report the inhibitory effect of nornidulin, a fungus-derived metabolite, on TMEM16A-dependent, Ca2+-activated Cl− secretion in a human airway epithelial cell line (Calu-3 cells). Electrophysiological analyses of TMEM16A-mediated airway epithelial Cl− transport and intracellular Ca2+ measurements indicated that the effect of nornidulin on TMEM16A inhibition occurs via a Ca2+-independent mechanism. Furthermore, nornidulin suppressed mucin secretion in both airway epithelial cells and in vivo allergic asthma model. Notably, nornidulin (N9), an inhibitor of TMEM16A, did not modulate inflammatory processes in the OVA-challenged allergic asthma model. Importantly, this study revealed a potential remedy for nornidulin in airway mucus hypersecretion in asthma, at least in part, by targeting TMEM16A.
To date, several TMEM16A inhibitors have been identified including T16Ainh-A01, MONNA, and Benzbromarone. Some have been tested to mitigate disease phenotypes such as cancer proliferation, hypertension, and diarrhea in vitro and in vivo.52 In normal airways, TMEM16A is considered a minor component of CaCC.20 Conversely, immunostaining of airway epithelial tissues revealed that TMEM16A was predominantly upregulated in IL-4-primed cultured human bronchial epithelial cells and Muc5AC-positive mucus-producing cells in OVA-challenged asthma mouse and guinea pig models, as well as in asthmatic patients.19,22,50 Consistently, inhibition of TMEM16A-mediated Cl− secretion by Benzbromarone or T16Ainh-A01 energetically abolished OVA-induced and ATP-induced mucin secretion in Th2 cytokine-treated human bronchial epithelial cells, respectively.19,21,22 Therefore, TMEM16A has been recognized as a potential drug target for attenuating mucin hypersecretion in asthma In our in vitro model using IL-4-primed Calu-3 cell monolayers, the Ca2+-activated Cl− current induced by UTP was higher than that induced by IL-4-free Calu-3 cell monolayers, suggesting an increase in functional CaCCs in the inflamed airway epithelial cells. We also found that a specific inhibitor of TMEM16A (MONNA) completely abrogated the UTP-induced CaCC activity in IL-4-primed Calu-3 monolayers. This result confirms that TMEM16A is a major CaCC in inflamed airway epithelial cells, consistent with previous findings.19 Indeed, our in vitro model recapitulates the airway epithelia in patients with asthma, which is associated with the state of airway goblet cell metaplasia.50 In this study, we searched for bioactive compounds from fungal metabolites that can act as TMEM16A inhibitors. Indeed, there are tremendous fungal species.53,54 The fungi produce diverse secondary metabolites.55 Importantly, fungal metabolites can be used for large-scale production by the pharmaceutical industry.55–57 Therefore, fungal metabolites are fascinating sources for drug discovery.36,38,56,58 Our recent study identified zearalenone as a fungal metabolite that inhibits cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-activated Cl− channel.59 Here, we identified two depsidone derivatives including N9 (nornidulin) and N15 (2-chlorounguinol) from the soil-derived fungus A. unguis PSU-RSPG204 that inhibited TMEM16A activity in Calu-3 cells. However, the inhibitory potency of N15 against TMEM16A was poor, with 60% inhibition being the maximal effect. Therefore, nornidulin was selected for further analysis of its mechanism of action. Inhibition of TMEM16A-mediated apical Cl− secretion may be due to either cell death or epithelial barrier dysfunction. Interestingly, nornidulin, at concentrations found to inhibit TMEM16A-mediated Cl− secretion (0.1 µM to 10 µM), is not cytotoxic to airway epithelial cell monolayers, but higher concentration (20 µM) should be concerned about its impacts on cell viability and barrier function.
Moreover, we determined the effect of nornidulin on P2Y receptors and intracellular Ca2+ signaling. P2Y is a receptor for nucleotides including UTP.60,61 Since nornidulin suppressed UTP-induced TMEM16A activity, it is possible that nornidulin might either antagonize P2Y receptor or suppress [Ca2+]i increase (eg, stimulation of Ca2+ ATPase-mediated Ca2+ uptake into endoplasmic reticulum). To test these hypotheses, thapsigargin and ionomycin were used to directly increase [Ca2+]i, resulting in enhanced TMEM16A-mediated Cl− movement in the Calu-3 monolayers via a P2Y receptor-independent mechanism. Thapsigargin is used to inhibit Ca2+ ATPase-mediated Ca2+ uptake on the membrane surface of the endoplasmic reticulum, which can lead to the elevation of [Ca2+]i. On the other hand, ionomycin is a Ca2+-specific ionophore that mediates extracellular-to-intracellular Ca2+ transport. Both thapsigargin and ionomycin mediate TMEM16A-dependent Cl− secretion.62,63 Nornidulin suppressed thapsigargin- and ionomycin-induced TMEM16A-mediated Cl− secretion. Interestingly, nornidulin had little effect on [Ca2+]i which does not correspond to its effect on TMEM16A-mediated Cl− secretion. Therefore, the effect of nornidulin on TMEM16A function is not mediated by P2Y- or Ca2+-dependent mechanisms. Furthermore, CaMKII, a negative regulator of TMEM16A, was unaffected by nornidulin. Therefore, nornidulin might directly inhibit TMEM16A expression. Nornidulin consistently suppressed TMEM16A-mediated Cl− efflux induced by Eact, a putative TMEM16A activator. However, molecular structural analyses of the nornidulin/TMEM16A interaction need to be performed in the future to prove this hypothesis and identify the nornidulin-binding site on TMEM16A. In addition, based on our data, we cannot exclude the effect of nornidulin on other mechanisms involving Ca2+-independent negative regulators of TMEM16A, including phosphatases PP1 and PP2A.64
Of particular importance, nornidulin suppressed mucus in vitro and extended mucus production in vivo. This disparity might be due to the longer exposure time of the lungs to nornidulin for additional reduction of mucus production, which was not observed in Calu-3 cell monolayers. We used mucin 5AC (MUC5AC), a pathogenic asthma-associated mucin, and PAS staining in lung tissues as markers for the detection of mucus secretion and mucus production, respectively.65 Unlike dexamethasone (a corticosteroid used as a standard treatment of asthma), nornidulin did not suppress inflammation in airway tissue, or eosinophil infiltration which is the key pathogenesis of asthma (Figure 8), suggesting that nornidulin antagonizes OVA-induced hypersecretion of mucin in an asthmatic mouse model with preserving inflammatory responses to defend against opportunistic infection. Regarding the persistence of Th2 inflammation, eosinophils play an important role in airway remodeling characterized by aberrant smooth muscle proliferation and fibroblast activation leading to impaired lung function.66 As TMEM16A is required for IL-13-induced cell proliferation,67 inhibition of TMEM16A by nornidulin may also alleviate airway remodeling caused by Th2 response and eosinophils. This requires further investigation on the effect of nornidulin on airway remodeling. This makes the compound attractive as it might reduce asthma symptoms resulted from mucus hypersecretion and airway remodeling without impairing defense mechanisms against pathogens such as parasites as seen in the adverse effects of corticosteroid. Therefore, the rate of respiratory infection in asthma treatment by nornidulin might be lower than that of other anti-inflammatory drugs. Additionally, we did not show that inhibition of TMEM16A by nornidulin is responsible for its action on mucus hypersecretion in vivo. To test this hypothesis, TMEM16A knockout mice may be necessary for further studies. In addition, the effects of nornidulin on mucus production require further investigation. Unfortunately, TMEM16A knockout mice can develop severe epithelial transport abnormalities, tracheomalacia, and death within a month of birth.33
TMEM16A is mainly expressed in inflamed airway epithelia and plays a key role in the hypersecretion of mucus. Inhibition of TMEM16A is an alternative way to treat asthma. In this study, we performed both in vitro and in vivo experiments to identify TMEM16A and its potential use in the suppression of mucus hypersecretion in an asthmatic mouse model. However, Calu-3 cells do not specialize in mucus production or secretion. The effect of nornidulin requires further investigation in mucus-producing cells such as rat tracheal epithelial (SPOC1) cells, humanized lung organoid models, or lung-on-A-chip models. Further studies are needed to delineate the effects of nornidulin on airway smooth muscle contraction, which is regulated by TMEM16A. To gain more knowledge, CRISPR-Cas9-mediated genome editing or site-directed mutagenesis of TMEM16A may be useful tools to know more about mechanisms of nornidulin on TMEM16A inhibition. For bench-to-bedside translation, the attention should be paid for drug metabolism and pharmacokinetic along with pharmacodynamic studies using both in vitro multi-organ chip and in vivo using mice or non-human primates before performing human study in clinical setting. In addition, undesirable adverse effects of nornidulin should be further taken into accounts.
Conclusion
In summary, nornidulin, a depsidone derivative from the soil-derived fungus Aspergillus unguis PSU-RSPG204, represents a novel TMEM16A inhibitor that holds therapeutic promise for the treatment of airway diseases associated with mucus hypersecretion, including asthma. Further research and development of nornidulin and its derivatives may provide a novel class of anti-asthmatic drugs.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Animal Care and Use Committee of the Faculty of Science, Mahidol University (Protocol No. MUSC62-028-492).
Data Sharing Statement
Data supporting the findings of this study are available from the corresponding authors.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Mahidol University (Basic Research Fund: fiscal year 2022), the NSTDA Chair Professor grant (Fourth Grant) of the Crown Property Bureau and the National Science and Technology Development Agency, the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (grant number B05F650041), and Tonkla Ramathibodi, Faculty of Medicine, Ramathibodi Hospital, Mahidol University.
Disclosure
The authors declare no conflict of interest.
References
1. Song P, Adeloye D, Salim H, et al. Global, regional, and national prevalence of asthma in 2019: a systematic analysis and modelling study. J Glob Health. 2022;12:04052. doi:10.7189/jogh.12.04052
2. Dharmage SC, Perret JL, Custovic A. Epidemiology of asthma in children and adults. Front Pediat. 2019;7:246. doi:10.3389/fped.2019.00246
3. Most recent national asthma data; 2023. Available from: https://www.cdc.gov/asthma/most_recent_national_asthma_data.htm.
4. Toskala E, Kennedy DW. Asthma risk factors. Nt Forum Allergy Rhinol. 2015;5:S11–S16.
5. Lajiness JA-O, Cook-Mills JM. Catching our breath: updates on the role of dendritic cell subsets in asthma. Advan Bio. 2023;7:2200296. doi:10.1002/adbi.202200296
6. Sinyor B, Concepcion Perez L. Pathophysiology of asthma; StatPearls; 2022.
7. Poddighe D, Mathias CB, Freyschmidt EJ, et al. Basophils are rapidly mobilized following initial aeroallergen encounter in naïve mice and provide a priming source of IL-4 in adaptive immune responses. J Biol Regul Homeost Agents. 2014;28(1):91–103.
8. Yoshimoto T, Bendelac A, Watson C, Hu-Li J, Paul WE. Role of NK1.1+ T cells in a TH2 response and in immunoglobulin E production. Science. 1995;270(5243):1845–1847. doi:10.1126/science.270.5243.1845
9. Rubin BK, Priftis KN, Schmidt HJ, Henke MO. Secretory hyperresponsiveness and pulmonary mucus hypersecretion. Chest. 2014;146(2):496–507. doi:10.1378/chest.13-2609
10. Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134(6):1019–1029. doi:10.1016/j.cell.2008.09.003
11. Caputo A, Caci E, Ferrera L, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322(5901):590–594. doi:10.1126/science.1163518
12. Yang YD, Cho H, Koo JY, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455(7217):1210–1215. doi:10.1038/nature07313
13. Rottgen TS, Nickerson AJ, Rajendran VM. Calcium-Activated Cl(-) Channel: insights on the molecular identity in epithelial tissues. Int J Mol Sci. 2018;19(5):1432. doi:10.3390/ijms19051432
14. Dam VS, Boedtkjer DM, Aalkjaer C, Matchkov V. The bestrophin- and TMEM16A-associated Ca(2+)- activated Cl(-) channels in vascular smooth muscles. Channels. 2014;8(4):361–369. doi:10.4161/chan.29531
15. Wang H, Zou L, Ma K, et al. Cell-specific mechanisms of TMEM16A Ca(2+)-activated chloride channel in cancer. Mol Cancer. 2017;16(1):152. doi:10.1186/s12943-017-0720-x
16. Matchkov VV, Boedtkjer DM, Aalkjaer C. The role of Ca(2+) activated Cl(-) channels in blood pressure control. Curr Opin Pharmacol. 2015;21:127–137. doi:10.1016/j.coph.2015.02.003
17. Zhang CH, Li Y, Zhao W, et al. The transmembrane protein 16A Ca(2+)-activated Cl- channel in airway smooth muscle contributes to airway hyperresponsiveness. Am J Respir Crit Care Med. 2013;187(4):374–381. doi:10.1164/rccm.201207-1303OC
18. Okuyama K, Yanamoto S. TMEM16A as a potential treatment target for head and neck cancer. J Exp Clin Cancer Res. 2022;41(1):196. doi:10.1186/s13046-022-02405-2
19. Huang F, Zhang H, Wu M, et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc Natl Acad Sci U S A. 2012;109(40):16354–16359. doi:10.1073/pnas.1214596109
20. Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286(3):2365–2374. doi:10.1074/jbc.M110.175109
21. Benedetto R, Cabrita I, Schreiber R, Kunzelmann K. TMEM16A is indispensable for basal mucus secretion in airways and intestine. FASEB J. 2019;33(3):4502–4512. doi:10.1096/fj.201801333RRR
22. Kondo M, Tsuji M, Hara K, et al. Chloride ion transport and overexpression of TMEM16A in a Guinea-pig asthma model. Clin Exp Allergy. 2017;47(6):795–804. doi:10.1111/cea.12887
23. Wang P, Zhao W, Sun J, et al. Inflammatory mediators mediate airway smooth muscle contraction through a G protein-coupled receptor-transmembrane protein 16A-voltage-dependent Ca(2+) channel axis and contribute to bronchial hyperresponsiveness in asthma. J Allergy Clin Immunol. 2018;141(4):1259–1268.e11. doi:10.1016/j.jaci.2017.05.053
24. Danielsson J, Kuforiji AS, Yocum GT, et al. Agonism of the TMEM16A calcium-activated chloride channel modulates airway smooth muscle tone. Am J Physiol Lung Cell Mol Physiol. 2020;318(2):L287–l295. doi:10.1152/ajplung.00552.2018
25. Centeio R, Ousingsawat J, Cabrita I, et al. Mucus release and airway constriction by TMEM16A may worsen pathology in inflammatory lung disease. Int J Mol Sci. 2021;22(15):7852. doi:10.3390/ijms22157852
26. Steinke JW, Borish L. Th2 cytokines and asthma. Interleukin-4: its role in the pathogenesis of asthma, and targeting it for asthma treatment with interleukin-4 receptor antagonists. Respir Res. 2001;2(2):66–70. doi:10.1186/rr40
27. Wang J, Haanes KA, Novak I. Purinergic regulation of CFTR and Ca(2+)-activated Cl(-) channels and K(+) channels in human pancreatic duct epithelium. Am J Physiol Cell Physiol. 2013;304(7):C673–84. doi:10.1152/ajpcell.00196.2012
28. Antonioli L, Blandizzi C, Pacher P, Hasko G. The purinergic system as a pharmacological target for the treatment of immune-mediated inflammatory diseases. Pharmacol Rev. 2019;71(3):345–382. doi:10.1124/pr.117.014878
29. Chavez J, Vargas MH, Martinez-Zuniga J, et al. Allergic sensitization increases the amount of extracellular ATP hydrolyzed by Guinea pig leukocytes. Purinergic Signal. 2019;15(1):69–76. doi:10.1007/s11302-019-09644-7
30. Gao ZG, Jacobson KA. Purinergic signaling in mast cell degranulation and asthma. Front Pharmacol. 2017;8(947). doi:10.3389/fphar.2017.00947
31. Jang Y, Oh U. Anoctamin 1 in secretory epithelia. Cell Calcium. 2014;55(6):355–361. doi:10.1016/j.ceca.2014.02.006
32. Ayon RJ, Hawn MB, Aoun J, et al. Molecular mechanism of TMEM16A regulation: role of CaMKII and PP1/PP2A. Am J Physiol Cell Physiol. 2019;317(6):C1093–C1106. doi:10.1152/ajpcell.00059.2018
33. Tian Y, Kongsuphol P, Hug M, et al. Calmodulin-dependent activation of the epithelial calcium-dependent chloride channel TMEM16A. FASEB J. 2011;25(3):1058–1068. doi:10.1096/fj.10-166884
34. Evans CM, Kim K, Tuvim MJ, Dickey BF. Mucus hypersecretion in asthma: causes and effects. Curr Opin Pulm Med. 2009;15(1):4–11. doi:10.1097/MCP.0b013e32831da8d3
35. Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod. 2016;79(3):629–661. doi:10.1021/acs.jnatprod.5b01055
36. Tsukada K, Shinki S, Kaneko A, et al. Synthetic biology based construction of biological activity-related library of fungal decalin-containing diterpenoid pyrones. Nat Commun. 2020;11(1):1830. doi:10.1038/s41467-020-15664-4
37. Ibrahim SRM, Mohamed GA, Al Haidari RA, El-Kholy AA, Zayed MF, Khayat MT. Biologically active fungal depsidones: chemistry, biosynthesis, structural characterization, and bioactivities. Fitoterapia. 2018;129:317–365. doi:10.1016/j.fitote.2018.04.012
38. Bernardini S, Tiezzi A, Laghezza Masci V, Ovidi E. Natural products for human health: an historical overview of the drug discovery approaches. Nat Prod Res. 2018;32(16):1926–1950. doi:10.1080/14786419.2017.1356838
39. Shamshuddin NSS, Mohd Zohdi R. Gelam honey attenuates ovalbumin-induced airway inflammation in a mice model of allergic asthma. J Tradit Complement Med. 2018;8(1):39–45. doi:10.1016/j.jtcme.2016.08.009
40. Phainuphong P, Rukachaisirikul V, Phongpaichit S, et al. Depsides and depsidones from the soil-derived fungus Aspergillus unguis PSU-RSPG204. Tetrahedron. 2018;74(39):5691–5699. doi:10.1016/j.tet.2018.07.059
41. Pongkorpsakol P, Yimnual C, Chatsudthipong V, Rukachaisirikul V, Muanprasat C. Cellular mechanisms underlying the inhibitory effect of flufenamic acid on chloride secretion in human intestinal epithelial cells. J Pharmacol Sci. 2017;134(2):93–100. doi:10.1016/j.jphs.2017.05.009
42. Pongkorpsakol P, Pathomthongtaweechai N, Srimanote P, Soodvilai S, Chatsudthipong V, Muanprasat C. Inhibition of cAMP-activated intestinal chloride secretion by diclofenac: cellular mechanism and potential application in cholera. PLoS Negl Trop Dis. 2014;8(9):e3119. doi:10.1371/journal.pntd.0003119
43. Muanprasat C, Sirianant L, Soodvilai S, Chokchaisiri R, Suksamrarn A, Chatsudthipong V. Novel action of the chalcone isoliquiritigenin as a cystic fibrosis transmembrane conductance regulator (CFTR) inhibitor: potential therapy for cholera and polycystic kidney disease. J Pharmacol Sci. 2012;118(1):82–91. doi:10.1254/jphs.11153FP
44. Pongkorpsakol P, Buasakdi C, Chantivas T, Chatsudthipong V, Muanprasat C. An agonist of a zinc-sensing receptor GPR39 enhances tight junction assembly in intestinal epithelial cells via an AMPK-dependent mechanism. Eur J Pharmacol. 2019;842:306–313. doi:10.1016/j.ejphar.2018.10.038
45. Yimnual C, Satitsri S, Ningsih BNS, Rukachaisirikul V, Muanprasat C. A fungus-derived purpactin A as an inhibitor of TMEM16A chloride channels and mucin secretion in airway epithelial cells. Biomed Pharmacother. 2021;139:111583. doi:10.1016/j.biopha.2021.111583
46. Meyerholz DK, Griffin MA, Castilow EM, Varga SM. Comparison of histochemical methods for murine eosinophil detection in an RSV vaccine-enhanced inflammation model. Toxicol Pathol. 2009;37(2):249–255. doi:10.1177/0192623308329342
47. Cabrita I, Benedetto R, Wanitchakool P, et al. TMEM16A mediates mucus production in human airway epithelial cells. Am J Respir Cell Mol Biol. 2021;64(1):50–58. doi:10.1165/rcmb.2019-0442OC
48. Cabrita I, Benedetto R, Schreiber R, Kunzelmann K. Niclosamide repurposed for the treatment of inflammatory airway disease. JCI Insight. 2019;4(15). doi:10.1172/jci.insight.128414
49. Genovese M, Borrelli A, Venturini A, et al. TRPV4 and purinergic receptor signalling pathways are separately linked in airway epithelia to CFTR and TMEM16A chloride channels. J Physiol. 2019;597(24):5859–5878. doi:10.1113/jp278784
50. Scudieri P, Caci E, Bruno S, et al. Association of TMEM16A chloride channel overexpression with airway goblet cell metaplasia. J Physiol. 2012;590(23):6141–6155. doi:10.1113/jphysiol.2012.240838
51. Ebrahim HY, Elsayed HE, Mohyeldin MM, et al. Norstictic acid inhibits breast cancer cell proliferation, migration, invasion, and in vivo invasive growth through targeting C-met. Phytother Res. 2016;30(4):557–566. doi:10.1002/ptr.5551
52. Hao A, Guo S, Shi S, et al. Emerging modulators of TMEM16A and their therapeutic potential. J Membr Biol. 2021;254(4):353–365. doi:10.1007/s00232-021-00188-9
53. Hawksworth DL, Lucking R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol Spectr. 2017;5(4). doi:10.1128/microbiolspec.FUNK-0052-2016
54. Blackwell M. The fungi: 1,2, 3. 5.1 million species? Am J Bot. 2011;98(3):426–438. doi:10.3732/ajb.1000298
55. Pham JV, Yilma MA, Feliz A, et al. A review of the microbial production of bioactive natural products and biologics. Front Microbiol. 2019;10:1404. doi:10.3389/fmicb.2019.01404
56. Keller NP. Fungal secondary metabolism: regulation, function and drug discovery. Nat Rev Microbiol. 2019;17(3):167–180. doi:10.1038/s41579-018-0121-1
57. Nielsen JC, Nielsen J. Development of fungal cell factories for the production of secondary metabolites: linking genomics and metabolism. Synth Syst Biotechnol. 2017;2(1):5–12. doi:10.1016/j.synbio.2017.02.002
58. Lenzi J, Costa TM, Alberton MD, Goulart JAG, Tavares LBB. Medicinal fungi: a source of antiparasitic secondary metabolites. Appl Microbiol Biotechnol. 2018;102(14):5791–5810. doi:10.1007/s00253-018-9048-8
59. Muangnil P, Satitsri S, Tadpetch K, et al. A fungal metabolite zearalenone as a CFTR inhibitor and potential therapy of secretory diarrheas. Biochem Pharmacol. 2018;150:293–304. doi:10.1016/j.bcp.2018.02.024
60. Braun OO, Lu D, Aroonsakool N, Insel PA. Uridine triphosphate (UTP) induces profibrotic responses in cardiac fibroblasts by activation of P2Y2 receptors. J Mol Cell Cardiol. 2010;49(3):362–369. doi:10.1016/j.yjmcc.2010.05.001
61. Schafer R, Sedehizade F, Welte T, Reiser G. ATP- and UTP-activated P2Y receptors differently regulate proliferation of human lung epithelial tumor cells. Am J Physiol Lung Cell Mol Physiol. 2003;285(2):L376–85. doi:10.1152/ajplung.00447.2002
62. Sun Y, Birnbaumer L, Singh BB. TRPC1 regulates calcium-activated chloride channels in salivary gland cells. J Cell Physiol. 2015;230(11):2848–2856. doi:10.1002/jcp.25017
63. Tian Y, Schreiber R, Wanitchakool P, et al. Control of TMEM16A by INO-4995 and other inositolphosphates. Br J Pharmacol. 2013;168(1):253–265. doi:10.1111/j.1476-5381.2012.02193.x
64. Hawn MB, Akin E, Hartzell HC, Greenwood IA, Leblanc N. Molecular mechanisms of activation and regulation of ANO1-Encoded Ca(2+)-Activated Cl(-) channels. Channels. 2021;15(1):569–603. doi:10.1080/19336950.2021.1975411
65. Bonser LR, Zlock L, Finkbeiner W, Erle DJ. Epithelial tethering of MUC5AC-rich mucus impairs mucociliary transport in asthma. J Clin Invest. 2016;126(6):2367–2371. doi:10.1172/JCI84910
66. Hough KP, Curtiss ML, Blain TJ, et al. Airway Remodeling in Asthma. Front Med. 2020;7:191. doi:10.3389/fmed.2020.00191
67. Bai W, Liu M, Xiao Q. The diverse roles of TMEM16A Ca(2+)-activated Cl(-) channels in inflammation. J Adv Res. 2021;33:53–68. doi:10.1016/j.jare.2021.01.013
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.