")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Discovery of 3-n-butyl-2,3-dihydro-1H-isoindol-1-one as a potential anti-ischemic stroke agent
Authors Lan Z, Xu X, Xu W, Li J, Liang Z, Zhang X, Lei M, Zhao C
Received 16 March 2015
Accepted for publication 30 April 2015
Published 30 June 2015 Volume 2015:9 Pages 3377—3391
DOI https://doi.org/10.2147/DDDT.S84731
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Zujian Lan, Xiaoyu Xu, Wenkai Xu, Jin Li, Zengrong Liang, Xuefei Zhang, Ming Lei, Chunshun Zhao
School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, People’s Republic of China
Abstract: To develop novel anti-ischemic stroke agents with better therapeutic efficacy and bioavailability, we designed and synthesized a series of 3-alkyl-2,3-dihydro-1H-isoindol-1-ones compounds (3a–i) derivatives, one of which (3d) exhibited the strongest inhibitory activity for the adenosine diphosphate-induced and arachidonic acid-induced platelet aggregation. This activity is superior to that of 3-n-butylphthalide and comparable with aspirin and edaravone. Meanwhile, 3d not only exhibited a potent activity in scavenging free radicals and improving the survival of HT22 cells against the reactive oxygen species-mediated cytotoxicity in vitro but also significantly attenuated the ischemia/reperfusion-induced oxidative stress in ischemic rat brains. Results from transient middle cerebral artery occlusion and permanent middle cerebral artery occlusion model, indicated that 3d could significantly reduce infarct size, improve neurobehavioral deficits, and prominently decrease attenuation of cerebral damage. Most importantly, 3d possessed a very high absolute bioavailability and was rapidly distributed in brain tissue to keep high plasma drug concentration for the treatment of ischemic strokes. In conclusion, our findings suggest that 3-alkyl-2,3-dihydro-1H-isoindol-1-ones, a novel series of compounds, might be candidate drugs for the treatment of acute ischemic strokes, and 3d may be a promising therapeutic agent for the primary and secondary prevention of ischemic stroke.
Keywords: stroke, platelet aggregation, ischemia/reperfusion, middle cerebral artery occlusion, 3-alkyl-2,3-dihydro-1H-isoindol-1-ones
Introduction
It has been reported that the ischemic stroke resulting from occlusion of arteries in the brain accounts for more than 80% of all strokes and is considered as one of the major causes of death and adult neurological disability in many countries.1,2 At present, the mortality of stroke patients is still increasing each year.3 Several evidences show that the platelet aggregation-related thrombosis is a key step in the pathophysiology of acute ischemic strokes.4,5 Therefore, the development of new drugs with antiplatelet aggregation and antithrombotic activities will be of great significance to the treatment of ischemic strokes. Many remarkable advances in the study of the mechanisms of ischemic brain injury have been made, and there are many pharmacological approaches to protect from injury caused by ischemic strokes.6–8 Unfortunately, some drugs are not ideal anti-ischemic agents because of unsatisfactory efficacy; only intravenous (iv) administration of the serine protease tissue-type plasminogen activator (t-PA) has been proven to be effective, and t-PA is the only drug approved by the US Food and Drug Administration for the treatment of acute ischemic strokes clinically. What is worse, about 90% of all stroke patients are excluded from t-PA treatment due to numerous contraindications and its extremely narrow therapeutic window.9,10
Previous studies have demonstrated that mechanisms leading to the development of ischemic damage include apoptosis, calcium ion [Ca2+] overload, necrosis, inflammation, immune modulation, and oxidative stress;10–13 specially oxidative stress because formation of reactive oxygen species (ROS) plays an important role in neuronal damage during cerebral ischemia. Overwhelming evidence shows a transient reduction of brain artery blood flow often initiates a large amount of free radicals after ischemia/reperfusion (I/R) injury, such as O2−, HO2, H2O2, and OH, which have been implicated in the pathogenesis of I/R injury because free radicals induce lipid peroxidation, protein oxidation, and DNA damage.14,15 Many methods have been adopted to reduce oxidative stress through formation of ROS and to block the ROS-mediated neuronal injury.16,17 Hence it will be particularly important for us to find novel agents with high activity in scavenging free radicals.
The 3-n-butylphthalide (NBP) is a new drug for treatment of ischemic strokes clinically, which was approved by the State Food and Drug Administration in People’s Republic of China at the end of 2002. Many clinical studies have proven that NBP is a potentially beneficial drug for treatment of ischemic strokes. It can inhibit platelet aggregation, reduce ischemia-induced oxidative damage, regulate energy metabolism, improve microcirculation, and reduce brain infarct size.6,18–20 However, the efficacy of NBP is limited because of its high hydrophobicity and low bioavailability,21–25 and thus its clinical use for patients suffering serious ischemic strokes is restricted. In addition, to improve its efficacy, it has to be simultaneously administered with antiplatelet and antioxidant drugs.26 Hence, the development of new anti-ischemic stroke drugs suitable for iv injection with remarkable antiplatelet aggregation and antioxidant activities for acute ischemic stroke treatment will be of great significance.
Previous studies have suggested that a class of compounds, isoindoline derivatives, exhibit a good activity in treating the central nervous systemic disease.27–34 Because the structure of isoindoline derivatives is very similar to that of NBP and its amide bond is more stable than ester bond of NBP in the gastrointestinal tract, 3-alkyl-2,3-dihydro-1H-isoindol-1-ones may exhibit better anti-ischemic stroke activity than NBP. Therefore, we designed and synthesized a novel class of compounds 3-alkyl-2,3-dihydro-1H-isoindol-1-ones by replacing the oxygen atom at the 2-position of phthalide with nitrogen and introducing various lengths of straight/branch chain alkyl into 3-position of phthalide. The resulting compound 3d (Figure 1) displayed potent antiplatelet aggregation, antioxidation activity, and anti-ischemic stroke activity, which was comparable with NBP and edaravone (Eda). More importantly, 3d displayed higher bioavailability than NBP, which will be beneficial to the clinical treatment of acute ischemic strokes.
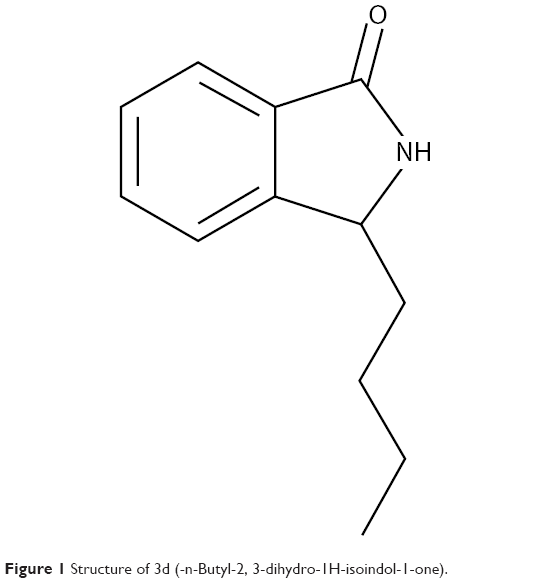 | Figure 1 Structure of 3d (-n-Butyl-2, 3-dihydro-1H-isoindol-1-one). |
Materials and methods
Chemistry
1H nuclear magnetic resonance (NMR) and 13C NMR spectral data were obtained from a Bruker Avance III 400 MHz spectrometer at 300 K using Tetramethylsilane as an internal standard. MS spectra were recorded on an Agi1lent 6120 mass spectrometer. The purities of the compounds were characterized by high-performance liquid chromatography (HPLC) analysis (L-2000 HPLC system consisting of 5110 pumps and 5430 diode array detector). The target compounds 3a–i with a purity of >95% were used for subsequent experiments.
Sprague Dawley rats (250–260 g, 350–400 g) and Kunming (KM) mice (18–22 g) were supplied by the Laboratory Animal Center of Sun Yat-sen University (Guangzhou, People’s Republic of China). All experimental protocols were approved and supervised by the Institutional Animal Care and Use Committee of Sun Yat-sen University (Guangzhou, People’s Republic of China).
Preparation of 3a–i
A Grignard reagent (3 mmol) was added to an ice bath-chilled solution of phthalimide (1 mmol) in CH2Cl2 (10 mL) under nitrogen atmosphere. The reaction mixture was stirred at the same temperature for 3 hours. Subsequently the reaction was quenched by a saturated aqueous solution of NH4Cl (6 mL). The resulting mixture was extracted with CH2Cl2 (3×10 mL), and the combined organic phases were washed with brine, then dried over anhydrous Na2SO4, filtered, and concentrated. The crude mixture was purified by a short silica gel column filtration (EtOAc-petroleum ether =2:1 v/v) to obtain 2a–i. Triethylsilane (10.0 mmol) and trifluoroboron etherate (3.0 mmol) were added successively to a solution of 3a–i in dry CH2Cl2 (10 mL) at -15°C under nitrogen atmosphere. The mixture was stirred at room temperature overnight. A saturated aqueous solution of NaHCO3 (3 mL) was added and followed by CH2Cl2 extraction (3×6 mL). The combined organic phases were washed with brine and then dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (EtOAc-petroleum ether =2:1 v/v) to give the title compounds.
(±)-3-Ethyl-2,3-dihydro-1H-isoindol-1-one (3a)
The title compound was obtained as white crystal, 1H NMR (400 MHz, CDCl3) δ/ppm: 7.85 (d, J=7.5 Hz, 1H, Ar), 7.56 (td, J=7.5, 1.2 Hz, 1H, Ar), 7.50–7.41 (m, 3H, Ar, NH), 4.61 (dd, J=7.0, 4.6 Hz, 1H, NCH), 2.07–1.98 (m, 1H, CH2), 1.84–1.65 (m, 1H, CH2), 0.97 (t, J=7.4 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ/ppm: 171.52(C), 147.52(C), 132.26(C), 131.69(CH), 127.99(CH), 123.66(CH), 122.38(CH), 58.15(CH), 27.35(CH2), 9.46(CH3). Mass spectrum (MS) (electronic spray ion [ESI], m/z): 162 (M + H+, 100), 323 [(2M + H)+, 25].
(±)-3-n-Propyl-2,3-dihydro-1H-isoindol-1-one (3b)
The title compound was obtained as white crystal, 1H NMR (400 MHz, CDCl3) δ/ppm: 7.85 (d, J=7.4 Hz, 1H, Ar), 7.56 (td, J=7.5, 1.2 Hz, 1H, Ar), 7.49–7.39 (m, 2H, Ar), 7.19 (s, 1H, NH), 4.63 (dd, J=7.8, 4.5 Hz, 1H, NCH), 1.98–1.89 (m, 1H, CH2CH2CH3), 1.69–1.59 (m, 1H, CH2CH2CH3), 1.53–1.38 (m, 2H, CH2CH3), 0.98 (t, J=7.3 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ/ppm: 171.33(C), 147.85(C), 131.97(C), 131.71(CH), 127.99(CH), 123.74(CH), 122.39(CH), 56.98(CH), 36.76(CH2), 18.97(CH2), 14.01(CH3). MS (ESI, m/z): 176 (M + H+, 100), 351 [(2M + H)+, 23].
(±)-3-Isopropyl-2,3-dihydro-1H-isoindol-1-one (3c)
The title compound was obtained as white crystal, 1H NMR (400 MHz, CDCl3) δ/ppm: 7.86 (d, J=7.5 Hz, 1H, Ar), 7.57 (td, J=7.5, 1.0 Hz, 1H, Ar), 7.46 (dd, J=15.2, 7.5 Hz, 2H, Ar), 7.06 (s, 1H, NH), 4.57 (d, J=3.5 Hz, 1H, NCH), 2.33–2.21 (m, 1H, CH(CH3)2), 1.10 (d, J=6.9 Hz, 3H, CH3), 0.74 (d, J=6.8 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ/ppm: 171.73(C), 146.65(C), 132.63(C), 131.61(CH), 127.98(CH), 123.68(CH), 122.66(CH), 62.40(CH), 31.76(CH), 19.56(CH3), 15.91(CH3). MS (ESI, m/z): 176 (M + H+, 100), 351 [(2M + H)+, 45].
(±)-3-n-Butyl-2,3-dihydro-1H-isoindol-1-one (3d)
The title compound was obtained as white crystal, 1H NMR (400 MHz, CDCl3) δ/ppm: 7.85 (d, J=7.5 Hz, 1H, Ar), 7.56 (td, J=7.5, 1.1 Hz, 1H, Ar), 7.45 (dd, J=15.3, 7.5 Hz, 2H, Ar), 7.03 (s, 1H, NH), 4.62 (dd, J=7.6, 4.5 Hz, 1H, NCH), 2.00–1.92 (m, 1H, CH2Pr), 1.70–1.60 (m, 1H, CH2Pr), 1.46–1.28 (m, 4H, 2CH2), 0.90 (t, J=7.1 Hz, 3H, CH3). 13C NMR (101 MHz, DMSO) δ/ppm: 169.34(C), 147.78(C), 132.31(C), 131.36(CH), 127.74(CH), 122.83(CH), 122.64(CH), 55.89(CH), 33.70(CH2), 26.75(CH2), 22.02(CH2), 13.77(CH3). MS (ESI, m/z): 190 (M + H+, 100), 379 [(2M + H)+, 20].
(±)-3-Isobutyl-2,3-dihydro-1H-isoindol-1-one (3e)
The title compound was obtained as white crystal, 1H NMR (400 MHz, CDCl3) δ/ppm: 7.85 (d, J=7.5 Hz, 1H, Ar), 7.56 (td, J=7.5, 1.1 Hz, 1H, Ar), 7.45 (dd, J=17.9, 7.5 Hz, 2H, Ar), 7.19 (s, 1H, NH), 4.66 (dd, J=9.5, 4.2 Hz, 1H, NCH), 1.92–1.82 (m, 1H, CH(CH3)2), 1.78–1.71 (m, 1H, CH2CH(CH3)2), 1.56–1.46 (m, 1H, CH2CH(CH3)2), 1.07 (d, J=6.5 Hz, 3H, CH3), 1.00 (d, J=6.6 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ/ppm: 171.09(C), 148.30(C), 131.78(C), 131.69(CH), 127.97(CH), 123.82(CH), 122.42(CH), 55.31(CH), 44.19(CH2), 25.83(CH), 23.47(CH3), 22.06(CH3). MS (ESI, m/z): 190 (M + H+, 100), 191 [(M + 2H)+, 12], 379 [(2M + H)+, 24].
(±)-3-n-Pentyl-2,3-dihydro-1H-isoindol-1-one (3f)
The title compound was obtained as white crystal, 1H NMR (400 MHz, CDCl3) δ/ppm: 7.86 (d, J=7.5 Hz, 1H, Ar), 7.57 (td, J=7.5, 1.1 Hz, 1H, Ar), 7.46 (dd, J=16.5, 7.8 Hz, 2H, Ar), 7.01 (s, 1H, NH), 4.63 (dd, J=7.7, 4.5 Hz, 1H, NCH), 2.02–1.91 (m, 1H, CH2), 1.70–1.60 (m, 1H, CH2), 1.52–1.44 (m, 1H, CH2), 1.38–1.27 (m, 5H, CH2), 0.89 (t, J=6.9 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ/ppm: 171.39(C), 147.90(C), 132.14(C), 131.65(CH), 127.94(CH), 123.68(CH), 122.39(CH), 57.15(CH), 34.55(CH2), 31.69(CH2), 25.14(CH2), 22.41(CH2), 13.92(CH3). MS (ESI, m/z): 204 (M + H+, 100), 205 [(M + 2H)+, 25], 407 [(2M + H)+, 31].
(±)-3-n-Hexyl-2,3-dihydro-1H-isoindol-1-one (3g)
The title compound was obtained as white crystal, 1H NMR (400 MHz, CDCl3) δ/ppm: 7.86 (d, J=7.5 Hz, 1H, Ar), 7.57 (td, J=7.5, 1.0 Hz, 1H, Ar), 7.46 (dd, J=16.6, 7.5 Hz, 2H, Ar), 6.90 (s, 1H, NH), 4.62 (dd, J=7.7, 4.5 Hz, 1H, NCH), 1.98–1.93 (m, 1H, CH2), 1.71–1.60 (m, 1H, CH2), 1.53–1.40 (m, 1H, CH2), 1.36–1.27 (m, 7H, CH2), 0.88 (t, J=6.9 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ/ppm: 171.22(C), 147.85(C), 132.03(C), 131.68(CH), 127.97(CH), 123.71(CH), 122.38(CH), 57.07(CH), 34.60(CH2), 31.57(CH2), 29.18(CH2), 25.46(CH2), 22.51(CH2), 13.99(CH3). MS (ESI, m/z): 218 (M + H+, 100), 219 [(M + 2H)+, 19], 435 [(2M + H)+, 27].
(±)-3-n-Butyl-2-methyl-2,3-dihydro-1H-isoindol-1-one (3h)
The title compound was obtained as light yellow oil, 1H NMR (400 MHz, CDCl3) δ/ppm: 7.82 (d, J=7.5 Hz, 1H, Ar), 7.52 (td, J=7.4, 1.1 Hz, 1H, Ar), 7.42 (dd, J=14.9, 7.5 Hz, 2H, Ar), 4.50–4.46 (m, 1H, NCH), 3.10 (s, 3H, NCH3), 2.05–1.86 (m, 2H, CH2), 1.25 (dd, J=14.8, 7.4 Hz, 2H, CH2), 1.09–0.93 (m, 2H, CH2), 0.81 (t, J=7.3 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ/ppm: 168.63(C), 145.11(C), 132.72(C), 131.18(CH), 127.96(CH), 123.39(CH), 121.88(CH), 61.50(CH), 30.26(CH2), 27.20(CH3), 24.44(CH2), 22.57(CH2), 13.82(CH3). MS (ESI, m/z): 204 (M + H+, 100), 205 [(M + 2H)+, 14], 408 [(2M + 2H)+, 5].
(±)-3-n-Butyl-2-ethyl-2,3-dihydro-1H-isoindol-1-one (3i)
The title compound was obtained as light yellow oil,1H NMR (400 MHz, CDCl3) δ/ppm: 7.83 (d, J=7.5 Hz, 1H, Ar), 7.52 (td, J=7.4, 1.2 Hz, 1H, Ar), 7.46–7.39 (m, 2H, Ar), 4.63 (dd, J=5.2, 3.5 Hz, 1H, NCH), 4.04 (dd, J=14.2, 7.2 Hz, 1H, NCH2), 3.19 (dd, J=14.1, 7.1 Hz, 1H, NCH2), 2.02–1.90 (m, 2H, CH2), 1.29–1.19 (m, 6H, CH3, CH2), 1.08–1.00 (m, 1H, CH2), 0.80 (d, J=7.4 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ/ppm: 168.23(C), 145.24(C), 132.96(C), 131.12(CH), 127.93(CH), 123.44(CH), 121.97(CH), 58.74(CH), 34.57(CH2), 30.23(CH2), 24.43(CH2), 22.63(CH2), 13.85(CH3), 13.64(CH3). MS (ESI, m/z): 218 (M + H+, 100), 435 [(2M + H)+, 37], 436 [(2M + 2H)+, 12].
Effect against platelet aggregation in vitro
Blood was collected from the Sprague Dawley rat abdominal aorta (350–400 g, n=60) and mixed with 3.8% sodium citrate (9:1, v/v). Then platelet-rich plasma (PRP) was separated by centrifugation at 110× g for 7 minutes at 25°C, and the residue was further centrifuged at 960× g for 20 minutes to collect platelet-poor plasma. We then measured the effects of individual compounds against platelet aggregation using Born’s method35 in vitro using a two-channel aggregometer (Chrono-log 700) within 2 hours. Briefly, rat PRP samples (250 μL) were preincubated in triplicate with vehicle dimethyl sulfoxide (DMSO), with the individual compounds (2.5 μL) and positive control drugs (aspirin [ASP], NBP, and Eda) at the same concentrations (1.02, 1.28, 1.60, 2.00, and 2.50 mM) for 5 minutes at 37°C, and then adenosine diphosphate (ADP) (10 μM; Sigma-Aldrich, St Louis, MO, USA) or arachidonic acid (AA) (0.8 mM; Sigma-Aldrich) induced platelet aggregation in individual PRP samples, followed by recording the maximal platelet aggregation within 5 minutes. The inhibition rate of individual drugs on platelet aggregation was determined according to the following formula: inhibition rate (%) =100% - the maximal platelet aggregation of compounds ×100%/the maximal platelet aggregation of DMSO (the same rat PRP), and then a dose achieving 50% inhibition of platelet aggregation (IC50) of individual compounds was calculated.
Model of carrageenan-induced thrombosis
KM mice (23–25 g) were randomly divided into seven groups (n=10 per group): 1) sham surgery group, 2) model group (50% propanediol), 3) 3d (20 mg/kg) group, 4) 3d (50 mg/kg) group, 5) NBP (50 mg/kg) group, 6) Eda (3.0 mg/kg) group, 7) ASP (50 mg/kg) group. Groups 3-7 were treated by gavage with the indicated dose of the compound in 50% propanediol daily for 2 consecutive days. Subsequently, the rats were injected by hypodermic injection of 0.2% carrageenan, as described previously.36 The black tail occurrence and black tail length of mice were, respectively, detected at 24 and 48 hours after carrageenan injection.
Transient middle cerebral artery occlusion
Male Sprague Dawley rats (250-260 g) were randomly divided into seven groups (n=6 per group): 1) sham-operation group, 2) middle cerebral artery occlusion (MCAO) model group (oil), 3) I/R + 3d (10 mg/kg) group, 4) I/R + 3d (20 mg/kg) group, 5) I/R + 3d (50 mg/kg) group, 6) I/R + NBP (20 mg/kg) group, and 7) I/R + Eda (3.0 mg/kg) group. Then rats were anesthetized by intraperitoneal (ip) injection of 10% chloral hydrate (300 mg/kg, ip) and underwent an MCAO surgery, as described previously, with few modifications. In brief, after a midline neck skin incision was made, the right common carotid artery, internal carotid artery (ICA), and external carotid artery of individual rats were surgically exposed free from surrounding tissue and nerves, and a 4-0 monofilament nylon suture (Beijing Sunbio Biotech, Beijing, People’s Republic of China), with its tips coated with poly-L-lysine, was introduced into the ICA through a small incision on the stump of the common carotid artery to occlude the origin of the middle cerebral artery (MCA). The suture was inserted into the ICA for 20–22 mm until a light obstruction was felt. After 2 hours of occlusion of MCA, reperfusion was restored with the suture withdrawn, and the warm light was used to maintain the rat’s core body temperature at 37°C±0.5°C during the surgical procedure.
Permanent middle cerebral artery occlusion
Male Sprague Dawley rats (250-260 g) were obtained from the Laboratory Animal Center of Sun Yat-sen University (Guangzhou, People’s Republic of China) and were housed for at least 1 week in the holding area before the start of experiments. Rats were randomly divided into five groups (n=9 per group): 1) sham-operation group, 2) permanent MCAO (pMCAO) model group (50% propanediol), 3) 3d (50 mg/kg) group, 4) NBP (50 mg/kg) group, and 5) Eda (3.0 mg/kg) group. The rest of the procedure was the same as transient MCAO (tMCAO) except that reperfusion was not restored.
Neurobehavioral testing
After MCAO surgery, neurological function of individual rats was assessed at 24 hours of reperfusion. The neurological deficits were graded on a 5-point scale by Longa’s method37 in a blinded manner: 0, no neurological deficit; 1, failure to extend the forepaw on lifting the animal by the tail; 2, circling to the left side; 3, falling to the contralateral side; and 4, inability to walk and absence of spontaneous activity.
Measurement of infarct size
At 24 hours of reperfusion, the animals were anesthetized with 10% chloral hydrate (300 mg/kg, ip) and killed, and their brains were then rapidly removed and sliced into five 2 mm thick slices. The brain sections were immediately immersed in 2% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich) for 30 minutes at 37°C, followed by fixing in 4% paraformaldehyde. The infarct area was white, whereas the live brain tissue was red. The white areas and the total area in individual sections were measured and analyzed using Image-Pro Plus, and infarct size was expressed as the ratio of the volume of the white area over that of the total brain area.
Histopathological assessment
At 24 hours postreperfusion, the rats (n=3 per group) were anesthetized with 10% chloral hydrate (300 mg/kg, ip), followed by perfusion with 70 mL 4% paraformaldehyde, and 100 mL normal saline until the liver was white. The infarcted tissue was separated and immersed into 4% paraformaldehyde, then the brain tissues were embedded in paraffin for hematoxylin and eosin (H&E) stain at 5 μm, and the cerebral cortex area was examined by a light microscope.
The activity of radical scavenging assay in vitro
The free radical scavenging activities of 3d, NBP, and Eda were determined against H2O2 against HT22 cells, which were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma-Aldrich) supplemented with 10% new-born calf serum (Life Technologies, Gibco, USA) at 37°C in a 5% CO2 atmosphere. Murine HT22 cells (20,000 cells/well) were cultured in 96-well plates for 24 hours during the exponential phase of growth. After that, the cells were cultured in quadruplicate with different concentrations (10-100 μM) of 3d, NBP, and Eda for 2 hours and were exposed to 500 μM H2O2 for 1 hour, which was then replaced with fresh medium. Finally, the cells were incubated for 12 hours, and cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
Evaluation of antioxidant stress activity in vivo
At 24 hours postreperfusion, all rats (n=10 per group) were anesthetized with 10% chloral hydrate (300 mg/kg, ip) and immediately sacrificed. The total brain of each rat was rapidly removed, rinsed with 0.9% cold saline and frozen for 10 minutes in a refrigerator at -4°C, and then the ischemic core and ischemic penumbra were separated according to previous reports and stored at -70°C.38 After protein concentrations of individual brain samples were quantified, glutathione peroxidase (GSH-Px), total superoxide dismutase (SOD), and nitric oxide (NO) activities and the levels of malondialdehyde (MDA) in brain tissue were determined using analysis kits (Jiancheng Institute of Biotechnology, Nanjing, People’s Republic of China) based on the manufacturer’s instruction.
Study on pharmacokinetics and biodistribution
Wistar rats (250-260 g) with half males and half females (1:1; male:female) were obtained from the Laboratory Animal Center of Sun Yat-sen University (Guangzhou, People’s Republic of China) and divided into two groups (n=6 per group); 10 mg/kg iv administration and 10 mg/kg orally (po) administration) of compound 3d, respectively. After administration, 0.5 mL blood samples were collected in centrifuge tubes containing heparin at 5, 10, 15, 20, 30, 45, 60, 90, 120, 180, 240, 360, 480, and 600 minutes and then centrifuged at 2,650× g for 10 minutes. 3d concentration was measured by liquid–liquid extraction; the addition of 50 μL internal standard n-butylbenzoic acid and 50 μL 40% acetonitrile to 100 μL plasma sample with 800 μL ethyl acetate and dichloromethane (v/v=4:1), and then 500 μL of supernatant was dried at 40°C under a stream of nitrogen gas after vortexed for 5 minutes. The residue was reconstituted into 200 μL 40% acetonitrile. After centrifuging at 13,000× g for 10 minutes, the supernatants (10 μL) were used for HPLC analysis (Hitachi Ltd., Tokyo, Japan). Pharmacokinetic parameters of 10 mg/kg iv administration and 10 mg/kg po administration were calculated according to the compartment model using the WinNonlin software. The po absolute bioavailability = area under the curve (AUC)po × plasma drug concentration of intravenous injection (Civ)/AUCiv × Cpo ×100%.
The KM mice (18–22 g) were divided into three groups randomly (n=6 per group), and individual mice were treated with an iv dose (10 mg/kg) of compound 3d. Each group of mice was sacrificed at 15, 45, and 90 minutes, respectively, after iv injection. Then, heart, liver, spleen, lung, kidney, and brain tissue were collected, rinsed in cold saline, and weighed. All tissue samples were homogenized in saline in an ice water bath (tissue:water=1:9, w/v). The tissue homogenate was subjected to the plasma processing method to measure the concentration of 3d.
Results and discussion
Chemistry
Our method for the synthesis of target compounds 3a–i, a flexible approach to 3-alkyl-2,3-dihydro-1H-isoindol-1-ones via the reductive-alkylation procedure,39 is displayed in Figure 2. An ice-bath chilled solution of phthalimide (10 mmol) in CH2Cl2 was subjected to a reaction with the Grignard reagents for 3 hours under ice-bath temperature to generate hydroxyl compounds 2a–i, which had a high yield of 97%. These compounds were converted to 3a–i which resulted in a 45%-69% yield via the reductive-alkylation procedure by reaction with triethylsilane and trifluoroboron etherate at -15°C under N2 atmosphere. The crude product was subjected to column chromatography purification on silica gel to get the pure target products, followed by MS, 1H NMR, 13C NMR, and HPLC to identify their structures. Compounds 3a–i in a purity of >95% were used for subsequent experiments.
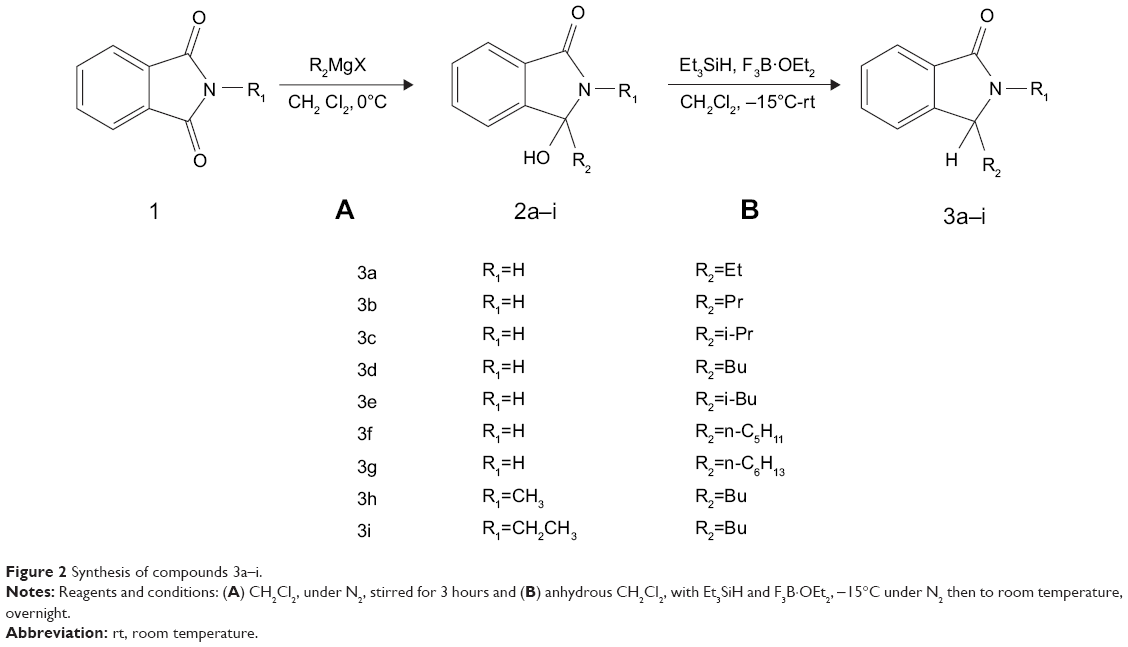 | Figure 2 Synthesis of compounds 3a–i. |
Pharmacology: study on antiplatelet aggregation effect in vitro
Unfortunately, the underlying mechanism of NBP for stroke remained unclear. Little was known about the mechanism of antiplatelet activity of NBP and 3-n-butyl-2,3-dihydro-1H-isoindol-1-one. The probable mechanism was not only related to AA-thromboxane A2 cascade but also involving NO-cyclic guanosine monophosphate signal path. Furthermore, NBP acted as an antiplatelet agent by disturbing the metabolic process of AA and improving cyclic adenosine monophosphate level.40–42
The antiplatelet aggregation activities of individual compounds in vitro were evaluated in rat PRP using Born’s turbidimetric method. The 10 μM ADP and 0.8 mM AA were used to induce platelet aggregation. ASP and Eda are inhibitor of AA-induced platelet aggregation and a free radical scavenger, respectively. There were five different concentrations of target compounds to be determined for calculating IC50.
As shown in Table 1, for the ADP-induced platelet aggregation, five compounds displayed significant inhibitory activities. The inhibitory effects of 3d, 3e, 3f, 3h, and 3i (IC50 of 1.38, 1.48, 1.43, 1.53, and 1.52 mM, respectively) were stronger than those of Eda (IC50=1.76 mM), ASP (IC50=1.58 mM), and NBP (IC50>1.79 mM). Compounds 3d and 3f (IC50 of 1.01 and 0.96 mM, respectively) inhibited AA-induced platelet aggregation more effectively than NBP (IC50= 1.10 mM) and less effectively than Eda (IC50=0.85 mM) and ASP (IC50=0.81 mM). In particular, as shown in Figure 3, the inhibitory effect of 1.28 mM 3d (71.69%) on the ADP-induced platelet aggregation was 1.71-, 2.22-, and 2.34-fold stronger than that of NBP (41.93%), ASP (32.25%), and Eda (30.64%), whereas its inhibition on the AA-induced platelet activation (83.33%) was 1.10- and 1.31-fold more potent than that of NBP (75.86%) and Eda (63.49%), comparable with that of ASP (95.43%). These data revealed that compounds with straight-chain alkyl had stronger inhibitory platelet aggregation activity than those with branch-chain alkyl. The results indicated that 3d had a potent antiplatelet aggregation activity in both the ADP- and AA-induced platelet aggregation. Thus, 3d was used for the following biological experiments.
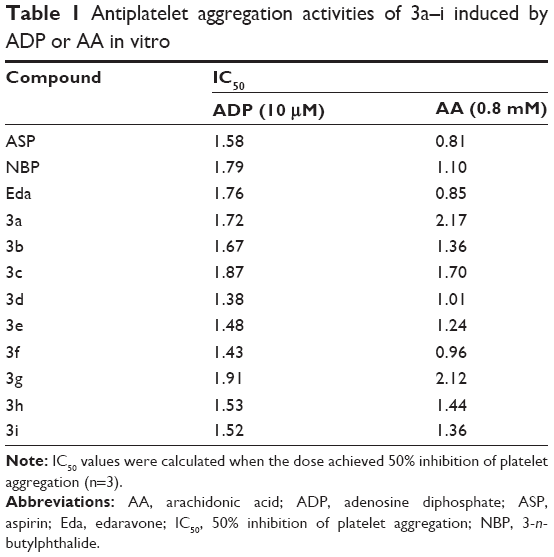 | Table 1 Antiplatelet aggregation activities of 3a–i induced by ADP or AA in vitro |
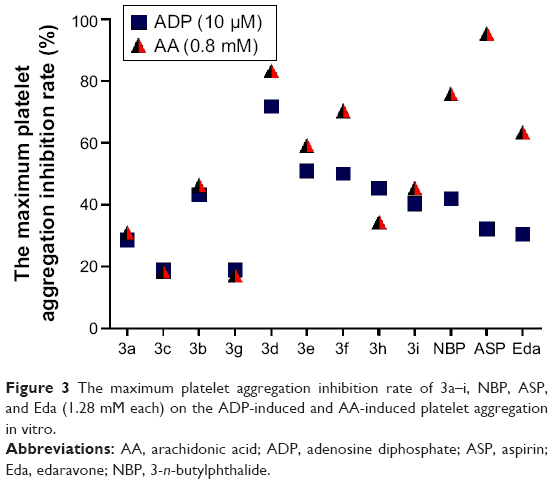 | Figure 3 The maximum platelet aggregation inhibition rate of 3a–i, NBP, ASP, and Eda (1.28 mM each) on the ADP-induced and AA-induced platelet aggregation in vitro. |
The antithrombotic activity assay in vivo
A red wine-colored section appeared in the tail tip of the rodent in the carrageenan-induced group, tail-thrombosis length increased and experienced typical dry necrosis. Our results indicated that 3d could inhibit thrombus formation in vivo; as shown in Table 2 and Figure 4, 3d (20 and 50 mg/kg) decreased the black tail occurrence and tail-thrombosis length, comparable with NBP (50 mg/kg) group and lower than ASP (50 mg/kg). However, Eda (3 mg/kg) did not decrease the black tail occurrence and tail-thrombosis length.
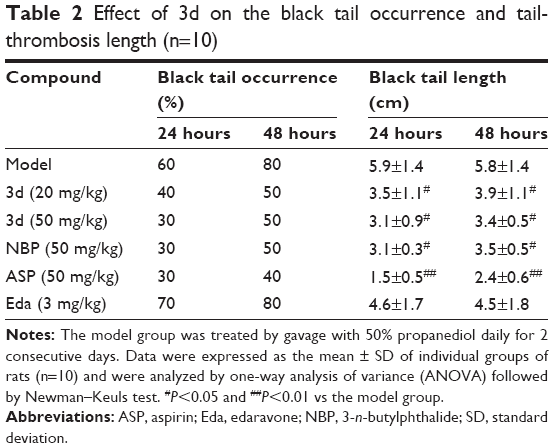 | Table 2 Effect of 3d on the black tail occurrence and tail-thrombosis length (n=10) |
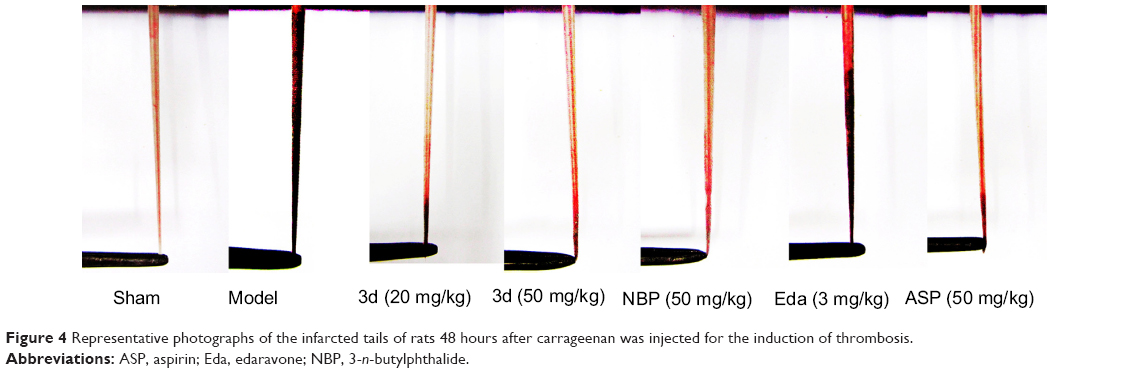 | Figure 4 Representative photographs of the infarcted tails of rats 48 hours after carrageenan was injected for the induction of thrombosis. |
The activity of anti-cerebral ischemia in tMCAO and pMCAO rats
The most common cause of ischemic strokes is occlusion of the MCA. Hence, we observed the effect of treatment with 3d on ischemic stroke injury by occlusion of MCAO for 2 hours followed by recirculation.43 Sprague Dawley rats were randomly divided into seven groups as follows: sham-operated group (just separated blood vessels), soybean oil group (negative control), NBP group (20 mg/kg), Eda group (3.0 mg/kg), and 3d group (10, 20, and 50 mg/kg). Fifteen minutes after the onset of MCAO, drugs or vehicle was administered to rats by ip injection, and the neurological deficits cores in rats were evaluated by Longa’s method 24 hours after reperfusion. The experiment result showed no obvious neurological deficit in the sham-operated group. However, the neurological deficit score in the group of soybean oil-treated rats significantly increased 24 hours after reperfusion. On the contrary, the neurobehavioral function score with 3d (10, 20, and 50 mg/kg) treatment significantly decreased, comparable with Eda-treated group, and treatment with 3d dose improved the neurological score 24 hours after ischemia in a dose-dependent manner in Figure 5. Meanwhile, we also observed the effect of treatment with 3d on ischemic stroke injury by occlusion of MCAO for 24 hours. Sprague Dawley rats were randomly divided into five groups as follows: sham-operated group (just separated blood vessels), 50% propanediol group (negative control), NBP group (50 mg/kg), Eda group (3.0 mg/kg), and 3d group (50 mg/kg). Two hours after the onset of MCAO, drugs or vehicle was administered to rats by ip injection, and the neurobehavioral function score with 3d (50 mg/kg) treatment also decreased (Figure 5).
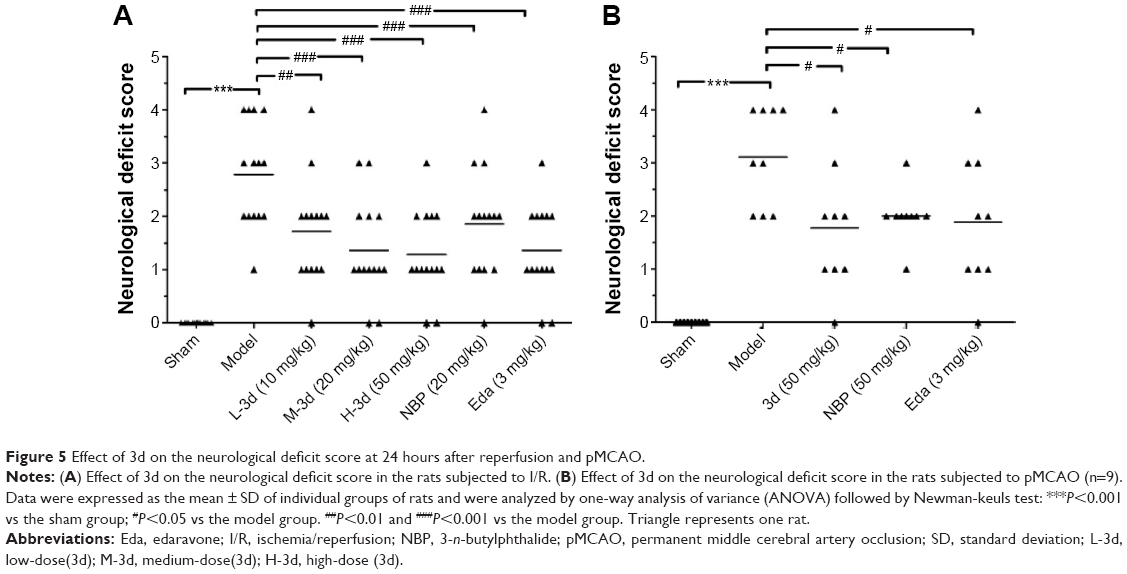 | Figure 5 Effect of 3d on the neurological deficit score at 24 hours after reperfusion and pMCAO. |
Infarct assessment
The ischemic infarct volume of each group of rats was assessed by the 2,3,5-triphenyltetrazolium chloride assay. As shown in Figure 6A and C, no infarct volume was observed in the sham-operated group, whereas the model group exhibited a white infarct area. From Figure 6B and D, we could see that 3d significantly reduced the infarct size. With doses of 10, 20, and 50 mg/kg in tMCAO rats, the infarct size in the 3d-treated groups was reduced by 30.22% (10 mg/kg), 63.84% (20 mg/kg), and 76.13% (50 mg/kg), respectively. In addition, the effect of moderate- and high-dose 3d was comparable with that of Eda (69.65%) and more potent than that of NBP (26.19%). Interestingly, 3d significantly reduced the infarct size at doses of 50 mg/kg in pMCAO rats; the effect of 3d (50 mg/kg) was more potent than that of NBP (50 mg/kg) and Eda (3 mg/kg).
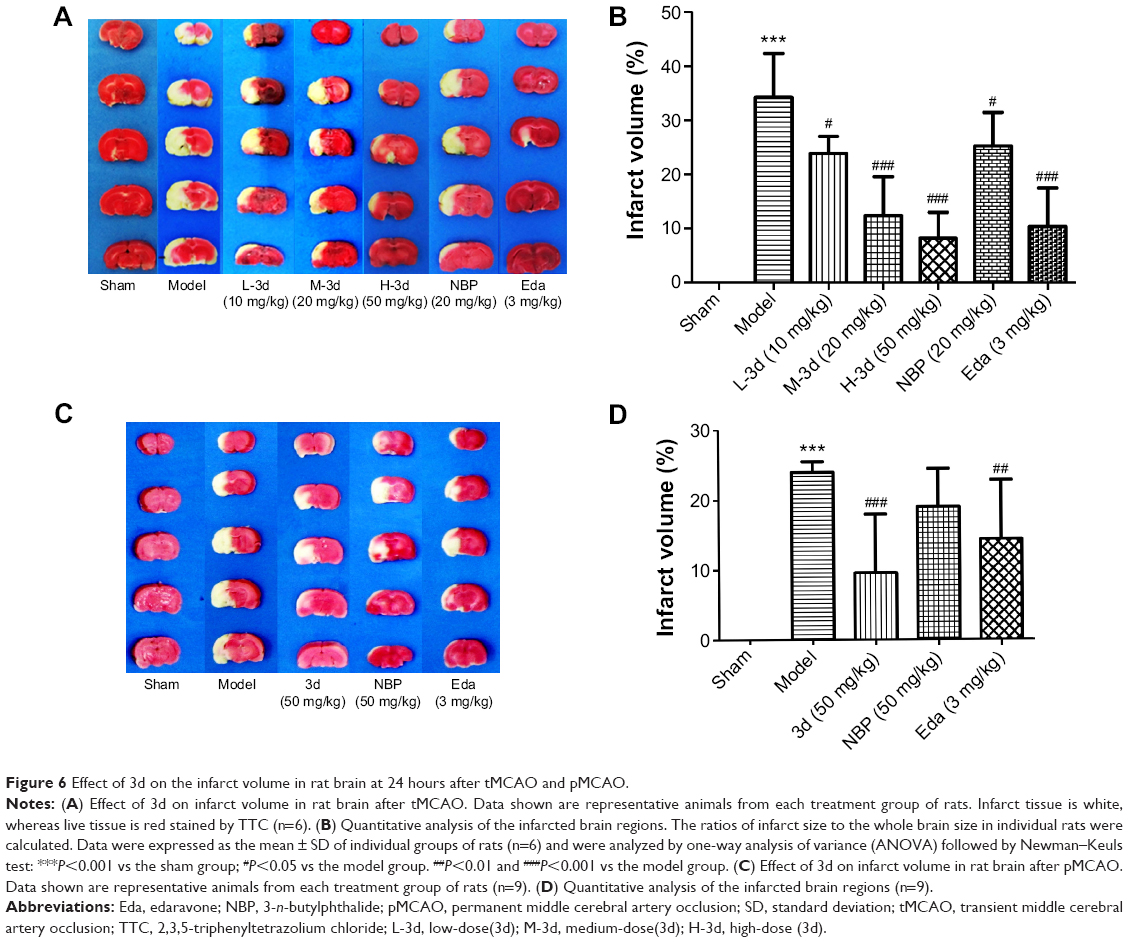 | Figure 6 Effect of 3d on the infarct volume in rat brain at 24 hours after tMCAO and pMCAO. |
Histopathological analysis
H&E staining analysis suggested that 3d could significantly attenuate cerebral damage. As shown in Figure 7, the H&E stain of the cerebral cortex in the model group showed a large area of infarction in the cerebral cortex, neuronal perikarya shrinkage, and microglial cells, in addition to degeneration of a large number of nerve cells in the vacuoles. However, compared with that in the model group, drug-treated groups exhibited minor infarction of cerebral cortex, and less neuronal perikarya shrinkage in the brains, particularly in those exposed to the highest dose of 3d.
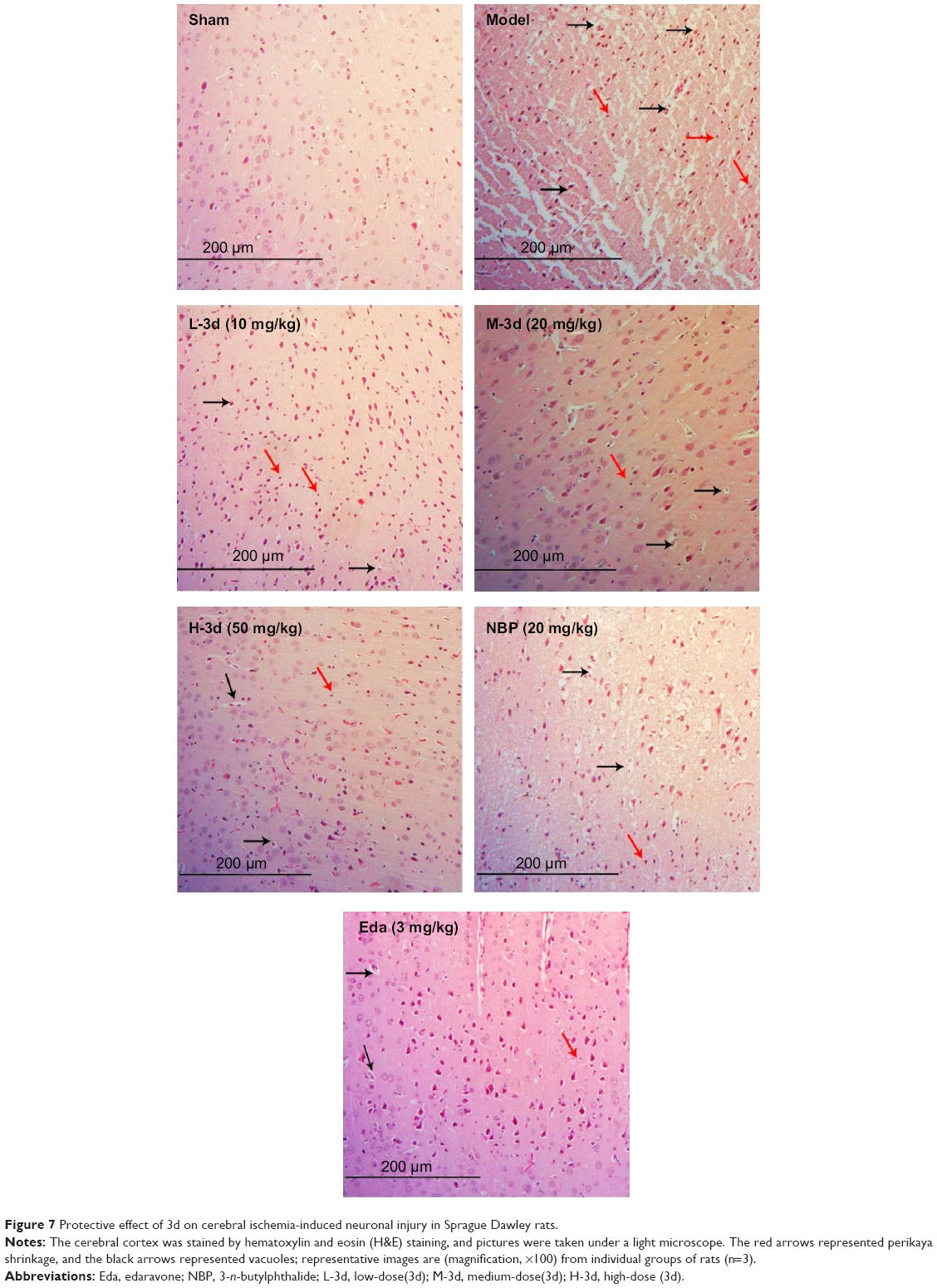 | Figure 7 Protective effect of 3d on cerebral ischemia-induced neuronal injury in Sprague Dawley rats. |
Free radical scavenging activity in HT22 cells
A growing body of evidence suggested that oxidative stress plays an important role in ischemic stroke reperfusion injury,44,45 which can exacerbate ischemic brain damage through the generation of ROS, such as superoxide anions (O2•−), hydroxyl radicals (OH•), and NO, and H2O2. Hydroxyl radicals (OH•) can damage DNA and have high cytotoxicity.46,47 NBP can reduce damage of oxidative stress. Thus, we hypothesized that 3d might have antioxidative stress effect. To verify this hypothesis, we investigated its scavenging activities in murine HT22 cells, which are commonly used for the analysis of diseases related to oxidative neuronal cell death.48–52 The influence of H2O2 on HT22 cell survival was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay and confirmed by microscopic evaluation. The 100 μM 3d remarkably increased the H2O2-treated HT22 viability percentage (74.25%), compared with 51.45% of only H2O2-treated HT22 cells. The neuroprotective effect of 3d exceeded that of NBP and Eda (71.55% and 69.87%, respectively) at the same dosage (Figure 8). The results implied that 3d could protect HT22 cells from H2O2-induced cytotoxicity damage and might be a free radical scavenger.
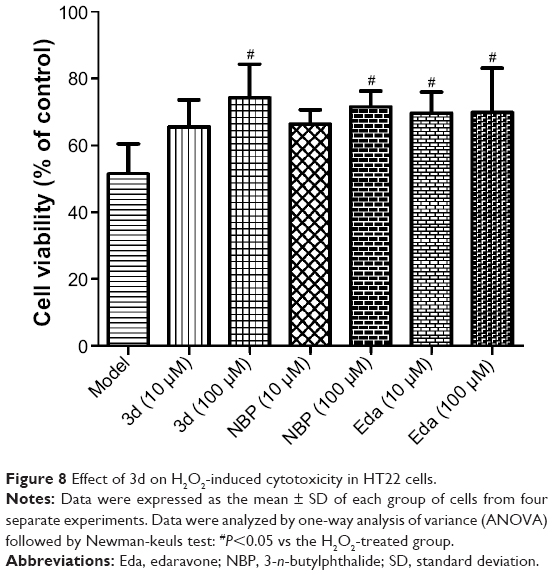 | Figure 8 Effect of 3d on H2O2-induced cytotoxicity in HT22 cells. |
Radical-scavenging activity in vivo
Postischemic reperfusion could enhance the formation of ROS in brain tissue,52–55 whereas endogenous antioxidants, including SOD, GSH-Px, and catalase, could scavenge ROS. Thus, to evaluate radical-scavenging activity of 3d in ischemic stroke rats, we evaluated the levels of GSH-Px, SOD, and the content of MDA (an indicator of lipid peroxidation) and NO in the brain tissues. Compared with the sham group, the content of SOD in the model group was decreased by 26.22% (from 88.66 to 65.41 U/mg), whereas the content of SOD in the 3d-treated group (10, 20, and 50 mg/kg), NBP-treated group, and Eda-treated group increased in comparison with the model group (Figure 9A). The GSH-Px activity in the ischemic core of rats in the model group was significantly decreased by 27.12% (from 30.17 to 21.98 nmol/mg), as shown in Figure 9B, compared with that in the sham group. However, the levels of GSH-Px activity in 3d-treated group (50 mg/kg) and Eda-treated group (3 mg/kg) were significantly higher than that in the model animals. Meanwhile, we measured the tissue content of MDA and the activity level of NO and found that these were significantly higher in the model group than in the sham group, whereas 3d treatment attenuated the increase in MDA and NO levels after reperfusion. Treatment with 3d (50 mg/kg) decreased the content of the cerebral cortex MDA by 26.16% (from 3.02 to 2.23 nmol/mg) and NO by 52.32% (from 1.51 to 0.72 U/mg), compared with that in the model group (as shown in Figure 9C and D). In addition, the effect of 3d on the brain MDA and NO levels was much stronger with the increase in dosage. The NO activity of the 20 mg/kg 3d-treated rats (1.01 U/mg) was comparable with that of the 20 mg/kg NBP-treated group (0.92 U/mg). Interestingly, after treatment with 3d in ischemic reperfusion rats, the levels of GSH-Px and SOD activity in ischemia penumbra were recovered to near the nonischemic control level and the content of MDA and NO activity remarkably decreased.
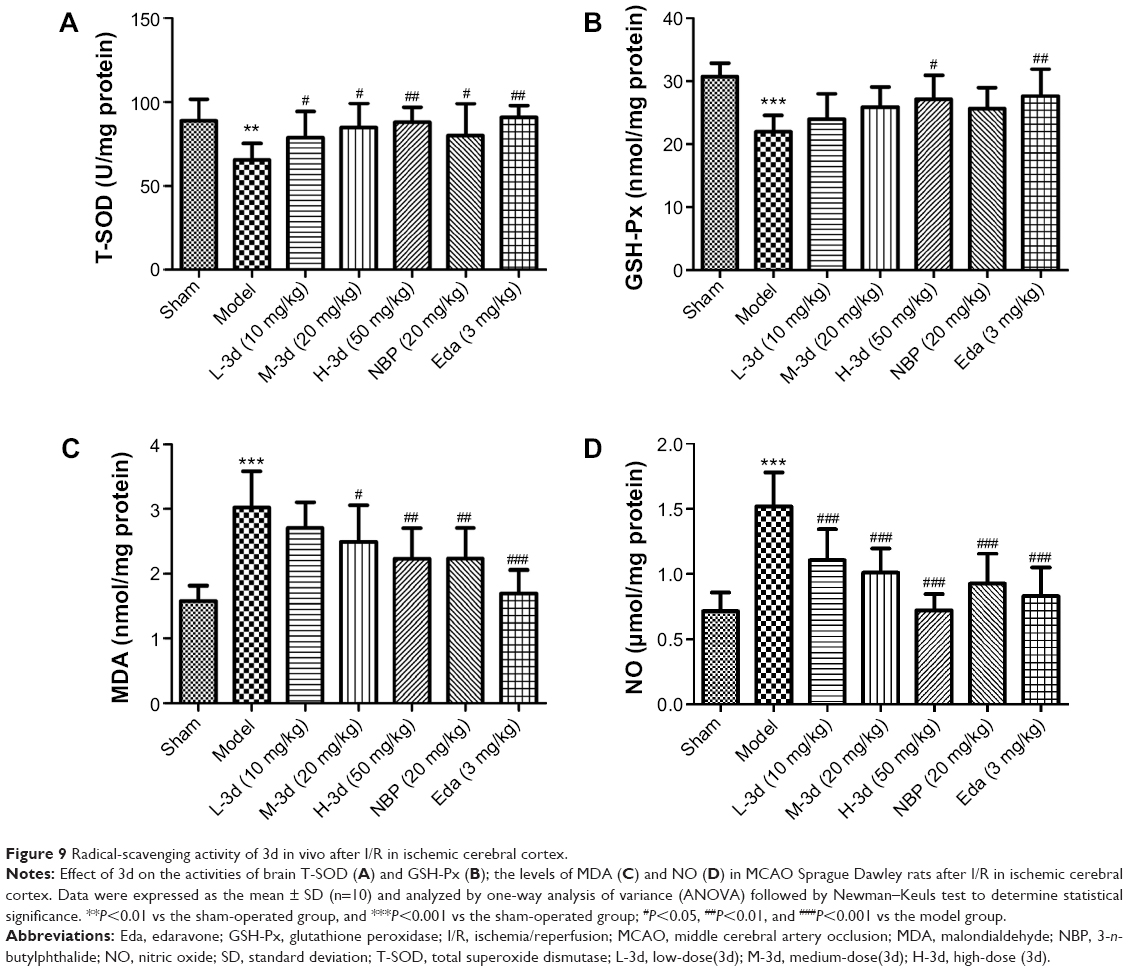 | Figure 9 Radical-scavenging activity of 3d in vivo after I/R in ischemic cerebral cortex. |
Study on the pharmacokinetics and biodistribution of 3d
An ideal anti-ischemic stroke drug should be quickly absorbed into blood circulation and permeate the cerebral infarction tissues following oral administration. The plasma concentration-time profiles of 3d are shown in Figure 10. These are obtained following oral administration or iv injection of 10 mg/kg of 3d in Wistar rats. As shown in Table 3, the AUC (min×μg/mL) of oral dose (10 mg/kg) and iv injection dose (10 mg/kg) were 770.6±19.6 and 821.5±41.9 min×μg/mL, respectively. The ~90% absolute bioavailability observed suggested that most of the compound 3d was absorbed into the intestine, which might make up for the deficiency of NBP with its low bioavailability. The pharmacokinetic parameters were calculated by different compartment model fitting using the WinNonlin software.
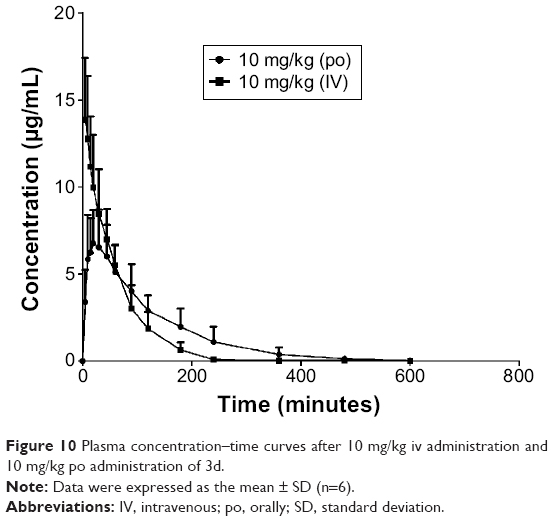 | Figure 10 Plasma concentration–time curves after 10 mg/kg iv administration and 10 mg/kg po administration of 3d. |
 | Table 3 Results of absolute bioavailability of 3d in rats (n=6) |
After tail vein administration of 3d at a dosage of 10 mg/kg to KM mice (18–22 g), the mean concentrations of 3d in heart, liver, spleen, lung, kidney, and brain tissues at 15, 45, and 90 minutes are shown in Figure 11. The figure showed that compound 3d distributed rapidly into various tissues after tail vein administration, the concentrations reaching the maxima in all organs at 15 minutes postdose, and the concentrations of 3d in brain rapidly increasing and then gradually declining, similar to that in liver. All these results demonstrated that 3d could be rapidly absorbed and penetrate the brain through blood–brain barrier in mice.
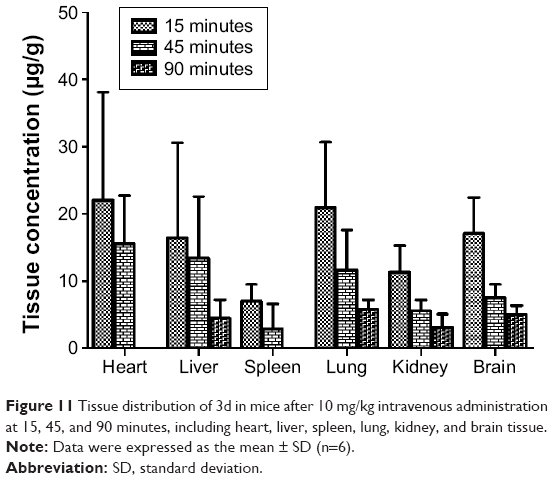 | Figure 11 Tissue distribution of 3d in mice after 10 mg/kg intravenous administration at 15, 45, and 90 minutes, including heart, liver, spleen, lung, kidney, and brain tissue. |
Conclusion
Antiplatelet aggregation activity screening results suggested that the target compounds 3a–i bearing all kinds of straight/branch-chained alkyl groups showed different antiplatelet aggregation activity. The straight-chained alkyl groups at the 3-position of isoindoline significantly affected its antiplatelet aggregation activity compared with the branch-chained alkyl groups; the inhibition of AA-induced platelet aggregation is as follows: 3b (46.42%) vs 3c (18.51%), 3d (83.33%) vs 3g (17.24%), and the inhibition of ADP-induced platelet aggregation is as follows: 3b (43.10%) vs 3c (18.86%), 3d (71.69%) vs 3g (18.86%) at 1.28 mM dosage. Interestingly, the antiplatelet aggregation activity of these compounds increased with the extension of alkyl side chains except for 3d, which exhibited remarkable antiplatelet aggregation activity. These results suggested that isoindoline by replacement of the oxygen atom at the 2-position of phthalide showed inhibitory activities against platelet aggregation.
In this study, a series of novel 3-alkyl-2,3-dihydro-1H-isoindol-1-ones compounds (3a–i) were designed and synthesized. Antiplatelet aggregation activity screening results showed that 3d significantly inhibited ADP- and AA-induced platelet aggregation in vitro and inhibited the formation of blood clots in vivo. Meanwhile, 3d exhibited a very high absolute bioavailability at 10 mg/kg dosage (93.7%) and rapidly distributed in brain tissue to keep high blood drug concentration for the treatment of ischemic strokes, which might make up for the deficiency of NBP. More importantly, 3d not only exhibited a potent activity in scavenging free radicals and improved the survival of HT22 cells against the ROS-mediated cytotoxicity in vitro but also significantly attenuated the I/R-induced oxidative stress in ischemic rat brain. For example, 3d could increase the content of brain antioxidant SOD and GSH and reduce the levels of brain MDA and NO. The ability of 3d to scavenge free radicals was comparable with that of clinical NBP or Eda. Most importantly, we observed that treatment of 3d significantly reduced infarct size, improved neurobehavioral deficits, and prominently decreased attenuation of cerebral damage in a rat model of transient and permanent MCA focal cerebral ischemia. Therefore, the results of our study demonstrated that 3-alkyl-2,3-dihydro-1H-isoindol-1-ones, a novel style of compounds, might be promising candidate drugs for the treatment of acute ischemic stroke, and that 3d may be a promising therapeutic agent for the prevention and treatment of clinical stroke. In addition, the neuroprotective effects of 3d might involve multiple mechanisms. Therefore, further studies are needed to reveal other pharmacological mechanisms of 3d for the treatment of ischemic stroke in the future.
Disclosure
The authors report no conflicts of interest in this work.
References
Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292. | ||
Khan M, Dhammu TS, Matsuda F, et al. Promoting endothelial function by S-nitrosoglutathione through the HIF-1α/VEGF pathway stimulates neurorepair and functional recovery following experimental stroke in rats. Drug Des Devel Ther. 2015;9:2233–2247. | ||
Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371:1612–1623. | ||
Kraft P, Schwarz T, Meijers JCM, Stoll G, Kleinschnitz C. Thrombin-activatable fibrinolysis inhibitor (TAFI) deficient mice are susceptible to intracerebral thrombosis and ischemic stroke. PLoS One. 2010;5:e11658. | ||
Mackman N. Triggers, Targets and treatments for thrombosis. Nature. 2008;451:914–918. | ||
Zhang Y, Wang L, Li J, Wang XL. 2-(1-Hydroxypentyl)-benzoate increases cerebral blood flow and reduces infarct volume in rats model of transient focal cerebral ischemia. J Pharmacol Exp Ther. 2006;317:973–979. | ||
Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. | ||
Pandya RS, Mao L, Zhou H, et al. Central nervous system agents for ischemic stroke: neuroprotection mechanisms. Cent Nerv Syst Agents Med Chem. 2011;11:81–97. | ||
Bouckaert M, Lemmens R, Thijs V. Reducing prehospital delay in acute stroke. Nat Rev Neurol. 2009;5:477–483. | ||
van den Berg JS, de Jong G. Why ischemic stroke patients do not receive thrombolytic treatment: results from a general hospital. Acta Neurol Scand. 2009;120:157–160. | ||
Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. | ||
Tuttolomondo A, Pecoraro R, Pinto A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: a review of the evidence to date. Drug Des Devel Ther. 2014;8:2221–2239. | ||
Harukuni I, Bhardwaj A. Mechanisms of brain injury after global cerebral ischemia. Neurol Clin. 2006;24:1–21. | ||
Wang T, Gu J, Wu PF, et al. Protection by tetrahydroxystilbene glucoside against cerebral ischemia: involvement of JNK, SIRT1, and NF-kappaB pathways and inhibition of intracellular ROS/RNS generation. Free Radic Biol Med. 2009;47:229–240. | ||
Candelario-Jalil E. Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neuro-therapeutics. Curr Opin Invesigt Drugs. 2009;10:644–654. | ||
Murakami K, Kondo T, Kawase M, et al. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. | ||
Suzuki K. Anti-oxidants for therapeutic use: why are only a few drugs in clinical use? Adv Drug Deliv Rev. 2009;61:287–289. | ||
Liu CL, Liao SJ, Zeng JS, et al. DL-3-n-butylphthalide prevents stroke via improvement of cerebral microvessels in RHRSP. J Neurol Sci. 2007;260:106–113. | ||
Peng Y, Zeng X, Feng Y, Wang X. Antiplatelet and antithrombotic activity of L-3-n-butyphthalide in rats. J Cardiovasc Pharmacol. 2004;43:876. | ||
Li L, Zhang B, Tao Y, et al. DL-3-n-butylphthalide protects endothelial cells against oxidative/nitrosative stress, mitochondrial damage and subsequent cell death after oxygen glucose deprivation in vitro. Brain Res. 2009;1290:91–101. | ||
Wang XL, Li Y, Zhao Q, et al. Design, synthesis and evaluation of nitric oxide releasing derivatives of 3-n-butylphthalide as antiplatelet and antithrombotic agents. Org Biomol Chem. 2011;9:5670–5681. | ||
Zhao C, He Z, Cui S, Zhang R. Determination of 3-n-butylphthalide in rabbit plasma by HPLC with fluorescence detection and its application in pharmacokinetic study. Biomed Chromatogr. 2003;17:391–395. | ||
Xu B, Zhao Z. Butylphthalide injection, an innovative drug originated in China for the treatment of ischemic stroke. Chin J New Drugs. 2011;20:11. | ||
Zhao C. Studies on Pharmacokinetics of 3-n-Butylphthalide in Rats and Rabbits and the Transport Mechanism in Brain [doctorate thesis]. Shenyang: Shenyang Pharmaceutical University; 2003. | ||
Lu R. The Comparative Study on Pharmacokinetics of R- and S-isomers of NBP in Beagle Dogs [master thesis]. Tianjin: Tianjin Medical University; 2006. | ||
Zhu Y, Tan S. Observation of DL-3-n-butylphthalide combined edarovone treating 36 patients with progressive cerebral infarction. Chin J Pract Nerv Dis. 2010;13:10–12. | ||
Belliotti TR, Brink WA, Kesten SR, et al. Isoindolinone enantiomers having affinity for the dopamine D4 receptor. Bioorg Med Chem Lett. 1998;8:1499–1502. | ||
Drutu I, Hurley DJ, Bandarage UK, et al, inventor; Vertex Pharmaceuticals Incorporated, USA., assignee. Spiropiperidine derivatives as modulators of muscarinic receptors and their preparation, pharmaceutical composition and use for treatment of muscarinic receptor-mediated diseases. United States patent US 20060270653. 2006 Nov 30. | ||
Greig NH, Holloway H, Brossi A, et al, inventor; The Secretary of the Department of Health and Human Services, USA., assignee. Preparation of thalidomide analogs as modulators of TNF-α activity and angiogenesis. United States patent US 20060211728. 2006 Sep 21. | ||
Carry JC, Mignani S, Bigot A, Ronan B, Kleemann HW, Hofmeister A, inventor; Aventis Pharma Sa, Fr, assignee. Preparation of isoindolones as NHE (sodium/hydrogen exchanger) inhibitors, and their pharmaceutical compositions used as cytoprotective agents. United States patent US 20040048916. 2004 Mar 11. | ||
Tweedie D, Giordano T, Yu QS. TNF-α synthesis inhibitors on the 3-phthalimidoglutarimide backbone as therapeutic candidates for neurodegenerative diseases. In: 7th International Conference on Alzheimer’s and Parkinson’s Disease; March 9–13, 2005; Sorrento, Italy. Bologna: Monduzzi Editore; 2005:77–86. | ||
Hagmann WK, Lin LS, Shah SK, et al, inventor; Merck & Co., Inc., USA., assignee. Preparation of substituted amides as antagonists and/or inverse agonists of the cannabinoid-1 receptor for therapy. United States patent US 20040058820. 2004 Mar 25. | ||
Sheridan JM, Heal JR, Hamilton WDO, Pike I, inventor; Electrophoretics Limited, UK., assignee. Casein kinase 1δ inhibitors for the treatment of neurodegenerative disorders. United States patent US 20140018540. 2014 Jan 16. | ||
Amberg W, Lange U, Ochse M, et al, inventor; AbbVie Inc., USA; AbbVie Deutschland GmbH & Co. KG., assignee. Preparation of isoindoline derivatives and their use in therapy. United States patent US 20130210880. 2013 Aug 15. | ||
Born GVR, Cross MJ. The aggregation of blood platelets. Physiology. 1963;168:178. | ||
Bertelli A, Giovannini L, Galmozzi G, Bertelli AA. Protective role of propionyl carnitine in vascular disorders experimentally induced by endothelin (ET-1) serotonin and K-carrageenin. Drugs Exp Clin Res. 1993;19:7–11. | ||
Longa EZ, Weinstein PR, Carlson S. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. | ||
Ashwal S, Tone B, Tian HR, Cole DJ, Pearce WJ. Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion. Stroke. 1998;29:1037–1046. | ||
Ruan YP, Chen MD, He MZ, Zhou X, Huang PQ. A practical two-step synthesis of 3-alkyl-2,3-dihydro-1H-isoindolin-1-ones. Synth Commun. 2004;34:853–861. | ||
Ye JQ, Zhai LL, Zhang SH, et al. DL-3-n-butylphthalide inhibits platelet activation via inhibition of cPLA2-mediated TXA2 synthesis and phosphodiesterase. Platelets. 2015;3:1–9. | ||
Ma FF, G Y, Q HL, H XJ, C JB. Antiplatelet activity of 3-butyl-6-bromo-1(3H)-isobenzofuranone on rat platelet aggregation. J Thromb Thrombolysis. 2012;33:64–73. | ||
Zhao WH, Luo C, Wang J, et al. 3-N-butylphthalide improves neuronal morphology after chronic cerebral ischemia. Neural Regen Res. 2014;9:719–726. | ||
Zhao Q, Zhang C, Wang XL, Chen L, Ji H, Zhang YH. (S)-ZJM-289, a nitric oxide-releasing derivative of 3-n-butylph-thalide, protects against ischemic neuronal injury by attenuating mitochondrial dysfunction and associated cell death. Neurochem Int. 2012;60:134–144. | ||
Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18:685–716. | ||
Moosmann B, Behl C. Antioxidants as treatment for neurodegenerative disorders. Expert Opin Investig Drugs. 2002;11:1407–1435. | ||
Iadecola C, Zhang FY, Casey R, Clark HB, Ross ME. Inducible nitric oxide synthase gene expression in vascular cells after transient focal cerebral ischemia. Stroke. 1996;27:1373–1380. | ||
Talalay P, Dinkova-Kostova AT, Holtzclaw WD. Importance of phase 2 gene regulation in protection against electrophile and reactive oxygen toxicity and carcinogenesis. Adv Enzyme Regul. 2003;4:121–134. | ||
Skutella T, Lezoualc’h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol. 1997;51:535–541. | ||
Dargusch R, Schubert D. Specificity of resistance to oxidative stress. J Neurochem. 2002;81:1394–1400. | ||
Moosmann B, Behl C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc Natl Acad Sci U S A. 1999;96:8867–8872. | ||
Sagara Y, Hendler S, Khoh-Reiter S, et al. Propofol hemisuccinate protects neuronal cells from oxidative injury. J Neurochem. 1999;73:2524–2530. | ||
Tan S, Somia N, Maher P, Schubert D. Regulation of antioxidant metabolism by translation initiation factor 2alpha. J Cell Biol. 2001;152:997–1006. | ||
Dirnagl U, Lindauer U, Them A, et al. Global cerebral ischemia in the rat: online monitoring of oxygen free radical production using chemiluminescence in vivo. J Cereb Blood Flow Metab. 1995;15:929–940. | ||
Lancelot E, Callebert J, Revaud ML, Boulu RG, Plotkine M. Detection of hydroxyl radicals in rat striatum during transient focal cerebral ischemia: possible implication in tissue damage. Neurosci Lett. 1995;197:85–88. | ||
Malinski T, Bailey F, Zhang ZG, Chopp M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1993;13:355–358. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.