")
Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 14
Diagnostic and Management Strategies of IgA Vasculitis Nephritis/Henoch-Schönlein Purpura Nephritis in Pediatric Patients: Current Perspectives
Received 5 December 2022
Accepted for publication 24 February 2023
Published 7 March 2023 Volume 2023:14 Pages 89—98
DOI https://doi.org/10.2147/PHMT.S379862
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Roosy Aulakh
Mario Sestan, Marija Jelusic
Department of Paediatrics, University of Zagreb School of Medicine, University Hospital Centre Zagreb, Zagreb, Croatia
Correspondence: Marija Jelusic, University of Zagreb School of Medicine, University Hospital Centre Zagreb, Kispaticeva 12, Zagreb, 10 000, Croatia, Tel +385 1 23 88 701, Email [email protected]
Abstract: IgA vasculitis (IgAV) or Henoch-Schönlein purpura (HSP) is the most common vasculitis in children, and nephritis (IgAVN or HSPN) is the most important and only chronic manifestation of the disease. Despite this, there are no diagnostic criteria and we rely on the European League Against Rheumatism/Paediatric Rheumatology International Trials Organization/Paediatric Rheumatology European Society-endorsed Ankara 2008 classification criteria in our daily practice. Basic investigations that should be done in every patient with IgAVN include blood pressure measurement, estimated glomerular filtration rate and urinalysis. Kidney biopsy is still the gold standard for the diagnosis of IgAVN since noninvasive confirmation of nephritis is still pending. According to the Single Hub and Access point for pediatric Rheumatology in Europe (SHARE) recommendations, the first-line treatment for with mild forms of IgAVN is oral glucocorticoids, for patients with moderate IgAVN parenterally administrated glucocorticoids in pulsed doses, while initial treatment for patients with the most severe forms of IgAVN include pulsed doses of glucocorticoids in combination with intravenous cyclophosphamide pulses. New therapeutic options are currently being tested, aiming to reduce the production of galactose-deficient IgA1 and autoantibodies or suppress the alternative or lectin complement pathway and blocking mesangial cell activation.
Keywords: IgA vasculitis nephritis, clinical manifestations, proteinuria, hematuria, treatment
Introduction
IgA vasculitis (IgAV), formerly known as Henoch-Schönlein purpura, represents the non-granulomatous systemic vasculitis, histologically characterised by infiltration of the walls of the blood vessels, mainly arterioles, capillaries and venules, by neutrophils with deposits of immune complexes containing predominantly IgA in the endothelium of small blood vessels in the skin, synovial membrane, intestines and urinary system.1,2 IgAV is the most common systemic vasculitis in childhood, clinically manifested as purpuric rash accompanied by either gastrointestinal symptoms, arthritis, and/or nephritis.2,3 The main attribute of the disease is palpable purpura, which affects the lower extremities and buttocks.1 By the 2012 revised Chapel Hill International Consensus Conference for Nomenclature of Vasculitides, the term HSP was replaced with IgAV.4
Despite the fact that the disease is most often self-limiting and lasts an average of up to four weeks, various acute and chronic complications are possible.5,6 Among the acute complications of the disease, the most frequent are those related to the gastrointestinal system, including intussusception, bowel perforation, and massive bleeding as the most serious ones.2,7 Nevertheless, the renal aspect of the disease is the most important chronic complication and the main cause of morbidity and mortality among children suffering from IgAV, and thus the main prognostic factor.2,8 Of note, it is necessary to distinguish IgA vasculitis nephritis (IgAVN) from IgA nephropathy (IgAN). Although these two diseases share many clinical, immunological and histological findings, there are also clinicopathological differences, not limited only the mere presence of extrarenal clinical signs in IgAV.9,10 This review article discusses the epidemiology, etiopathogenesis, clinical manifestations, with special emphasis on diagnosis and treatment of IgA vasculitis nephritis (IgAVN).
Epidemiology
The incidence of IgAV varies worldwide ranging from 3 to 27 cases per 100,000 children, while the prevalence varies between 6.1 and 20.4 per 100,000 children.1,11–13 IgAVN occurs in 20–60% of children suffering from IgAV.14,15 The highest occurrence of IgAV and IgAVN is found in East Asians, intermediate in Europeans, and the lowest in individuals of African ancestry.1,11 Furthermore, recently, it was demonstrated that both IgAV and IgAVN may not be randomly distributed in an area, but clustered more around cities.2,11 IgAVN showed linear clustering in the eastern part of Croatia, which follows the course of the Drava and Danube rivers, in the vicinity of areas of Balkan endemic nephropathy. The reasons for these geospatial variations of IgAV and IgAVN are not known, but it can be hypothesised that hotspot clusters appear where genetic and environmental factors substantially overlap.2 This is another important difference between IgAVN and IgA nephropathy, which is predominantly associated with different genetic variants of innate immunity genes and genes important for defense against parasitic infections.16 The median age of onset is around 6 years, and 90% of patients are younger than 10 years, with males being more frequently affected than females at a ratio of 1.5:1.3
Etiopathogenesis
The etiopathogenesis of IgAV and IgAVN is reflected in the interaction of genetic and environmental factors, with special emphasis on infections.12,17 Studies investigating the genetic background of the disease were expected to identify candidate genes associated with IgAV susceptibility, particularly IgAVN.2,18 However, genome-wide association studies (GWAS) have indicated so far the significance of the HLA class II genes.19,20 It is important to emphasise that there are currently no large GWAS for pediatric IgAV and IgAVN, and only two small studies have been published to date. The results summarise the complex pathogenesis of IgAV caused primarily by interactions among a number of different genes, with a limited role played by each individual gene in disease development.2 The first GWAS on IgAV susceptibility detected that the polymorphisms with the highest risk of developing disease were in the HLA-DQA1 and DQB1 intergenic zone and at the HLA-DRB1*11 and B1*13 loci.19 The second GWAS study found that haplotype DQA1*01:01/DQB1*05:01/DRB1*01:01 was associated with susceptibility to IgAV but not with other autoimmune diseases.20 The results of the GWAS to date classify IgAV as a prototype of a disease related to HLA class II loci, therefore sharing some features with giant cell arteritis and anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV).2
Although the exact etiology of the disease is not known, various infectious agents, drugs, vaccines, food allergens and insect bites may be precipitants triggering the onset of this disease in genetically predisposed individuals.7,17,21 In more than 75% of patients, upper respiratory tract or gastrointestinal infections precede the onset of disease. Multiple bacterial and viral infections have been described as triggers of IgAV: group A streptococcal infection (most common), infectious mononucleosis, subacute bacterial endocarditis, hepatitis, Mycoplasma pneumoniae infection, Campylobacter enteritis, Helicobacter pylori infection, Yersinia infection, Shigella infection, Salmonella infection, Brucellosis, Legionella species, Parvovirus, Adenovirus, Varicella-zoster virus infection, Rotavirus, etc.2,22,23 The role of infections in etiology and pathogenesis is further confirmed by observations of seasonal tendencies with fewer cases seen during the summer months and showing a peak in autumn and winter. Different non-infective triggers such as drugs (eg, ampicillin, erythromycin, penicillin, quinidine, quinine, losartan and cytarabine), vaccines, foods, cold temperatures and insect bites have been described to potentially be associated with a higher risk of developing IgAV.2,24
According to some authors, two multi-hit pathogenesis models for IgAV and IgAVN are described.25,26
In patients with IgAV, and especially in those patients who developed renal involvement, it seems that the aberrantly glycosylated IgA1 plays a key role in the pathogenesis.2 Specifically, IgA1 from most patients with IgAV lack galactose residues (Gd-IgA1). Aberrant glycosylation occurs predominantly due to decreased galactosyltransferase activity in the Golgi apparatus of IgA1-producing immune cells. Gd-IgA1 might be recognised as an autoantigen by IgA and IgG antibodies, leading to the formation of polymeric immune complexes (Gd-IgA1-IgA, Gd-IgA1-IgG and Gd-IgA1-sCD89, where sCD89 is a soluble IgA Fc alpha receptor).2 Circulating immune complexes may accumulate in the blood serum, resulting in their deposition in the endothelium of small blood vessels in the skin, synovial membrane, intestines and urinary system. It has been demonstrated that the serum level of Gd-IgA1 is higher in IgAVN patients compared to IgAV patients without nephritis.27 In IgAV patients who developed IgAVN, some of these complexes, Gd-IgA1-IgG, deposit in the kidneys, resulting in mesangial cell activation, release of inflammatory mediators and glomerular injury.21
Nonetheless, there is a second multi-hit hypothesis to explain the systemic symptoms of IgAV and IgAVN.26 In this model, it is proposed that infection with microorganisms that have similar antigenic structures, as human vessel walls or genetic influences, could lead to the production of cross-reactive anti-endothelial cell antibodies (IgA1-AECA). IgA1-AECA bind to small vessels and induce the production of interleukin-8, which is a potent chemoattractant for neutrophils. Neutrophils become activated and cause damage to the vascular endothelial cells.2
However, none of the proposed models can explain the IgAV pathogenesis in the proportion of IgAV patients (<10%) who do not have elevated Gd-IgA1 in serum or in biopsy specimens, nor can they explain the observation that the disease occurs only in some people with a IgA1 glycosylation defect, while in others with elevated Gd-IgA1 the disease does not occur.2,15,28
IgAVN and Other Clinical Manifestations
Renal involvement ranges from urinary abnormalities (including hematuria or/and proteinuria) through nephritic and nephrotic syndrome to chronic renal failure.2 It is typically mild and manifested only by pathological urine findings. However, chronic renal failure has been reported in 1 to 15% of children with IgAV who develop nephritis, and in the vast majority it is diagnosed within 6 months of disease onset.13,29,30
Cutaneous manifestations in the form of nonthrombocytopenic purpura or petechiae with lower limb predominance are the most common and are a characteristic sign of the disease.1 Alterations to the skin have proved to be a symptom in approximately 75% of patients, usually preceding other symptoms.31 They are present in all patients, but atypical distributions of skin changes are also possible, affecting the head and neck area, involving the upper extremities more than the lower extremities, sparing the lower extremities totally or otherwise leaving diffusely distributed lesions.2 In the most severe cases, bullae, ulcerations and necrotic lesions can be seen. Such skin alterations developed in 2–3% of patients with IgAV and recently it was shown that with increasing severity and duration of cutaneous manifestations in IgAV, the risk of developing IgAVN increases, making the prognosis worse with a greater likelihood to have required more aggressive treatment.32 Furthermore, it was found that the presence of ulcerations and necroses, persistent purpura (more than 1 month) and age were significant predictors of IgAVN, while persistent purpura, male gender and age were predictors of persistent IgAVN (hematuria and/or proteinuria more than 3 months).32
Musculoskeletal manifestations are the second most common feature and up to 70–90% of patients will have arthralgia or arthritis.31 The affected joints have painful ranges of movement due to periarticular swelling, but the characteristic findings of warmth, erythema and effusion are often absent.
Gastrointestinal manifestations are present in more than 50% of children with IgAV, and about 10–20% of patients with gastrointestinal involvement may develop serious complications such as intussusception, bowel perforation and massive bleeding.7 The abdominal pain is the most frequent sign of gastrointestinal involvement, and it is characteristically colicky and localised to the periumbilical and epigastric regions. According to newer study the patients with IgAV started with gastrointestinal symptoms, older children with severe gastrointestinal symptoms (severe abdominal pain, intussusception, hematochezia and/or massive gastrointestinal bleeding) of IgAV were a particularly high-risk group for developing IgAVN.33
In 10–20% of boys, orchitis may develop. Other rare manifestations include lung involvement, neurological manifestations and multiple organ involvement.34
Diagnostic Considerations
There are currently no diagnostic criteria for IgAV nor biomarkers in routine clinical use for IgAV and IgAVN in order to stratify patients with respect to the risk of developing kidney disease progression and contribute to the earlier diagnosis of renal involvement. For this reason, in our daily clinical practice we rely on classification criteria defined by the European League Against Rheumatism (EULAR), Paediatric Rheumatology International Trials Organization (PRINTO) and Paediatric Rheumatology European Society (PRES).1 The criteria provide sensitivity and specificity in the classification of IgAV (using other forms of vasculitis as controls) at 100% and 87%, respectively. The mandatory criterion is palpable purpura in association with at least one of the following: diffuse abdominal pain, arthritis or arthralgia, renal involvement (hematuria and/or proteinuria), and IgA deposition in biopsy specimens (skin, intestinal tract and kidney). A skin biopsy is not obligatory, but it is required in the event of atypically distributed skin change and is recommended if the rash is severe, in order to exclude other forms of vasculitis (Figure 1). The finding of a leucocytoclastic vasculitis associated with IgA deposition in a skin biopsy can help to accurately diagnose IgAV, but absence of IgA staining on biopsy does not exclude the diagnosis of IgAV.35,36
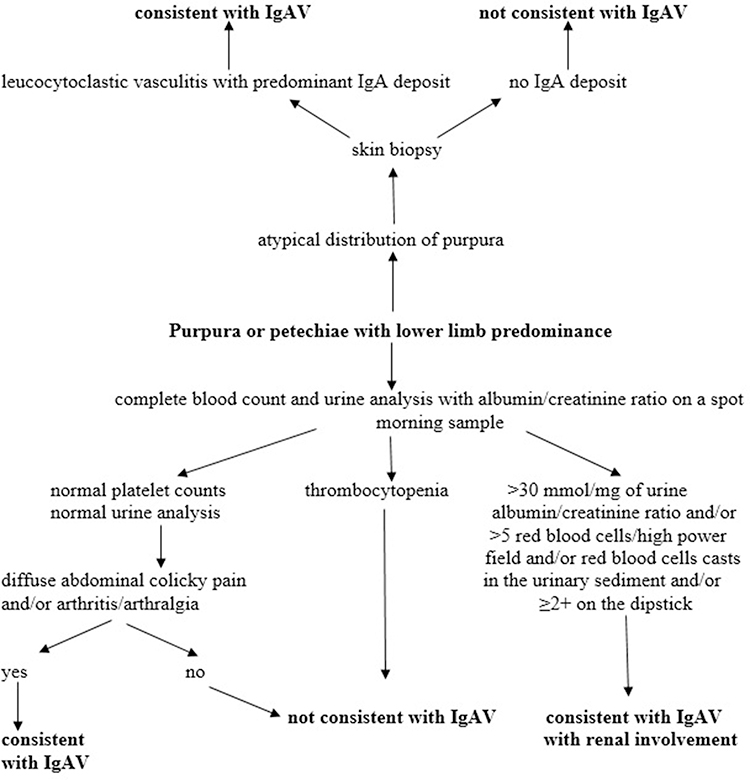 |
Figure 1 Diagnostic approach to IgAV and IgAVN. Data from Ozen et al.35 Adapted from Jelusic M, Sestan M, Giani T, Cimaz R. New insights and challenges associated with IgA vasculitis and IgA vasculitis with nephritis-is it time to change the paradigm of the most common systemic vasculitis in childhood? Front Pediatr. 2022;10:853724. Creative commons.36 |
Recently, the European initiative SHARE (Single Hub and Access point for pediatric Rheumatology in Europe) developed European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis.35
In all patients with IgAV blood pressure should be measured and renal involvement should be investigated using estimated glomerular filtration rate (eGFR) and urinalysis that includes assessment of hematuria and proteinuria with urine protein: urine creatinine ratio or urine albumin: urine creatinine ratio in an early morning urine sample.35 If there is hypertension, macroscopic haematuria or significant proteinuria, investigations to consider are complete blood count with platelet count and differential, urea, serum creatinine and electrolyte levels, urinary protein–creatinine ratio, albumin and coagulation studies.
Kidney biopsy is still the gold standard for the diagnosis of IgAVN.2 According to the SHARE guidelines, renal biopsy should be performed in patients with severe proteinuria defined as urine protein: urine creatinine ratio >250 mg/mmol for at least 4 weeks and persistent moderate proteinuria defined as urine protein: urine creatinine ratio 100–250 mg/mmol for at least 3 months or >50 mg/mmol for 6 months.35 Impaired eGFR (<80 mL/min/1.73 m2) is also absolute indication for kidney biopsy. Relative indications include nephritic and/or nephrotic syndrome and/or worsening of renal function (rapidly progressive glomerulonephritis).
Several histological classifications are used in the analysis of renal biopsy findings in IgAVN, but it remains unknown which one has the strongest association with the severity and outcome.2,29 According to the analysis of histological classifications used in different parts of the world for IgAVN,29 the most commonly used is that of the International Study of Kidney Disease in Children (ISKDC),37,38 followed by the revised Oxford classification.39 In some parts of the world, histologic classification of IgA nephropathy of Haas and the modified semi-quantitative classification developed by a Finnish group of authors are used.40,41 Although the treatment recommendations of SHARE are based on the ISKDC classification, this classification is not without limitations, the most important of which is focusing only on glomeruli and active inflammation, neglecting vascular and tubulointerstitial changes.10,42 Meanwhile, the revised Oxford classification is increasingly being used. However, the working group of Oxford classification does not recommend its use in IgAVN since patients with IgAVN were not included in the validation cohort, and recent study showed that the Oxford classification could not be fully validated in IgAVN.39,43 On the other side, Finnish group of authors published the results according to which their modified semi-quantitative classification, the most complicated one, showed promising results with the need to include a larger number of patients to properly validate the classification.41 Most recently, international study on histologic classifications in IgAVN, which compared four previously mentioned classifications, demonstrated that the modified semi-quantitative classification followed by the Oxford classification are the best classifications to be used in renal biopsy analysis in patients with IgAVN.44
There are currently no biomarkers in routine clinical use for IgAV and IgAVN; noninvasive confirmation of nephritis is still pending. Many potential biomarkers are currently under investigation, but an increased serum level of Gd-IgA1 is still the most consistent finding in patients with IgAV.45 Regarding urinary biomarkers, which have an advantage over blood biomarkers, because they are easier to collect, without using invasive procedures, IgA and IgM performed best in the study conducted by Pillebout et al27 Many promising biomarkers in predicting nephritis are on the horizon: kidney injury molecule-1 (KIM-1), monocyte chemotactic protein-1 (MCP-1), N-acetyl-β-glucosaminidase (NAG), and angiotensinogen (AGT).46 Further studies are needed.
New approaches in the detection of potential biomarkers include the application of proteomics and metabolomics. Proteomics investigates protein interaction networks and identifies their biological functions. Using liquid chromatography-tandem mass spectrometry, Fang et al detected two proteins in the urine of IgAVN patients, integrin beta-1 and tenascin, that participate in multiple pathways in disease.47 However, since only the level of tenascin was different among IgAVN children and IgAV children, they concluded that tenascin might be a novel biomarker of early renal damage in IgAVN. Metabolomics is a newly emerged field involved in the identification and quantification of metabolites in biofluids, cells and tissues. Several studies have applied metabolomics to reveal molecular mechanism of IgAVN pathogenesis and define potential biomarkers.48–50 Sun et al identified (S)-3-hydroxyisobutyric acid, p-Cresol sulfate, and 3-carboxy-4-methyl-5-pentyl-2-furanpropanoic acid that with high sensitivity and specificity may predict development of IgAVN among IgAV patients.48 Demir et al suggest that DHAP (18:0), prostaglandin D2/I2, porphobilinogen, 5-methyltetrahydrofolic acid, and N-Acetyl-4-O-acetylneuraminic acid/N-Acetyl-7-O-acetylneuraminic acid may serve as biomarkers for predicting renal involvement in IgAV considering these metabolites were increased in IgAVN patients before the onset of renal involvement.49 Zhang et al found that choline and cis-vaccenic acid could serve as biomarkers to predict the progression of IgAVN.50 Further biological validation is expected.
In addition to urine analysis, renal and skin biopsy, other investigations may be required to rule out differentials if the diagnosis is unclear: blood and urine cultures, abdominal imaging, antistreptolysin O titer and anti-DNAse B while antinuclear antibodies (ANA), anti-double stranded DNA antibodies (dsDNA), ANCA, C3 and C4 may be necessary if there is significant renal involvement with an unclear diagnosis.51 In patients with IgAV leukocytosis with eosinophilia and a left shift may be observed and thrombocytosis is present in many of the patients. Erythrocyte sedimentation rate may be mildly elevated in the most patients. Elevation of blood urea nitrogen and creatinine indicate decreased kidney function. Stool guaiac test may reveal occult blood. It is possible to find coagulation disorders: D-dimer may be substantially increased, while prothrombin time and activated partial thromboplastin time may be reduced. Serum IgA may be increased during the acute phase of illness but not in all patients.51
There are limited data for vasculitis activity and damage assessment in children with IgAV.2 Since disease activity and damage have been recognised as the key components of outcome measures in patients with various forms of vasculitis for a standardised comparison of patient cohorts for clinical trials, as well for observing the course of an individual patient’s disease, it is important to validate the available assessment tools adjusted for the pediatric population.2,52 Therefore, for the purpose of determining the disease activity and degree of kidney damage in patients with IgAV/IgAVN, two clinical questionnaires are used: the Paediatric Vasculitis Activity Score (PVAS) and Paediatric Vasculitis Damage Index (PVDI).
PVAS is a set of 64 clinical variables (symptoms or signs of disease) which are divided into 9 organ systems and it is assessed on which of them are new or have deteriorated in the previous 4 weeks or have persisted, but for no longer than a period of 3 months. The presence of each of the variables is evaluated with a certain number of points which are then added together. The total number of points is in the range from 0 to 63 points and represents the activity of the disease at the time of scoring.52
PVDI represents a set of 72 clinical variables which are divided into 9 organ systems and an “other” section. Damage is defined by the duration of symptoms or signs lasting a period of at least 3 months, which have occurred at any time since the onset of the disease.53
All patients with IgAV should be followed-up for at least 6–12 months even if the initial blood pressure measurements and urinalysis are normal.35 The minimum set of tests includes blood pressure measurement and urinalysis for the presence of haematuria and quantification of albuminuria and/or proteinuria. It has not yet been defined whether the follow-up time should be extended for some groups of patients, ie, older children with the onset of gastrointestinal symptoms before other IgAV symptoms and severe GI form of IgAV, as well as those who develop ulcerations and necroses and persistent purpura, since they may be at higher risk for the later development of nephritis.32,33
There are different suggestions for monitoring patients. According to one strategy, the initial urinalysis should be repeated three times.54 In the case of normal findings, during the first month of follow-up, the urinalysis must be repeated once a week, then once a month during the next 6 months, and then once every 6 months. According to the second strategy, blood pressure measuring and urinalysis should be repeated weekly during the first month of the disease, then every 2 weeks in the second and third month, every month from the third to sixth month and then at 9th and 12th month.55 Special attention should be paid to pregnant women with IgAVN with onset in childhood, because in these patients disease may complicate by hypertension and/or proteinuria.56
Treatment Strategies
In the vast majority of patients with IgAV, during the self-limited nature of the disease, specific treatment is not required.2 The optimal way to treat patients with severe skin manifestation is not known, although the majority of these patients are treated with systemic glucocorticoids, sometimes in combination with dapsone or azathioprine.32 Musculoskeletal involvement is usually treated with rest and analgesia, while other treatment options are rarely indicated. However, the total opposite can be recommended in patients with severe gastrointestinal manifestations, renal involvement or those who develop other complications such as lung involvement, neurological manifestations and multiple organ involvement.2 There are two recommendations that can be used in the treatment with IgAVN (Table 1). The Kidney Disease: Improving Global Outcomes (KDIGO) practice guideline on glomerulonephritis is older and in one chapter provide recommendations for the treatment of IgAVN in children and adults.57 European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis developed by the SHARE initiative are newer ones.35 The latter also provide information about the treatment of severe gastrointestinal manifestations. In patients with severe abdominal pain or gastrointestinal hemorrhage, glucocorticoids should be considered: orally, or with pulsed glucocorticoids if the oral route is not an option or if they fail to respond. Second-line treatments may include mycophenolate mofetil, cyclophosphamide, intravenous immunoglobulin, rituximab, methotrexate, colchicine and hydroxychloroquine.31 Other supportive treatment may be required: nasogastric decompression, parenteral nutrition and antibiotics.2
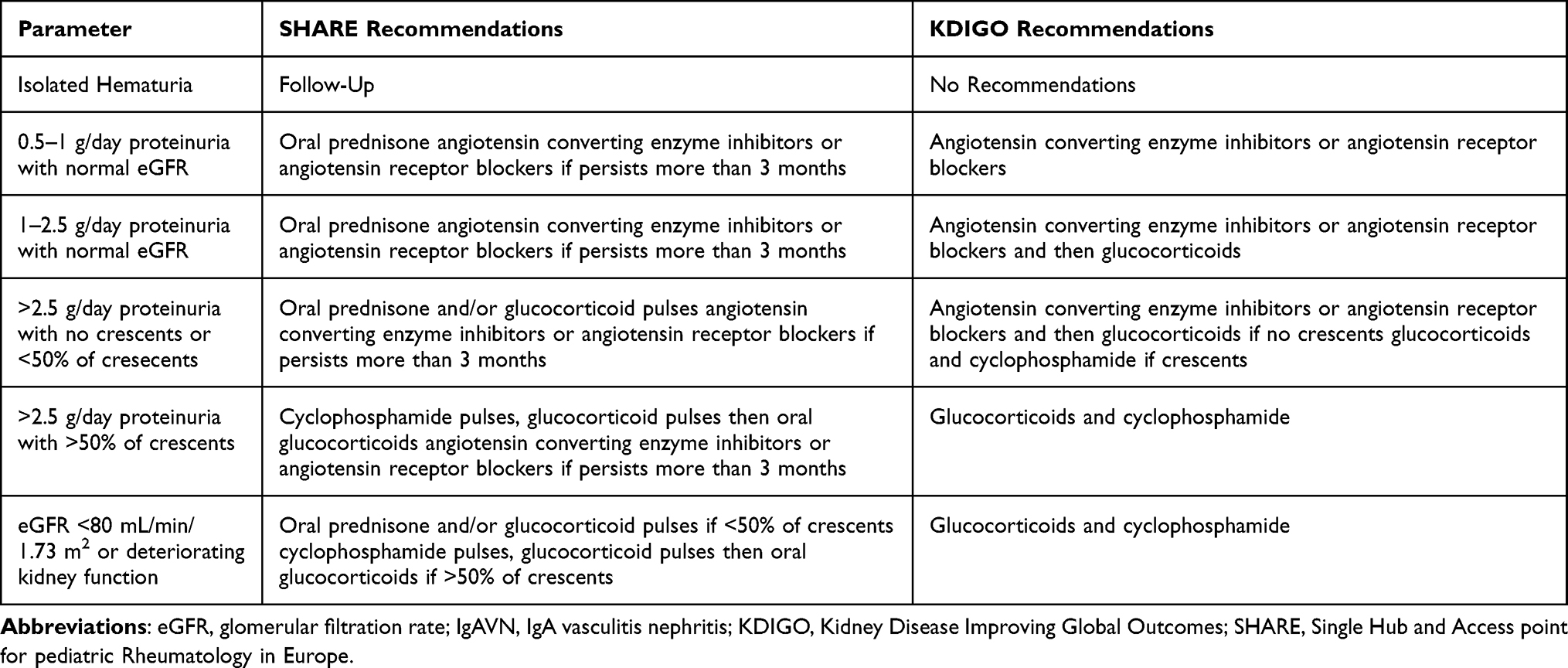 |
Table 1 Comparison of the SHARE and KDIGO Recommendations for Treatment of IgAVN |
According to the SHARE management algorithm (Figure 2), IgAVN is divided into three categories, using three parameters: proteinuria, estimated glomerular filtration rate and percentage of crescents on a renal biopsy.35 Children without renal dysfunction or proteinuria usually do not require any specific therapeutic intervention. First-line therapy in patients with mild forms of IgAVN, defined as ≤2.5 g/day of proteinuria in 24 h urine collection with normal estimated glomerular filtration rate, is oral glucocorticoids which are usually sufficient, and in the event of persistence of proteinuria, second-line drugs may be used: azathioprine, mycophenolate mofetil or glucocorticoid pulses. In the treatment of moderate IgAVN, defined as <50% crescents on a renal biopsy and an impaired estimated glomerular filtration rate (<80 mL/min/1.73 m2) or severe persistent proteinuria (>2.5 g/day of proteinuria in 24 h urine collection for more than 4 weeks), the preferred option is to use glucocorticoids, which usually need to be administered parenterally and in pulsed doses, and in the absence of any effect, second-line drugs are added: azathioprine, mycophenolate mofetil or cyclophosphamide parenterally. Treatment of the most severe forms of IgAVN consists of induction using pulsed doses of glucocorticoids in combination with intravenous cyclophosphamide pulses, and continues as a maintenance therapy with lower doses of glucocorticoids in combination with immunomodulators: azathioprine or mycophenolate mofetil.2,35 Angiotensin converting enzyme inhibitors or angiotensin receptor blockers are recommended to prevent or limit secondary glomerular damage in patients with IgAVN who have persistent proteinuria (lasting a period of more than 3 months).
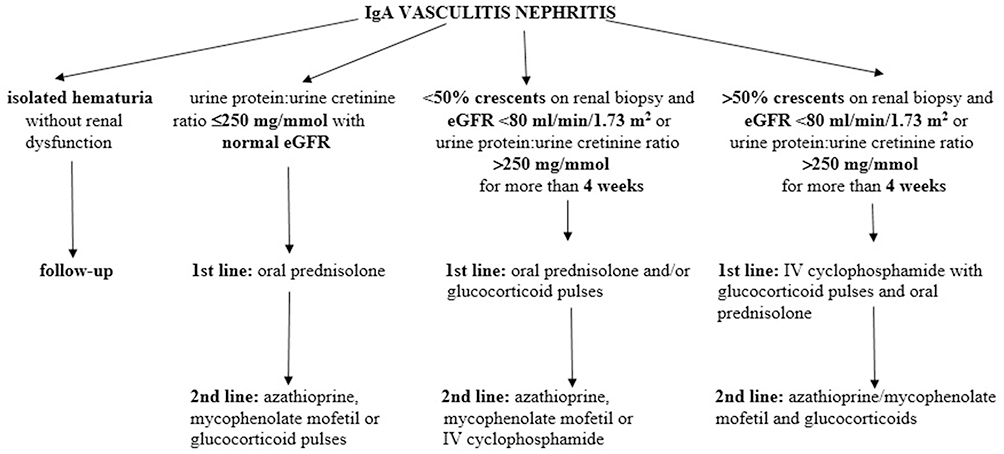 |
Figure 2 The SHARE recommendations for IgAVN treatment. Data from Ozen et al.35 Jelusic M, Sestan M, Giani T, Cimaz R. New insights and challenges associated with IgA vasculitis and IgA vasculitis with nephritis-is it time to change the paradigm of the most common systemic vasculitis in childhood? Front Pediatr. 2022;10:853724. Creative Commons.36 |
For the most severe, unresponsive cases, there is an option of plasma exchange which demonstrated an efficacy in one study, while there is not enough evidence regarding the use of rituximab (although it is used in severe cases, and there are case reports and case series concerning its usage) or intravenous immunoglobulins.31,58
According to the KDIGO recommendations, patients with IgAVN are also divided into three categories, but they are defined differently.2,57 The mildest category is represented by patients with proteinuria 0.5–1 g/day per 1.73 m2, for which treatment with angiotensin converting enzyme inhibitors or angiotensin receptor blockers is suggested. The second group consists of patients with proteinuria >1 g/day per 1.73 m2. These patients should receive a 6-month course of glucocorticoid therapy after a trial of angiotensin converting enzyme inhibitors or angiotensin receptor blockers. Glucocorticoids and cyclophosphamide should be used in patients with crescentic IgAVN with nephrotic syndrome and/or deteriorating kidney function.
Knowledge of new pathogenetic mechanisms of IgAVN enables the development of new treatment strategies. They can be aimed at reducing production of Gd-IgA1 and autoantibodies or on suppression of the alternative or lectin complement pathway and blocking mesangial cell activation.21 For now, these new therapeutic strategies are being tested mainly in IgA nephropathy, which some consider to be pathogenetically close to IgAV. Therapeutic options directed on the production of Gd-IgA1 include oral budesonide, which can target Peyer’s patches in the ileum, that are thought to be origin of Gd-IgA1 production.59 Another option targeting the similar mechanism is a proteasome inhibitor, bortezomib, which acts as a plasma cell depleting agent affecting the production of IgG autoantibodies.60 Rituximab is a monoclonal antibody against surface cell marker on B cells, CD20, which may suppress the production of autoantibodies and reduce the synthesis of Gd-IgA1. A systematic review from 2020 described clinical improvement and remission in significant percentage of patients with refractory IgAV.61 Complement inhibitors such as eculizumab and experimental drugs APL-2, CCX168, LNP023, and OMS721 represent new potential therapeutic targets and may be promising therapeutic interventions in the future.62 Recently, the spleen tyrosine kinase inhibitors demonstrated successful inhibition of pro-inflammatory responses in rat model of vasculitis with significant improvement of renal pathology and preserved renal function.63
Conclusions
Although IgAV is the most common systemic vasculitis in children, and nephritis is its most important chronic complication, there are still open questions related to the diagnostic approach and treatment. The most important future steps will be to find non-invasive biomarkers that will correlate with the severity and prognosis of nephritis and to find new therapeutic options that will be based on the pathogenetic mechanisms of the disease.
Acknowledgments
This work has been supported in part by Croatian Science Foundation under the project IP-2019-04-8822. This work has been based on the PhD thesis: M. Sestan. Contribution of the whole exome sequencing in the identification of genetic variants associated with childhood-onset systemic lupus and IgA vasculitis. PhD [dissertation]. Zagreb: University of Zagreb School of Medicine; 2022. Available from https://urn.nsk.hr/urn:nbn:hr:105:954607.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ozen S, Pistorio A, Iusan SM, et al. EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. part II: final classification criteria. Ann Rheum Dis. 2010;69(5):798–806. doi:10.1136/ard.2009.116657
2. Sestan M Contribution of the Whole Exome Sequencing in the Identification of Genetic Variants Associated With Childhood-Onset Systemic Lupus and IgA Vasculitis PhD [dissertation]. Zagreb: University of Zagreb School of Medicine; 2022. Available from: https://urn.nsk.hr/urn:nbn:hr:105:954607.
3. Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360:1197–1202.
4. Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1–11.
5. Olson JC, Kelly KJ, Pan CG, Wortmann DW. Pulmonary disease with hemorrhage in Henoch-Schöenlein purpura. Pediatrics. 1992;89:1177–1181.
6. Reid-Adam J. Henoch-Schonlein purpura. Pediatr Rev. 2014;35:447–449.
7. Trnka P. Henoch-Schönlein purpura in children. J Paediatr Child Health. 2013;49:995–1003.
8. Davin JC, Coppo R. Henoch-Schönlein purpura nephritis in children. Nat Rev Nephrol. 2014;10:563–573.
9. Davin JC, Ten Berge IJ, Weening JJ. What is the difference between IgA nephropathy and Henoch-Schönlein purpura nephritis? Kidney Int. 2001;59:823–834.
10. Davin J-C, Coppo R. Pitfalls in recommending evidence-based guidelines for a protean disease like Henoch–Schönlein purpura nephritis. Pediatr Nephrol. 2013;28(10):1897–1903. doi:10.1007/s00467-013-2550-4
11. Piram M, Maldini C, Biscardi S, et al. Incidence of IgA vasculitis in children estimated by four-source capture–recapture analysis: a population-based study. Rheumatology. 2017;56(8):1358–1366. doi:10.1093/rheumatology/kex158
12. Park SJ, Suh J-S, Lee JH, et al. Advances in our understanding of the pathogenesis of Henoch-Schönlein purpura and the implications for improving its diagnosis. Expert Rev Clin Immunol. 2013;9(12):1223–1238. doi:10.1586/1744666X.2013.850028
13. Sapina M, Frkovic M, Sestan M, et al. Geospatial clustering of childhood IgA vasculitis and IgA vasculitis-associated nephritis. Ann Rheum Dis. 2021;80(5):610–616. doi:10.1136/annrheumdis-2020-218649
14. Brogan P, Eleftheriou D, Dillon M. Small vessel vasculitis. Pediatr Nephrol. 2010;25(6):1025–1035. doi:10.1007/s00467-009-1317-4
15. Pohl M. Henoch–Schönlein purpura nephritis. Pediatr Nephrol. 2015;30(2):245–252. doi:10.1007/s00467-014-2815-6
16. Kiryluk K, Li Y, Sanna-Cherchi S, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012;8(6):e1002765. doi:10.1371/journal.pgen.1002765
17. Rigante D, Castellazzi L, Bosco A, Esposito S. Is there a crossroad between infections, genetics, and Henoch–Schönlein purpura? Autoimmun Rev. 2013;12(10):1016–1021. doi:10.1016/j.autrev.2013.04.003
18. Jelusic M, Sestan M. IgA vasculitis or Henoch-Schönlein purpura: genetics and beyond. Pediatr Nephrol. 2021;36(8):2149–2153. doi:10.1007/s00467-021-04987-z
19. Lopez-Mejias R, Carmona FD, Castaneda S, et al. A genome-wide association study suggests the HLA class II region as the major susceptibility locus for IgA vasculitis. Sci Rep. 2017;7(1):5088. doi:10.1038/s41598-017-03915-2
20. Koskela M, Nihtilä J, Ylinen E, et al. HLA-DQ and HLA-DRB1 alleles associated with Henoch-Schönlein purpura nephritis in Finnish pediatric population: a genome-wide association study. Pediatr Nephrol. 2021;36(8):2311–2318. doi:10.1007/s00467-021-04955-7
21. Hastings MC, Rizk DV, Kiryluk K, et al. IgA vasculitis with nephritis: update of pathogenesis with clinical implications. Pediatr Nephrol. 2022;37(4):719–733. doi:10.1007/s00467-021-04950-y
22. Hwang HH, Lim IS, Choi B, Yi DY. Analysis of seasonal tendencies in pediatric Henoch–Schönlein purpura and comparison with outbreak of infectious diseases. Medicine. 2018;97(36):e12217. doi:10.1097/MD.0000000000012217
23. Weiss PF, Klink AJ, Luan X, et al. Temporal association of streptococcus, staphylococcus, and parainfluenza pediatric hospitalizations and hospitalized cases of Henoch-Schönlein purpura. The Journal of Rheumatology. 2010;37(12):2587–2594. doi:10.3899/jrheum.100364
24. Gonzalez LM, Janniger CK, Schwartz RA. Pediatric Henoch-Schönlein purpura. Int J Dermatol. 2009;48(11):1157–1165. doi:10.1111/j.1365-4632.2009.04162.x
25. Suzuki H, Raska M, Yamada K, et al. Cytokines alter IgA1 O-glycosylation by dysregulating C1GalT1 and ST6GalNAc-II enzymes. J Biol Chem. 2014;289(8):5330–5339. doi:10.1074/jbc.M113.512277
26. Heineke MH, Ballering AV, Jamin A, Ben Mkaddem S, Monteiro RC, Van Egmond M. New insights in the pathogenesis of immunoglobulin A vasculitis (Henoch-Schönlein purpura). Autoimmun Rev. 2017;16:1246–1253.
27. Pillebout E, Jamin A, Ayari H, et al. Biomarkers of IgA vasculitis nephritis in children. PLoS One. 2017;12:e0188718.
28. Chen JY, Mao JH. Henoch-Schönlein purpura nephritis in children: incidence, pathogenesis and management. World J Pediatr. 2015;11:29–34.
29. Jelusic M, Sestan M, Cimaz R, Ozen S. Different histological classifications for Henoch-Schönlein purpura nephritis: which one should be used? Pediatr Rheumatol Online J. 2019;17:10.
30. Goldstein AR, White RHR, Akuse R, Chantler C. Long-term follow-up of childhood Henoch-Schönlein nephritis. Lancet. 1992;339:280–282.
31. Oni L, Sampath S. Childhood IgA vasculitis (Henoch Schonlein purpura) - advances and knowledge gaps. Front Pediatr. 2019;7:257.
32. Sestan M, Srsen S, Kifer N, et al. Persistence and severity of cutaneous manifestations in IgA vasculitis is associated with development of IgA vasculitis nephritis in children. Dermatology. 2022;238:340–346.
33. Sestan M, Kifer N, Frkovic M, et al. Gastrointestinal involvement and its association with the risk for nephritis in IgA vasculitis. Ther Adv Musculoskelet Dis. 2021;13:1759720X211024828.
34. Rajagopala S, Shobha V, Devaraj U, D’Souza G, Garg I. Pulmonary hemorrhage in Henoch-Schönlein purpura: case report and systematic review of the English literature. Semin Arthritis Rheum. 2013;42:391–400.
35. Ozen S, Marks SD, Brogan P, et al. European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-the SHARE initiative. Rheumatology. 2019;58:1607–1616.
36. Jelusic M, Sestan M, Giani T, Cimaz R. New insights and challenges associated with IgA vasculitis and IgA vasculitis with nephritis-is it time to change the paradigm of the most common systemic vasculitis in childhood? Front Pediatr. 2022;10:853724.
37. Counahan R, Winterborn MH, White RH, et al. Prognosis of Henoch-Schonlein nephritis in children. BMJ. 1977;2:11–14.
38. Haas M. IgA nephropathy and Henoch-Schönlein Purpura nephritis. In: Jennette JC, Olson JC, Schwartz MM, Silva FG, editors. Heptinstall’s Pathology of the Kidney. Philadelphia: Lippincott, Williams & Wilkins; 2007:423–486.
39. Trimarchi H, Barratt J, Cattran DC, et al. Oxford classification of IgA nephropathy 2016: an update from the IgA nephropathy classification working group. Kidney Int. 2017;91:1014–1021.
40. Haas M. Histologic subclassification of IgA nephropathy: a clinicopathologic study of 244 cases. Am J Kidney Dis. 1997;29:829–842.
41. Koskela M, Ylinen E, Ukonmaanaho E-M, et al. The ISKDC classification and a new semiquantitative classification for predicting outcomes of Henoch-Schönlein purpura nephritis. Pediatr Nephrol. 2017;32:1201–1209.
42. Davin JC. Henoch-Schönlein Purpura nephritis: pathophysiology, treatment, and future strategy. Clin J Am Soc Nephrol. 2011;6:679–689.
43. Yu B, Shi S, Hou W, et al. Evaluation of the Oxford classification in immunoglobulin A vasculitis with nephritis: a cohort study and meta-analysis. Clin Kidney J. 2020;14:516–525.
44. Kifer N, Bulimbasic S, Sestan M, et al. SQC and Oxford classifications predict poor renal outcome better than ISKDC and Haas in patients with IgAV nephritis: a multicenter study. J Nephrol. 2022;2022:1.
45. Brogan P, Eleftheriou D. Vasculitis update: pathogenesis and biomarkers. Pediatr Nephrol. 2018;33:187–198.
46. Williams CEC, Toner A, Wright RD, Oni L. A systematic review of urine biomarkers in children with IgA vasculitis nephritis. Pediatr Nephrol. 2021;36:3033–3044.
47. Fang X, Wu H, Lu M, et al. Urinary proteomics of Henoch-Schönlein purpura nephritis in children using liquid chromatography-tandem mass spectrometry. Clin Proteomics. 2020;17:10.
48. Sun L, Xie B, Zhang Q, et al. Biomarkers identification by a combined clinical and metabonomics analysis in Henoch-Schonlein purpura nephritis children. Oncotarget. 2017;8:114239–114250.
49. Demir S, Kaplan O, Celebier M, et al. Predictive biomarkers of IgA vasculitis with nephritis by metabolomic analysis. Semin Arthritis Rheum. 2020;50:1238–1244.
50. Zhang Q, Lai LY, Cai YY, et al. Serum-urine matched metabolomics for predicting progression of Henoch-Schonlein purpura nephritis. Front Med. 2021;8:657073.
51. Bhimma R, Nandlal L. Henoch-Schonlein Purpura (IgA Vasculitis). Available from: https://emedicine.medscape.com/article/984105-overview.
52. Dolezalova P, Price-Kuehne FE, Özen S, et al. Disease activity assessment in childhood vasculitis: development and preliminary validation of the Paediatric Vasculitis Activity Score (PVAS). Ann Rheum Dis. 2013;72:1628–1633.
53. Dolezalova P, Wilkinson N, Brogan PA, et al. SAT0286 Paediatric vasculitis damage index: a new tool for standardised disease assessment. Ann Rheum Dis. 2014;73:696.
54. Peruzzi L, Coppo R. IgA vasculitis nephritis in children and adults: one or different entities? Pediatr Nephrol. 2021;36:2615–2625.
55. Abu-Zaid MH, Salah S, Lotfy HM, et al. Consensus evidence-based recommendations for treat-to-target management of immunoglobulin A vasculitis. Ther Adv Musculoskelet Dis. 2021;13:1759720X211059610.
56. Ronkainen J, Nuutinen M, Koskimies O. The adult kidney 24 years after childhood Henoch-Schonlein purpura: a retrospective cohort study. Lancet. 2002;360:666–670.
57. Radhakrishnan J, Cattran DC. The KDIGO practice guideline on glomerulonephritis: reading between the (guide) lines–application to the individual patient. Kidney Int. 2012;82:840–856.
58. Crayne CB, Eloseily E, Mannion ML, et al. Rituximab treatment for chronic steroid dependent Henoch-Schonlein purpura: 8 cases and a review of the literature. Pediatr Rheumatol Online J. 2018;16:71.
59. Fellström BC, Barratt J, Cook H, et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): a double-blind, randomised, placebo-controlled phase 2b trial. Lancet. 2017;389:2117–2127.
60. Van de Perre E, Smith RM, Bardsley V, Crawley C, Willcocks LC, Jayne DR. Successful outcome using bortezomib in adult refractory IgA vasculitis: a case report. Rheumatology. 2016;55:2089–2091.
61. Hernández-Rodríguez J, Carbonell C, Mirón-Canelo JA, Diez-Ruiz S, Marcos M, Chamorro AJ. Rituximab treatment for IgA vasculitis: a systematic review. Autoimmun Rev. 2020;19:102490.
62. Rizk DV, Maillard N, Julian BA, et al. The emerging role of complement proteins as a target for therapy of IgA nephropathy. Front Immunol. 2019;10:504.
63. McAdoo SP, Prendecki M, Tanna A, et al. Spleen tyrosine kinase inhibition is an effective treatment for established vasculitis in a pre-clinical model. Kidney Int. 2020;97:1196–1207.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.