")
Back to Journals » ImmunoTargets and Therapy » Volume 13
Dermatomyositis: Practical Guidance and Unmet Needs
Authors Cassard L , Seraly N, Riegert M, Patel A, Fernandez AP
Received 1 December 2023
Accepted for publication 13 February 2024
Published 6 March 2024 Volume 2024:13 Pages 151—172
DOI https://doi.org/10.2147/ITT.S381472
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Lydia Cassard,1 Noelle Seraly,2 Maureen Riegert,3 Aditi Patel,4 Anthony P Fernandez5
1Cleveland Clinic Lerner College of Medicine at Case Western Reserve University, Cleveland, OH, USA; 2Department of Dermatology, Cleveland Clinic, Cleveland, OH, USA; 3Department of Dermatology, Rush University, Chicago, IL, USA; 4Department of Rheumatology, Cleveland Clinic, Cleveland, OH, USA; 5Departments of Dermatology and Pathology, Cleveland Clinic, Cleveland, OH, USA
Correspondence: Anthony P Fernandez, Departments of Dermatology and Pathology, Cleveland Clinic, 9500 Euclid Avenue; A61, Cleveland, OH, 44195, USA, Email [email protected]
Abstract: Dermatomyositis is a heterogeneous idiopathic inflammatory myopathy associated with various cutaneous manifestations and variable presence of myositis, interstitial lung disease, and other visceral organ involvement. An accurate diagnosis of dermatomyositis requires correlating clinical examination findings with serological and histological findings. Familiarity with pathognomonic and common cutaneous manifestations of dermatomyositis, which are highlighted here, can be especially helpful in making an accurate diagnosis. Additionally, evaluating patients for presence of myositis-specific autoantibodies can further support or refute a dermatomyositis diagnosis. When present, myositis-specific autoantibodies can also help guide workups for various dermatomyositis-associated manifestations, as each is associated with relatively distinct clinical characteristics. Evaluating patients for various systemic manifestations often relies on expert opinion recommendations; however, societal guideline statements concerning the evaluation of some manifestations have recently been described. Although malignancy-associated dermatomyositis is a well-accepted subtype, there is limited evidence to support extensive malignancy screening has a favorable benefit–risk ratio in most dermatomyositis patients. However, recent research has uncovered novel associations between dermatomyositis and malignancy, suggesting the possibility of identifying high-risk subsets of dermatomyositis patients in whom malignancy screening may have a high value. Treatment for dermatomyositis has remained largely unchanged over the past several decades. Although many dermatomyositis patients can be effectively treated with current options, either as monotherapy or with combination regimens, there is a need for more targeted and effective DM therapies, in general, and for MDA5(+) dermatomyositis-associated rapidly progressive interstitial lung disease. Fortunately, significant current and emerging research activities evaluating various novel medications for dermatomyositis provide hope for exciting future advances in patients with this intriguing immune-mediated disease.
Keywords: idiopathic inflammatory myopathy, myositis, autoantibody, malignancy, MDA-5, treatment
Introduction
Dermatomyositis (DM) is an idiopathic inflammatory myopathy (IIM) with characteristic skin features and heterogeneous systemic manifestations. Incidence and prevalence of DM vary geographically, with the highest adult incidence rate seen in the United States (US; 15.2/million person-years) and highest prevalence rates seen in the US (21.4/100,000 people) and Spain (19/100,000).1 DM incidence demonstrates a bimodal distribution, with juvenile DM commonly diagnosed between ages 4–14 years and adult DM commonly diagnosed between ages 40–60 years.2
The pathogenesis of DM is multifactorial and not completely understood, with contributions from genetic, environmental, and immunological factors. Genetic contributions are supported by observed associations with major histocompatibility complex (MHC) polymorphisms and human leukocyte antigen alleles.3 Additionally, potential environmental triggers have been identified, including ultraviolet radiation, viral infections, medications, and pollutants, all of which may lead to immune dysregulation in genetically susceptible individuals.4
Once immune dysregulation occurs, substantial evidence supports that type I interferons (IFNs), which regulate both innate and adaptive immune responses, play a key role in DM pathogenesis.5,6 The role of type I IFNs in DM pathogenesis is supported by numerous studies demonstrating upregulation of type I IFN pathways and cytokines in lesional skin, muscle, and peripheral blood of DM patients, correlation between type I IFN signature levels and DM cutaneous disease activity, and induction of DM by IFN immunotherapy.5–7 Furthermore, changes in blood type I IFN scores correlate with disease activity changes in skin, muscle, and possibly other organs.7
Practical Guidance
Cutaneous Manifestations
Cutaneous manifestations represent key clinical features of DM and can be categorized as pathognomonic, characteristic, compatible, or rare. Pathognomonic cutaneous manifestations of DM include Gottron papules, Gottron’s sign, and heliotrope rash (Figure 1A–D). Gottron papules present as erythematous to violaceous papules/plaques overlying interphalangeal joints of the hands, feet, elbows, and knees. Gottron’s sign refers to non-palpable erythematous macules/patches overlying extensor surfaces of similar joints, most commonly elbows and knees. Heliotrope rash is characterized by periorbital violaceous erythema and edema.
 |
Figure 1 Pathognomonic Cutaneous Manifestations of Dermatomyositis. (A) A 69-year-old man with violaceous periorbital erythema and edema (heliotrope rash) related to TIF1γ(+) DM. He was subsequently diagnosed with diffuse large B-cell lymphoma. This patient provided written consent to use this image for publication. (B) A 60-year-old woman with TIF1γ autoantibodies and erythematous-to-violaceous papules overlying the interphalangeal joints of the hands (Gottron’s papules) related to TIF1γ(+) DM. (C) A 55-year-old woman with erythematous-to-violaceous patches on the knees (Gottron’s sign) related to TIF1γ(+) DM. (D) A 52-year-old woman with erythematous-to-violaceous patches on the elbows (Gottron’s sign) related to SAE(+) DM. |
Cutaneous manifestations considered characteristic of DM, but not pathognomonic, include Shawl sign, V-neck sign, Holster sign, scalp erythema, poikiloderma, and proximal nail fold (PNF) changes (Figures 2A–D and 3A–D). Shawl and V-neck signs present as erythematous-to-violaceous patches/plaques involving the posterior shoulders, neck, and upper back, and anterior neck and upper chest, respectively.8 The Holster sign refers to symmetric erythema involving the lateral hips and thighs. Scalp involvement in DM patients is also characteristic and may present as violaceous erythema with fine scale or psoriasiform plaques with thicker scale. Scalp involvement is typically accompanied by alopecia and significant pruritus. Poikiloderma is characterized by hypo- and hyperpigmentation, telangiectasia, and atrophy and typically occurs in areas of long-standing DM inflammation. Lastly, characteristic PNF changes include periungual erythema, dilated capillary loops, dystrophic cuticles, and hemorrhagic infarcts.
 |
Figure 2 Common Cutaneous Manifestations of Dermatomyositis. (A) A 55-year-old woman with erythematous patches and plaques involving the upper back and shoulders (shawl sign) related to TIF1γ(+) DM; (B) A 54-year-old man with erythematous patches and plaques involving the lateral Hip (holster sign) related to DM. He declined MSA testing; (C) A 78-year-old man with erythematous patches involving the anterior neck and upper chest (V-neck erythema) related to TIF1γ(+) DM; (D) A 66-year-old woman with erythematous-to-violaceous patches and plaques with thick, psoriasiform scale and associated alopecia involving the scalp in relation to TIF1γ(+) DM. |
Less common cutaneous DM manifestations are often associated with specific DM subtypes. Widespread erythema/erythroderma has been associated with coexisting malignancy. Wong-type DM may present with follicular hyperkeratosis on extensor surfaces; flagellate erythema has been associated with anti-Mi2-associated DM; Oral mucosal ovoid palatal patches are characteristically seen in anti-transcription intermediary factor 1-γ (TIF1γ)-associated DM; acral hyperkeratotic papules/plaques and fissures (mechanic’s hands) (Figure 3C) are typically associated with anti-synthetase syndrome (ASTS); cutaneous ulcerations and panniculitis are often associated with anti-melanoma differentiation-associated gene 5 (MDA5)-associated DM.2,9,10 Raynaud’s phenomenon also occurs in up to 40% of DM patients, especially those with ASTS. Finally, calcinosis cutis occurs in ~4% of adult DM patients, especially those with anti-nuclear matrix protein-2 (NXP2) antibodies (Figure 3B).11
 |
Figure 3 Other Cutaneous Manifestations of Dermatomyositis. (A) A 68-year-old woman with long-standing DM and skin atrophy, telangiectasia, and dyspigmentation (poikiloderma) in a V-neck distribution; (B) A 54-year-old man with long-standing DM and firm, white nodules of calcinosis cutis (arrows) involving his scalp; (C) A 42-year-old woman with Jo1(+) Antisynthetase syndrome with significant fissuring and hyperkeratosis of her fingers (Mechanic’s hands); (D) A 68-year-old woman with TIF1γ autoantibodies with (arrows) dilated proximal nailfold capillary loops, capillary dropout, and focal cuticular hemorrhage related to TIF1γ(+) DM. |
Cutaneous DM manifestations can sometimes overlap with those of other connective tissue diseases, making diagnosis challenging. We occasionally see DM patients who have been misdiagnosed with lupus erythematosus (LE) due to photodistributed cutaneous lesions that, when biopsied, have histologic features similar to DM. Awareness of hallmark cutaneous clues can help accurately diagnose DM in these patients. For example, DM facial erythema tends to involve nasolabial folds, which are spared in LE.8,10 Additionally, the scleroderma PNF pattern of low-density capillaries, hemorrhage, and dilated capillary loops is present significantly more often in DM than in LE.
Musculoskeletal Manifestations
Patients with both skin and muscle involvement are considered to have classic DM (CDM). Clinical evidence of myositis is found in ~80% of DM patients.2 The characteristic myopathy manifests as symmetric progressive proximal muscle weakness affecting the upper and lower extremities without associated muscle pain. With more prominent myositis activity, involvement of pharyngeal and esophageal striated muscles occurs, presenting as dysphagia and dysphonia. Involvement of neck extensor muscles may lead to difficulty holding the head up. In severe myositis, weakness of the diaphragm and accessory muscles of breathing may contribute to respiratory insufficiency and aspiration pneumonia risk.12
Notably, ~20% of patients develop amyopathic DM (ADM).13 ADM is defined as patients with hallmark clinical and biopsy-proven cutaneous DM manifestations who have no clinical, serologic, or other evidence of myositis for ≥6 months in the absence of systemic immunomodulatory therapy.13 Alternatively, some patients are best categorized as having hypomyopathic DM. These patients have no clinical evidence of myositis; however, ancillary studies such as magnetic resonance imaging (MRI), needle electromyogram (EMG), or muscle biopsy results provide evidence of subclinical myositis. Together, these two subsets are often referred to as clinically amyopathic DM (CADM). Notably, CADM patients can develop clinically evident myositis later in the disease course, highlighting importance of continued monitoring for this manifestation.14
In patients affected by DM, chronic inflammation, treatment with glucocorticoids, and immobilization may precipitate impaired bone health. Adult women with DM are more significantly likely to develop osteoporosis of the spine and femoral neck when compared to healthy controls.15 In one longitudinal study, nearly half of patients with myositis were found to have vertebral fractures, with prior vertebral fracture incurring a five-times higher risk of subsequent fracture.16,17 Low bone-mineral density (BMD) and vertebral fractures are associated with reduced quality of life and increased disability and pain in DM and other IIM patients.18 As glucocorticoids are a major contributor to impaired bone health in DM patients, it is paramount for physicians to follow published guidelines to prevent and treat glucocorticoid-induced osteoporosis in DM patients.19
Diagnosis
Initial evaluation for suspected DM should begin with a thorough history and physical examination, including total body skin examination and muscle strength testing of extremities and neck flexors. Historically, DM was clinically classified by the Bohan and Peter criteria, which divided IIMs into five groups: polymyositis (PM), DM, DM/PM associated with cancer, childhood DM/PM, and PM/DM with associated collagen-vascular disease.20 They described five major criteria to define DM: (1) symmetric weakness of limb-girdle muscles and anterior neck flexors; (2) muscle biopsy evidence of myositis; (3) elevation in serum skeletal-muscle enzymes; (4) a triad of needle EMG findings; and (5) presence of dermatologic features including heliotrope rash, Gottron’s sign, and erythematous dermatitis involving the face, neck, and upper torso. According to the Bohan and Peter criteria, a DM diagnosis is considered “definite” if ≥3 criteria and characteristic rash are present, “probable” if 2 criteria and rash are present, and “possible” if 1 criterion and rash are present.20 Notably, the Bohan and Peter criteria did not recognize amyopathic patients as qualifying for DM diagnosis.
Despite these criteria being the most widely used for DM diagnosis over several decades, critiques arose surrounding inclusion of non-specific myositis features, vague exclusionary criteria, and unclear definitions of DM skin manifestations. As a result, several new classification systems were proposed in following decades.21 Although these proposals represented respectable efforts to improve IIM classification criteria, it became clear that a dedicated effort by IIM experts to develop classification criteria with consensus and validation was needed. Subsequently, an expert group was assembled in 2004, and new, validated IIM classification criteria were published in 2017 and endorsed by the European League Against Rheumatism and American College of Rheumatology (EULAR/ACR).22 Two models were proposed, one with and one without muscle biopsy results, using 16 criteria which are assigned various point values to determine probability of IIM. Patients are required to have certain numbers of total points to meet thresholds for “possible”, “probable”, and “definite” IIM. When muscle biopsy results are included, EULAR/ACR criteria demonstrate sensitivity and specificity for probable IIM diagnosis of 93% and 88%, respectively. Without muscle biopsy data, sensitivity and specificity are 87% and 82%, respectively.22 Although by definition “classification criteria”, the EULAR/ACR criteria are increasingly being used in research and clinical trial settings to establish and prove DM diagnosis.
Once a patient meets EULAR/ACR criteria for IIM, presence of heliotrope rash, Gottron papules, or Gottron’s sign accurately subclassifies patients with DM. Objective muscle involvement, defined as abnormal manual muscle testing or other objective strength testing, further categorizes CDM patients. One of the unique aspects of the EULAR/ACR classification criteria compared with the Bohan and Peter criteria is that patients with pathognomonic DM skin manifestations are no longer required to exhibit clinical or histological signs of myositis for diagnosis. Thus, these criteria recognize CADM as a DM subtype, for which a skin biopsy revealing characteristic histological findings is recommended to further support the diagnosis.22
Although EULAR/ACR classification criteria are considered to represent a major improvement over previous criteria, they are not without limitations. For example, anti-Jo-1 autoantibodies but not other myositis-specific autoantibodies (MSAs) are included in the laboratory measurement scoring assessment section. Additionally, interstitial lung disease (ILD) is not included as a variable that contributes points used to reach an IIM diagnostic threshold.23 Furthermore, although these criteria recognize CADM as a DM subtype, there are only three skin manifestations (Gottron’s sign, Gottron papules, and heliotrope rash) included in the scoring assessments, and patients without all three may not meet the scoring threshold for “probable” CADM/IIM. In a study that applied EULAR/ACR classification criteria to a cohort of 110 CADM patients, 26.3% did not meet the minimum probability cutoff for DM classification.24 Thus, EULAR/ACR criteria may be strengthened by expanding scored variables to include a wider range of common cutaneous DM features (such as V-neck erythema and shawl sign), additional MSAs (such as TIF1γ and NXP2), cutaneous histopathological features, and ILD.
Skin Biopsy
Due to requirement of skin manifestations for DM diagnosis and easy accessibility of the skin, one of the most common and valuable procedures in a diagnostic workup is a lesional cutaneous punch biopsy. The classic histologic findings in DM skin biopsies include vacuolar interface dermatitis with keratinocyte dyskeratosis, increased reticular dermal mucin, superficial dermal vascular dilatation, and variably dense perivascular lymphohistiocytic inflammation (Figure 4A and B).25 Notably, 95% of 228 DM skin biopsies in one study demonstrated ≥1 of the following: basal vacuolization, dermal mucin deposition, or perivascular inflammation. Furthermore, 91% of biopsies demonstrated either basilar vacuolization, dyskeratotic keratinocytes, or both.26 Importantly, this data also highlights that the absence of vacuolar changes or increased dermal mucin in skin biopsies does not exclude a DM diagnosis. Thus, correlation between histology and clinical findings is critical for arriving at an accurate diagnosis. Additionally, treatment with glucocorticoids and/or immunomodulatory medications at time of biopsy may affect presence/severity of hallmark histopathological features in skin biopsies.26
 |
Figure 4 Characteristic Histologic Features of Lesional Dermatomyositis Skin Biopsies. (A) A photomicrograph (Hematoxylin and Eosin staining; 100X magnification) of a lesional punch biopsy from the upper back (shawl area) of a 55-year-old Caucasian woman with TIF1γ DM revealing vacuolar interface dermatitis affecting the epidermis, mild superficial dermal lymphocytic inflammation, and abundant mucin deposition throughout the reticular dermis denoted by areas of blue discoloration (arrows) between collagen bundles. (B) At higher power (Hematoxylin and Eosin staining; 400X magnification), dyskeratotic keratinocytes (the hallmark feature of interface dermatitis) are seen as cells along the basilar epidermis with pyknotic nuclei and condensed eosinophilic cytoplasm (white arrow) and eosinophilic globules just below the basilar epidermis (black arrow). |
Routine Serologic Studies
Five muscle enzymes, creatine phosphokinase (CK), aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase (LDH), and aldolase, are released into the serum when skeletal muscle is damaged. Of these, CK is the most sensitive and specific for skeletal muscle damage.27 CK levels can be elevated up to 50X the upper normal limit in 70–80% of CDM patients.27 During muscle injury, transaminase elevation levels are tightly correlated with CK levels, typically with AST > ALT.28 As CK and transaminase levels can be elevated in hepatic disorders, checking for elevated gamma-glutamyl transferase (GGT) levels can help differentiate hepatic versus skeletal muscle CK origin. Importantly, CK levels can also be elevated because of myocardial injury. Thus, if cardiac origin or contribution to CK elevation is suspected, checking for elevated troponin I levels can be helpful.
Baseline testing for elevated serum muscle enzymes is of particular importance for diagnosing patients with hypomyopathic DM who have laboratory findings supporting myositis but lack subjective muscle weakness. Alternatively, muscle enzyme levels may be normal in patients with DM, even when active myositis is present. Therefore, clinicians may need to utilize other diagnostic tools (detailed below) to further assess muscle involvement.29 The above enzymes can also be useful in monitoring DM response to treatment. After successful therapy, serum CK levels decline over 2-8-weeks, followed by a sequential decline in transaminase, LDH, and aldolase levels.30
A complete blood count is useful at presentation to assess for possible anemia and white blood cell count alterations.30,31 Specifically, active DM is associated with lymphopenia and platelet counts have been correlated with disease activity.31 Finally, there are reports of complement C4 deficiencies with active myositis, suggesting that evaluating C3 and C4 levels may be beneficial.32
Myositis Autoantibodies
IIMs are associated with autoantibodies against nuclear and cytoplasmic antigens and are classified as myositis-associated autoantibodies (MAAs) and MSAs. MAAs are autoantibodies found in IIMs and other autoimmune conditions, such as anti-nuclear antibodies (ANA) and anti-SSA (Ro). MSAs are autoantibodies that are specific to patients with IIMs (Table 1). Commercial MSA antibody panels often include DM-specific autoantibodies (Mi2, TIF1γ, MDA5, NXP2, and SAE), ASTS autoantibodies (Jo-1, PL-7, PL-12, etc.), and immune-mediated necrotizing myositis autoantibodies (SRP and HMGCR).
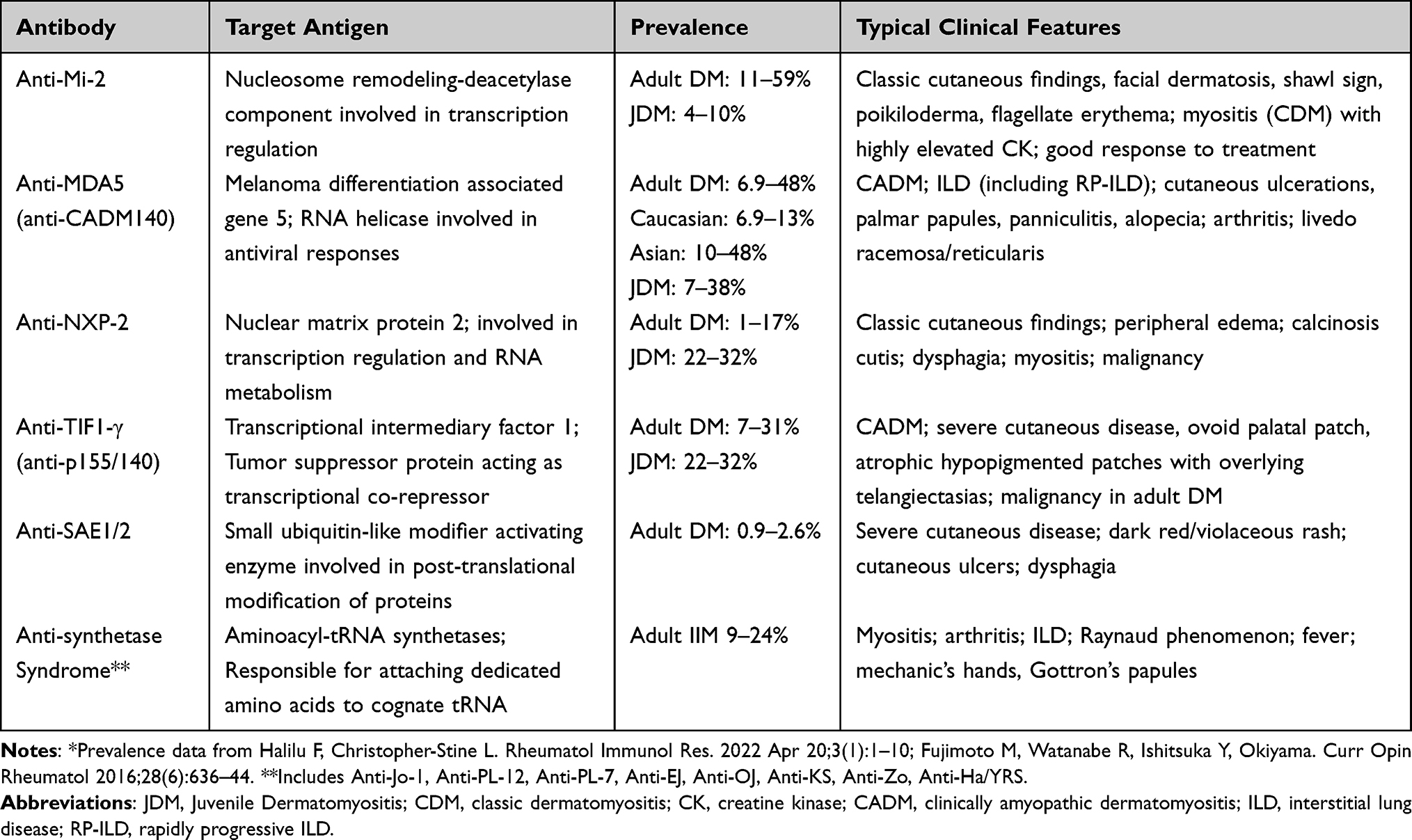 |
Table 1 Myositis-Specific Autoantibodies* |
Patients suspected of having DM should be screened for MAAs, including ANA and anti-extractable nuclear antigen (ENA) antibodies such as anti-SSA (Ro). ANA testing by immunofluorescence in human epithelial (Hep-2) cells should be used because specific cytoplasmic or nucleolar patterns may be missed by alternative methods. Distinctive Hep-2 patterns with negative ANA can be observed when some myositis antibodies are present (cytoplasmic for anti-MDA5, for example). ANA is positive in 24–60% of patients with DM.33 Positive anti-ENA antibodies may indicate alternative autoimmune diseases or DM overlap syndromes.
MSAs are detected in >60% of DM patients, and incidence may approach 80–90% with appropriate assays.34 Importantly, presence of >1 MSA in the same individual is rare, increasing their usefulness as biomarkers for specific DM phenotypes.35,36 For example, patients with TIF1γ autoantibodies tend to have skin-predominant, amyopathic disease and are less likely to have Raynaud’s phenomenon and arthralgias/arthritis compared to other DM patients.37 Thus, MSAs help guide subsequent work-up for systemic manifestations. Also, serum levels of MSAs may relate to disease severity, highlighting their potential utility as prognostic markers.34–36
Evaluation for MSAs is particularly useful in facilitating DM diagnosis in cases where EULAR/ACR criteria may have limitations. Specifically, presence of MSAs can aid in CADM diagnosis when some pathognomonic skin findings are absent. One study demonstrated appropriate classification of DM and other IIMs using only clinical manifestations and MSAs.38 While benefits of testing for MSAs are clear, testing is not standardized, and commercially available tests use variable methodologies.39 When comparing commercial line-blot and dot-blot assays to immunoprecipitation assay results, accuracy has been found to highly depend upon MSA specificity. Specifically, commercial line/dot-blot assays demonstrated poor performance in detecting anti-TIF1γ, anti-Mi-2 and rarer ASTS antibodies.40,41 In contrast, commercial enzyme-linked immunosorbent assays (ELISA) have demonstrated agreement with immunoprecipitation in detecting several MSAs.42,43
Challenging Cases
If clinical suspicion for DM is high, but clinical features and/or serologic data concerning myositis remain equivocal, EMG studies may be considered and show abnormalities supporting diagnosis in ~70-90% of DM cases.27,33,44 Diagnostic evaluation may also benefit from T2-weighted MRI in areas of muscle weakness. Bilateral thigh MRI has demonstrated utility for patients with subjective muscle weakness but normal or only mildly elevated muscle enzymes.45 Additionally, T1-weighted MRI images can be useful for detecting changes in muscle architecture suggestive of DM, particularly fatty infiltration.46 If MRI changes suggestive/consistent with myositis are identified, a targeted muscle biopsy for diagnostic confirmation can be considered on a case-by-case basis.44
Although muscle biopsy has classically been a key diagnostic procedure in patients with suspected DM, its invasive nature coupled with the discovery of MSAs, predictive value of serologic muscle inflammation markers, pathognomonic cutaneous manifestations, and hallmark cutaneous histologic features have made it less commonly utilized. Approximately 6% of DM patients have no or subtle skin involvement (dermatomyositis sine dermatitis) and may particularly benefit from muscle biopsy to aid in diagnosis.12
Although highly specific for DM, sensitivity of muscle biopsies showing the hallmark feature of perifascicular atrophy is only 47%.47 Other histologic features include complement deposition on endomysial capillaries, decreased capillary density, and perifascicular expression of MHC I.48 Both perimysial and perivascular mixed inflammation are also common.2 A recent study suggested immunohistochemical staining for myxovirus resistance protein A (MxA), a marker for type I IFN activity, in myofibers of muscle biopsies is a better indicator of DM muscle involvement than conventional histologic markers.47
Evaluation for Systemic Manifestations
Once a DM diagnosis is made, it is critical to assess and follow patients for presence/development of visceral organ involvement and malignancy, as these contribute greatly to morbidity and mortality.
Pulmonary Disease
ILD is the most common systemic manifestation of DM and occurs in ~15-30% of patients.49–51 Early symptoms suggestive of ILD include dry cough and progressive dyspnea. Severity can range from mild to rapidly progressive, and ILD is a leading cause of hospitalization and death in DM patients.49,50 Median time to ILD onset is 16.9–18 months after DM diagnosis, though ILD can develop anytime during the disease course and may occur despite treatment.51
Presence of particular MSAs may help determine ILD risk.52 ILD is most strongly associated with anti-MDA5 and ASTS MSAs.53 Furthermore, presence of MDA-5 antibodies is associated with rapidly progressive ILD (RP-ILD) and poor prognosis.54 In patients with other MSAs, ILD prevalence varies, with estimates ranging from 18% with anti-TIF1γ to 40% with anti-SAE1 antibodies.55
Although dry cough and dyspnea may suggest presence of ILD, using symptoms as ILD markers may be unreliable.50 In DM, impaired ventilation and clinical signs suggestive of ILD may occur secondary to myositis affecting the diaphragm and accessory respiratory muscles.48 The ACR recently published a summary of 2023 guidelines for screening and monitoring of ILD in patients with systemic autoimmune rheumatologic disorders (SARDS), including DM.56 This guideline statement conditionally recommends screening SARDS patients with both pulmonary function tests (PFTs; spirometry, lung volumes, and diffusion capacity) and high-resolution chest computed tomography (HRCT) scan at time of diagnosis. Importantly, these guidelines recommend against screening with chest radiography, 6-minute walking test, ambulatory desaturation testing, bronchoscopy, and lung biopsy.
For patients diagnosed with ILD, the 2023 ACR guidelines conditionally recommend monitoring with PFTs every 3–6 months during the first year, then less frequently once stable; ambulatory desaturation testing every 3–12 months, and HRCT as needed. If ILD is not identified on initial screening in DM patients, clinicians should continue monitoring for signs/symptoms of ILD onset. A high index of clinical suspicion for ILD development should especially persist in patients with anti-MDA5 and ASTS antibodies. The 2023 ACR guidelines suggest retesting if signs/symptoms of ILD develop and/or considering annual re-screening in high-risk patients.
Although pulmonary arterial hypertension (PAH) is most often seen in patients with ASTS, it has rarely been reported in patients with DM.57 In such cases, it has been hypothesized that PAH is the result of microvascular disease.57 Importantly, such patients may develop PAH without evidence of significant ILD.57
Cardiac Disease
Cardiac involvement in DM is associated with poor prognosis. Depending on consideration of clinical versus subclinical cardiac involvement, prevalence estimates range from 9% to 72% in DM patients.58 Manifestations may include subclinical diastolic dysfunction, arrhythmias, myocarditis, pericarditis, pericardial effusion/tamponade, cardiomyopathy, myocardial fibrosis, and congestive heart failure (CHF). No MSAs have consistently shown close correlation with cardiac involvement.59 Although previous studies have associated anti-SRP antibodies with increased risk of cardiac involvement, this is controversial in current literature.60,61
The most common subclinical cardiac manifestations are arrhythmias and conduction defects.62,63 Observed electrocardiogram (ECG) abnormalities include bundle branch blocks, nonspecific ST-T wave changes, atrial or ventricular tachycardia, atrial fibrillation, atrioventricular conduction block, and QTc prolongation.62 Although often asymptomatic, some of these carry great clinical significance, as pathologically prolonged QTc intervals and complete atrioventricular block may lead to fatal arrhythmias and sudden cardiac death.
Clinically apparent cardiac involvement is uncommon in DM.63 The most frequently reported manifestation is CHF, occurring in ~10-15% of patients and presenting with typical clinical symptoms of dyspnea on exertion, orthopnea, and nocturnal dyspnea.62 Additionally, multiple factors including systemic inflammation, glucocorticoid use, and presence of underlying traditional cardiovascular risk factors in DM patients may lead to increased risk of cardiovascular events such as acute myocardial infarction, ischemic stroke, deep venous thromboses, and pulmonary emboli, which account for 15–55% of DM deaths.62–64
Currently, evidence-based screening guidelines for cardiac involvement are lacking and recommendations are based on expert opinions. Unfortunately, most cases are subclinical, and patients may not be appropriately evaluated for cardiac disease.62 Gupta et al recommend screening all DM patients with a cardiac history and ECG irrespective of cardiac symptoms.65 Basic cardiac labs such as Troponin I and pro-BNP should be considered in patients who report symptoms or have ECG abnormalities, and they should be referred to cardiology for further evaluation.
Gastrointestinal Disease
Gastrointestinal involvement primarily manifests as dysphagia secondary to active myositis affecting pharyngeal and upper esophageal skeletal muscle and occurs in ~18-20% of DM patients.66 Symptoms range from mild-to-severe dysphagia, the latter often associated with choking and risk of aspiration pneumonia, and sometimes requiring nasogastric and/or percutaneous feeding tube placement.48 Presence of chronic diarrhea (lasting >4 weeks) in DM patients should prompt consideration of microscopic colitis.67 Relative risk of microscopic colitis in DM patients was found to be 14.1 in a relatively large cohort; CADM patients conferred a relative risk of 28.6.67 Other gastrointestinal manifestations are usually mild and typically manifest as motility disorders.68 Rarely, severe and life-threatening gastrointestinal manifestations may occur, including ulcerations, hemorrhage, perforation, and intestinal ischemia/infarction.68
Currently, there are no evidence-based screening guidelines for gastrointestinal involvement in DM patients. Aside from inquiring about dysphagia, Casal-Dominguez et al suggested screening for functional esophageal abnormalities in DM patients.66 Importantly, esophageal motility abnormalities have been detected with high-resolution manometry in absence of symptoms in DM patients. Other concerning gastrointestinal symptoms should prompt gastroenterology referral. Diagnosis of microscopic colitis requires colonoscopy with confirmatory colonic biopsy.
Other Visceral Organ Involvement
Renal dysfunction is uncommonly reported in DM patients and may present as acute kidney injury secondary to rhabdomyolysis-induced kidney damage.69 Autoimmune chronic glomerulonephritis can also occur, with clinical features that include hematuria, proteinuria, and declining kidney function. Diagnosis requires renal biopsy. Cucchiari and Angelini advocate close monitoring for kidney function abnormalities, proteinuria, and urinary sediment in all DM patients.69
Several hepatic manifestations in DM patients have been described, including non-alcoholic fatty liver disease, primary biliary cholangitis, and autoimmune hepatitis.70 Elevated transaminase levels are commonly noted in DM. However, as previously mentioned, evaluating GGT levels can distinguish between skeletal muscle and hepatic origin. Notably, there is a strong correlation between skeletal muscle ALT and CK levels.71 If ALT levels are inappropriately high compared to CK levels, GGT levels are elevated, or there are other signs of liver abnormalities, further investigation for liver disease is warranted.
Malignancy
DM is associated with an increased risk of malignancy, with an estimated incidence of 15–27%.72,73 The first year following DM diagnosis appears to be the highest-risk period for malignancy diagnosis. However, cancer-associated DM is typically defined as DM occurring within ± 3 years of a malignancy, and increased risk may persist for >5 years after diagnosis.74,75 A large meta-analysis revealed decreasing malignancy risk in DM patients over time, with a standardized incidence ratio of 17.29 in the first year following DM diagnosis, 2.70 from 2 to 5 years, and 1.37 beyond 5 years.73–75 Another meta-analysis identified several risk factors associated with malignancy, the strongest being presence of TIF1γ autoantibodies, conferring a four-fold increased risk.76 Other risk factors included age >45 years at DM diagnosis, male sex, dysphagia, and cutaneous ulceration.75,76 However, malignancy screening practices in DM patients vary greatly among clinicians and are a topic of debate.
All DM patients should be up to date on age-appropriate cancer screenings recommended by United States Preventative Services Task Force (USPSTF) and American Cancer Society (ACS) guidelines. However, a retrospective DM cohort study revealed 8 of 17 cancers identified by blind screening tests would have gone undetected based on ACS and USPSTF guidelines.77 In light of this, the authors of this study suggested age-appropriate malignancy screenings may not be aggressive enough in DM patients.75
Several groups have reported that chest/abdomen/pelvis CT imaging discovers a high percentage of cancers.76,77 Whereas one prospective study found whole-body [(18)F] fluorodeoxyglucose positron emission tomography/CT (FDG-PET/CT) yielded similar results as CT scans, a retrospective cohort study found FDG-PET/CT was not more sensitive than conventional screening for detecting malignancies in DM patients and may be associated with increased numbers of subsequent biopsies.78,79
Notable DM Subtypes
Anti-MDA5 DM
Anti-MDA5 DM is typically amyopathic and characterized by high risk of ILD and distinctive cutaneous features (Figures 5A–C, 6A and B). The unique anti-MDA5 DM mucocutaneous phenotype is thought to reflect underlying occlusive vasculopathy. One of the most characteristic features is cutaneous ulceration, developing in up to 82% of anti-MDA5 patients.80 Cutaneous ulcers are strongly associated with ILD development and serve as an independent risk factor for pneumomediastinum in anti-MDA5 DM patients.81,82 Ulcers in anti-MDA5 patients are typically deep and punched-out and usually occur overlying interphalangeal joints and elbows, digital pulp, and/or periungual areas.71 However, they may also occur on the chest, back, and other sun-exposed sites (Figure 6A). They are often associated with surrounding violaceous erythema and livedo reticularis, which are indicative of vasculopathy.
 |
Figure 5 Characteristic Cutaneous Manifestations of MDA5-associated Dermatomyositis. (A) A 52-year-old man with MDA5(+) DM and deep, punched out ulcers involving the palmar and lateral aspects of numerous fingers. Note the associated violaceous discoloration suggestive of underlying vasculopathy. (B) A 42-year-old woman with MDA5(+) DM-associated palmar papules with central ivory-white discoloration and surrounding violaceous erythema near the creases of interphalangeal joints. (C) A 67-year-old man with MDA5(+) DM-associated crusted erosions and ulcers and violaceous erythema involving his elbow and forearm. |
 |
Figure 6 MDA5(+) Dermatomyositis-associated Rapidly Progressive Interstitial Lung Disease. A 58-year-old woman admitted with shortness-of-breath and a rash consisting of (A) crusted ulcerations involving the V-neck area and (B) violaceous-to-hyperpigmented papules overlying the interphalangeal joints of her hands. A lesional punch biopsy from the V-neck area revealed vacuolar interface dermatitis and (C) CT chest at baseline revealed patchy opacities throughout the lungs and focal areas of fibrosis. Despite aggressive treatment her pulmonary opacities and fibrotic CT changes progressed and pulmonary function decreased over (D) 2 months and (E) 7 months and she eventually expired from her disease. |
Palmar papules are another unique cutaneous feature of anti-MDA5 DM. Presenting as macules/papules on the palms and palmar aspects of fingers, these lesions are often painful and cause significant morbidity (Figure 5B). Other anti-MDA5 DM mucocutaneous features include diffuse alopecia, calcinosis, oral ulcers, and panniculitis.80,83 Other characteristic anti-MDA5 DM systemic manifestations include seronegative rheumatoid arthritis (RA)-like arthropathy, fever, and fatigue.80
The key systemic manifestation of anti-MDA5 DM is ILD, with an estimated prevalence of 42–100%.80 Higher ILD prevalence has been reported among anti-MDA5 DM patients of Asian descent (~92-100%).84 Anti-MDA5-associated ILD is heterogeneous and ranges from smoldering to rapidly progressive (RP-ILD) disease linked with considerable mortality (Figure 6C–E). Odds of developing RP-ILD are >20 times higher in anti-MDA5-positive DM patients than in anti-MDA5-negative patients.85
Independent risk factors for RP-ILD development in anti-MDA5 DM patients include age >50 years, serum LDH >300 IU/L, hyperferritinemia, elevated carcinoembryonic antigen, lymphopenia, and neutrophil-to-lymphocyte ratio >7 at diagnosis.86,87 Additionally, patients with higher anti-MDA5 antibody titers appear more likely to develop RP-ILD and less likely to respond to therapy. Alternatively, presence of arthralgias has been identified as protective against RP-ILD development.87
Anti-Synthetase Syndrome (ASTS)
ASTS is categorized by autoantibodies against aminoacyl-tRNA synthetases, including Jo-1, PL-7, PL-12, OJ, EJ, KS, Zo, and Ha.39 Anti-Jo-1 antibodies are most common, accounting for up to 75% of all anti-aminoacyl-tRNA antibodies.88 ASTS presents with a triad of myositis, arthritis, and ILD in up to 90% of patients. Raynaud’s phenomenon, fever, and rashes are variably present. Mechanics’ hands are a characteristic cutaneous manifestation found in 16–21% of ASTS patients and present as fissured, hyperkeratotic papules/plaques on the palmoplantar and lateral edges of hands/fingers/feet/toes (Figure 3C). Some studies have associated mechanic’s hands with increased ILD rates, particularly in those with anti-Jo-1 antibodies.89 Arthralgias and arthritis are seen in 54–70% of ASTS patients, most commonly presenting as a symmetric polyarticular arthritis.90 Isolated arthritis is the presenting symptom in ~25% of patients and may be misdiagnosed as RA.91
The hallmark pulmonary manifestation of ASTS is ILD, with an estimated prevalence of 70–95% and representing the major contributor to morbidity and mortality.89,92 Patients with anti-PL-7 and anti-PL-12 antibodies more frequently have ILD than patients with anti-Jo-1 antibodies (90% vs 60%, respectively, in a relatively large ASTS cohort), and it seems to be more severe and rapidly progressive.93 Although ILD prevalence is high, ASTS patients tend to have better prognoses, improved therapeutic responses, and lower mortality compared to those with anti-MDA5 antibodies.85,89 Also reported in ASTS is PAH, with a prevalence of 8–21% and occurring more commonly in those without anti-Jo-1 antibodies.89,94 When present, pulmonary hypertension is independently associated with decreased survival.89,94
Of note, the spectrum of ASTS is heterogeneous, and the typical clinical triad of manifestations may not be present at disease onset.95 Importantly, presence of all typical clinical manifestations is not necessary for making a diagnosis of ASTS. However, establishing an accurate diagnosis based on limited manifestations at initial presentation can be challenging. With that said, recognizing that initial symptoms can correlate with specific MSA, such as isolated arthritis with anti-Jo-1 antibodies, isolated myositis with anti-OJ antibodies, and isolated ILD with anti-PL-7, anti-PL-12, or anti-EJ antibodies can increase clinical suspicion for ASTS in astute clinicians.95 Other clinical manifestations of ASTS may emerge gradually over time, underscoring the importance of persistent monitoring of patients following initial presentation.
Dermatomyositis Treatment
Treatment approaches to DM depend on multiple factors, including presence/severity of myositis, cutaneous lesion type (non-vasculopathic vs vasculopathic), extent of cutaneous involvement, cutaneous symptoms, visceral organ involvement, presence of underlying malignancy, and type of MSA. There is significant overlap in treatment options for CDM and CADM. Systemic glucocorticoids are the mainstay treatment for DM, especially when myositis is present. These are typically prescribed at doses of 0.5–1.0 mg/kg/day initially in an outpatient setting, with tapering based on treatment response. When patients are hospitalized because of more severe myositis, higher doses of IV glucocorticoids are often administered in attempts to promptly halt further skeletal muscle damage. Not uncommonly, pulse methylprednisolone therapy (1000mg daily for 3–4 consecutive days) is given with this goal in mind.
Typically, steroid-sparing immunomodulatory agents are immediately prescribed along with glucocorticoids at time of diagnosis with the goal of minimizing cumulative glucocorticoid exposure because DM is a chronic disease and most steroid-sparing immunomodulatory agents require several months before optimal effects can be expected. The most commonly prescribed drugs include methotrexate, azathioprine, mycophenolate mofetil (MMF), and hydroxychloroquine. Calcineurin inhibitors, tacrolimus and cyclosporine, and repository corticotropin injections may also be effective.96
For skin manifestations, all patients should be educated about strict photoprotection, as ultraviolet light exposure may exacerbate disease activity.97 Patients are typically offered a topical medication regimen that includes either glucocorticoids or calcineurin inhibitors. However, in our experience, currently available topical medications are largely ineffective for DM, especially for treating significant pruritus. Thus, most patients require systemic therapy to control cutaneous disease activity.
The most widely accepted first-line systemic treatments for moderate-to-severe cutaneous disease are MMF and methotrexate, although other previously mentioned therapies may also be used.98,99 A single-center cohort study demonstrated that MMF monotherapy was associated with increased odds of achieving clinical cutaneous disease remission (OR 6.00) compared to other therapies.100 Notably, most patients who achieved clinical remission were receiving 3g of MMF daily. Methotrexate has been demonstrated to be effective in clinical trials and may be preferred due to cost and once-weekly dosing schedules.98 Unfortunately, skin disease in both CDM and CADM often tends to be resistant to treatment with these standard therapies, even when myositis is well-controlled, necessitating combination systemic immunomodulatory medication regimens.100
Intravenous immunoglobulin (IVIG) has become a favored treatment for severe/refractory DM.98,101,102 In patients with severe myositis, especially when dysphagia is present, IVIG is typically initiated immediately and often along with pulse glucocorticoid therapy. In other cases, however, IVIG therapy is typically reserved for those with refractory muscle, skin, or other DM manifestations due to cost, limited supply, and logistical inconveniences of infusions. Importantly, IVIG has recently achieved FDA approval in the US for adult DM treatment based on results of the ProDERM clinical trial.103 In the ProDERM trial, the primary endpoint (total improvement score ≥20 at 16 weeks) was met in 79% of patients who received 2g/kg IVIG every 4 weeks, compared to 44% in the placebo group. There was also significant improvement in cutaneous disease activity in the IVIG versus placebo group based on Cutaneous Dermatomyositis Activity and Severity Index Activity (CDASI-A) scores. A retrospective study demonstrated that 83% of DM patients with refractory skin disease improved after initiating IVIG, regardless of disease subtype, and 80% were able to decrease/discontinue immunosuppressive therapies.101
Rituximab is another option for refractory DM. A randomized, placebo-controlled trial demonstrated myositis improvement in 83% of children and adults, but differences between rituximab and placebo arms were not statistically significant.104 Utility of rituximab for treating cutaneous DM has been met with mixed results.105,106 However, rituximab has shown efficacy in treating ILD and vasculopathic lesions in anti-MDA5(+) patients.107,108
The choice of therapy may depend on patient comorbidities and/or DM manifestations. For example, MMF has also shown efficacy in treating ILD and is often the preferred initial therapy.109 Cyclosporine and tacrolimus have also shown efficacy in ILD and may be appropriate treatment options in these patients, although their use is limited by adverse effects and intensive monitoring requirements.110
Antimalarials are most often used as adjuvant agents in DM patients with cutaneous disease or arthritis.111 Previously, HCQ was considered a first-line systemic agent for DM. However, recent data suggest that it has limited efficacy as monotherapy and is associated with an increased risk of severe cutaneous allergic reactions in DM patients, especially those with anti-SAE antibodies.111–113
Vasculopathic cutaneous lesions, as observed in patients with MDA5-associated DM, are challenging to treat. In addition to abovementioned medications, concomitant treatment with vasodilatory/vasoactive agents such as nifedipine, sildenafil, pentoxifylline, and bosentan can be particularly useful for healing ulcers and palmar papules in both studies and our experience.80,114
Unmet Needs
Malignancy Screening
Although DM diagnosis typically triggers an extensive screening work-up to search for an underlying malignancy, a mortality benefit, which is the gold standard for supporting screening procedures, has never been demonstrated.115 Thus, the benefit–risk ratio of extensive malignancy screening in all DM patients is questionable. It is extremely important for clinicians to recognize the unproven utility of malignancy screening, and the fact that screening tests have potential for causing patient harm.75
Newly published DM malignancy screening guidelines based on an International Myositis Assessment and Clinical Studies Group (IMACS) initiative represent a significant advancement towards evidence-based screening; however, much work, including demonstration of real-world utility, is still needed.116 Arguably, the best current strategy for addressing malignancy screening in DM patients is to use shared decision-making strategies, which represents our approach. Shared decision-making takes into account patients’ experiences, priorities, and goals to help them make informed decisions.117 A shared decision-making approach first involves clinicians discussing risks/benefits of malignancy screening with DM patients, which is not trivial. In one study, nearly all patients reported that clinicians addressed benefits of cancer screening, but only ~25% reported receiving information about risks.118 Additionally, a systematic review of beliefs concerning benefits/harms of medical interventions, including screening tests, found that clinicians more often overestimated benefits and underestimated harms.119
In 2015, the High-Value Care Task Force of the American College of Physicians (HVCTF-ACP) published a framework for cancer screening with high-value care.120 In their framework, the HVCTF-ACP emphasized need to identify a high-risk patient subset in whom malignancy screening is likely to have a favorable benefit–risk ratio. Recently, novel research concerning malignancy in DM patients has inspired the future possibility of identifying high-risk DM patient subsets.
In 2022, Fiorentino et al evaluated for the presence of additional autoantibodies in serum from DM patients with anti-TIF1γ autoantibodies.121 Four autoantibodies with frequencies >6.5% in two TIF1γ-positive DM cohorts, with anti-cell division cycle and apoptosis regulator protein-1 (anti-CCAR1) antibodies being the most prevalent. Interestingly, presence of anti-CCAR1 antibodies was negatively associated with cancer emergence within 3 years of DM onset. Additionally, a statistically significant inverse dose–response relationship was found between number of additional autoantibodies and cancer emergence. Although 30% of patients were diagnosed with cancer within 3 years of DM diagnosis when anti-TIF1γ autoantibodies alone were present, only 15% developed cancer within 3 years when one additional autoantibody was present, and no patients developed cancer when >2 additional autoantibodies were present. Furthermore, patients with anti-CCAR1 autoantibodies who eventually developed cancer were typically diagnosed later after DM diagnosis and at an earlier stage (89% TNM stage 0/1) than those without anti-CCAR1 antibodies (42% TNM stage 0/1).
Fiorentino et al proposed that a cancer–DM relationship can be explained within the framework of cancer immunoediting. During cancer development and progression, neoantigens are exposed to the host’s immune system as mutations accumulate. Fiorentino et al’s data are consistent with a model supporting the breadth of the immune response to cancer dictates whether the cancer will emerge or remain quiescent. In patients with relatively poor immune responses against cancers, TIF1γ autoantibodies alone are generated, DM develops, cancers easily escape the poor immune response, and are diagnosed early after DM diagnosis, typically at advanced stages. In patients with more robust immune responses against cancers, multiple autoantibodies develop (anti-TIF1γ/CCAR1/possibly others), DM develops, but more robust immune responses effectively keep cancers in a subclinical equilibrium state; over time cancers eventually escape the immune responses, but they are diagnosed relatively late after DM diagnosis and at earlier stages than in patients with TIF1γ autoantibodies alone. Finally, in patients with the most vigorous immune responses against cancers, numerous autoantibodies develop (anti-TIF1γ/CCAR1/numerous others). DM still develops, but no cancers emerge due to either their elimination or persistent control in an equilibrium state by effective immune responses.
Although validation is still required, this model suggests that a higher percentage of patients than previously thought may develop DM due to underlying cancer; however, whether cancers manifest clinically depends upon the robustness of host immune responses against them. Importantly, this model also suggests that testing for the presence of additional autoantibodies, such as anti-CCAR1 and anti-Sp4, may be helpful in identifying a high-risk, TIF1γ-positive DM subset in whom malignancy screening would be expected to have a high benefit–risk ratio.122 Despite much future work being needed, this concept may represent a significant breakthrough in our understanding of cancer-associated DM, and one can imagine ordering a myositis autoantibody panel in the future whose results can stratify malignancy risk in DM patients to help determine who should undergo malignancy screening and who can forego it.
Treatment of MDA-5-Associated RP-ILD
The refractory nature of RP-ILD in anti-MDA5 DM patients makes development of effective treatment strategies extremely important.123 Although typical DM treatments have been attempted for MDA5-associated RP-ILD, these are variably effective, and better treatments represent a significant unmet need. There is also a lack of well-designed randomized controlled trials for MDA5-associated RP-ILD treatment, making it difficult to develop useful treatment algorithms.80
Treatment for RP-ILD is guided by severity at time of diagnosis. First-line treatment for milder RP-ILD with MMF is suggested in several studies.80,124 In addition to addressing inflammation, MMF decreases expression of profibrotic cytokines and may attenuate the RP-ILD course.125,126 In a large cohort study, MMF resulted in stable or improved lung function in most at 2.5 years.125
Tofacitinib has also been successfully used to treat MDA5-associated RP-ILD. A retrospective study of MDA5-associated RP-ILD patients revealed improved 6-month and 1-year all-cause mortality in those receiving tofacitinib compared to tacrolimus.127 Additionally, an open-label study revealed improved survival rates in MDA5-associated RP-ILD patients treated with tofacitinib (100% survival) compared to those treated with conventional immunosuppressants (78% survival).128 Although these are small studies, their results show promise for Janus Kinase (JAK) inhibitors as potentially effective RP-ILD treatments.
Despite possible effectiveness of single agents, most patients with MDA5-associated RP-ILD presently require combination immunomodulatory regimens. Numerous studies have evaluated treatment of MDA5-associated RP-ILD with either a combination of glucocorticoids and calcineurin inhibitors or triple therapy with glucocorticoids, calcineurin inhibitors, and IV cyclophosphamide.123,129,130 A systematic review suggested a survival benefit for patients who received initial combination therapy with pulse-dose glucocorticoids, calcineurin inhibitors and/or IV cyclophosphamide compared to step-up therapy.123 Nakashima et al demonstrated a 75% survival rate in patients who received triple therapy versus 28.6% receiving step-up therapy.129 In a multicenter prospective study, overall survival rate of MDA5-associated RP-ILD patients receiving initial triple therapy was 85% at 12 months versus 33% receiving step-up therapy.130
Addition of rituximab and/or IVIG to various treatment regimens has also been reported to be effective for MDA5-associated RP-ILD in small cohorts.107,108 In one retrospective study, 5 of 8 MDA-5-associated RP-ILD patients demonstrated clinical improvement in PFTs, chest imaging, and ferritin levels following treatment with rituximab.108 For IVIG, a retrospective study demonstrated decreased mortality (22.6%) in IVIG-treated patients compared to non-IVIG-treated patients (52.9%).131
Plasma exchange may also be a potential salvage therapy for anti-MDA5-positive patients with refractory RP-ILD.132,133 In a retrospective study of 10 patients with RP-ILD refractory to intensive immunosuppressive therapy, the 1-year survival rate was 100% in the plasma exchange group versus 25% in the non-plasma exchange group.132 High ferritin levels, older age, pulmonary dysfunction severity, and higher HRCT scores may predict benefits from plasma exchange.133 More recently, intensive induction therapy consisting of triple therapy, rituximab, tofacitinib, and plasma exchange showed significant survival benefits in MDA5-associated RP-ILD patients.134 However, this heavily immunosuppressive regimen increased risk of adverse events. Finally, extracorporeal membrane oxygenation (ECMO) bridging to lung transplantation may serve as a life-saving salvage therapy for MDA5-associated DM patients with refractory RP-ILD based on several case reports and series.124,135
Reports supporting several emerging therapies for MDA5-associated RP-ILD have also been reported. Basiliximab, an IL-2 receptor/CD25 monoclonal antibody that targets activated lymphocytes, was effective in 3 of 4 patients with refractory RP-ILD.136 Daratumumab, an anti-CD38 antibody, has also been effective in several patients with severe and refractory RP-ILD.137 Well-designed prospective, randomized clinical trials are needed to better evaluate these and other emerging therapies for MDA5-associated RP-ILD.
Novel DM Treatments
While disease activity can be controlled in many patients with currently available DM treatments, most are broadly immunosuppressive, and there is a tremendous need for more targeted and effective treatments. Although recently FDA-approved in July 2021, IVIG has long been utilized to effectively treat skin, musculoskeletal, and other DM manifestations. Luckily, there has recently been great interest in bringing novel medications to patients suffering from DM (Table 2). An anti-IFNβ monoclonal antibody (PF-06823859, Dazukibart; Pfizer) is one of the most promising emerging DM therapies. Recently, results of a Phase 2 trial evaluating efficacy, safety, and tolerability of PF-06823859 in patients with skin-predominant DM were reported.138 In this trial, PF-06823859 150 mg and 600 mg IV every 4 weeks both met the primary endpoint of significantly decreasing CDASI-A scores at 12 weeks compared with placebo. A minimal clinically significant change in CDASI-A score was observed in >96% of patients and a decrease in CDASI-A score of >40%, which has been correlated with improved quality of life, was observed in >80% of patients.138,139 Myositis parameters also improved in a cohort of CDM patients.140 Based on these exciting results, a multicenter Phase 3 trial is underway.141
 |
Table 2 Completed, Active and Planned Clinical Trials Evaluating Novel Medications for Dermatomyositis |
JAK inhibitors are also currently being explored as therapeutic agents for DM. Currently, there are two active phase 3 multicenter trials evaluating efficacy and safety of JAK inhibitors for treating adult DM. The VALOR trial is evaluating efficacy and safety of brepocitinib, a dual TYK2/JAK1 inhibitor, for adult DM.142 The BIRD trial is evaluating efficacy and safety of baricitinib, a dual JAK1/JAK2 inhibitor, for adult DM.143 GALARISSO is a phase 2 trial evaluating GLPG3667, a selective TYK2 inhibitor, for adult DM.144 Although the pan-JAK inhibitor, tofacitinib, was shown to be efficacious for adult DM in a small, open-label trial, future plans are unclear.145
In addition to the above trials, numerous other medications are currently being studied or have completed Phase 1–3 clinical trials for the treatment of adult DM. Other medications currently/already studied include CAR-T therapy, ravulizumab, IMO-8400, GLPG3667, Enpatoran, and abatacept.146–151 Thus, the next decade may represent an exciting period for DM patients marked by approval of novel medications that can better control disease or even favorably alter disease course.
Conclusions
DM is a heterogeneous immune-mediated disease associated with various cutaneous manifestations and variable presence of myositis and visceral organ involvement. Discovery of MSAs has allowed for characterization of associated clinical phenotypes that can help guide work-up for various DM-associated manifestations. Although malignancy-associated DM is well accepted, little evidence currently supports extensive malignancy screening in all patients has a favorable benefit–risk ratio. However, future research may lead to identification of high-risk DM patient subsets for which malignancy screening may have high value. Finally, there is a need for more targeted and effective therapies for DM, in general, and for MDA5-associated RP-ILD. Fortunately, significant research evaluating various novel medications for DM provides hope for exciting future advances in patients suffering from this intriguing disease.
Acknowledgments
The authors thank Janine Sot, MBA, for her expertise and help in preparing the figures.
Disclosure
APF: Investigator for Pfizer, Corbus, Mallinckrodt, Alexion, Priovant, and Novartis receives personal research support from Mallinckrodt and Novartis; honorarium from AbbVie, BMS, Biogen, Novartis, and UCB for consultation and advisory board participation; and honorarium from AbbVie, Novartis, BMS, Kyowa Kirin, and Mallinckrodt for teaching and speaking. The other authors have no conflicts of interest to declare for this work.
References
1. Khoo T, Lilleker JB, Thong BY-H, Leclair V, Lamb JA, Chinoy H. Epidemiology of the idiopathic inflammatory myopathies. Nat Rev Rheumatol. 2023;19(11):695–712. doi:10.1038/s41584-023-01033-0
2. DeWane ME, Waldman R, Lu J. Dermatomyositis: clinical features and pathogenesis. J Am Acad Dermatol. 2020;82(2):267–281. doi:10.1016/j.jaad.2019.06.1309
3. Rothwell S, Cooper RG, Lundberg IE, et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann Rheum Dis. 2016;75(8):1558–1566. doi:10.1136/annrheumdis-2015-208119
4. Bax CE, Maddukuri S, Ravishankar A, Pappas-Taffer L, Werth VP. Environmental triggers of dermatomyositis: a narrative review. Ann Transl Med. 2021;9(5):434. doi:10.21037/atm-20-3719
5. Hall JC, Rosen A. Type I interferons: crucial participants in disease amplification in autoimmunity. Nat Rev Rheumatol. 2010;6(1):40–49.
6. Wenzel J, Tüting T. An IFN-associated cytotoxic cellular immune response against viral, self-, or tumor antigens is a common pathogenetic feature in “interface dermatitis. J Invest Dermatol. 2008;128(10):2392–2402.
7. Tabata MM, Hodgkinson LM, Wu TT, et al. The type i interferon signature reflects multiple phenotypic and activity measures in dermatomyositis. Arthritis Rheumatol. 2023;75(10):1842–1849. doi:10.1002/art.42526
8. Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allerg Immunol. 2016;51(3):293–302. doi:10.1007/s12016-015-8496-5
9. Bernet LL, Lewis MA, Rieger KE, Casciola-Rosen L, Fiorentino DF. Ovoid palatal patch in dermatomyositis: a novel finding associated with anti-TIF1γ (p155) antibodies. JAMA Dermatol. 2016;152(9):1049–1051. doi:10.1001/jamadermatol.2016.1429
10. Connolly A, Gordon PA, Hannah J, Creamer D. The chameleon rash: a review of the polyphenotypic dermatoses of dermatomyositis. Clin Exp Dermatol. 2021;46(6):1016–1022. doi:10.1111/ced.14689
11. Ichimura Y, Konishi R, Shobo M, et al. Anti-nuclear matrix protein 2 antibody-positive inflammatory myopathies represent extensive myositis without dermatomyositis-specific rash. Rheumatology. 2022;61(3):1222–1227. doi:10.1093/rheumatology/keab518
12. Yang S-H, Chang C, Lian Z-X. Polymyositis and dermatomyositis – challenges in diagnosis and management. J Transl Autoimmun. 2019;2:100018. doi:10.1016/j.jtauto.2019.100018
13. Gerami P, Schope JM, McDonald L, Walling HW, Sontheimer RD. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis siné myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54(4):597–613. doi:10.1016/j.jaad.2005.10.041
14. Bailey EE, Fiorentino DF. Amyopathic dermatomyositis: definitions, diagnosis, and management. Curr Rheumatol Rep. 2014;16(12):465. doi:10.1007/s11926-014-0465-0
15. de Andrade DCO, de Magalhães Souza SC, de Carvalho JF, et al. High frequency of osteoporosis and fractures in women with dermatomyositis/polymyositis. Rheumatol Int. 2012;32(6):1549–1553. doi:10.1007/s00296-011-1821-2
16. Gupta L, Lawrence A, Edavalath S, Misra R. Prevalence and predictors of asymptomatic vertebral fractures in inflammatory myositis. Int J Rheum Dis. 2018;21(3):725–731. doi:10.1111/1756-185X.13257
17. Ganguly S, Lawrence A, Gupta L. Prevalent vertebral fractures incur high risk of future fractures in inflammatory myositis. Clin Rheumatol. 2021;40(4):1431–1436. doi:10.1007/s10067-020-05365-0
18. Cox M, Sandler RD, Matucci-Cerinic M, Hughes M. Bone health in idiopathic inflammatory myopathies. Autoimmun Rev. 2021;20(4):102782. doi:10.1016/j.autrev.2021.102782
19. Humphrey MB, Russell L, Danila MI, et al. 2022 American college of rheumatology guideline for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Rheumatol. 2023;75(12):2088–2102. doi:10.1002/art.42646
20. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292(7):344–347. doi:10.1056/NEJM197502132920706
21. Leclair V, Lundberg IE. New myositis classification criteria-what we have learned since Bohan and Peter. Curr Rheumatol Rep. 2018;20(4):18. doi:10.1007/s11926-018-0726-4
22. Lundberg IE, Tjärnlund A, Bottai M, et al. EULAR/ACR classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. 2017;76(12):1955–1964. doi:10.1136/annrheumdis-2017-211468
23. Barsotti S, Dastmalchi M, Notarnicola A, et al. Performance of the new EULAR/ACR classification criteria for idiopathic inflammatory myopathies (IIM) in a large monocentric IIM cohort. Semin Arthritis Rheum. 2020;50(3):492–497. doi:10.1016/j.semarthrit.2019.12.001
24. Patel B, Khan N, Werth VP. Applicability of EULAR/ACR classification criteria for dermatomyositis to amyopathic disease. J Am Acad Dermatol. 2018;79(1):77–83.e1. doi:10.1016/j.jaad.2017.12.055
25. Janis JF, Winkelmann RK. Histopathology of the skin in dermatomyositis. A histopathologic study of 55 cases. Arch Dermatol. 1968;97(6):640–650. doi:10.1001/archderm.1968.01610120030004
26. Wolstencroft PW, Rieger KE, Leatham HW, Fiorentino DF. Clinical factors associated with cutaneous histopathologic findings in dermatomyositis. J Cutan Pathol. 2019;46(6):401–410. doi:10.1111/cup.13442
27. Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic inflammatory myopathies: clinical approach and management. Front Neurol. 2016;7:64. doi:10.3389/fneur.2016.00064
28. Mathur T, Manadan AM, Thiagarajan S, Hota B, Block JA. Serum transaminases are frequently elevated at time of diagnosis of idiopathic inflammatory myopathy and normalize with creatine kinase. J Clin Rheumatol. 2014;20(3):130–132. doi:10.1097/RHU.0000000000000038
29. Connolly CM, Paik JJ. Clinical pearls and promising therapies in myositis. Expert Rev Clin Immunol. 2023;19:797–811. doi:10.1080/1744666X.2023.2212162
30. Rider LG, Miller FW. Laboratory evaluation of the inflammatory myopathies. Clin Diagn Lab Immunol. 1995;2(1):1–9. doi:10.1128/cdli.2.1.1-9.1995
31. Li M, Yan S, Dong R, Xiang W, Ma Z, Yang Q. Elevated platelet-to-lymphocyte ratio and neutrophil-to-lymphocyte ratio in patients with polymyositis/dermatomyositis: a retrospective study. Clin Rheumatol. 2023;42(6):1615–1624. doi:10.1007/s10067-023-06542-7
32. Zhou D, King EH, Rothwell S, et al. Low copy numbers of complement C4 and C4A deficiency are risk factors for myositis, its subgroups and autoantibodies. Ann Rheum Dis. 2023;82(2):235–245. doi:10.1136/ard-2022-222935
33. Amato AA, Barohn RJ. Evaluation and treatment of inflammatory myopathies. J Neurol Neurosurg Psychiatry. 2009;80(10):1060–1068. doi:10.1136/jnnp.2008.169375
34. Hodgkinson LM, Wu TT, Fiorentino DF. Dermatomyositis autoantibodies: how can we maximize utility? Ann Transl Med. 2021;9(5):433. doi:10.21037/atm-20-5175
35. Satoh M, Tanaka S, Ceribelli A, Calise SJ, Chan EKL. A comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allergy Immunol. 2017;52(1):1–19. doi:10.1007/s12016-015-8510-y
36. Betteridge Z, Tansley S, Shaddick G, et al. Frequency, mutual exclusivity and clinical associations of myositis autoantibodies in a combined European cohort of idiopathic inflammatory myopathy patients. J Autoimmun. 2019;101:48–55. doi:10.1016/j.jaut.2019.04.001
37. Fiorentino DF, Kuo K, Chung L, Zaba L, Li S, Casciola-Rosen L. Distinctive cutaneous and systemic features associated with antitranscriptional intermediary factor-1γ antibodies in adults with dermatomyositis. J Am Acad Dermatol. 2015;72(3):449–455. doi:10.1016/j.jaad.2014.12.009
38. Mariampillai K, Granger B, Amelin D, et al. Development of a new classification system for idiopathic inflammatory myopathies based on clinical manifestations and myositis-specific autoantibodies. JAMA neurol. 2018;75(12):1528–1537. doi:10.1001/jamaneurol.2018.2598
39. Halilu F, Christopher-Stine L. Myositis-specific antibodies: overview and clinical utilization. Rheumatol Immunol Res. 2022;3(1):1–10. doi:10.2478/rir-2022-0001
40. Hamaguchi Y, Kuwana M, Takehara K. Performance evaluation of a commercial line blot assay system for detection of myositis- and systemic sclerosis-related autoantibodies. Clin Rheumatol. 2020;39(11):3489–3497. doi:10.1007/s10067-020-04973-0
41. Tansley SL, Li D, Betteridge ZE, McHugh NJ. The reliability of immunoassays to detect autoantibodies in patients with myositis is dependent on autoantibody specificity. Rheumatology. 2020;59(8):2109–2114. doi:10.1093/rheumatology/keaa021
42. Mulhearn B, Li D, McMorrow F, Lu H, McHugh NJ, Tansley SL. A commercial anti-TIF1γ ELISA is superior to line and dot blot and should be considered as part of routine myositis-specific antibody testing. Front Immunol. 2022;13:804037. doi:10.3389/fimmu.2022.804037
43. Loganathan A, McMorrow F, Lu H, et al. The use of ELISA is comparable to immunoprecipitation in the detection of selected myositis-specific autoantibodies in a European population. Front Immunol. 2022;13:975939. doi:10.3389/fimmu.2022.975939
44. Iaccarino L, Ghirardello A, Bettio S, et al. The clinical features, diagnosis and classification of dermatomyositis. J Autoimmun. 2014;48–49:122–127. doi:10.1016/j.jaut.2013.11.005
45. Sigmund EE, Baete SH, Luo T, et al. MRI assessment of the thigh musculature in dermatomyositis and healthy subjects using diffusion tensor imaging, intravoxel incoherent motion and dynamic DTI. Eur Radiol. 2018;28(12):5304–5315. doi:10.1007/s00330-018-5458-3
46. Malartre S, Bachasson D, Mercy G, et al. MRI and muscle imaging for idiopathic inflammatory myopathies. Brain Pathol. 2021;31(3):e12954. doi:10.1111/bpa.12954
47. Uruha A, Nishikawa A, Tsuburaya RS, et al. Sarcoplasmic MxA expression: a valuable marker of dermatomyositis. Neurology. 2017;88(5):493–500. doi:10.1212/WNL.0000000000003568
48. Schmidt J. Current classification and management of inflammatory myopathies. J Neurom Dis. 2018;5(2):109–129.
49. Won Huh J, Soon Kim D, Keun Lee C, et al. Two distinct clinical types of interstitial lung disease associated with polymyositis-dermatomyositis. Respir Med. 2007;101(8):1761–1769. doi:10.1016/j.rmed.2007.02.017
50. Hallowell RW, Paik JJ. Myositis-associated interstitial lung disease: a comprehensive approach to diagnosis and management. Clin Exp Rheumatol. 2022;40(2):373–383. doi:10.55563/clinexprheumatol/brvl1v
51. Trallero-Araguás E, Grau-Junyent JM, Labirua-Iturburu A, et al. Clinical manifestations and long-term outcome of anti-Jo1 antisynthetase patients in a large cohort of Spanish patients from the GEAS-IIM group. Semin Arthritis Rheum. 2016;46(2):225–231. doi:10.1016/j.semarthrit.2016.03.011
52. Tomonaga M, Sakamoto N, Ishimatsu Y, et al. Comparison of pulmonary involvement between patients expressing anti-PL-7 and anti-Jo-1 antibodies. Lung. 2015;193(1):79–83. doi:10.1007/s00408-014-9665-7
53. Connors GR, Christopher-Stine L, Oddis CV, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years? Chest. 2010;138(6):1464–1474.
54. Hall JC, Casciola-Rosen L, Samedy L-A, et al. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res. 2013;65(8):1307–1315. doi:10.1002/acr.21992
55. Teel A, Lu J, Park J, Singh N, Basharat P. The role of myositis-specific autoantibodies and the management of interstitial lung disease in idiopathic inflammatory myopathies: a systematic review. Semin Arthritis Rheum. 2022;57:152088. doi:10.1016/j.semarthrit.2022.152088
56. ACR Board of Directors. American College of Rheumatology (ACR) guideline for the screening and monitoring of interstitial lung disease in people with systemic autoimmune rheumatic disease; 2023. Available from: https://rheumatology.org/interstitial-lung-disease-guideline.
57. Sanges S, Yelnik CM, Sitbon O, et al. Pulmonary arterial hypertension in idiopathic inflammatory myopathies: data from the French pulmonary hypertension registry and review of the literature. Medicine. 2016;95(39):e4911. doi:10.1097/MD.0000000000004911
58. Lu Z, Guo‐chun W, Li M, Ning Z. Cardiac involvement in adult polymyositis or dermatomyositis: a systematic review. Clin Cardiol. 2012;35(11):685–691. doi:10.1002/clc.22026
59. Liu X-H, Feng X-J, Shi J-Y, et al. The quest for diagnostic approaches of cardiac involvement in polymyositis and dermatomyositis. Ann Palliat Med. 2020;9(4):2256–2270. doi:10.21037/apm-19-650
60. Targoff IN, Johnson AE, Miller FW. Antibody to signal recognition particle in polymyositis. Arthritis Rheum. 1990;33(9):1361–1370. doi:10.1002/art.1780330908
61. Hengstman GJD, ter Laak HJ, Vree Egberts WTM, et al. Anti-signal recognition particle autoantibodies: marker of a necrotising myopathy. Ann Rheum Dis. 2006;65(12):1635–1638. doi:10.1136/ard.2006.052191
62. Jayakumar D, Zhang R, Wasserman A, Ash J. Cardiac manifestations in idiopathic inflammatory myopathies: an overview. Cardiol Rev. 2019;27(3):131–137. doi:10.1097/CRD.0000000000000241
63. Lundberg IE. The heart in dermatomyositis and polymyositis. Rheumatology. 2006;45:iv18–21.
64. Xiong A, Hu Z, Zhou S, et al. Cardiovascular events in adult polymyositis and dermatomyositis: a meta-analysis of observational studies. Rheumatology. 2022;61(7):2728–2739.
65. Gupta R, Wayangankar SA, Targoff IN, Hennebry TA. Clinical cardiac involvement in idiopathic inflammatory myopathies: a systematic review. Int J Cardiol. 2011;148(3):261–270. doi:10.1016/j.ijcard.2010.08.013
66. Casal-Dominguez M, Pinal-Fernandez I, Mego M, et al. High-resolution manometry in patients with idiopathic inflammatory myopathy: elevated prevalence of esophageal involvement and differences according to autoantibody status and clinical subset. Muscle Nerve. 2017;56(3):386–392. doi:10.1002/mus.25507
67. Pearson DR, Werth VP. Relative risk of microscopic colitis in dermatomyositis. J Am Acad Dermatol. 2019;81(5):1188–1190. doi:10.1016/j.jaad.2019.03.045
68. Matas-Garcia A, Milisenda JC, Espinosa G, et al. Gastrointestinal involvement in dermatomyositis. Diagnostics (Basel). 2022;12(5):1200. doi:10.3390/diagnostics12051200
69. Cucchiari D, Angelini C. Renal involvement in idiopathic inflammatory myopathies. Clin Rev Allergy Immunol. 2017;52(1):99–107.
70. Gadiparthi C, Hans A, Potts K, Ismail MK. Gastrointestinal and hepatic disease in the inflammatory myopathies. Rheum Dis Clin North Am. 2018;44(1):113–129. doi:10.1016/j.rdc.2017.09.006
71. Edge K, Chinoy H, Cooper RG. Serum alanine aminotransferase elevations correlate with serum creatine phosphokinase levels in myositis. Rheumatology. 2006;45(4):487–488. doi:10.1093/rheumatology/kel009
72. Sigurgeirsson B, Lindelöf B, Edhag O, Allander E. Risk of cancer in patients with dermatomyositis or polymyositis. A population-based study. N Engl J Med. 1992;326(6):363–367.
73. Olazagasti JM, Baez PJ, Wetter DA, Ernste FC. Cancer risk in dermatomyositis: a meta-analysis of cohort studies. Am J Clin Dermatol. 2015;16(2):89–98. doi:10.1007/s40257-015-0120-1
74. Vaughan H, Rugo HS, Haemel A. Risk-based screening for cancer in patients with dermatomyositis: toward a more individualized approach. JAMA Dermatol. 2022;158(3):244–247. doi:10.1001/jamadermatol.2021.5841
75. Khanna U, Galimberti F, Li Y, Fernandez AP. Dermatomyositis and malignancy: should all patients with dermatomyositis undergo malignancy screening? Ann Transl Med. 2021;9(5):432. doi:10.21037/atm-20-5215
76. Oldroyd AGS, Allard AB, Callen JP, et al. A systematic review and meta-analysis to inform cancer screening guidelines in idiopathic inflammatory myopathies. Rheumatology. 2021;60(6):2615–2628. doi:10.1093/rheumatology/keab166
77. Leatham H, Schadt C, Chisolm S, et al. Evidence supports blind screening for internal malignancy in dermatomyositis: data from 2 large US dermatology cohorts. Medicine. 2018;97(2):e9639. doi:10.1097/MD.0000000000009639
78. Selva-O’Callaghan A, Grau JM, Gámez-Cenzano C, et al. Conventional cancer screening versus PET/CT in dermatomyositis/polymyositis. Am J Med. 2010;123(6):558–562. doi:10.1016/j.amjmed.2009.11.012
79. Maliha PG, Hudson M, Abikhzer G, Singerman J, Probst S. 18F-FDG PET/CT versus conventional investigations for cancer screening in autoimmune inflammatory myopathy in the era of novel myopathy classifications. Nucl Med Commun. 2019;40(4):377–382. doi:10.1097/MNM.0000000000000981
80. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78(4):776–785. doi:10.1016/j.jaad.2017.12.010
81. Narang NS, Casciola-Rosen L, Li S, Chung L, Fiorentino DF. Cutaneous ulceration in dermatomyositis: association with anti–melanoma differentiation–associated gene 5 antibodies and interstitial lung disease. Arthritis Care Res. 2015;67(5):667–672. doi:10.1002/acr.22498
82. Ma X, Chen Z, Hu W, et al. Clinical and serological features of patients with dermatomyositis complicated by spontaneous pneumomediastinum. Clin Rheumatol. 2016;35(2):489–493. doi:10.1007/s10067-015-3001-3
83. Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65(1):25–34. doi:10.1016/j.jaad.2010.09.016
84. Hamaguchi Y, Kuwana M, Hoshino K, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. 2011;147(4):391–398. doi:10.1001/archdermatol.2011.52
85. Chen Z, Cao M, Plana MN, et al. Utility of Anti–Melanoma Differentiation–Associated Gene 5 antibody measurement in identifying patients with dermatomyositis and a high risk for developing rapidly progressive interstitial lung disease: a review of the literature and a meta-analysis. Arthritis Care Res. 2013;65(8):1316–1324. doi:10.1002/acr.21985
86. So J, So H, Wong VT-L, et al. Predictors of rapidly progressive interstitial lung disease and mortality in patients with autoantibodies against melanoma differentiation-associated protein 5 dermatomyositis. Rheumatology. 2022;61(11):4437–4444. doi:10.1093/rheumatology/keac094
87. Zuo Y, Ye L, Chen F, et al. Different multivariable risk factors for rapid progressive interstitial lung disease in Anti-MDA5 positive dermatomyositis and anti-synthetase syndrome. Front Immunol. 2022;13:845988.
88. Dalakas MC. Inflammatory muscle diseases. N Engl J Med. 2015;372(18):1734–1747. doi:10.1056/NEJMra1402225
89. Huang K, Aggarwal R. Antisynthetase syndrome: a distinct disease spectrum. J Scleroderma Relat Disord. 2020;5(3):178–191. doi:10.1177/2397198320902667
90. González-Gay MA, Montecucco C, Selva-O’Callaghan A, et al. Timing of onset affects arthritis presentation pattern in antisyntethase syndrome. Clin Exp Rheumatol. 2018;36(1):44–49.
91. Cavagna L, Nuño L, Scirè CA, et al. Serum Jo-1 autoantibody and isolated arthritis in the antisynthetase syndrome: review of the literature and report of the experience of AENEAS collaborative group. Clin Rev Allergy Immunol. 2017;52(1):71–80.
92. Shi J, Li S, Yang H, et al. Clinical profiles and prognosis of patients with distinct antisynthetase autoantibodies. J Rheumatol. 2017;44(7):1051–1057. doi:10.3899/jrheum.161480
93. Marie I, Josse S, Decaux O, et al. Comparison of long-term outcome between anti-Jo1- and anti-PL7/PL12 positive patients with antisynthetase syndrome. Autoimmun Rev. 2012;11(10):739–745. doi:10.1016/j.autrev.2012.01.006
94. Marco JL, Collins BF. Clinical manifestations and treatment of antisynthetase syndrome. Best Pract Res Clin Rheumatol. 2020;34(4):101503. doi:10.1016/j.berh.2020.101503
95. Opinc AH, Makowska JS. Antisynthetase syndrome - much more than just a myopathy. Semin Arthritis Rheum. 2021;51(1):72–83.
96. Fernandez AP, Gallop J, Polly S, Khanna U. Efficacy and safety of repository corticotropin injection for refractory cutaneous dermatomyositis: a prospective, open-label study. Rheumatology. 2023;kead595. doi:10.1093/rheumatology/kead595
97. Dourmishev L, Meffert H, Piazena H. Dermatomyositis: comparative studies of cutaneous photosensitivity in lupus erythematosus and normal subjects. Photod Photoiml Phot. 2004;20(5):230–234. doi:10.1111/j.1600-0781.2004.00115.x
98. Waldman R, DeWane ME, Lu J. Dermatomyositis: diagnosis and treatment. J Am Acad Dermatol. 2020;82(2):283–296.
99. Olivo Pallo PA, de Souza FHC, Miossi R, Shinjo SK. Mycophenolate mofetil in patients with refractory systemic autoimmune myopathies: case series. Adv Rheumatol. 2018;58(1):34. doi:10.1186/s42358-018-0035-7
100. Wolstencroft PW, Chung L, Li S, Casciola-Rosen L, Fiorentino DF. Factors associated with clinical remission of skin disease in dermatomyositis. JAMA Dermatol. 2018;154(1):44–51. doi:10.1001/jamadermatol.2017.3758
101. Galimberti F, Kooistra L, Li Y, Chatterjee S, Fernandez AP. Intravenous immunoglobulin is an effective treatment for refractory cutaneous dermatomyositis. Clin Exp Dermatol. 2018;43(8):906–912. doi:10.1111/ced.13607
102. Danieli MG, Calcabrini L, Calabrese V, Marchetti A, Logullo F, Gabrielli A. Intravenous immunoglobulin as add on treatment with mycophenolate mofetil in severe myositis. Autoimmun Rev. 2009;9(2):124–127. doi:10.1016/j.autrev.2009.04.003
103. Aggarwal R, Charles-Schoeman C, Schessl J, et al. Trial of Intravenous Immune Globulin in Dermatomyositis. N Engl J Med. 2022;387(14):1264–1278. doi:10.1056/NEJMoa2117912
104. Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65(2):314–324. doi:10.1002/art.37754
105. Chung L, Genovese MC, Fiorentino DF. A pilot trial of rituximab in the treatment of patients with dermatomyositis. Arch Dermatol. 2007;143(6):763–767. doi:10.1001/archderm.143.6.763
106. Aggarwal R, Loganathan P, Koontz D, Qi Z, Reed AM, Oddis CV. Cutaneous improvement in refractory adult and juvenile dermatomyositis after treatment with rituximab. Rheumatology. 2017;56(2):247–254. doi:10.1093/rheumatology/kew396
107. So H, Wong VTL, Lao VWN, Pang HT, Yip RML. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37(7):1983–1989. doi:10.1007/s10067-018-4122-2
108. Ge Y, Li S, Tian X, He L, Lu X, Wang G. Anti-melanoma differentiation-associated gene 5 (MDA5) antibody-positive dermatomyositis responds to rituximab therapy. Clin Rheumatol. 2021;40(6):2311–2317. doi:10.1007/s10067-020-05530-5
109. Hayashi M, Kikuchi T, Takada T. Mycophenolate mofetil for the patients with interstitial lung diseases in amyopathic dermatomyositis with anti-MDA-5 antibodies. Clin Rheumatol. 2017;36(1):239–240. doi:10.1007/s10067-016-3443-2
110. Shimojima Y, Ishii W, Matsuda M, Kishida D, Ikeda S-I. Effective use of calcineurin inhibitor in combination therapy for interstitial lung disease in patients with dermatomyositis and polymyositis. J Clin Rheumatol. 2017;23(2):87–93. doi:10.1097/RHU.0000000000000487
111. Anyanwu CO, Chansky PB, Feng R, Carr K, Okawa J, Werth VP. The systemic management of cutaneous dermatomyositis: results of a stepwise strategy. Int J Womens Dermatol. 2017;3(4):189–194. doi:10.1016/j.ijwd.2017.05.001
112. Pelle MT, Callen JP. Adverse cutaneous reactions to hydroxychloroquine are more common in patients with dermatomyositis than in patients with cutaneous lupus erythematosus. Arch Dermatol. 2002;138(9):1231–1233. doi:10.1001/archderm.138.9.1231
113. Wolstencroft PW, Casciola-Rosen L, Fiorentino DF. Association between autoantibody phenotype and cutaneous adverse reactions to hydroxychloroquine in dermatomyositis. JAMA Dermatol. 2018;154(10):1199–1203. doi:10.1001/jamadermatol.2018.2549
114. Combalia A, Giavedoni P, Tamez L, Grau-Junyent JM, Mascaró JM. Bosentan for cutaneous ulcers in anti-MDA5 dermatomyositis. JAMA Dermatol. 2018;154(3):371–373. doi:10.1001/jamadermatol.2017.5462
115. Prasad V, Lenzer J, Newman DH. Why cancer screening has never been shown to “save lives”--and what we can do about it. BMJ. 2016;352:1.
116. Oldroyd AGS, Callen JP, Chinoy H, et al. International guideline for idiopathic inflammatory myopathy-associated cancer screening: an international myositis assessment and clinical studies group (IMACS) initiative. Nat Rev Rheumatol. 2023;19:805–817. doi:10.1038/s41584-023-01045-w
117. Rabi DM, Kunneman M, Montori VM. When guidelines recommend shared decision-making. JAMA. 2020;323(14):1345–1346. doi:10.1001/jama.2020.1525
118. Harris RP, Sheridan SL, Lewis CL, et al. The harms of screening: a proposed taxonomy and application to lung cancer screening. JAMA Intern Med. 2014;174(2):281–285. doi:10.1001/jamainternmed.2013.12745
119. Hoffmann TC, Del Mar C. Clinicians’ expectations of the benefits and harms of treatments, screening, and tests: a systematic review. JAMA Intern Med. 2017;177(3):407–419. doi:10.1001/jamainternmed.2016.8254
120. Harris RP, Wilt TJ, Qaseem A. High value care task force of the American college of physicians. A value framework for cancer screening: advice for high-value care from the American college of physicians. Ann Intern Med. 2015;162(10):712–717. doi:10.7326/M14-2327
121. Fiorentino DF, Mecoli CA, Rosen MC, et al. Immune responses to CCAR1 and other dermatomyositis autoantigens are associated with attenuated cancer emergence. J Clin Invest. 2022;132(2):e150201. doi:10.1172/JCI150201
122. Hosono Y, Sie B, Pinal-Fernandez I, et al. Coexisting autoantibodies against transcription factor Sp4 are associated with decreased cancer risk in patients with dermatomyositis with anti-TIF1γ autoantibodies. Ann Rheum Dis. 2023;82(2):246–252. doi:10.1136/ard-2022-222441
123. McPherson M, Economidou S, Liampas A, Zis P, Parperis K. Management of MDA-5 antibody positive clinically amyopathic dermatomyositis associated interstitial lung disease: a systematic review. Semin Arthritis Rheum. 2022;53:151959. doi:10.1016/j.semarthrit.2022.151959
124. Huang K, Vinik O, Shojania K, et al. Clinical spectrum and therapeutics in Canadian patients with anti-melanoma differentiation-associated gene 5 (MDA5)-positive dermatomyositis: a case-based review. Rheumatol Int. 2019;39(11):1971–1981. doi:10.1007/s00296-019-04398-2
125. Fischer A, Brown KK, Du Bois RM, et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J Rheumatol. 2013;40(5):640–646. doi:10.3899/jrheum.121043
126. Guo H, Leung JCK, Chan LYY, Lui SL, Tsang AWL, Lai KN. Modulation of intra-pulmonary TGF-beta expression by mycophenolate mofetil in lupus prone MRL/lpr mice. Lupus. 2005;14(8):583–592. doi:10.1191/0961203305lu2170oa
127. Fan L, Lyu W, Liu H, et al. A retrospective analysis of outcome in melanoma differentiation-associated gene 5-related interstitial lung disease treated with tofacitinib or tacrolimus. J Rheumatol. 2022;49(12):1356–1364. doi:10.3899/jrheum.220367
128. Chen Z, Wang X, Ye S. Tofacitinib in amyopathic dermatomyositis-associated interstitial lung disease. N Engl J Med. 2019;381(3):291–293. doi:10.1056/NEJMc1900045
129. Nakashima R, Hosono Y, Mimori T. Clinical significance and new detection system of autoantibodies in myositis with interstitial lung disease. Lupus. 2016;25(8):925–933. doi:10.1177/0961203316651748
130. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72(3):488–498. doi:10.1002/art.41105
131. Wang L-M, Yang Q-H, Zhang L, et al. Intravenous immunoglobulin for interstitial lung diseases of anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Rheumatology. 2022;61(9):3704–3710. doi:10.1093/rheumatology/keab928
132. Abe Y, Kusaoi M, Tada K, Yamaji K, Tamura N. Successful treatment of anti-MDA5 antibody-positive refractory interstitial lung disease with plasma exchange therapy. Rheumatology. 2020;59(4):767–771. doi:10.1093/rheumatology/kez357
133. Shirakashi M, Nakashima R, Tsuji H, et al. Efficacy of plasma exchange in anti-MDA5-positive dermatomyositis with interstitial lung disease under combined immunosuppressive treatment. Rheumatology. 2020;59(11):3284–3292. doi:10.1093/rheumatology/keaa123
134. Shirai T, Machiyama T, Sato H, Ishii T, Fujii H. Intensive induction therapy combining tofacitinib, rituximab and plasma exchange in severe anti-melanoma differentiation-associated protein-5 antibody-positive dermatomyositis. Clin Exp Rheumatol. 2023;41(2):291–300. doi:10.55563/clinexprheumatol/8kulbf
135. Lian Q-Y, Chen A, Zhang J-H, et al. Lung transplantation for anti-MDA5-positive dermatomyositis-associated rapid progressive interstitial lung disease: report of two cases and review of the literature. Clin Rheumatol. 2023;42(3):941–947. doi:10.1007/s10067-022-06422-6
136. Zou J, Li T, Huang X, Chen S, Guo Q, Bao C. Basiliximab may improve the survival rate of rapidly progressive interstitial pneumonia in patients with clinically amyopathic dermatomyositis with anti-MDA5 antibody. Ann Rheum Dis. 2014;73(8):1591–1593. doi:10.1136/annrheumdis-2014-205278
137. Holzer M-T, Nies JF, Oqueka T, Huber TB, Kötter I, Krusche M. Successful rescue therapy with daratumumab in rapidly progressive interstitial lung disease caused by MDA5-positive dermatomyositis. Chest. 2023;163(1):e1–5. doi:10.1016/j.chest.2022.08.2209
138. Dermatomyositis: data Support IFN-beta as therapeutic target. Medscape; 2023. Available from: https://www.medscape.com/viewarticle/989902.
139. Ahmed S, Chakka S, Concha J, Krain R, Feng R, Werth VP. Evaluating important change in cutaneous disease activity as an efficacy measure for clinical trials in dermatomyositis. Br J Dermatol. 2020;182(4):949–954. doi:10.1111/bjd.18223
140. Aggarwal R, Domyslawska I, Carreira P, et al.. Pos1207 Efficacy And Safety Of Anti-Ifnβ-Specific Monoclonal Antibody, Pf-06823859, On Myositis: phase 2 Study In Patients With Moderate-To-Severe Dermatomyositis. In Scientific Abstracts. BMJ Publishing Group Ltd and European League Against Rheumatism; 2023:936–937.
141. Pfizer. A phase 3, multicenter, double-blind, randomized, placebo-controlled study to evaluate the efficacy and safety of Pf-06823859 in participants with active idiopathic inflammatory myopathies (including participants with active dermatomyositis or polymyositis). clinicaltrials.gov; 2023. Available from: https://clinicaltrials.gov/study/NCT05895786.
142. Priovant Therapeutics, Inc. A phase 3, randomized, double-blind, placebo-controlled study to investigate the efficacy and safety of oral brepocitinib in adults with dermatomyositis. clinicaltrials.gov; 2023. Available from: https://clinicaltrials.gov/study/NCT05437263.
143. Assistance Publique - Hôpitaux de Paris. Baricitinib in patients with relapsing or naïve dermatomyositis: a multicenter randomized controlled Trial (BIRD). clinicaltrials.gov; 2022. Available from: https://clinicaltrials.gov/study/NCT04972760.
144. Galapagos NV. A randomized, double-blind, placebo-controlled, multi-center study to evaluate the efficacy, safety, tolerability, pharmacokinetics, and pharmacodynamics of orally administered GLPG3667 once daily for 24 weeks in adult subjects with dermatomyositis. clinicaltrials.gov; 2023. Available from: https://clinicaltrials.gov/study/NCT05695950.
145. Paik JJ, Casciola-Rosen L, Shin JY, et al. Study of tofacitinib in refractory dermatomyositis: an open-label pilot study of ten patients. Arthritis Rheumatol. 2021;73(5):858–865. doi:10.1002/art.41602
146. Alexion. A Phase 2/3, double-blind, randomized, placebo-controlled, parallel group, multicenter study to evaluate the efficacy and safety of ravulizumab in adult participants with dermatomyositis. clinicaltrials.gov; 2023. Available from: https://clinicaltrials.gov/study/NCT04999020.
147. Kim YJ, Schiopu E, Dankó K, et al. A phase 2, double-blinded, placebo-controlled trial of toll-like receptor 7/8/9 antagonist, IMO-8400, in dermatomyositis. J Am Acad Dermatol. 2021;84(4):1160–1162. doi:10.1016/j.jaad.2020.07.122
148. EMD Serono Research & Development Institute, Inc. A phase iia, randomized, parallel, double-blind, placebo controlled study to evaluate the efficacy and safety of enpatoran in dermatomyositis and polymyositis participants receiving standard of care (NEPTUNIA). clinicaltrials.gov; 2023. Available from: https://clinicaltrials.gov/study/NCT05650567.
149. Horizon Therapeutics Ireland DAC. A phase 2, randomized, double-blind, placebo-controlled, efficacy and safety study of daxdilimab subcutaneous injection in adult participants with inadequately controlled dermatomyositis or anti-synthetase inflammatory myositis. clinicaltrials.gov; 2023. Available from: https://clinicaltrials.gov/study/NCT05669014.
150. Tjärnlund A, Tang Q, Wick C, et al. Abatacept in the treatment of adult dermatomyositis and polymyositis: a randomised, phase IIb treatment delayed-start trial. Ann Rheum Dis. 2018;77(1):55–62. doi:10.1136/annrheumdis-2017-211751
151. Müller F, Boeltz S, Knitza J, et al. CD19-targeted CAR T cells in refractory antisynthetase syndrome. Lancet. 2023;401(10379):815–818. doi:10.1016/S0140-6736(23)00023-5
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.