")
Back to Journals » OncoTargets and Therapy » Volume 9
Dendritic cell immunotherapy versus bevacizumab plus irinotecan in recurrent malignant glioma patients: a survival gain analysis
Authors Artene S, Turcu-Stiolica A , Hartley R, Ciurea ME, Daianu O, Brindusa C, Alexandru O, Tataranu LG, Purcaru SO, Dricu A
Received 16 May 2016
Accepted for publication 30 July 2016
Published 4 November 2016 Volume 2016:9 Pages 6669—6677
DOI https://doi.org/10.2147/OTT.S112842
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Stefan-Alexandru Artene,1 Adina Turcu-Stiolica,2 Richard Hartley,1 Marius Eugen Ciurea,3 Oana Daianu,1 Corina Brindusa,1 Oana Alexandru,4 Ligia Gabriela Tataranu,5 Stefana Oana Purcaru,1 Anica Dricu1
1Unit of Biochemistry, 2Department of Biostatistics, 3Department of Plastic and Reconstructive Surgery, 4Department of Neurology, University of Medicine and Pharmacy of Craiova, Craiova, 5Department of Neurosurgery, “Bagdasar–Arseni” Emergency Hospital, Bucharest, Romania
Background: The bevacizumab and irinotecan protocol is considered a standard treatment regimen for recurrent malignant glioma. Recent advances in immunotherapy have hinted that vaccination with dendritic cells could become an alternative salvage therapy for the treatment of recurrent malignant glioma.
Methods: A search was performed on PubMed, Cochrane Library, Web of Science, ScienceDirect, and Embase in order to identify studies with patients receiving bevacizumab plus irinotecan or dendritic cell therapy for recurrent malignant gliomas. The data obtained from these studies were used to perform a systematic review and survival gain analysis.
Results: Fourteen clinical studies with patients receiving either bevacizumab plus irinotecan or dendritic cell vaccination were identified. Seven studies followed patients that received bevacizumab plus irinotecan (302 patients) and seven studies included patients that received dendritic cell immunotherapy (80 patients). For the patients who received bevacizumab plus irinotecan, the mean reported median overall survival was 7.5 (95% confidence interval [CI] 4.84–10.16) months. For the patients who received dendritic cell immunotherapy, the mean reported median overall survival was 17.9 (95% CI 11.34–24.46) months. For irinotecan + bevacizumab group, the mean survival gain was -0.02±2.00, while that for the dendritic cell immunotherapy group was -0.01±4.54.
Conclusion: For patients with recurrent malignant gliomas, dendritic cell immunotherapy treatment does not have a significantly different effect when compared with bevacizumab and irinotecan in terms of survival gain (P=0.535) and does not improve weighted survival gain (P=0.620).
Keywords: malignant glioma, irinotecan, bevacizumab, dendritic cell, systematic analysis
Introduction
Glioblastoma multiforme (GBM) is the malignant form of glial tissue/cells and is the most common and aggressive primary brain tumor, accounting for 50% of all adult gliomas.1,2 Treating GBM (World Health Organization grade IV astrocytoma) proves challenging due to its highly diffuse nature that makes complete surgical resection difficult and its location behind the blood–brain barrier provides protection against several chemotherapeutic agents. Despite recent advances, prognosis remains poor with the median survival being ~9–15 months and 2-year survival rate being between 9% and 26%.2–5 Standard first-line treatment for GBM involves surgical resection of the tumor bulk followed by 6 weeks of focalized fractional radiotherapy, alongside chemotherapy with an oral alkylating agent, such as temozolomide.1,2 This particular drug regimen, along with others tried previously, does not produce an effective method of treatment in clinical practice as is seen in laboratory testing. This discrepancy has been highlighted in a number of in vitro studies,6 which have consistently shown much more promising results than is observed in vivo.4,7,8 The most commonly cited reason for the laboratory findings not translating well into clinical practice is a large reduction in drug efficacy.6 This reduction in potency is attributed to a variety of structural and molecular pathways that work together to hinder the action of the drug.9,10 The first major difference between the theoretical models, in vitro and in vivo, is the protection that the surrounding central nervous system (CNS) provides to the tumor, which simply does not exist in the other testing methodologies. The first obstacle is the blood–brain barrier, which blocks or reduces the transmission of many drugs (including those used in the treatment of GBM) from the bloodstream into the CNS.11 Furthermore, GBM is known to produce highly vascularized tumors, which enables them to sustain their rapid development. This increased level of vasculature would be a positive factor for drug uptake, if it were not for the irregular and tortuous nature of the vessels that GBM create. These malformed vessels result in chaotic and stagnant blood flow across different regions of the tumor, which results in inadequate drug perfusion.11–13 This effect is exacerbated by a large number of breaks in the tight junctions and fenestrations of tumor capillaries. This improper formation of vasculature leads to an increase in capillary permeability and raises interstitial pressure, further preventing drug access into the tumor bulk.12,13 So, for an aggressive cancer such as GBM to grow and spread as fast as it does, it requires a more substantial supply of blood. To achieve this, the malignant tumor stimulates angiogenesis through upregulation of several angiogenic growth factors, such as basic fibroblast growth factor (bGFG), platelet derived growth factor (PDGF), and vascular epithelial growth factor (VEGF).14 Inhibition of VEGF-mediated angiogenesis by the monoclonal antibody bevacizumab is already used in breast cancer to improve progression-free survival15 and may prove a necessary supplement to anti-EGFR targeting drugs.16
Active immunotherapy involves the introduction of antigen-presenting dendritic cells containing a tumor-associated antigen, which can train the host immune system to recognize tumor cells as a pathogen to be eliminated. Immunotherapeutic treatment has proven to be a success in other aggressive cancers17–19 and could potentially provide similar results for GBM.
The idea that the immune system can be utilized to treat GBM has a lot of merit, the reason being that even though GBM is a highly aggressive and invasive tumor, it rarely metastasizes outside the CNS. A study by Lee et al20 showed that GBM could form metastases in immune-deficient mice, but only when both adaptive and innate immune systems were inactive. This study also demonstrated the ability of the innate and adaptive immune systems to detect and destroy GBM tumor cells after they have crossed the blood–brain barrier and had invaded other systems – notably subcutaneous and lung tissue.20 The CNS lies behind the blood–brain barrier, in an immune-privileged site. This protection is thought to allow the rapid growth associated with GBM; however, increasing numbers of studies demonstrate that the protection that this barrier provides is not absolute, and that cells of the immune system may permeate under specific circumstances.20 When GBM typically arises, the small number of antigen-presenting cells that could potentially detect the tumor and illicit an immune response (as seen outside the CNS) are inhibited by the expression of high levels of MHC class I (HLA-ABC) molecules on GBM tumor cell surfaces. These two forms of protection work in conjunction to prevent tumor cells from being targeted and eliminated by cytotoxic T-cells or natural killer cells, as is seen outside of the blood–brain barrier. There have been quite a few approaches to immune therapy in malignant glioma patients in recent years. Targeting immunosuppressive checkpoint pathways such as CTLA-4, TIGIT, PD-1, LAG-3, and TIM-321–25 have proven successful in other types of cancers such as brain-metastasizing melanoma.26,27 Currently, a trial that uses ipilimumab for patients with GMB is recruiting patients (NCT02017717). Another therapeutic approach used in glioblastoma is called adoptive immunotherapy, which relies on autologous T-cell transfer and chemeric antigen receptor (CAR) T-cells. Two preclinical studies based on CAR T-cells have shown encouraging responses against malignant glioma xenografts in immunodeficient mice.28,29 Two clinical trials using anti-EGFRvIII CAR T-cells in patients with gliomas expressing the EGFRvIII mutation are currently recruiting participants to determine the safety and response to this treatment. (NCT02442297, NCT01454596).
A more studied immunotherapeutic approach in GBM is based on dendritic cell vaccination. Initially, immature dendritic cells were used but with limited results.30 This method relied on the intrinsic nature of these antigen-presenting cells to detect cancerous tissue and initiate an immune response. However, it was soon discovered that in vitro maturation of dendritic cells using various inflammatory modulators, such as tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), interleukin-1 (IL1), and IL-4 alone or in combination yielded better results.30 Once harvested and isolated, the dendritic cells are mixed with the desired tumor-associated antigen. A variety of antigen sources may be used, such as a GBM tumor lysate, synthetic or autologous tumor proteins (eg, EGFRvIII/delta, a common mutation found in GBM), mRNA from apoptosed tumor cells, and radiologically inactivated tumor cells.30
The antigen-primed dendritic cells may be implanted into subcutaneous or subdermal tissue, intranodally into the lymphatics, or directly into the tumor; combined intravenous and intracranial injections were found to be most effective.31 Additionally, those who responded best to the vaccination produced greater quantities of IFN-γ post-vaccination.32 Furthermore, the same study demonstrated that this elevated IFN-γ improved the effectiveness of subsequent chemotherapy cycles by sensitizing the tumor to chemotherapeutic agents. The vaccination produces a delayed-type hypersensitivity reaction, leading to tumor infiltration of primarily CD8+ cytotoxic T lymphocytes, CD4+ T helper lymphocytes, as well as natural killer cells.30,33 The effectiveness of vaccination, which depends on its ability to increase T-cell infiltration of the tumor bulk, is reflected in the overall survival (OS) of GBM patient. Additionally, greater lymphocyte infiltration leads to a decrease in intratumoral transforming growth factor-beta (TGF-β) levels, which further inhibits tumor growth.32
Systematic analysis of nine Phase I and II trials, covering 409 patients and analyzing the clinical efficacy of dendritic cell treatment for high-grade gliomas, by Cao et al34 demonstrated that dendritic cell immunotherapy significantly improves patient OS and progression-free survival, but with no significant difference in Karnofsky performance status (quality of life assessment) when compared to control groups. Furthermore, no dose-related toxicity has been found to be associated with dendritic cell vaccination.30 The results reported by Cao et al show that in these selected trials, the 1- and 5-year OS rates of patients improved from 63% and 1%, respectively, to 82% and 14%,34 and the 1- and 4-year progression-free survival improved from 32% and 1%, respectively, to 70% and 27%. These results strongly favor the use of dendritic cell immunotherapy to supplement the current gold standard of tumor resection, radio- and chemotherapies.
Bevacizumab had initially shown promising results in treating recurrent high-grade gliomas,35 while irinotecan, a topoisomerase I inhibitor, has proved to be ineffective when treating this particular malignancy even though it displayed high blood–brain barrier permeability.36,37 The idea that the combination of these two agents would provide improvement in patients suffering from high-grade gliomas was first presented in two independent studies by Vredenburgh et al38 and Chen et al39 in 2007. However, two subsequent meta-analyses have shown that the combination of bevacizumab and irinotecan has no benefit over single-therapy bevacizumab,40,41 and more evidence is required to determine which therapeutic approach is superior. The aim of our study was to determine if the more recent, dendritic cell therapy is an improvement over the older, more studied therapeutic regimen based on bevacizumab and irinotecan, which is the preferred bevacizumab-based approach by the majority of authors.40,41
Methods
Search strategy and study selection
A systematic literature search was performed in February 2016 through PubMed, Cochrane Library, Web of Science, ScienceDirect, and Embase in order to identify clinical studies reporting the effect of bevacizumab plus irinotecan or dendritic cell therapy on patients suffering from recurrent malignant gliomas. The literature search was performed by two independent authors. After the initial selection by each author, the lists were compared and any disagreement was resolved by either consulting the third author or by mutual discussion. The search terms used to identify titles and abstracts were “malignant glioma”, “recurrent”, “bevacizumab”, “irinotecan”, and “dendritic cell”. No restrictions were applied to the language or publication date.
Eligibility criteria
Published full-text articles were considered for review if they met the following criteria: 1) reported outcome of interest was patient OS; 2) patients were 18 years or older when they were histopathologically diagnosed with malignant gliomas according to WHO standards; 3) all the patients suffered from recurrent high-grade gliomas for which they initially received treatment before observed therapeutic failure and/or tumor progression; and 4) treatment with either bevacizumab and irinotecan or dendritic cell therapy. Case reports, reviews, nonpeer-reviewed studies, conference abstracts, commentaries, and letters were excluded.
Data analysis
The database was analyzed using SPSS 17.0 (SPSS Inc., Chicago, IL, USA). Descriptive statistics were used to summarize and describe the data. The outcome was influenced by number of patients, sex, patients’ age, and histological grade. The median overall survival (mOS) time was recorded for each study. We could not quantify the response rate in the same way. Multiple regression analysis was used to describe the association of mOS and response rate with the basic characteristics of the patients. All the predictions were based on observed patient information contained within the clinical trial. The data were weighted by the square route of the patient numbers reported in each study. This information allowed the calculation of the predicted mOS. No information with regard to treatment was used in calculating the predicted OS. The influence of the treatment (bevacizumab and irinotecan or dendritic cell immunotherapy) was characterized by survival gain, which is defined and validated as the difference between observed and predicted mOS. The weighted survival gain was considered as the product of survival gain and the square root of the number of patients. The survival gain and weighted survival gain obtained in the two different treatments were compared using the Mann–Whitney–Wilcoxon test. The P-values were calculated at the two-tailed level. A P-value <0.05 was regarded as statistically significant.
Results
An initial inquiry into online literature yielded 574 titles and abstracts. After excluding duplicates, a total of 112 titles and abstracts were eligible for reviewing. After reviewing the full text of the articles, only 27 studies were considered for the eligibility criteria. Of these, only 14 studies met the eligibility criteria and were included in our systematic analysis (Figure 1). Seven studies described patients who received bevacizumab plus irinotecan, while seven studies described patients who received dendritic cell immunotherapy.
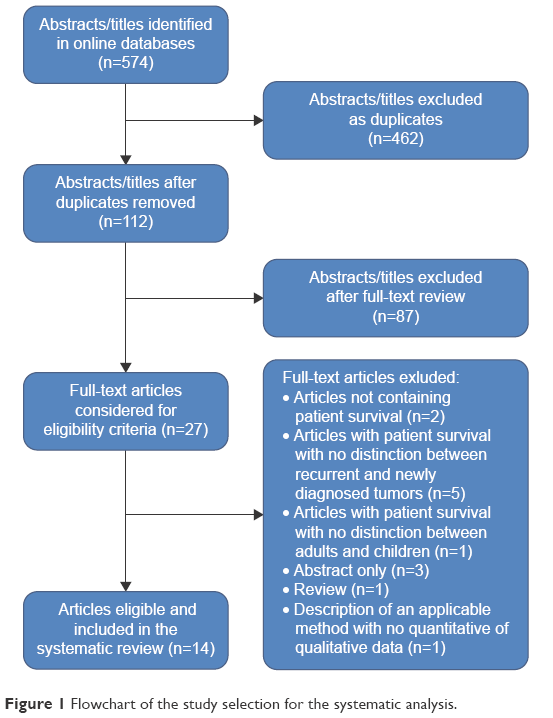 | Figure 1 Flowchart of the study selection for the systematic analysis. |
Patient characteristics for each study, such as number of patients, median age, sex, tumor histology, and treatment protocol, are described in Tables 1 and 2, while treatment responses (mOS, predicted mOS, survival gain, and weighted gain) are presented in Tables 3 and 4. OS was defined as the time from enrolment in the study until death (due to any cause). A total of 381 patients were included in our systematic review with 302 (79.26%) of them receiving bevacizumab plus irinotecan and 79 (20.74%) receiving dendritic cell vaccination. The study included 243 (63.78%) males and 138 (36.22%) females. The median age for the bevacizumab plus irinotecan group was 49 years (95% confidence interval [CI] 45.13–52.87) and for the dendritic cell therapy group was 43 years (95% CI 38.92–47.08). In terms of histology, 268 (70.34%) patients suffered from grade IV gliomas, while 113 (29.63%) were diagnosed with nongrade IV malignant gliomas. In addition, 233 (77.15%) patients receiving bevacizumab plus irinotecan were diagnosed with GBM, while only 69 (22.85%) patients were diagnosed as having grade III gliomas. In the dendritic cell vaccination group, 58 (73.41%) patients had GB while 21 (26.59%) patients had grade III gliomas.
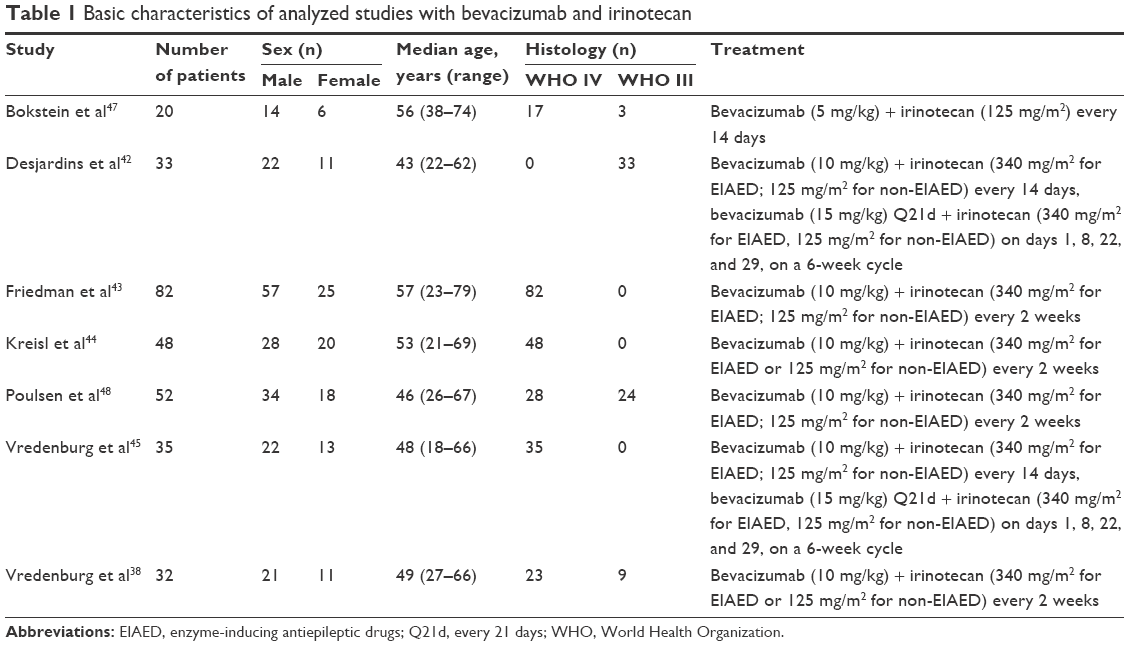 | Table 1 Basic characteristics of analyzed studies with bevacizumab and irinotecan |
 | Table 2 Basic characteristics of analyzed studies with dendritic cell immunotherapy |
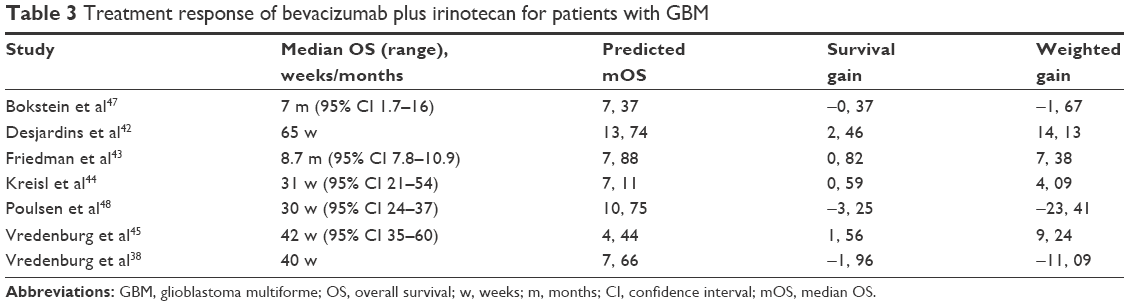 | Table 3 Treatment response of bevacizumab plus irinotecan for patients with GBM |
 | Table 4 Treatment response of dendritic cell immunotherapy for patients with GBM |
In terms of treatment outcome, mean reported mOS was 7.5 (95% CI 4.84–10.16) months for patients receiving bevacizumab and irinotecan. For patients receiving dendritic cell vaccination, mean reported mOS was 17.9 (95% CI 11.34–24.46) months. In terms of survival gain and weighted survival gain, four of the seven42–45 studies using bevacizumab and irinotecan reported positive results for patients included in the studies. Furthermore, only two of the seven29,46 studies which used dendritic cell vaccination presented a positive survival gain for the patients included in the study. In comparison, the studies following the bevacizumab and irinotecan protocol reported a mean survival gain of −0.02±2.00, while the mean survival gain was −0.01±4.54 for dendritic cell immunotherapy group. Overall, we observed that compared with the bevacizumab and irinotecan treatment protocol, dendritic cell vaccination does not have a statistically different effect on OS time (P=0.535) and does not statistically improve weighted survival gain (P=0.620) for patients with malignant gliomas. The survival gain distributions of the two groups are shown in Figure 2.
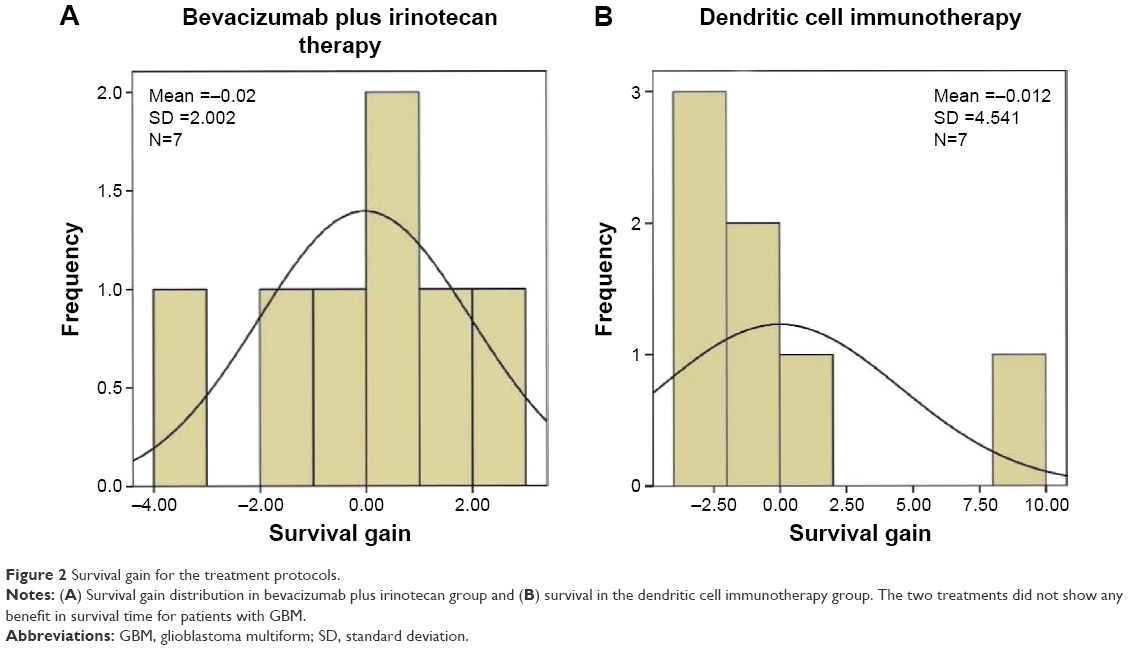 | Figure 2 Survival gain for the treatment protocols. |
Discussion
Malignant gliomas represent one the most challenging tumors from a therapeutic and clinical point of view. Due to the rapid pace at which cells multiply and invade the surrounding tissue and the number of factors which determine its resistance to standard therapeutic protocols, tumor recurrences are generally the rule for this this type of tumor. A large number of therapeutic approaches have been tried in the treatment of malignant gliomas, but with limited results so far. One of these approaches was the use of bevacizumab, a VEGF-blocking agent with antiangiogenic properties which yielded limited results when administered as both single therapy39–41,53–55 and in combination with temozolomide and radiotherapy.56,57 A more recent approach is the use of immune therapy, which is based on turning the body’s own immune system against the tumor cells.58–61 One variant of immune therapy is the use of the patient’s dendritic cells in order to trigger an immune response against malignant glioma cells.62
In this study, we compared the effects of bevacizumab and irinotecan against the effects of dendritic cell therapy in patients with recurrent malignant gliomas. A total of 381 patients from 14 studies were included in our study. Three hundred and two patients received bevacizumab with irinotecan, while 79 patients received dendritic cell vaccination. According to our results, the newer dendritic cell therapy did not have a significant influence on patients’ survival in comparison to the older, more studied bevacizumab plus irinotecan treatment protocol. While mOS was significantly longer in patients receiving dendritic cell vaccination, this was not reflected in the survival gain of the patients. Of the seven studies that involved patients undergoing dendritic cell therapy, only two studies presented a positive survival gain, making dendritic cell therapy similar to other salvage treatment protocols used in recurrent malignant gliomas. However, interpretation of our study should be met with caution due to the limitations of our study and lack of randomized control trials directly comparing the effects of these two treatment protocols. Several limitations can also influence the outcome of this study and must be taken into consideration. First, systematic analysis studies remain a controversial method of comparing different therapeutic protocols because of the large number of variables pertaining to the patients involved in each study, making the overall patient pool very heterogeneous. Our study focused on the therapeutic combination of bevacizumab and irinotecan rather than bevacizumab in single therapy in recurrent high-grade gliomas because of the significantly larger number of studies using the first option in comparison to the second. We chose not to include both types of therapies under the same group because it would only increase the discrepancy between the groups receiving either bevacizumab-based regimens or dendritic cell immunotherapy in terms of patient numbers and characteristics, leading to a highly uneven comparison. In addition, the relatively small number of studies (14) involved in our analysis does not cover a sufficient number of patients to account for all the different variables, which could influence the outcome of each individual study. The patients who received bevacizumab and irinotecan also notably outnumber those who received dendritic cell immunotherapy, potentially influencing the outcome of our systematic analysis. Second, most of the studies in our systematic review referred to malignant gliomas as a unique entity in terms of treatment and outcome. We acknowledge the difference, in terms of outcome and treatment, between malignant glioma types, especially between GBM and grade III malignant gliomas. Some of the studies encompass patients strictly with GBM or with grade III malignant gliomas, while the distribution of grade III and grade IV malignant gliomas between the two therapeutic groups is not evenly matched. This difference in tumor grading can influence the outcome of the mOS in the two groups. Third, dendritic cell immune vaccination encompasses different protocols involving a heterogeneous population of cells. However, due to the limited amount of information available in literature regarding the differences in terms of patient outcome between different dendritic cell immunotherapies, we considered them as a singular therapeutic entity.
Conclusion
The survival gain analysis demonstrated that there is no real clinical benefit for patients undergoing dendritic cell vaccination in comparison to those receiving bevacizumab and irinotecan for the treatment of recurrent malignant gliomas.
Acknowledgment
The study was funded by Executive Agency for Higher Education, Research, Development and Innovation Funding, Romania (Grant number: PN-II-ID-PCE-2011-3-1041). All authors contributed equally to this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. | ||
Peereboom DM, Shepard DR, Ahluwalia MS, et al. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J Neurooncol. 2010;98(1):93–99. | ||
Joshi AD, Loilome W, Siu IM, Tyler B, Gallia GL, Riggins GJ. Evaluation of tyrosine kinase inhibitor combinations for glioblastoma therapy. PloS One. 2012;7(10):e44372. | ||
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | ||
Stewart LA. Chemotherapy in adult high-grade glioma: a systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet. 2002;359(9311):1011–1018. | ||
Tortora G, Bianco R, Daniele G, et al. Overcoming resistance to molecularly targeted anticancer therapies: rational drug combinations based on EGFR and MAPK inhibition for solid tumours and haematologic malignancies. Drug Resist Updat. 2007;10(3):81–100. | ||
Gunther W, Pawlak E, Damasceno R, Arnold H, Terzis AJ. Temozolomide induces apoptosis and ence in glioma cells cultured as multicellular spheroids. Br J Cancer. 2003;88(3):463–469. | ||
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. | ||
Cohen AL, Colman H. Glioma biology and molecular markers. Cancer Treat Res. 2015;163:15–30. | ||
Alentorn A, Duran-Pena A, Pingle SC, Piccioni DE, Idbaih A, Kesari S. Molecular profiling of gliomas: potential therapeutic implications. Expert Rev Anticancer Ther. 2015;15(8):955–962. | ||
Taylor TE, Furnari FB, Cavenee WK. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr Cancer Drug Targets. 2012;12(3):197–209. | ||
Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–124. | ||
Krakstad C, Chekenya M. Survival signalling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Mol Cancer. 2010;9:135. | ||
Reardon DA, Wen PY. Therapeutic advances in the treatment of glioblastoma: rationale and potential role of targeted agents. Oncologist. 2006;11(2):152–164. | ||
Andre F, Deluche E, Bonnefoi H. Bevacizumab: the phoenix of breast oncology? Lancet Oncol. 2015;16(6):600–601. | ||
Sathornsumetee S, Desjardins A, Vredenburgh JJ, et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro Ooncol. 2010;12(12):1300–1310. | ||
Hillerdal V, Essand M. Chimeric antigen receptor-engineered T cells for the treatment of metastatic prostate cancer. BioDrugs. 2015;29(2):75–89. | ||
Leavy O. Immunotherapy: a triple blow for cancer. Nat Rev Cancer. 2015;15(5):258–259. | ||
Whiteside TL, Demaria S, Rodriguez-Ruiz ME, Zarour HM, Melero I. Emerging opportunities and challenges in cancer immunotherapy. Clin Cancer Res. 2016;22(8):1845–1855. | ||
Lee SJ, Kang WY, Yoon Y, et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer. 2015;15(1):1011. | ||
Joller N, Hafler JP, Brynedal B, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol. 2011;186(3):1338–1342. | ||
Driessens G, Kline J, Gajewski TF. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev. 2009;229(1):126–144. | ||
Jackson C, Ruzevick J, Phallen J, Belcaid Z, Lim M. Challenges in immunotherapy presented by the glioblastoma multiforme microenvironment. Clin Dev Immunol. 2011;2011:732413. | ||
Saikali S, Avril T, Collet B, et al. Expression of nine tumour antigens in a series of human glioblastoma multiforme: interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. J Neurooncol. 2007;81(2):139–148. | ||
Granucci F, Lutz MB, Zanoni I. The nature of activatory and tolerogenic dendritic cell-derived signal 2. Front Immunol. 2014;5:42. | ||
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | ||
Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459–465. | ||
Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160–9166. | ||
Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64(14):4973–4979. | ||
Xu X, Stockhammer F, Schmitt M. Cellular-based immunotherapies for patients with glioblastoma multiforme. Clin Dev Immunol. 2012;2012:764213. | ||
Yamanaka R, Homma J, Yajima N, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005;11(11):4160–4167. | ||
Wheeler CJ, Black KL, Liu G, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008;68(14):5955–5964. | ||
Wehner R, Lobel B, Bornhauser M, et al. Reciprocal activating interaction between 6-sulfo LacNAc+ dendritic cells and NK cells. Int J Cancer. 2009;124(2):358–366. | ||
Cao JX, Zhang XY, Liu JL, et al. Clinical efficacy of tumor antigen-pulsed DC treatment for high-grade glioma patients: evidence from a meta-analysis. PloS One. 2014;9(9):e107173. | ||
Stark-Vance V. Bevacizumab and CPT-11 in the treatment of relapsed malignant glioma. Neuro Oncol. 2005;7:369. Abstract 342. | ||
Prados MD, Lamborn K, Yung WK, et al. A phase 2 trial of irinotecan (CPT-11) in patients with recurrent malignant glioma: a North American Brain Tumor Consortium study. Neuro Oncol. 2006;8(2):189–193. | ||
Prados MD, Yung WK, Jaeckle KA, et al. Phase 1 trial of irinotecan (CPT-11) in patients with recurrent malignant glioma: a North American Brain Tumor Consortium study. Neuro Oncol. 2004;6(1):44–54. | ||
Vredenburgh JJ, Desjardins A, Herndon JE 2nd, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13(4):1253–1259. | ||
Chen W, Delaloye S, Silverman DH, et al. Predicting treatment response of malignant gliomas to bevacizumab and irinotecan by imaging proliferation with [18F] fluorothymidine positron emission tomography: a pilot study. J Clin Oncol. 2007;25(30):4714–4721. | ||
Zhang G, Huang S, Wang Z. A meta-analysis of bevacizumab alone and in combination with irinotecan in the treatment of patients with recurrent glioblastoma multiforme. J Clin Neurosci. 2012;19(12):1636–1640. | ||
Wong ET, Gautam S, Malchow C, Lun M, Pan E, Brem S. Bevacizumab for recurrent glioblastoma multiforme: a meta-analysis. J Natl Compr Canc Netw. 2011;9(4):403–407. | ||
Desjardins A, Reardon DA, Herndon JE 2nd, et al. Bevacizumab plus irinotecan in recurrent WHO grade 3 malignant gliomas. Clin Cancer Res. 2008;14(21):7068–7073. | ||
Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–4740. | ||
Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27(5):740–745. | ||
Vredenburgh JJ, Desjardins A, Herndon JE 2nd, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25(30):4722–4729. | ||
Yamanaka R, Abe T, Yajima N, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer. 2003;89(7):1172–1179. | ||
Bokstein F, Shpigel S, Blumenthal DT. Treatment with bevacizumab and irinotecan for recurrent high-grade glial tumors. Cancer. 2008;112(10):2267–2273. | ||
Poulsen HS, Grunnet K, Sorensen M, et al. Bevacizumab plus irinotecan in the treatment patients with progressive recurrent malignant brain tumours. Acta Oncol. 2009;48(1):52–58. | ||
Prins RM, Soto H, Konkankit V, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17(6):1603–1615. | ||
Chang CN, Huang YC, Yang DM, et al. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci. 2011;18(8):1048–1054. | ||
Sakai K, Shimodaira S, Maejima S, et al. Dendritic cell-based immunotherapy targeting Wilms’ tumor 1 in patients with recurrent malignant glioma. J Neurosurg. 2015;123(4):989–997. | ||
Iwami K, Shimato S, Ohno M, et al. Peptide-pulsed dendritic cell vaccination targeting interleukin-13 receptor alpha2 chain in recurrent malignant glioma patients with HLA-A*24/A*02 allele. Cytotherapy. 2012;14(6):733–742. | ||
Narita Y. Bevacizumab for glioblastoma. Ther Clin Risk Manag. 2015;11:1759–1765. | ||
Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699–708. | ||
Wang Y, Xing D, Zhao M, Wang J, Yang Y. The role of a single angiogenesis inhibitor in the treatment of recurrent glioblastoma multiforme: a meta-analysis and systematic review. PloS One. 2016;11(3):e0152170. | ||
Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):709–722. | ||
van Linde ME, Verhoeff JJ, Richel DJ, et al. Bevacizumab in combination with radiotherapy and temozolomide for patients with newly diagnosed glioblastoma multiforme. Oncologist. 2015;20(2):107–108. | ||
Weathers SP, Gilbert MR. Current challenges in designing GBM trials for immunotherapy. J Neurooncol. 2015;123(3):331–337. | ||
Rolle CE, Sengupta S, Lesniak MS. Challenges in clinical design of immunotherapy trials for malignant glioma. Neurosurg Clin N Am. 2010;21(1):201–214. | ||
Liebelt BD, Finocchiaro G, Heimberger AB. Principles of immunotherapy. Handb Clin Neurol. 2016;134:163–181. | ||
Binder DC, Davis AA, Wainwright DA. Immunotherapy for cancer in the central nervous system: current and future directions. Oncoimmunology. 2016;5(2):e1082027. | ||
Garg AD, Vandenberk L, Koks C, et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med. 2016;8(328):328ra327. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.