")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Dabigatran etexilate retards the initiation and progression of atherosclerotic lesions and inhibits the expression of oncostatin M in apolipoprotein E-deficient mice
Authors Preusch MR, Ieronimakis N, Wijelath ES, Cabbage S, Ricks J, Bea F, Reyes M, van Ryn J, Rosenfeld ME
Received 20 April 2015
Accepted for publication 17 July 2015
Published 10 September 2015 Volume 2015:9 Pages 5203—5211
DOI https://doi.org/10.2147/DDDT.S86969
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Michael R Preusch,1,2 Nicholas Ieronimakis,1 Errol S Wijelath,3 Sara Cabbage,1 Jerry Ricks,1 Florian Bea,2 Morayma Reyes,1 Joanne van Ryn,4 Michael E Rosenfeld1,5
1Department of Pathology, University of Washington, Seattle, WA, USA; 2Department of Internal Medicine, University of Heidelberg, Heidelberg, Germany; 3Department of Surgery, University of Washington, Seattle, WA, USA; 4Department of CardioMetabolic Disease Research, Boehringer Ingelheim Pharma GmbH & Co KG, Biberach, Germany; 5Department of Environmental and Occupational Health Sciences, University of Washington, Seattle, WA, USA
Objective: Thrombin has multiple proatherogenic effects including platelet activation and the induction of inflammatory processes. Recently, the cytokine oncostatin M has been shown to have proinflammatory effects. This study was designed to investigate the effects of thrombin inhibition on the initiation and progression of atherosclerosis and on the expression of oncostatin M.
Methods: Apolipoprotein E-deficient mice at different ages were fed the thrombin inhibitor dabigatran etexilate. The mean lesion area was measured in the aortic sinus and in the innominate artery. CD45-positive cells within the aortic tissue were measured by flow cytometry. Oncostatin M expression was measured in the tissue sections by immunocytochemistry.
Results: Treatment with dabigatran etexilate resulted in a significant reduction of the mean area of atherosclerotic lesions in the aortic sinus in both the young mice (11,176±1,500 µm2 (control) versus 3,822±836 µm2 (dabigatran etexilate), P<0.05) and selectively in the older mice at 28 weeks (234,099±13,500 µm2 (control) versus 175,226±16,132 µm2 (dabigatran etexilate), P<0.05). There were also fewer CD45-positive cells within the aortas of the dabigatran-treated mice and enhanced NO production in endothelial cells pretreated with dabigatran. In addition, the expression of oncostatin M was reduced in the lesions of dabigatran etexilate-treated mice.
Conclusion: Inhibition of thrombin by dabigatran retards the development of early lesions and the progression of some established lesions in ApoE-/- mice. It improves endothelial function and retards macrophage accumulation within the vascular wall. Dabigatran also inhibits the expression of oncostatin M, and this suggests that oncostatin M may play a role in the initiation and progression of atherosclerosis.
Keywords: macrophages, thrombin, coagulation, inflammation
Introduction
The coagulation cascade is a very complex system involving cells and several clotting factors. More recently, it has also been linked to the immune system and directly to vascular disease.1 The serine protease thrombin plays a pivotal role in both coagulation and inflammation and thus has proatherogenic effects.2 The commercial launch of oral thrombin inhibitors for prevention of stroke and venous embolism now offers new therapeutic options for targeting vascular inflammation.3,4 Thrombin activates G-protein-coupled receptors (the protease-activated receptors [PARs]), and there are a number of steps in the atherogenic process that may be stimulated by PAR activation. For example, it has been shown that thrombin mediates vasoconstriction due to an inhibition of the activation of endothelial nitric oxide synthase (eNOS).5–7 There are additional reports of thrombin-triggered oxidative stress due to increased synthesis of reactive oxygen species,8 and thrombin induced recruitment of monocytes and T-cells into the vascular wall due to increased expression of adhesion molecules on endothelial cells.9,10 Thrombin has also been reported to induce the expression of the proinflammatory chemokine MCP-1 as well as matrix metalloproteinases.11,12 Using a transgenic ApoE−/− mouse model with a hypercoagulable phenotype, Borissoff et al demonstrated a neutrophil-dependent increase in atherosclerotic plaque burden that was inhibited by dabigatran etexilate.13 We have previously reported on the anti-atherogenic effect of thrombin inhibition with melagatran in a mouse model of advanced atherosclerosis,14 and another recent study reported a reduction in early lesions in ApoE−/− mice treated with dabigatran etexilate.15,16
There is evidence of involvement of the cytokine oncostatin M (OSM) in atherosclerosis.17 OSM induces tissue factor expression but also exerts other proinflammatory effects.18,19 Kastl et al demonstrated that thrombin stimulates OSM expression in human carotid plaque macrophages.20 Furthermore, the expression of proatherogenic and plaque-destabilizing factors like MMPs and MCP-1 are stimulated by OSM.21,22
The aim of this study was to evaluate the effect of oral thrombin inhibition on different steps in atherosclerotic lesion development and its effect on OSM expression within atherosclerotic lesions in a mouse model of atherosclerosis.
Materials and methods
Animals and drug administration
Male ApoE−/− mice with a C57Bl/6 background were bred within the Brotman animal care facility of the University of Washington (Seattle, WA, USA) and maintained in a temperature-controlled room on a 12-hour light cycle. Three different groups of mice at 4 (n=14), 8 (n=39), and 20 (n=38) weeks of age were included in the study. Half of the mice in each group received regular chow supplemented with dabigatran etexilate (40 mg dabigatran etexilate/mouse/day); the other half were fed standard chow and served as controls. The dose of dabigatran was based on the low bioavailability of dabigatran in mice (about 6.5%) necessitating a relatively high dose for oral administration.22 All animal procedures were approved by the University of Washington Institutional Animal Care and Use Committee.
Animal sacrifice and preparation of tissues
The 4-week-old mice were sacrificed after either 5 weeks (histology for early lesions) or 9 weeks (for flow cytometry) on the dabigatran etexilate-supplemented diet, while the remaining animals were sacrificed after 20 weeks of drug administration. Mice were heavily sedated (ketamine/xylazine, intraperitoneal). Blood was collected from the inferior vena cava, and the animals were sacrificed by exsanguination. The animals were perfused via the left ventricle with either 10 mL phosphate-buffered saline (PBS) for frozen sections or 4% buffered formalin for paraffin sections. The entire heart, the aorta, and the innominate artery from each animal were dissected out. Hearts of the 9-week-old mice were embedded in Tissue-Tek®, and frozen sections (15 μm) were generated. The aortic sinuses and innominate arteries of the remaining mice were embedded in paraffin and serially sectioned (5 μm). Every fifth section was stained with a modified Movat’s pentachrome stain.24
Digestion of tissue and flow cytometric analyses
The aortas and perivascular adventitial tissue from the mice sacrificed at 13 weeks of age for flow cytometric analysis were minced and enzymatically digested with collagenase type IV and dispase II (Sigma-Aldrich Co., St Louis, MO, USA) as previously described.22 Tissues were incubated for a total of 45 minutes at 37°C and stirred every 15 minutes during the digestion. The digestion was then stopped with F10 medium plus 15% horse serum and filtered through 40 μM cell strainers. The isolated cells were then incubated with purified rat anti-CD16 and anti-CD32 IgG (Fc receptor block; eBioscience, eBioscience, San Diego, CA, USA) for 10 minutes on ice at a concentration of 1 μg/106 cells and then incubated for 1 hour on ice with directly conjugated rat anti-CD45, anti-CD11b, anti-F4/80, and anti-CD31 monoclonal antibodies (all from eBioscience) for the respective FACS analysis (gating strategy, Figure S1).25,26 For analysis of nitric oxide production, mononuclear cells were incubated for 3 hours at 37°C in PBS with 10% fetal calf serum, 0.3% BSA, and a final concentration of 100 μM arginine and 5 μM DAF2-DA (a fluorescent probe for nitric oxide production).27,28 Data were acquired using a FACS Aria II with Diva software and post-analyzed with FlowJo v 7.5.5.
Determination of plasma cholesterol and cell counts
Total plasma cholesterol levels were determined at the time of sacrifice with a colorimetric kit (Diagnostic Chemicals Ltd., Charlottetown, PEI, Canada) with cholesterol standards (Sigma-Aldrich Co.). Whole blood (EDTA) was analyzed for the percentage of leukocytes, platelets, and red blood cells with a Cell Dyn 3,500 hemocounter (Abbott).
Evaluation of atherosclerotic lesion size and composition
The cross-sectional area (μm2) of atherosclerotic lesions was determined in the Movat’s pentachrome-stained sections by computer-assisted morphometry (Image Pro; Media Cybernetics, Silver Spring, Rockville, MD, USA). We further evaluated each section for several characteristic features of plaque composition: 1) thickness of the fibrous cap (data given in μm); and 2) size of the necrotic core (a large necrotic core was defined as occupying more than 50% of the plaque’s volume and was also measured by morphometry and reported as μm2).
Immunohistochemistry
Detection of monocytes/macrophages within lesions was performed using monoclonal rat anti-mouse Mac-2 antibody (anti-Mac-2; Cedarlane Laboratories Ltd, Burlington, NC, USA), while detection of OSM used a polyclonal goat anti-mouse antibody (R&D Systems, Inc., Minneapolis, MN, USA). Sections were incubated with a biotinylated secondary antibody, rinsed three times with PBS, and incubated for 10 minutes with HRP–streptavidin at room temperature. Sections were developed using the Immpress reagent and Impact DAB kits (Vector Labs, Burlingame, CA, USA) and counterstained with hematoxylin. Controls consisted of normal IgG or isotype-matched monoclonal antibodies. The extent of positive staining within the lesions was determined using computer-assisted morphometry (Image Pro).
Statistical analysis
All data were expressed as mean ± standard error of the mean. Differences between means in plasma lipid profiles and FACS data were determined with the two-tailed unpaired Student’s t-test. For analysis of plaque morphometry and areas of positive staining, groups were compared using the Mann–Whitney U-test. For evaluation of plaque morphology, groups were compared using the chi-square test. P-values <0.05 were considered as statistically significant. Statistical analyses and graphing were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Effect of dabigatran etexilate on plasma lipid levels and body weight
At the time of necropsy, there were no significant differences in total plasma cholesterol or body weight between groups (Table 1). There were also no differences in white blood cell count, red blood cell count, and platelets (data not shown).
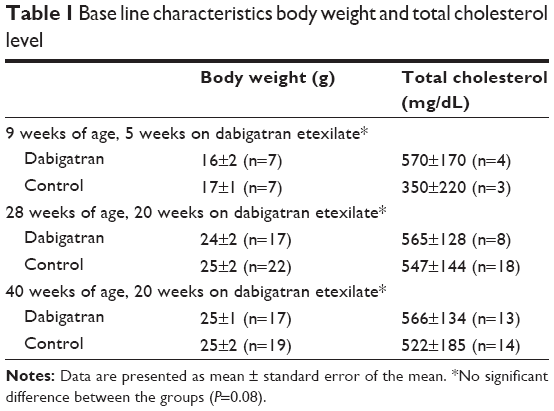 | Table 1 Base line characteristics body weight and total cholesterol level |
Effect of dabigatran etexilate on maximum lesion area within the aortic sinus
Early lesions
After 5 weeks on the drug-supplemented diet (at 9 weeks of age), dabigatran etexilate treatment resulted in a significant reduction of the mean atherosclerotic lesion area within the aortic sinus 3,822±836 μm2 (dabigatran etexilate) versus 11,176±1,500 μm2 (control), p= 0.004 (Figure 1A).
 | Figure 1 Effects of dabigatran etexilate on atherosclerotic lesion size within the aortic sinus. |
Intermediate lesions
After 20 weeks of drug administration (at 28 weeks of age), dabigatran etexilate also resulted in a reduction of mean atherosclerotic lesion area in the aortic sinus 175,226±16,132 μm2 (dabigatran etexilate) versus 234,099±13,500 μm2 (control), p= 0.014 (Figure 1B).
Established lesions
There were no statistically significant differences in average lesion area in the aortic sinus in mice at 40 weeks of age, after 20 weeks of dabigatran etexilate treatment (405,292± 22,777 μm2 (dabigatran etexilate) versus 450,975±18,215 μm2 (control) p= 0.1).
Maximum lesion area within the innominate artery
Intermediate lesions (innominate artery)
After 20 weeks of drug administration, dabigatran etexilate had no effect on the average lesion area within the innominate artery in mice at 28 weeks of age (dabigatran etexilate [n=10] 60,084±7,966 μm2 versus control [n=11] 74,314±8,959 μm2, P=0.4).
Established lesions (innominate artery)
There also were no statistically significant differences in the average lesion area in mice at 40 weeks of age, after 20 weeks of dabigatran etexilate treatment (dabigatran etexilate [n=18] 170,684±7,800 μm2 versus control [n=16] 197,774±13,478 μm2, P=0.1).
Features of lesion composition
Size of the necrotic core within the aortic sinus
At 28 weeks of age, dabigatran etexilate treatment (20 weeks) resulted in a reduction of the necrotic core area within the aortic sinus, presented as the ratio of necrotic core area/total lesion area (dabigatran etexilate 0.31±0.03 versus control 0.39±0.02, P=0.02) (Figure 2A).
 | Figure 2 Effects of dabigatran etexilate on atherosclerotic lesion composition in the aortic sinus following 20 weeks of treatment with dabigatran etexilate. |
Thickness of fibrous caps
Microscopic evaluation in 28-week-old mice showed a trend towards thicker fibrous caps in mice treated with dabigatran etexilate compared to controls (25±2 μm versus 19±2 μm, P=0.06) (Figure 2B).
Macrophage content within the aortic sinus
There were no differences in the area of positive immunostaining for macrophages within the atherosclerotic lesions at any time points.
Mice treated with dabigatran etexilate showed reduced expression of OSM
Expression of OSM was markedly reduced within atherosclerotic lesions of mice treated with dabigatran etexilate at 28 as well as 40 weeks of age (ratio of positive staining/total lesion area: 0.062±0.012 versus 0.11±0.014, P=0.04 [Figure 3A] and 0.05±0.014 versus 0.1±0.012, P=0.01 [Figure 3B]).
 | Figure 3 Effects of dabigatran etexilate on the lesion content of oncostatin M (OSM) in the aortic sinus. |
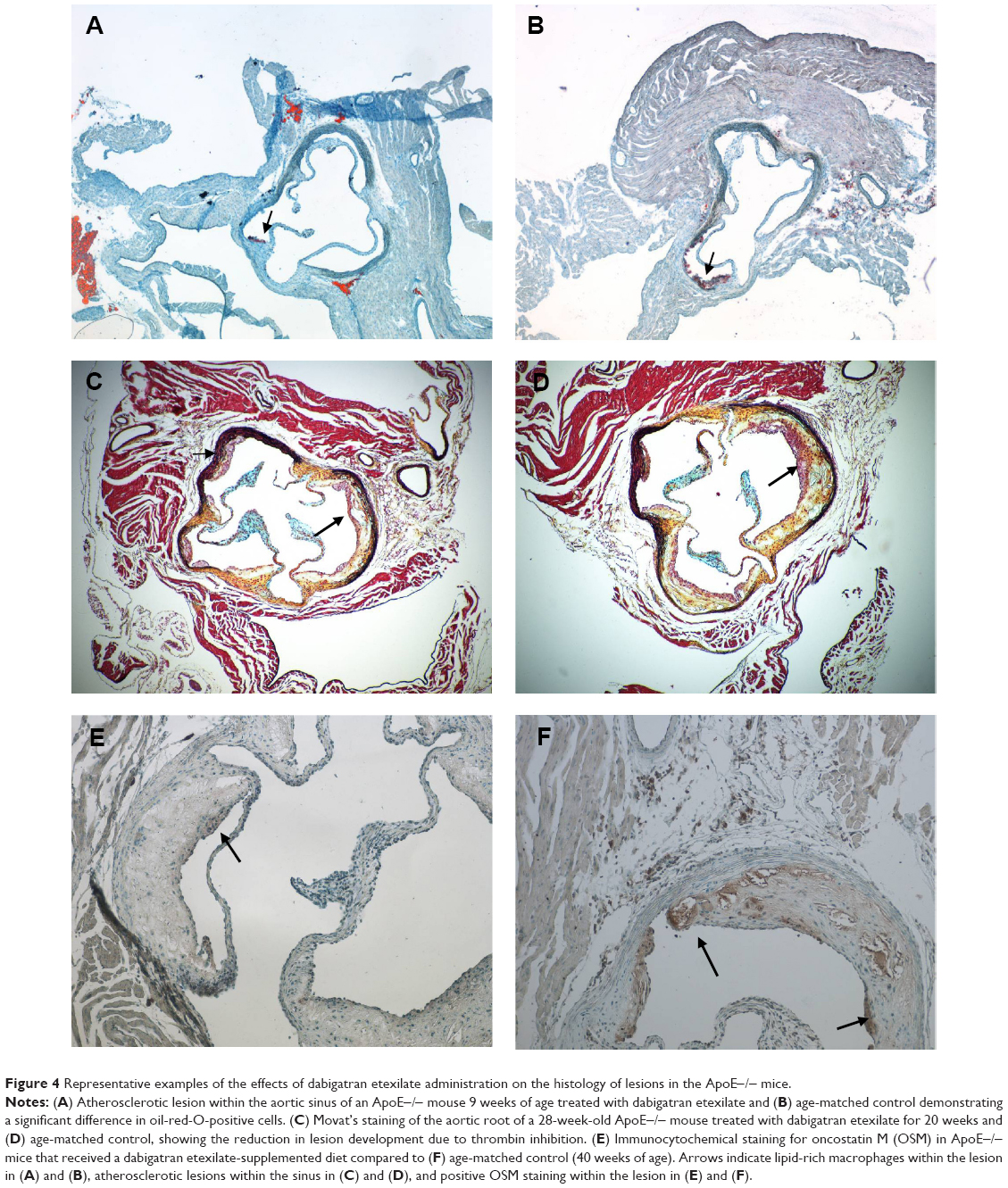 | Figure 4 Representative examples of the effects of dabigatran etexilate administration on the histology of lesions in the ApoE−/− mice. |
CD45-positive cells were significantly reduced within the aortic wall but not in the perivascular tissue in mice that received dabigatran etexilate
The proportion of cells positive for the common leukocyte antigen CD45 that is expressed by inflammatory and hematopoietic cells was significantly reduced in the aortas from the 14-week-old mice (9 weeks of dabigatran etexilate treatment). (ApoE−/− males treated with dabigatran [n=3] versus placebo [n=3]; 4.3%±0.14% versus 6.58%±0.38%, P=0.03.) There were no differences in the numbers of CD45+ cells in the adventitia. There were also no differences in the percentage of cells positive for either CD11b or F4/80 in both vascular compartments.
Increase of NO production by aortic endothelial cells after in vivo exposure to dabigatran etexilate
FACS analysis of the same samples utilizing the nitric oxide fluorescent indicator DAF2-DA showed an increase in the percentage of NO-producing aortic endothelial cells in the dabigatran etexilate-treated mice (identified as CD45−, CD31+ cells; 14.45%±2.19% versus 9.48%±1.96%, P=0.02).
Discussion
The development of selective thrombin inhibitors may offer new therapeutic options for both coagulation and atherosclerosis. We have previously reported a reduction of established atherosclerotic lesion progression in ApoE−/− mice with oral administration of the thrombin inhibitor melagatran.14 However, the drug has been taken off the market because of potential serious liver damage. Recently, Kadoglou et al as well as Pingel et al demonstrated a reduction in the area of early lesions in the aortic arch of ApoE−/− mice treated with dabigatran etexilate.15,16 These investigators started drug treatment in mice at the age of 8 and 12 weeks, coupled with a Western-type high-fat diet. Thus, the focus of these two studies was on lesion initiation rather than progression of established lesions. In the current study, we focused on both the early development and progression of established atherosclerotic lesions in ApoE−/− mice following treatment with dabigatran etexilate. We also avoided inducing non-physiologically high plasma cholesterol levels by feeding a standard mouse chow rather than a Western-type high-fat diet, which can have off-target proinflammatory effects.29,30
The present data show a significant reduction of atherosclerotic lesion area in the aortic sinus in mice at 9 weeks of age after dabigatran etexilate treatment for 5 weeks. This is consistent with the observations of the two aforementioned studies of the effects of dabigatran on lesion initiation.15,16 There is already monocyte adhesion at 5–6 weeks of age and significant lipid accumulation by 6–10 weeks of age in ApoE−/− mice.30 There is evidence of procoagulant activity within atherosclerotic lesions, and this activity is associated with the stages of plaque development, with more procoagulant activity in early lesions as compared to advanced plaques.31 This might also help explain the protective effects in the younger animals. Furthermore, thrombin has been shown to induce the transcription of several proinflammatory genes, like tissue factor in monocytes, and early lesions are primarily composed of newly recruited monocyte-derived macrophages.13,32
We also show a reduction in lesion area in the aortic sinus in mice at 28 weeks of age following 20 weeks of drug treatment, again demonstrating an effect on lesion progression. However, in contrast to our previous study with melagatran,14 we did not detect any significant reduction of lesion area in either the aortic sinus or innominate arteries in mice at 40 weeks of age, after 20 weeks of dabigatran etexilate treatment. Nevertheless, our observation that, in the mice at 28 weeks, there was a reduction in the size of the necrotic core as well as a thickening of fibrous caps in the dabigatran etexilate-treated mice is in agreement with our previous study with melagatran and with the study of Kadoglou et al.14,15
Besides lesion-dependent differences, data demonstrated a more procoagulant stage in early lesions compared to advanced plaques.31 This might explain the distinct effects in younger animals. Thrombin has been shown to induce the transcription of several proinflammatory genes, like tissue factor in monocytes, which are known to be a pivotal cell type for atherosclerotic lesion development.32 Furthermore, inhibition of thrombin rescued the initiation of plaque formation in a mouse model of hypercoagulability and therefore punctuates the role of thrombin in the early onset of atherosclerosis.13
As noted, the dosing for oral administration of dabigatran etexilate in the mice was based on the low bioavailability of approximately 6.5% and the fact that dabigatran etexilate is less potent in rats than in humans.23 It was also based on the recent data of Lee et al wherein the dose-dependent prolongation of thrombin clotting time was measured in dabigatran etexilate-treated ApoE−/− mice.33 It is not clear why there were protective effects in the aortic sinus and not the innominate artery following 20 weeks of drug treatment. However, it is well known that interventions in lesion development in ApoE−/− mice can be site-specific. Witting et al described an increase in lesions in the aortic root in ApoE−/− mice but also documented a significant decrease in the arch and parts of the thoracic and abdominal aorta with probucol treatment.34 Reardon et al described a lesion reduction in the aortic sinus but not the innominate artery in ApoE−/−/RAG−/− mice deficient in mature B- and T-cells with administration of polyunsaturated fatty acids.35 Susceptibility to atherosclerotic lesion formation at different sites may also be genetically determined.36
It is also not clear why the immunostaining for macrophage content did not show any differences, whereas flow cytometric analysis of cells released from the digested aortas demonstrated a reduction of CD45-positive cells in mice treated with dabigatran etexilate. It may be that the enzyme digestion released intact and newly recruited cells, and this is in keeping with thrombin’s known effects on inducing MCP-1 expression and monocyte and lymphocyte recruitment. However, there were also no changes in the percentage of cells that were positive for the monocyte and macrophage markers CD11b and F4/80. This suggests that dabigatran may reduce the infiltration of some hematopoietic cells such as lymphocytes while not reducing recruitment of other myeloid populations.37
The effects of the thrombin inhibition on reducing early and intermediate lesions may also be explained by the known effects of thrombin on endothelial dysfunction and is supported by our data showing increased endothelial NO production with dabigatran treatment.5,38 This is also in line with the study of Pingel et al showing a dabigatran etexilate-related reduction in oxidative stress.16 Endothelial dysfunction is one of the initial steps in the development of atherosclerosis. It has been shown that thrombin induces smooth muscle contraction both directly and via endothelial-dependent mechanisms.5,6 For example, endothelial NOS activity is regulated by thrombin activation of PARs,39 and thrombin inhibits eNOS expression in endothelial cells.7 Thus, the observed increase in NO production suggests that dabigatran etexilate may restore endothelial-mediated vasodilation which is reduced in ApoE−/− mice.40
Recently, the cytokine OSM has been linked to the pathogenesis of atherosclerosis, and it has also been shown that thrombin stimulates OSM expression in macrophages.17,20 The present study demonstrates for the first time that inhibition of thrombin leads to a reduced expression of OSM in atherosclerotic lesions. OSM is known to trigger MMP-9 expression in smooth muscle cells, and thus the inhibition of OSM expression may help explain the observed increase in the thickness of the fibrous cap and the decreased size of the necrotic core with the dabigatran treatment.21
Taken together, the present study further supports the link between thrombin and atherosclerosis. In contrast to previous studies, we have shown the anti-atherosclerotic effects of thrombin inhibition at both early and intermediate stages of atherosclerotic lesion development. Furthermore, this is the first study that shows a reduction of OSM expression in an atherosclerotic mouse model due to thrombin inhibition.
Acknowledgments
We would like to thank Alec Selby for expert technical assistance.
Disclosure
This study was financially supported by Boehringer Ingelheim Pharma GmbH & Co KG, Department of CardioMetabolic Disease Research, Biberach, Germany. The authors report no further conflicts of interest in this work.
References
Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131(4):417–430. | ||
Borissoff JI, Spronk HM, Heeneman S, ten Cate H. Is thrombin a key player in the ‘coagulation-atherogenesis’ maze? Cardiovasc Res. 2009;82(3):392–403. | ||
Eriksson BI, Dahl OE, Rosencher N, et al; RE-NOVATE Study Group Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet. 2007;370(9591):949–956. | ||
Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139–1151. | ||
Ku DD, Zaleski JK. Receptor mechanism of thrombin-induced endothelium-dependent and endothelium-independent coronary vascular effects in dogs. J Cardiovasc Pharmacol. 1993;22(4):609–616. | ||
Derkach DN, Ihara E, Hirano K, Nishimura J, Takahashi S, Kanaide H. Thrombin causes endothelium-dependent biphasic regulation of vascular tone in the porcine renal interlobar artery. Br J Pharmacol. 2000;131(8):1635–1642. | ||
Eto M, Barandiér C, Rathgeb L, et al. Thrombin suppresses endothelial nitric oxide synthase and upregulates endothelin-converting enzyme-1 expression by distinct pathways: role of Rho/ROCK and mitogen-activated protein kinase. Circ Res. 2001;89(7):583–590. | ||
Brandes RP, Viedt C, Nguyen K, et al. Thrombin-induced MCP-1 expression involves activation of the p22phox-containing NADPH oxidase in human vascular smooth muscle cells. Thromb Haemost. 2001;85(6):1104–1110. | ||
Kaplanski G, Fabrigoule M, Boulay V, et al. Thrombin induces endothelial type II activation in vitro: IL-1 and TNF-alpha-independent IL-8 secretion and E-selectin expression. J Immunol. 1997;158(11):5435–5441. | ||
Kaplanski G, Marin V, Fabrigoule M, et al. Thrombin-activated human endothelial cells support monocyte adhesion in vitro following expression of intercellular adhesion molecule-1 (ICAM-1; CD54) and vascular cell adhesion molecule-1 (VCAM-1; CD106). Blood. 1998;92(4):1259–1267. | ||
Kawanami D, Matoba K, Kanazawa Y, Ishizawa S, Yokota T, Utsunomiya K. Thrombin induces MCP-1 expression through Rho-kinase and subsequent p38MAPK/NF-κB signaling pathway activation in vascular endothelial cells. Biochem Biophys Res Commun. 2011;411(4):798–803. | ||
Chang CJ, Hsu LA, Ko YH, et al. Thrombin regulates matrix metalloproteinase-9 expression in human monocytes. Biochem Biophys Res Commun. 2009;385(2):241–246. | ||
Borissoff JI, Otten JJ, Heeneman S, et al. Genetic and pharmacological modifications of thrombin formation in apolipoprotein e-deficient mice determine atherosclerosis severity and atherothrombosis onset in a neutrophil-dependent manner. PLoS One. 2013;8(2):e55784. | ||
Bea F, Kreuzer J, Preusch M, et al. Melagatran reduces advanced atherosclerotic lesion size and may promote plaque stability in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26(12):2787–2792. | ||
Kadoglou NP, Moustardas P, Katsimpoulas M, et al. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E-deficient mice: dabigatran etexilate and atherosclerosis. Cardiovasc Drugs Ther.2012;26(5):367–374. | ||
Pingel S, Tiyerili V, Mueller J, Werner N, Nickenig G, Mueller C. Thrombin inhibition by dabigatran attenuates atherosclerosis in ApoE deficient mice. Arch Med Sci. 2014;10(1):154–160. | ||
Albasanz-Puig A, Murray J, Preusch M, et al. Oncostatin M is expressed in atherosclerotic lesions: a role for oncostatin M in the pathogenesis of atherosclerosis. Atherosclerosis. 2011;216(2):292–298. | ||
Fritz DK, Kerr C, Fattouh R, et al. A mouse model of airway disease: oncostatin M-induced pulmonary eosinophilia, goblet cell hyperplasia, and airway hyperresponsiveness are STAT6 dependent, and interstitial pulmonary fibrosis is STAT6 independent. J Immunol. 2011;186(2):1107–1118. | ||
Rychli K, Kaun C, Hohensinner PJ, et al. The inflammatory mediator oncostatin M induces angiopoietin 2 expression in endothelial cells in vitro and in vivo. J Thromb Haemost. 2010;8(3):596–604. | ||
Kastl SP, Speidl WS, Katsaros KM, et al. Thrombin induces the expression of oncostatin M via AP-1 activation in human macrophages: a link between coagulation and inflammation. Blood. 2009;114(13):2812–2818. | ||
Nagata T, Kai H, Shibata R, Koga M, Yoshimura A, Imaizumi T. Oncostatin M, an interleukin-6 family cytokine, upregulates matrix metalloproteinase-9 through the mitogen-activated protein kinase kinase-extracellular signal-regulated kinase pathway in cultured smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23(4):588–593. | ||
Langdon C, Leith J, Smith F, Richards CD. Oncostatin M stimulates monocyte chemoattractant protein-1- and interleukin-1-induced matrix metalloproteinase-1 production by human synovial fibroblasts in vitro. Arthritis Rheum. 1997;40(12):2139–2146. | ||
Wienen W, Stassen JM, Priepke H, Ries UJ, Hauel N. In-vitro profile and ex-vivo anticoagulant activity of the direct thrombin inhibitor dabigatran and its orally active prodrug, dabigatran etexilate. Thromb Haemost. 2007;98(1):155–162. | ||
Movat HZ. Demonstration of all connective tissue elements in a single section; pentachrome stains. AMA Arch Pathol. 1955;60(3):289–295. | ||
Ieronimakis N, Balasundaram G, Reyes M. Direct isolation, culture and transplant of mouse skeletal muscle derived endothelial cells with angiogenic potential. PLoS One. 2008;3(3):e0001753. | ||
Ieronimakis N, Hays A, Reyes M. Bone marrow-derived cells do not engraft into skeletal muscle microvasculature but promote angiogenesis after acute injury. Exp Hematol. 2012;40(3):238–249.e3. | ||
Kojima H, Sakurai K, Kikuchi K, et al. Development of a fluorescent indicator for nitric oxide based on the fluorescein chromophore. Chem Pharm Bull (Tokyo). 1998;46(2):373–375. | ||
Navarro-Antolin J, Lamas S. Nitrosative stress by cyclosporin A in the endothelium: studies with the NO-sensitive probe diaminofluorescein-2/diacetate using flow cytometry. Nephrol Dial Transplant. 2001;16 Suppl 1:6–9. | ||
Reardon CA, Getz GS. Mouse models of atherosclerosis. Curr Opin Lipidol. 2001;12(2):167–173. | ||
Jawień J, Nastałek P, Korbut R. Mouse models of experimental atherosclerosis. J Physiol Pharmacol. 2004;55(3):503–517. | ||
Borissoff JI, Heeneman S, Kilinç E, et al. Early atherosclerosis exhibits an enhanced procoagulant state. Circulation. 2010;122(8):821–830. | ||
López ML, Bruges G, Crespo G, et al. Thrombin selectively induces transcription of genes in human monocytes involved in inflammation and wound healing. Thromb Haemost. 2014;112(5):992–1001. | ||
Lee IO, Kratz MT, Schirmer SH, Baumhäkel M, Böhm M. The effects of direct thrombin inhibition with dabigatran on plaque formation and endothelial function in apolipoprotein E-deficient mice. J Pharmacol Exp Ther. 2012;343(2):253–257. | ||
Witting PK, Pettersson K, Letters J, Stocker R. Site-specific antiatherogenic effect of probucol in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20(8):E26–E33. | ||
Reardon CA, Blachowicz L, Gupta G, Lukens J, Nissenbaum M, Getz GS. Site-specific influence of polyunsaturated fatty acids on atherosclerosis in immune incompetent LDL receptor deficient mice. Atherosclerosis. 2006;187(2):325–331. | ||
Bennett BJ, Wang SS, Wang X, Wu X, Lusis AJ. Genetic regulation of atherosclerotic plaque size and morphology in the innominate artery of hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2009;29(3):348–355. | ||
Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. | ||
Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91(3A):7A–11A. | ||
Watts VL, Motley ED. Role of protease-activated receptor-1 in endothelial nitric oxide synthase-Thr495 phosphorylation. Exp Biol Med (Maywood). 2009;234(2):132–139. | ||
d’Uscio LV, Baker TA, Mantilla CB, et al. Mechanism of endothelial dysfunction in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21(6):1017–1022. | ||
Brown DL, Hibbs MS, Kearney M, Loushin C, Isner JM. Identification of 92-kD gelatinase in human coronary atherosclerotic lesions. Association of active enzyme synthesis with unstable angina. Circulation. 1995;91(8):2125–2131. |
Supplementary material
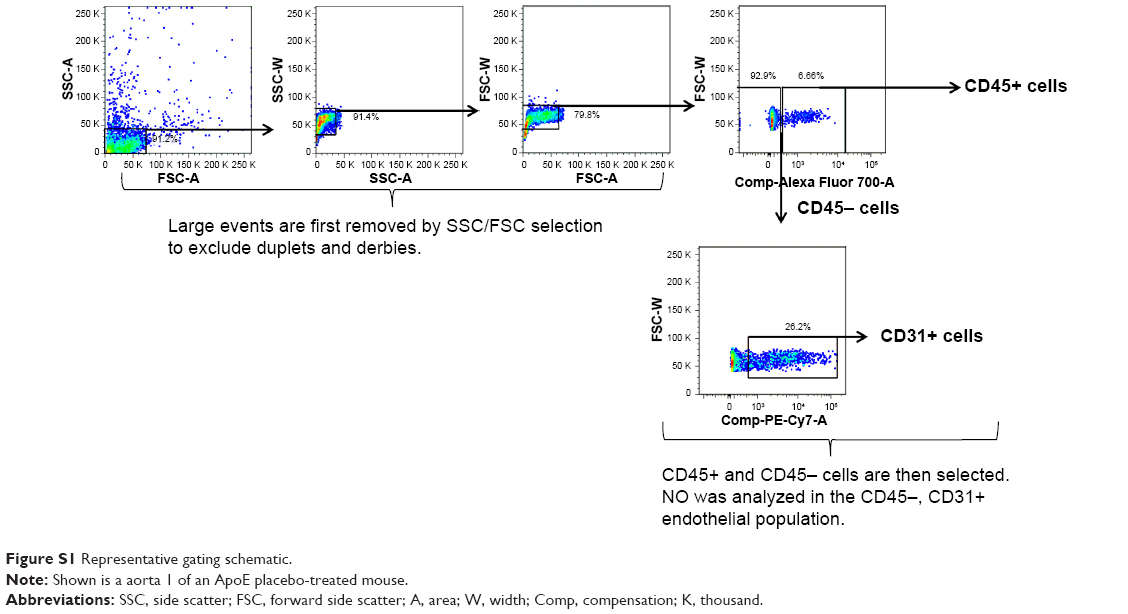 | Figure S1 Representative gating schematic. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.