")
Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 14
Current Screening Strategies for the Diagnosis of Adrenal Insufficiency in Children
Authors Bowden SA
Received 15 August 2022
Accepted for publication 21 March 2023
Published 6 April 2023 Volume 2023:14 Pages 117—130
DOI https://doi.org/10.2147/PHMT.S334576
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Roosy Aulakh
Sasigarn A Bowden
Division of Endocrinology, Department of Pediatrics, Nationwide Children’s Hospital/The Ohio State University College of Medicine, Columbus, OH, USA
Correspondence: Sasigarn A Bowden, Nationwide Children’s Hospital, Division of Endocrinology, 700 Children’s Drive, Columbus, OH, 43205, USA, Tel +1 614-722-4118, Fax +1 614-722-4440, Email [email protected]
Abstract: Adrenal insufficiency can arise from a primary adrenal disorder, secondary to adrenocorticotropic hormone deficiency, or by suppression of hypothalamic-pituitary-adrenal axis due to exogenous glucocorticoids. Diagnosis of adrenal insufficiency is usually delayed because the initial presentation is often subtle and nonspecific. Clinician awareness and recognition is crucial for timely diagnosis to avoid adrenal crisis. Current screening strategies for the diagnosis of adrenal insufficiency in children in various clinical situations are discussed in this review.
Keywords: adrenal gland, adrenal insufficiency, cortisol, glucocorticoids, hydrocortisone, fludrocortisone, adrenocorticotrophic hormone, aldosterone, renin
Introduction
Adrenal Insufficiency (AI) is a condition characterized by a deficiency of adrenal cortical hormone production. It is caused by disorders or pathology affecting the hypothalamic-pituitary adrenal (HPA) axis. AI is classified to 2 main groups: primary and secondary AI, based on the level of defects of the HPA axis. Primary AI is caused by destruction or dysfunction of the adrenal gland (primary adrenal gland disorders) where all three zones of the adrenal cortex are damaged by a disease process, which leads deficiency of cortisol, mineralocorticoids (aldosterone), and androgen hormones. Secondary AI is caused by deficient adrenocorticotrophic hormone (ACTH) secretion from the pituitary gland or deficient cortisol releasing hormone (CRH) from the hypothalamus (tertiary AI), collectively called central AI. Central AI is typically caused by intracranial pathology and may be isolated deficiency of ACTH or CRH, or in combination with other pituitary hormonal deficiencies (hypopituitarism). Iatrogenic tertiary AI caused by suppression of the hypothalamic-pituitary adrenal (HPA) axis secondary to chronic glucocorticoid treatment is the most common cause of central AI.1 Treatment of primary AI includes hydrocortisone (10–12.5 mg/m2/day) and fludrocortisone (0.05–0.2 mg/day) as maintenance therapy, while treatment of secondary AI includes only hydrocortisone. The daily maintenance hydrocortisone dose needs to be increased by 3–5 times for mild to moderate stress, or up to 10 times for major stress.2–4 Chronic and acute AI is a potentially life-threatening condition associated with significant morbidity and mortality, especially when the diagnosis is delayed. Clinicians must maintain high index of suspicion for AI when encountering different clinical case scenarios and recognize when and how to screen for AI for early diagnosis to prevent overt AI or adrenal crisis. This article focuses on the screening strategies to help clinicians recognize, diagnose, differentiate the causes of AI and treat in a timely manner.
Etiology and Clinical Presentation of Adrenal Insufficiency
The clinical presentation of AI ranges from subtle and nonspecific to hypotension or shock, depending on the rate and degree of loss of adrenal function and on the degree of physiological stress. Hallmark clinical features in both primary and central AI are profound fatigue, anorexia, unintentional weight loss, postural hypotension, muscle and abdominal pain.5 The signs and symptoms also depend on whether mineralocorticoid hormone production is preserved (as is the case in central AI), or deficient (as is usually the case in primary AI). AI is often insidious in onset and may go undetected until an intercurrent illness or stress precipitates a crisis.
Primary Adrenal Insufficiency
Primary AI results from destruction or dysfunction of all 3 zones of the adrenal cortex. The causes of primary AI are extensive in children including genetic disorders with enzyme deficiencies in the steroidogenesis pathway, peroxisomal defects, systemic diseases such as autoimmune pathology, infections, or hemorrhage of the adrenal glands (Table 1). Congenital adrenal hyperplasia (CAH) is the most common cause of primary AI in children, followed by autoimmune adrenalitis or Addison’s disease.
 |
Table 1 Conditions Associated with Adrenal Insufficiency (AI) in Children |
Infants with acute primary adrenal insufficiency during the first weeks of life present with acute dehydration, poor weight gain, poor feeding, lethargy, hypotension, and hypoglycemia. Infants with a gradual onset of primary AI may present with failure to thrive, hyperpigmentation, in fasting hypoglycemia. The most common cause of AI in the first 2–3 weeks of life is the salt-wasting form of CAH. Characteristic electrolyte abnormalities include hyponatremia, hyperkalemia, mild metabolic acidosis and mild hypoglycemia (Table 2).
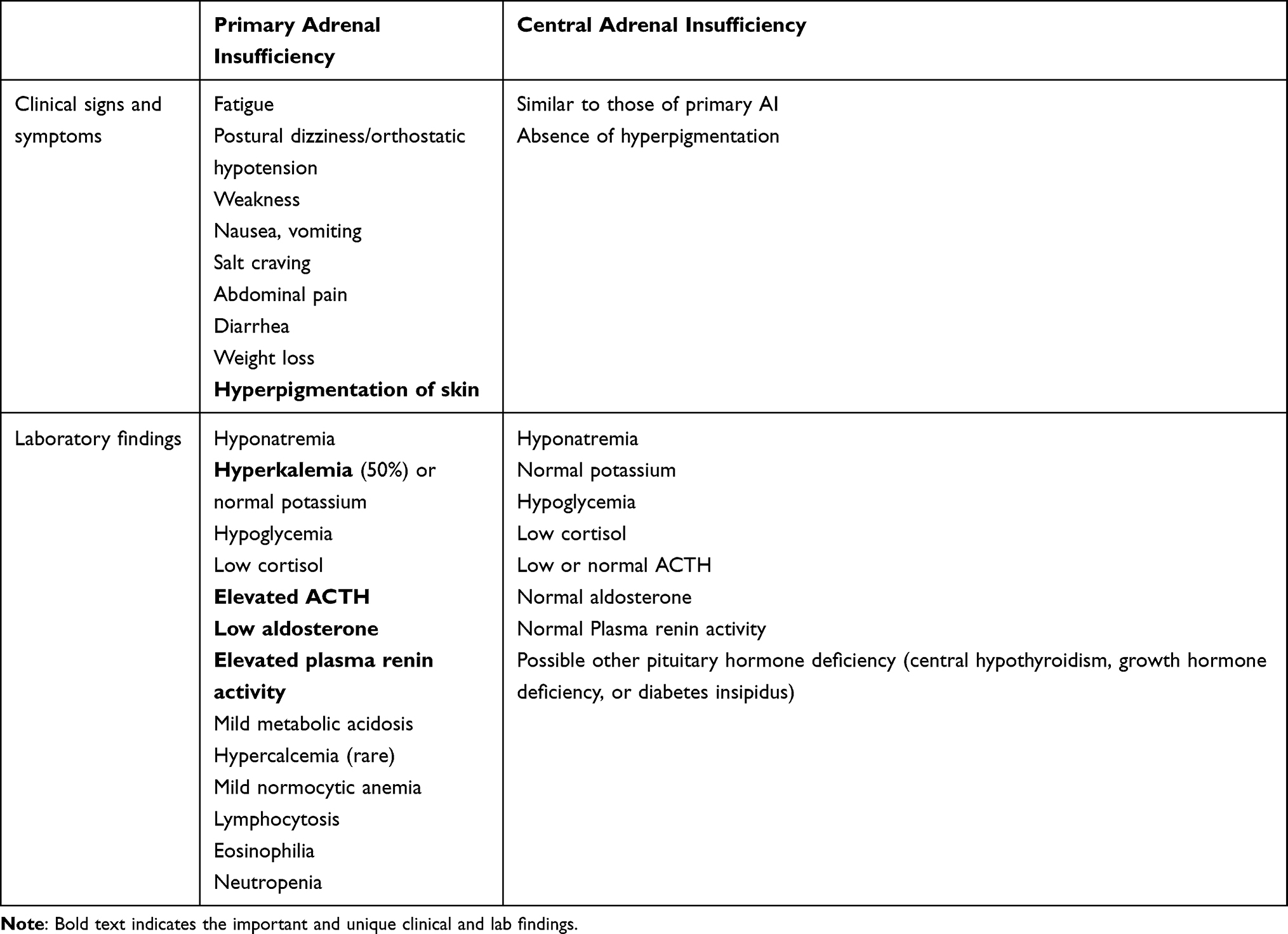 |
Table 2 Clinical and Laboratory Findings for Adrenal Insufficiency |
In older children and adolescents, the onset of clinical manifestations is usually gradual as clinical signs and symptoms do not become manifest until at least 90% of the adrenal cortex is destroyed. The initial phase of partial glucocorticoid deficiency results only in an insufficient cortisol rise in response to physiologic stress. Manifestations of complete glucocorticoid deficiency include fatigue, weakness, decreased sense of well-being, abdominal pain, nausea, vomiting, diarrhea and decline in linear growth rate. Inadequate secretion of aldosterone results in reduced renal reabsorption of sodium and decreased renal potassium and hydrogen ion excretion, which leads to electrolyte abnormalities including hyponatremia, hyperkalemia and metabolic acidosis, volume depletion, hypotension and dehydration. Symptoms of aldosterone deficiency include salt craving, anorexia, dizziness, hypotension, dehydration and weight loss. Cortisol deficiency also results in reduced peripheral vascular adrenergic tone which can lead to vascular collapse and shock. Mild metabolic acidosis contributes to the hyperkalemia by permitting potassium to shift from the intracellular to the extracellular space. Lack of physiological feedback mechanism due to cortisol deficiency leads to increased hypothalamic CRH, which stimulates release of pituitary ACTH and α-melanocyte-stimulating hormone (αMSH), both of which are derived from proopiomelanocortin (POMC). Elevated ACTH and αMSH cause mucocutaneous hyperpigmentation (Figure 1) involving skin creases, areolae, axillae, gingival, groin, and scars.
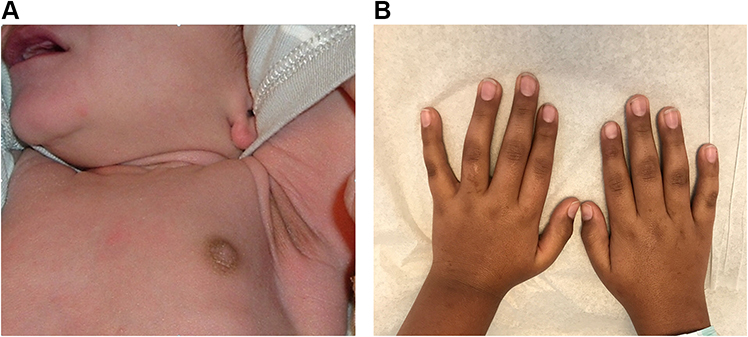 |
Figure 1 Skin hyperpigmentation in primary adrenal insufficiency. (A) Hyperpigmentation at axillary and nipples in a 10-day-old male infant with salt-wasting congenital adrenal hyperplasia due to 21-hydroxylase deficiency. (B) Skin hyperpigmentation in a 10-year-old Caucasian male with autoimmune adrenalitis (Addison’s disease). Notes: (A) Reprinted from Bowden SA, Henry R. Pediatric adrenal insufficiency: diagnosis, management, and new therapies. Int J Pediatr. 2018;2018;1,739,831. Creative Commons.5 |
Adrenal androgen deficiency is evident only in females, as decreased pubic and axillary hair development. Males derive most of their androgens from the testes which result in normal pubarche.
Central Adrenal Insufficiency
Central AI is characterized by deficient production of adrenal cortisol due to hypothalamic-pituitary dysfunction with pituitary ACTH deficiency and/or hypothalamic CRH deficiency (Table 1). ACTH deficiency may be isolated, or more frequently in combination with other pituitary hormone deficiencies (hypopituitarism). Clinical presentation may vary considerably, depending on the number and severity of other associated pituitary hormone deficiencies. Clinical presentation may include hypoglycemia that may cause seizure, especially when ACTH deficiency occurs in conjunction with growth hormone deficiency. Other symptoms include lethargy, poor feeding, nausea, vomiting, or prolonged jaundice in neonates (Table 2). Since aldosterone secretion is intact in central AI, as it is regulated by the renin-aldosterone pathway, salt wasting and hyperkalemia are not present. However, mild hyponatremia can occur occasionally in association with central AI. The mechanism is due to increased vasopressin secretion with water retention from lack of cortisol to inhibit the effect of vasopressin6,7 and impaired electrolyte-free water excretion in the kidney.8 Because ACTH is not elevated in central AI, therefore, skin hyperpigmentation is absent in central AI.
Adrenal Crisis
Acute adrenal crisis, the most severe manifestation of AI, is a life-threatening medical emergency that requires prompt diagnosis and treatment. It is defined as an acute deterioration in health status associated with an acute hemodynamic disturbance.9 Adrenal crisis usually manifests as hypotension in a previously undiagnosed patient with primary AI who has encountered a major acute stress event, or in a patient with established adrenal AI who does not increase glucocorticoid replacement to meet the physiological stress demand during the times of severe infection, surgery, or trauma, or cannot retain oral glucocorticoid medication because of persistent vomiting due to viral gastroenteritis or other causes. In addition to hypotension, patients often have other non-specific symptoms (similar to chronic AI symptoms) such as nausea, vomiting abdominal pain, weakness, fatigue, or postural dizziness (Table 2). Clinical signs of circulatory decompensation that are distinct for an adrenal crisis include sinus tachycardia or delayed capillary refill.9 Impaired consciousness may occur during adrenal crisis, which can progress from lethargy or confusion to delirium, obtundation, or coma. Abdominal pain, nausea, vomiting and diarrhea may lead to misdiagnosis of gastroenteritis. Fever is often present, usually caused by a precipitating infection, or may be secondary to the hypocortisolemia. The abdominal tenderness and fever may lead to an erroneous diagnosis of an acute surgical abdomen and potentially catastrophic surgical exploration without corticosteroid replacement. Marked electrolyte abnormalities include hyponatremia, hyperkalemia (in acute primary AI), and hypoglycemia. Hyponatremia can be profound as low as 111 mmol/L.10 It is important to note that hyperkalemia is present in approximately 50% of children with primary AI.11,12 Therefore, the absence of hyperkalemia in the presence of severe hyponatremia cannot rule out adrenal crisis or primary AI in children.
Patients with primary AI may have higher risk of adrenal crisis than patients with central AI, due to mineralocorticoid deficiency with greater risk of salt-losing hypovolemia and dehydration.
Rarely, hypercalcemia may occur in both chronic AI and adrenal crisis. Mechanisms for hypercalcemia in AI are thought to be due to decreased renal excretion of calcium and increased bone resorption.8
It is important to note that in patients with central hypothyroidism and AI (as part of hypopituitarism) or primary autoimmune AI in conjunction with primary hypothyroidism (as in autoimmune polyendocrine syndrome type II), treatment initiation with levothyroxine without hydrocortisone replacement first may precipitate an acute adrenal crisis because thyroid hormone increases cortisol metabolism.13,14 Clinicians must be aware of this and initiate hydrocortisone treatment before thyroid hormone replacement.
Screening Strategies for Adrenal Insufficiency
Clinical Evaluation
Clinicians must have high index of suspicion of AI and a low threshold for testing for AI in any patients presenting with unexplained collapse, dehydration, hypotension, vomiting or diarrhea, hyponatremia, hyperkalemia, acidosis and hypoglycemia (Table 2). Hyperkalemia is present only in primary AI, and can be masked by vomiting. As most symptoms are non-specific, clinical clues that are most suggestive for AI should be looked for. One distinct clinical clue for primary AI is hyperpigmentation which is a manifestation of increased ACTH level in response to primary adrenal failure. In newborns, the nipples, axilla, umbilicus, and the genitalia should be inspected, whereas, in children, palmar creases, axilla, and gingival borders should be examined for hyperpigmentation.
Neonates born with atypical genitalia should alert suspicion for CAH with symptomatic salt wasting and adrenal insufficiency that may manifest in the first 1–3 weeks of life.
Other important clinical history to help delineating the etiology of AI includes family history of consanguinity which may provide the clue to an autosomal recessive inherited disorder. Sex of the patient is also an important clinical clue for X-linked recessive disorders that affect mainly males. These are adrenoleukodystrophy and congenital adrenal hypoplasia. Young age onset below 2 years suggests CAH, congenital adrenal hypoplasia, or familial glucocorticoid deficiency. Childhood onset AI includes autoimmune adrenalitis, infective etiology, and adrenoleukodystrophy.
If there is a concern for impending acute adrenal crisis, the patient should receive hydrocortisone stress dose immediately without delay or waiting for results of diagnostic tests. Blood samples for cortisol and ACTH measurement should be obtained prior to treatment. If this is not possible, diagnosis can still be established later, even after hydrocortisone treatment.
Laboratory Evaluation
Baseline Laboratory Studies
Once AI is suspected, endocrine laboratory testing is required to confirm the diagnosis. A morning (around 8 AM) cortisol level ≤3 ug/dL (83 nmol/L) is indicative of AI, whereas serum cortisol concentration ≥18 ug/dL (497 nmol/L) virtually rules out AI.15,16 Inappropriately low cortisol level at the time of stress or adrenal crisis indicates adrenal insufficiency. Basal plasma ACTH is used to differentiate between primary and central AI, with high ACTH (typically >100 pg/mL or 22.2 pmol/L) suggestive of primary AI. If the results are equivocal, an ACTH stimulation test should be performed (see below section). Elevated plasma renin activity with low serum aldosterone confirms aldosterone deficiency. Patients with mild or early stage of AI or central hypoadrenalism would often require additional dynamic testing as described below.
Diagnostic approach for patients with suspected primary AI is outlined in Figure 2. Additional testing for differentiation of the causes of primary adrenal insufficiency include adrenal autoantibodies (or 21-hydroxylase antibodies) with positive results indicating autoimmune AI or Addison’s disease, elevated serum 17-hydroxy progesterone establishing diagnosis of CAH. Plasma very long chain fatty acids should be obtained in all males diagnosed with primary AI without evidence of autoimmunity to rule out X-linked adrenoleukodystrophy.
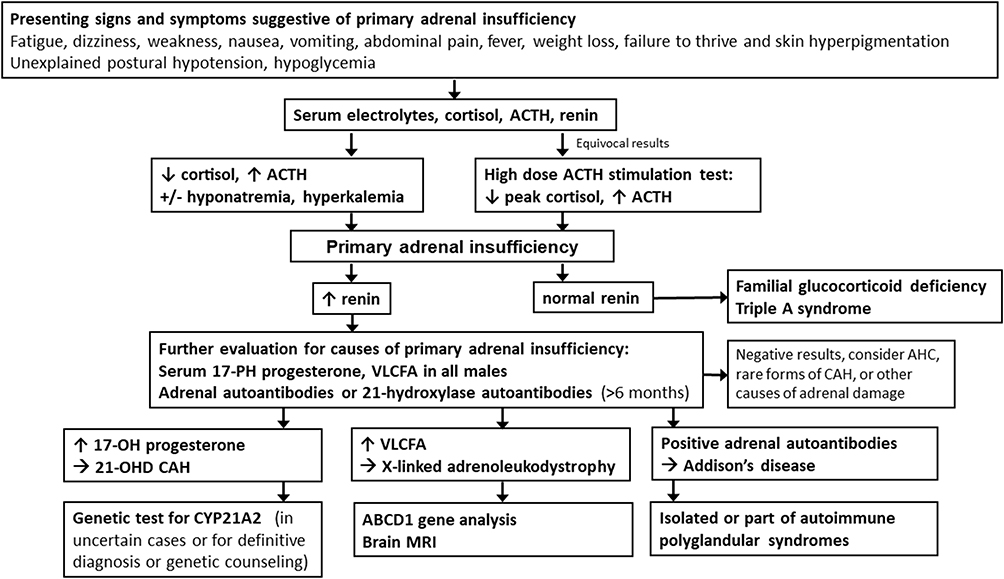 |
Figure 2 Diagnostic approach for patients with suspected primary adrenal insufficiency. Abbreviations: ACTH, adrenocorticotropic hormone; VLCFA, very long chain fatty acids; CAH, congenital adrenal hyperplasia; 21-OHD CAH, congenital adrenal hyperplasia due to 21-hydroxylase deficiency; AHC, adrenal hypoplasia congenita. |
Dynamic Tests of Hypothalamic-Pituitary-Adrenal Function
ACTH Stimulation Test
ACTH stimulation test using either 1 µg (low-dose) or 250 µg (high-dose or standard test) of cosyntropin (1–24 ACTH; Cortrosyn) to directly stimulate adrenal cortisol release is the most common diagnostic test to evaluate the HPA axis. Baseline cortisol and ACTH are obtained prior to administration of cosyntropin. The rise in cortisol is measured at 30 and 60 minutes. A normal response to ACTH stimulation test is a peak serum cortisol ≥18 µg/dL (497 nmol/L). In patients with acute ACTH deficiency or recent onset central AI, such as the patient who has just had a suprasellar tumor resection with hypophysectomy, the high-dose cosyntropin stimulation test may produce a normal cortisol response. This is because the test exposes the adrenal glands to overwhelming amount of ACTH and the adrenal reserve may still be adequate to produce a normal cortisol response to exogenous ACTH, but there is no endogenous ACTH release in response to stress.17 The low-dose ACTH stimulation test is more sensitive in detecting mild AI or recent onset central AI than the standard high-dose test.18,19 Technical difficulties with dilution error or loss of ACTH through long tubing when using low dose 1 ug cosyntropin may influence diagnostic accuracy resulting in false-positive tests or low cortisol response.20 Suboptimal cortisol response does not distinguish between the primary and central AI. Baseline ACTH concentration is used to differentiate between primary and central AI. High ACTH level indicates primary AI, whereas low or low normal ACTH level indicates central AI. When the diagnosis of ACTH deficiency or central AI is made, assessment for other pituitary hormone deficiencies must be performed because isolated ACTH deficiency is rare.
Clinicians need to be aware of different cortisol cutoff values that are assay-specific, when evaluating patients for AI. The newer highly specific cortisol assays with lower cross-reactivity to other steroid metabolites produce approximately 20–39% lower cortisol thresholds than the older, less-specific radioimmunoassays.21–23 Therefore, the cortisol cutoff values can be lowered to 14–15 ug/dL (386–413 nmol/L) when using newer assays.21 When using the classic threshold of cortisol cutoff <18 ug/dL (497 nmol/L) with these newer assays, many more patients would have been given the biochemical diagnosis of AI and may have been treated with hydrocortisone unnecessarily.
In neonates with congenital hypopituitarism, initial ACTH stimulation test performed during the first 1–2 weeks of life to diagnose ACTH deficiency may produce a normal cortisol response even in the presence of central AI.24 This is explained by the fact that neonates with ACTH deficiency have a normal upregulation of fetal steroidogenic enzymes and normal fetal adrenal maturation and steroidogenesis under the placental CRH stimulation,25 therefore, have sufficient adrenal reserve that allows temporary normal cortisol response to exogenous ACTH administered shortly after birth. Lack of pituitary ACTH secretion overtime will eventually cause the adrenocortical zonae fasciculata and reticularis to undergo functional and anatomic atrophy, at which time a failed cosyntropin stimulation test will become evident. Clinicians must recognize this phenomenon and repeat cosyntropin stimulation test again within a month when there are clinical concerns for central AI in these at risk infants even after initial normal adrenal testing.24
Insulin-Induced Hypoglycemia
Hypoglycemia produces a major stress response, with increases in counter-regulatory hormones, including ACTH, cortisol, and growth hormone, therefore, is used to assess the integrity of the HPA axis. Serum cortisol is obtained at baseline, 30, 45, 60, 90 and 120 minutes after regular insulin given intravenously at 0.1 unit/kg. This test was once considered the gold standard for diagnosing AI, but is not recommended for use in children because of the risk of hypoglycemic seizures.15,26
Glucagon Stimulation Test
Glucagon stimulation test is a safe alternative to insulin-induced hypoglycemia in evaluating central AI. Serum glucose and cortisol levels are assessed at 60, 90, 120, and 150 minutes after subcutaneous administration of glucagon at 0.03 mg/kg (maximum 1 mg). The physiological mechanism by which glucagon induces cortisol release is believed to be due to initial blood glucose rise after glucagon administration, which then evokes an endogenous insulin secretion, with a subsequent decrease in blood glucose, which stimulates a counter-regulatory hormone response including growth hormone and cortisol.27 Common side effects of glucagon include nausea, vomiting and abdominal cramps. Glucagon stimulation test generates peak cortisol levels similar to those by insulin tolerance test,27 but may have a high false-positive rate (23.7%) in children (low cortisol on the glucagon test but normal peak cortisol on the ACTH stimulation test).28 In children, peak glucagon-stimulated cortisol levels were inversely correlated with age28,29 and sex.28 Lower peak cortisol cutoff for glucagon stimulation test has been suggested in adults.30,31
Metyrapone Test
Metyrapone inhibits the conversion from 11-deoxycortisol to cortisol by blocking the 11β-hydroxylase enzyme. This causes decreased cortisol level, with a subsequent rise in ACTH and 11-deoxycortisol levels, which are measured at 8 AM in the morning following oral metyrapone administration 8 hours prior (at midnight). The metyrapone dose at 30 mg/kg, up to a maximum of 2 grams, is given with a snack to decrease the side effect of nausea. A normal response is the increase in plasma 11-deoxycortisol to >7 µg/dL.32 Lack of increase in ACTH and 11-deoxycortisol levels after metyrapone administration indicates ACTH deficiency. The metyrapone test is rarely performed because of the risk of precipitating an adrenal crisis or AI symptoms (hypotension and nausea) for several hours and other difficulties with obtaining metyrapone or performing ACTH and 11-deoxycortisol (compound S) assays, the results of which may not be rapidly available.15,33
Screening Considerations for Common Etiologies of Adrenal Insufficiency
- Congenital adrenal hyperplasia (CAH).
CAH is the most common cause of primary AI in children, with a prevalence of 1:10,000–1:15,000 newborns. CAH is an autosomal recessive disorder, most commonly caused by a deficiency of the 21-hydroxylase enzyme (>90% of cases), leading to adrenal insufficiency and androgen excess. The cortisol synthetic block leads to increased ACTH which stimulates the adrenal cortex, with accumulation of adrenal hormone precursors that are diverted to excess androgen hormone synthesis.34 There are 3 clinical phenotypes of 21-hydroxylase deficiency: classical salt wasting, classical simple virilizing, and nonclassical CAH. Classical salt-wasting CAH is the most severe phenotype, accounting for 75% of classic cases. A cardinal feature of classic or severe virilizing CAH in newborn females is genital ambiguity, which should alert clinicians to evaluate for CAH. Due to aldosterone deficiency secondary to severe 21-hydroxylase enzyme deficiency, neonatal salt loss with life-threatening hypovolemia, hyponatremia, hyperkalemia, weight loss may occur within the first 1–3 weeks of life, which may cause death, without early diagnosis or treatment. Simple virilizing CAH, accounting for 25% of the classic cases, presents with early virilization but no salt loss because there is 1–2% of the 21-hydroxylase enzyme activity present. These patients have cortisol deficiency, but aldosterone production is sufficient to maintain normal sodium balance. Nonclassical CAH or late-onset CAH has about 20–50% of the 21-hydroxylase enzyme activity present; therefore, aldosterone production is normal and ACTH-stimulated cortisol levels are near normal or slightly subnormal.35 Affected patients have mild androgen excess presenting with early adrenarche, growth acceleration and advanced bone age.
When CAH is clinically suspected in newborns present with ambiguous genitalia, serum 17-hydroxy progesterone is obtained to confirm or exclude diagnosis.34 Currently, all 50 states in the United States and >50 countries worldwide have newborn screening program for 21-hydroxylase deficiency, enabling early diagnosis and prevention of morbidity and mortality due to adrenal crisis.36 The newborn screening is particularly important and beneficial in affected boys whose diagnosis is frequently delayed without newborn screening owing to the lack of genital ambiguity. Abnormal newborn screening should prompt confirmatory serum 17-hydroxy progesterone and electrolytes. Newborns with classical CAH typically have markedly elevated serum 17-hydroxy progesterone levels (always >1000 ng/dL or 30 nmol/L, usually >3500 ng/dL or 105 nmol/L; levels increase with time in infants with CAH to >10,000 ng/dL or 300 nmol/L). A single basal measurement is all that is required to make the diagnosis and treatment with hydrocortisone and fludrocortisone can be initiated. For indeterminate 17-hydroxy progesterone levels (200–1000 ng/dL or 6–30 nmol/L), high-dose cosyntropin stimulation test (125 µg in neonates; 250 µg in children) is needed. Samples should be obtained at baseline and 60 minutes after cosyntropin. Low cortisol and elevated 17-hydroxy progesterone levels confirm CAH. Classical CAH typically has stimulated 17-hydroxy progesterone >10,000 ng/dL (300 nmol/L), with simple virilizing CAH having stimulated level between 10,000 and 30,000 ng/dL (300–900 nmol/L), and salt-wasting CAH much greater than 30,000 ng/dL (900 nmol/L). Nonclassical CAH has stimulated 17-hydroxy progesterone between 1000 and 10,000 ng/dL (30–300 nmol/L).37,38
T1D is an autoimmune disease that is often associated with other autoimmune diseases. These include autoimmune thyroid disease (hyper- or hypothyroidism) with frequency of 15–30%, celiac disease (4–9%), and autoimmune adrenal failure or Addison’s disease (0.5%).39 Up to 3% of patients with type 1 DM have positive adrenal autoantibodies that are detected prior to the development of symptomatic AI.40 A previous natural history study indicates that 40% of patients with positive adrenal autoantibodies developed Addison’s disease.41 Currently, there are no specific guidelines for routine screening of Addison’s disease in asymptomatic patients with type 1 diabetes. Clinicians should assess and screen for Addison’s disease based on clinical ground. Aside from typical symptoms of AI including fatigue, weight loss, salt craving, and hyperpigmentation of skin, unique symptoms to look for in patients with type 1 DM are unexplained recurrent hypoglycemia or decreased insulin dose requirement.42 Cortisol deficiency results in enhanced insulin sensitivity which leads to an increased peripheral glucose utilization, and reduced glycogenesis and gluconeogenesis.39 Baseline morning cortisol and ACTH should be obtained as a screening test. ACTH stimulation test may be obtained to confirm diagnosis.
Adrenoleukodystrophy is an X-linked neurodegenerative disease caused by mutations in the ABCD1 gene, encoding protein that transports very long chain fatty acids (VLCFAs) to the peroxisome. The mutations in this gene cause peroxisomal membrane protein deficiency leading to toxic accumulation of VLCFAs in the plasma, white matter, spinal cord, and adrenal cortex, resulting in primary AI, neurologic deterioration and death. There are 3 main clinical phenotypes: primary AI, cerebral ALD (characterized by progressive white matter demyelination) and adrenomyeloneuropathy (spastic paresis, gait instability, and incontinence). ALD affects predominantly males with an incidence of 1/15,000–20,000 live male births.43 In males, the first manifestation of ALD is usually AI, which has been reported to occur as young as 5 months of age.44 Cerebral ALD can occur at any age, with earliest age onset of 3 years. Majority of boys with ALD will demonstrate biochemical or clinical evidence of primary AI prior to the onset of neurologic symptoms, with lifetime prevalence of 80%.45 Therefore, all males diagnosed with primary AI should have plasma VLCFAs obtained to rule out X-linked ALD. Early diagnosis of ALD is the key to prevent mortality, as early intervention with life-saving hematopoietic cell transplant can arrest cerebral ALD progression, while delayed diagnosis of ALD after onset of primary AI is associated with poor survival and neurological outcomes.46 ALD newborn screening allows early diagnosis with prospective monitoring for adrenal function and the onset of cerebral ALD. ALD newborn screening has been developed by measuring VLCFA levels in dried blood spots, was first initiated in New York in 2013, added to the Recommended Uniform Screening Panel in 2016.43,47 Currently, newborn screening for ALD has been implemented in 30 states and Washington DC with continuing expansion. Once ALD is diagnosed by newborn screening, all male infants should be referred to pediatric endocrinologists for evaluation for adrenal dysfunction which should be initiated in the first 6 months of life by measuring morning serum cortisol and ACTH levels. If these are abnormal, then ACTH stimulation test is indicated. Counseling regarding risk for AI should be provided. Adrenal testing should be performed when AI symptoms manifest; otherwise, routine adrenal testing should be done every 4–6 months until age 10 years (because the risk of AI is highest in the first decade of life, 47%), and once yearly thereafter until age 40 years.45,48 Once AI is diagnosed, stress hydrocortisone should be given at the time of stress and prior to sedated magnetic resonance imaging of the brain. Maintenance corticosteroids may also be given as clinically indicated.
Glucocorticoids are widely used for their anti-inflammatory and immunosuppressive actions. Prolonged glucocorticoid use can cause a possible unwanted effect which is the suppression of the HPA axis, leading to secondary AI. The Pediatric Endocrine Society recommends that adrenal suppression be considered in all children who have received supraphysiologic doses of oral daily glucocorticoids (>12 mg/m2/day hydrocortisone or equivalent) for ≥2 weeks.15 Adrenal suppression may also occur after long-term use of inhaled glucocorticoids or high potency topical corticosteroids.49,50 Glucocorticoid-induced AI is considered to be the most common causes of AI. Although it is not possible to predict which patients will develop AI, the long duration, high glucocorticoid dosage and potency, long half-life, and time of administration increase the risk.1 AI can occur after abrupt discontinuation of long-term glucocorticoid therapy, either inadvertently (ie, patients have surgery and do not receive stress dose hydrocortisone coverage peri-operatively) or during gastrointestinal illnesses with vomiting that inhibits normal absorption of glucocorticoids.
AI can also occur after discontinuation of long-term glucocorticoid therapy. There are no evidence-based guidelines on how to best taper glucocorticoid.51,52 A gradual tapering is generally recommended with close monitoring for signs and symptoms of AI, especially when the glucocorticoid dose is near physiologic dose (12 mg/m2/day hydrocortisone or equivalent), at which time switching to short-acting hydrocortisone is advisable. Clinicians should have a low threshold to test and treat for AI in patients experiencing AI symptoms during the tapering process or after cessation of glucocorticoids. Morning cortisol should be assessed as the initial screening in patients at high-risk for AI when the glucocorticoid dose is reaching half the physiologic dose or less to guide management.52 The HPA axis is intact when morning cortisol is >18 ug/dL (>500 nmol/L) (assay dependent) and glucocorticoids can be discontinued (Figure 3). When morning cortisol values 7–18 ug/dL (193–500 nmol/L), adrenal function is likely adequate for non-stressed state, but may be inadequate at the time of stress. Therefore, daily hydrocortisone is discontinued but stress hydrocortisone coverage at time of stress is continued until ACTH stimulation test is performed to document recovery of adrenal function. Glucocorticoid-induced AI is possible in patients with morning cortisol <7 ug/dL (<193 nmol/L). Daily hydrocortisone with stress-dose coverage should be continued and adrenal function should be reassessed over time.53 Clinicians should educate patients about the risk and symptoms of AI and “steroid-induced adrenal suppression” should be on the “Problem List” in the patient’s medical records, to alert other health-care providers caring for patients in the urgent care or emergency department for the risk of AI.
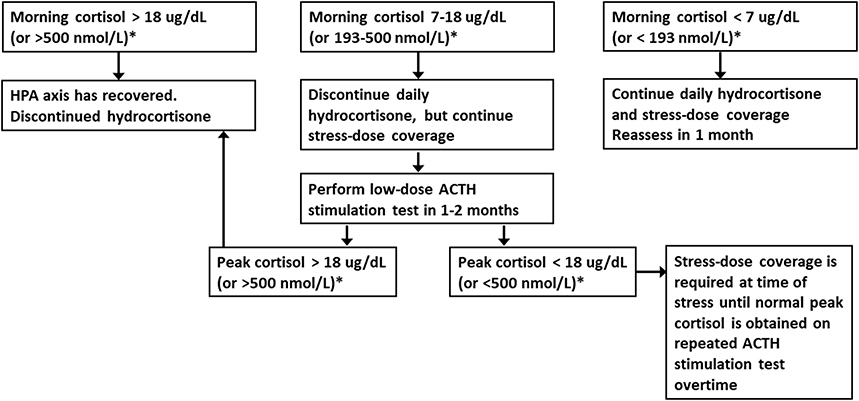 |
Figure 3 Screening and assessment of recovery of the hypothalamic-pituitary-adrenal axis after corticosteroid withdrawal. After reaching half the physiologic dose of hydrocortisone (≤5 mg/m2/day), obtain morning serum cortisol to determine if daily and stress dose hydrocortisone can be discontinuation. *Serum cortisol cutoff values are assay dependent and may be lower with newer cortisol assays. Reprinted from Bowden SA, Connolly AM, Kinnett K, Zeitler PS. Management of adrenal insufficiency risk after long-term systemic glucocorticoid therapy in Duchenne muscular dystrophy: clinical practice recommendations. J Neuromuscular Dis. 2019;6(1):31–41, with permission from IOS Press. The publication is available at IOS Press through http://dx.doi.org/doi: 10.3233/JND-180346.53 |
Autoimmune AI or Addison’s disease is one of the common causes of primary AI and can be isolated or in association with other endocrine autoimmune diseases. Clinicians managing patients with single endocrine autoimmune diseases need to consider the possibility of development of other autoimmune endocrine disorders. Autoimmune polyglandular syndromes (APSs) comprise a group of organ-specific autoimmune diseases. APS type 1 and type 2, the two common types of APS, are associated with Addison’s disease. APS type 1 is characterized by the presence of two of the following: mucocutaneous candidiasis, hypoparathyroidism, and Addison’s disease. Candidiasis is usually the first manifestation, followed by hypoparathyroidism. Addison’s disease tends to be the third disease to appear, usually before age 15 years.54 APS type 1 diagnosis can be confirmed by mutational analysis of the AIRE gene. APS type 2 is defined by the combination of chronic autoimmune AI (Addison’s disease) with autoimmune thyroid disease and/or type 1 diabetes mellitus. Other autoimmune diseases associated with APS type 1 and 2 include keratitis, autoimmune hepatitis, pernicious anemia, celiac disease, vitiligo, or primary ovarian insufficiency. After diagnosis of APS type 1 without AI is made, adrenal autoantibodies should be obtained as a screening test for AI. If adrenal autoantibodies are present, yearly morning cortisol and ACTH should be obtained; if abnormal, ACTH stimulation test should be performed for early detection of AI.55
Conclusion
Early recognition and diagnosis of AI is crucial to prevent morbidity and mortality associated with adrenal crisis. Clinicians must be aware of the extensive list of conditions or precipitating factors associated with AI and various clinical scenarios that different causes of AI may present (Table 1). Improved screening strategies as well as clinician education for timely diagnosis and treatment of AI is central to the management of this life-threatening medical emergency.
Funding
This project was completed with no specific funding.
Disclosure
The author has nothing to disclose for this work.
References
1. Broersen LH, Pereira AM, Jørgensen JOL, Dekkers OM. Adrenal insufficiency in corticosteroids use: systematic review and meta-analysis. J Clin Endocrinol Metab. 2015;100(6):2171–2180. doi:10.1210/jc.2015-1218
2. Lamberts SW, Bruining HA, De Jong FH. Corticosteroid therapy in severe illness. N Eng J Med. 1997;337(18):1285–1292. doi:10.1056/NEJM199710303371807
3. Kehlet H. A rational approach to dosage and preparation of parenteral glucocorticoid substitution therapy during surgical procedures: a short review. Acta Anaesthesiol Scand. 1975;19(4):260–264. doi:10.1111/j.1399-6576.1975.tb05182.x
4. Woodcock T, Barker P, Daniel S, et al. Guidelines for the management of glucocorticoids during the peri‐operative period for patients with adrenal insufficiency: guidelines from the Association of Anaesthetists, the Royal College of Physicians and the Society for Endocrinology UK. Anaesthesia. 2020;75(5):654–663. doi:10.1111/anae.14963
5. Bowden SA, Henry R. Pediatric adrenal insufficiency: diagnosis, management, and new therapies. Int J Pediatr. 2018;2018:87.
6. Kamoi K, Tamura T, Tanaka K, Ishibashi M, Yamaji T. Hyponatremia and osmoregulation of thirst and vasopressin secretion in patients with adrenal insufficiency. J Clin Endocrinol Metab. 1993;77(6):1584–1588.
7. Diederich S, Franzen N-F, Bahr V, Oelkers W. Severe hyponatremia due to hypopituitarism with adrenal insufficiency: report on 28 cases. Eur J Endocrinol. 2003;148(6):609–618. doi:10.1530/eje.0.1480609
8. Puar TH, Stikkelbroeck NM, Smans LC, Zelissen PM, Hermus AR. Adrenal crisis: still a deadly event in the 21st century. Am J Med. 2016;129(3):339.e1–e9... doi:10.1016/j.amjmed.2015.08.021
9. Rushworth RL, Torpy DJ, Falhammar H. Adrenal crisis. N Eng J Med. 2019;381(9):852–861. doi:10.1056/NEJMra1807486
10. Thompson MD, Kalmar E, Bowden SA. Severe hyponatraemia with absence of hyperkalaemia in rapidly progressive Addison’s disease. BMJ Case Rep. 2015;2015:bcr2015209903. doi:10.1136/bcr-2015-209903
11. Hsieh S, White PC. Presentation of primary adrenal insufficiency in childhood. J Clin Endocrinol Metab. 2011;96(6):E925–E8. doi:10.1210/jc.2011-0015
12. Gagnon RF, Halperin ML. Possible mechanisms to explain the absence of hyperkalaemia in Addison’s disease. Nephrology Dialysis Transpl. 2001;16(6):1280–1284. doi:10.1093/ndt/16.6.1280
13. Lakhani OJ, Tripathi S, Indu K, Desai M. Levothyroxine replacement before glucocorticoid replacement leading to adrenal crisis in a case of autoimmune polyendocrine syndrome type II (Schmidt syndrome). Thyroid Res Practice. 2015;12(3):116. doi:10.4103/0973-0354.157932
14. Fonseca V, Brown R, Hochhauser D, Ginsburg J, Havard C. Acute adrenal crisis precipitated by thyroxine. Br Med J. 1986;292(6529):1185. doi:10.1136/bmj.292.6529.1185
15. Shulman DI, Palmert MR, Kemp SF. Drug and Therapeutic Committee. Adrenal insufficiency: still a cause of morbidity and death in childhood. Pediatrics. 2007;119(2):e484–e94. doi:10.1542/peds.2006-1612
16. Hägg E, Asplund K, Lithner F. Value of basal plasma cortisol assays in the assessment of pituitary‐adrenal insufficiency. Clin Endocrinol (Oxf). 1987;26(2):221–226. doi:10.1111/j.1365-2265.1987.tb00780.x
17. Kazlauskaite R, Evans AT, Villabona CV, et al. Corticotropin tests for hypothalamic-pituitary-adrenal insufficiency: a metaanalysis. J Clin Endocrinol Metab. 2008;93(11):4245–4253. doi:10.1210/jc.2008-0710
18. Rose SR, Lustig RH, Burstein S, Pitukcheewanont P, Broome DC, Burghen GA. Diagnosis of ACTH deficiency. Horm Res Paediatr. 1999;52(2):73–79. doi:10.1159/000023438
19. Dickstein G. Commentary to the Article: comparison of low and high dose corticotropin stimulation tests in patients with pituitary disease. J Clin Endocrinol Metab. 1998;83(12):4531–4532. doi:10.1210/jcem.83.12.5322-5
20. Wade M, Baid S, Calis K, Raff H, Sinaii N, Nieman L. Technical details influence the diagnostic accuracy of the 1 microg ACTH stimulation test. Eur J Endocrinol. 2010;162(1):109–113. doi:10.1530/EJE-09-0746
21. Javorsky BR, Raff H, Carroll TB, et al. New cutoffs for the biochemical diagnosis of adrenal insufficiency after ACTH stimulation using specific cortisol assays. J Endocrine Soci. 2021;5(4):bvab022. doi:10.1210/jendso/bvab022
22. Kline G, Buse J, Krause R. Clinical implications for biochemical diagnostic thresholds of adrenal sufficiency using a highly specific cortisol immunoassay. Clin Biochem. 2017;50(9):475–480. doi:10.1016/j.clinbiochem.2017.02.008
23. El‐Farhan N, Pickett A, Ducroq D, et al. Method‐specific serum cortisol responses to the adrenocorticotrophin test: comparison of gas chromatography‐mass spectrometry and five automated immunoassays. Clin Endocrinol (Oxf). 2013;78(5):673–680. doi:10.1111/cen.12039
24. Coshway LK, Indyk JA, Bowden SA. Repeating ACTH stimulation test is necessary to diagnose ACTH deficiency in neonatal hypopituitarism with initial false negative result. Global Pediatric Health. 2014;1:2333794X14563385. doi:10.1177/2333794X14563385
25. Sirianni R, Rehman KS, Carr BR, Parker JCR, Rainey WE. Corticotropin-releasing hormone directly stimulates cortisol and the cortisol biosynthetic pathway in human fetal adrenal cells. J Clin Endocrinol Metab. 2005;90(1):279–285. doi:10.1210/jc.2004-0865
26. Binder G, Bosk A, Gass M, Ranke MB, Heidemann PH. Insulin tolerance test causes hypokalaemia and can provoke cardiac arrhythmias. Horm Res Paediatr. 2004;62(2):84–87. doi:10.1159/000079539
27. Ach T, Yosra H, Jihen M, et al. Cortisol cut-points for the glucagon stimulation test in the evaluation of hypothalamic pituitary adrenal axis. Endocr J. 2018;65(9):935–942. doi:10.1507/endocrj.EJ18-0147
28. Tenenbaum A, Phillip M, de Vries L. The intramuscular glucagon stimulation test does not provide good discrimination between normal and inadequate ACTH reserve when used in the investigation of short healthy children. Horm Res Paediatr. 2014;82(3):194–200. doi:10.1159/000365190
29. Johnstone HC, Cheetham TD. GH and cortisol response to glucagon administration in short children. Horm Res. 2004;62(1):27–32. doi:10.1159/000078722
30. Simsek Y, Karaca Z, Tanriverdi F, Unluhizarci K, Selcuklu A, Kelestimur F. A comparison of low-dose ACTH, glucagon stimulation and insulin tolerance test in patients with pituitary disorders. Clin Endocrinol (Oxf). 2015;82(1):45–52. doi:10.1111/cen.12528
31. Hamrahian AH, Yuen KC, Gordon MB, Pulaski-Liebert KJ, Bena J, Biller BM. Revised GH and cortisol cut-points for the glucagon stimulation test in the evaluation of GH and hypothalamic–pituitary–adrenal axes in adults: results from a prospective randomized multicenter study. Pituitary. 2016;19(3):332–341. doi:10.1007/s11102-016-0712-7
32. Fiad TM, Kirby JM, Cunningham SK, McKenna TJ. The overnight single‐dose metyrapone test is a simple and reliable index of the hypothalamic‐pituitary‐adrenal axis. Clin Endocrinol (Oxf). 1994;40(5):603–609. doi:10.1111/j.1365-2265.1994.tb03011.x
33. Chanson P, Guignat L, Goichot B, et al. Group 2: adrenal insufficiency: screening methods and confirmation of diagnosis. Ann d’endocrinologie. 2017;78(6):495–511. doi:10.1016/j.ando.2017.10.005
34. Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(9):4133–4160. doi:10.1210/jc.2009-2631
35. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. 2015;50(1):32–50. doi:10.1007/s12020-015-0656-0
36. Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, et al. Congenital adrenal hyperplasia—Current insights in pathophysiology, diagnostics, and management. Endocr Rev. 2022;43(1):91–159. doi:10.1210/endrev/bnab016
37. New MI, Lorenzen F, Lerner AJ, et al. Genotyping steroid 21-hydroxylase deficiency: hormonal reference data. J Clin Endocrinol Metab. 1983;57(2):320–326. doi:10.1210/jcem-57-2-320
38. New MI, Wilson RC. Steroid disorders in children: congenital adrenal hyperplasia and apparent mineralocorticoid excess. Proce National Acad Sci. 1999;96(22):12790–12797. doi:10.1073/pnas.96.22.12790
39. Krzewska A, Ben-Skowronek I. Effect of associated autoimmune diseases on type 1 diabetes mellitus incidence and metabolic control in children and adolescents. Biomed Res Int. 2016;2016:1–12. doi:10.1155/2016/6219730
40. Warncke K, Fröhlich-Reiterer EE, Thon A, et al. Polyendocrinopathy in children, adolescents, and young adults with type 1 diabetes: a multicenter analysis of 28,671 patients from the German/Austrian DPV-Wiss database. Diabetes Care. 2010;33(9):2010–2012. doi:10.2337/dc10-0404
41. Betterle C, Scalici C, Presotto F, et al. The natural history of adrenal function in autoimmune patients with adrenal autoantibodies. J Endocrinol. 1988;117(3):467–475. doi:10.1677/joe.0.1170467
42. Vinci F, d’Annunzio G, Napoli F, et al. Type 1 Diabetes and Addison’s Disease: when the diagnosis is suggested by the continuous glucose monitoring system. Children. 2021;8(8):702. doi:10.3390/children8080702
43. Turk BR, Theda C, Fatemi A, Moser AB. X‐linked adrenoleukodystrophy: pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Int J Dev Neurosci. 2020;80(1):52–72. doi:10.1002/jdn.10003
44. Eng L, Regelmann MO. Early onset primary adrenal insufficiency in males with adrenoleukodystrophy: case series and literature review. J Pediatr. 2019;211:211–214. doi:10.1016/j.jpeds.2019.04.021
45. Huffnagel IC, Laheji FK, Aziz-Bose R, et al. The natural history of adrenal insufficiency in X-linked adrenoleukodystrophy: an international collaboration. J Clin Endocrinol Metab. 2019;104(1):118–126. doi:10.1210/jc.2018-01307
46. Polgreen LE, Chahla S, Miller W, et al. Early diagnosis of cerebral X-linked adrenoleukodystrophy in boys with Addison’s disease improves survival and neurological outcomes. Eur J Pediatr. 2011;170(8):1049–1054. doi:10.1007/s00431-011-1401-1
47. Moser AB, Seeger E, Raymond GV. Newborn screening for X-linked adrenoleukodystrophy: past, present, and future. Int J Neonatal Screening. 2022;8(1):16. doi:10.3390/ijns8010016
48. Regelmann MO, Kamboj MK, Miller BS, et al. Adrenoleukodystrophy: guidance for adrenal surveillance in males identified by newborn screen. J Clin Endocrinol Metab. 2018;103(11):4324–4331. doi:10.1210/jc.2018-00920
49. Ahmet A, Kim H, Spier S. Adrenal suppression: a practical guide to the screening and management of this under-recognized complication of inhaled corticosteroid therapy. Allergy Asthma Clin Immunol. 2011;7:1–12. doi:10.1186/1710-1492-7-13
50. Levin C, Maibach HI. Topical corticosteroid-induced adrenocortical insufficiency: clinical implications. Am J Clin Dermatol. 2002;3:141–147. doi:10.2165/00128071-200203030-00001
51. Richter B, Neises G, Clar C. Glucocorticoid withdrawal schemes in chronic medical disorders: a systematic review. Endocrinol Metab Clin. 2002;31(3):751–778. doi:10.1016/S0889-8529(02)00008-7
52. Liu D, Ahmet A, Ward L, et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin Immunol. 2013;9(1):1–25. doi:10.1186/1710-1492-9-30
53. Bowden SA, Connolly AM, Kinnett K, Zeitler PS. Management of adrenal insufficiency risk after long-term systemic glucocorticoid therapy in Duchenne muscular dystrophy: clinical practice recommendations. J Neuromuscular Dis. 2019;6(1):31–41. doi:10.3233/JND-180346
54. Betterle C, Dal Pra C, Mantero F, Zanchetta R. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev. 2002;23(3):327–364. doi:10.1210/edrv.23.3.0466
55. Eisenbarth GS, Gottlieb PA. Autoimmune polyendocrine syndromes. N Eng J Med. 2004;350(20):2068–2079. doi:10.1056/NEJMra030158
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.