")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Cordycepin Inhibits Human Gestational Choriocarcinoma Cell Growth by Disrupting Centrosome Homeostasis
Authors Wang CY , Tsai SW, Chien HH, Chen TY, Sheu SY, So EC , Huang BM
Received 4 March 2020
Accepted for publication 6 July 2020
Published 27 July 2020 Volume 2020:14 Pages 2987—3000
DOI https://doi.org/10.2147/DDDT.S252401
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Yan Zhu
Chia-Yih Wang,1,2,* Shih-Wei Tsai,3,* Han-Hsiang Chien,1 Ting-Yu Chen,2,4 Shi-Yuan Sheu,5,6 Edmund Cheung So,7,8 Bu-Miin Huang1,9
1Department of Cell Biology and Anatomy, College of Medicine, National Cheng Kung University, Tainan, Taiwan; 2Institute of Basic Medical Sciences, College of Medicine, National Cheng Kung University, Tainan, Taiwan; 3Department of Obstetrics and Gynecology, An Nan Hospital, China Medical University, Tainan, Taiwan; 4Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan; 5School of Chinese Medicine for Post-Baccalaureate, I-Shou University, Kaohsiung, Taiwan; 6Department of Chinese Medicine, E-Da Cancer Hospital, Kaohsiung, Taiwan; 7Department of Anesthesia & Medical Research, An Nan Hospital, China Medical University, Tainan, Taiwan; 8Graduate Institute of Medical Sciences, Chang Jung Christian University Tainan, Tainan, Taiwan; 9Institute of Medical Research, China Medical University, Taichung, Taiwan
*These authors contributed equally to this work
Correspondence: Edmund Cheung So; Bu-Miin Huang Tel +886-6-3553111 ext.1517
; +886-6-2353535 ext. 5337
Fax +886-6-3553111
; +886-6-2093007
Email [email protected]; [email protected]
Introduction: Human gestational choriocarcinoma, a type of gestational trophoblastic disease, occurs after miscarriage, abortion, ectopic pregnancy, or molar pregnancy. Despite recent advances in the mechanism of anticancer drugs that induce human gestational choriocarcinoma apoptosis or block its growth, new therapeutic approaches are needed to be established. Cordycepin is an active anti-cancer component extracted from Cordyceps sinensis. It prevents cell proliferation both in vitro and in vivo.
Materials and Methods: Here, we examined cell growth by counting cell numbers, and performing a flow cytometry assay and EdU incorporation assay. Centrosome and cytoskeleton-related structures were observed by immunofluorescence assay. The DNA damage-related signaling was examined by Western blot assay.
Results: Here, we showed that cordycepin inhibited human gestational choriocarcinoma cell proliferation and induced cell death. In addition, treatment with cordycepin activated DNA-PK and ERK, thus inducing centrosome amplification and aberrant mitosis. These amplified centrosomes also disrupted microtubule arrays and actin networks, thus leading to defective cell adhesion. Furthermore, cordycepin induced autophagy for triggering cell death.
Conclusion: Thus, our study demonstrates that cordycepin inhibits cell proliferation and disrupts the cytoskeleton by triggering centrosome amplification.
Keywords: human gestational choriocarcinoma, cordycepin, centrosome, cell growth, microtubule
Introduction
Human gestational choriocarcinoma, which is a type of gestational trophoblastic disease, occurs after miscarriage, abortion, ectopic pregnancy, or molar pregnancy.1 It is a fast-growing cancer that occurs in a woman’s uterus. Human gestational choriocarcinoma is often treated with diverse therapeutic strategies, such as radiation therapy, surgery, or chemotherapy. Several therapeutic drugs including methotrexate, etoposide, and actinomycin D are used most often to treat human gestational choriocarcinoma. The effects of anticancer drugs on human gestational choriocarcinoma have been studied extensively. However, treating choriocarcinoma requires high doses of systemic chemotherapeutic agents, leading to nonspecific drug effects and severe toxicity. Thus, a new therapeutic approach is needed to be established.
Extracts of Cordyceps sinensis have been widely used in Chinese traditional medicine. Among these, cordycepin (3ʹ-deoxyadenosine) is known as a very active compound possessing several biological activities.2 The therapeutic potential and proposed pharmacological activities of cordycepin have been demonstrated in several studies. In addition, chemotherapy combined with cordycepin preclinical treatment has been conducted to test its therapeutic effects.2 For example, it can inhibit fungal and bacterial growth.3 In addition, cordycepin inhibits LPS-induced inflammation via suppressing the NF-κB signaling pathway.4 Recent studies also show that cordycepin promotes steroid hormone production in the adrenal gland and testis.5,6 More importantly, cordycepin is now considered as an anti-cancer compound and has been examined in several cancers, such as testicular, blood, liver, and brain tumors.7,8 Cordycepin induces apoptosis by activating JNK, ERK, p38, and Akt signaling or inhibits cell proliferation by reducing expression of cyclin A or cyclin E and CDK2 activity.7,9–11 Despite no available data from clinical trials, cordycepin inhibits tumor cell growth without causing acute toxicity as demonstrated by rat and mouse xenograft models in vivo.12 These results support that cordycepin functions as a novel and safe anti-cancer compound.
Autophagy maintains metabolic homeostasis by degrading and reutilizing the old organelles or proteins via the lysosomal-degradation process in cells.13 When cells are suffering from metabolic starvation, DNA damage, or hypoxia, the autophagic flux is activated. In addition, autophagy either promotes or protects cancer cells from apoptosis upon anti-cancer drug treatment. Thus, clarifying the dual roles of autophagy in cell death during chemotherapy is an important issue to develop a new therapeutic strategy.14–17 Autophagy is regulated by several steps. The initiation of autophagy requires the formation of a double-membraned vesicle, the autophagosome. Once it is formed, the autophagosome fuses with a lysosome to form an autolysosome; lysosomal hydrolases degrade the contents within this acidic compartment.13,18 The autophagosome formation is regulated by microtubule-associated protein l light chain 3 (LC3).19 Atg4 activates LC3 by exposing the Gly116 residue, known as LC3-I. Then, LC3-I is conjugated to phosphatidylethanolamine (PE) and becomes LC3-II. LC3-II associates with and promotes autophagosome formation. Thus, the ratio of LC3-II to -I is used for examining the autophagic flux. Cordycepin affects autophagic flux;20,21 however, the physiological significance and the inter-correlation between autophagy and cell survival remain unclear.
The centrosome is composed of a pair of centrioles (mother and daughter centrioles) and the surrounding pericentriolar materials.22 During interphase, the centrosome is the microtubule-organizing center for proper directional migration. Microtubule-oriented centrosome reposition provides directional migration by facing the centrosome to the leading edge juxtaposed to the nucleus.23 Meanwhile, during centrosome reposition, Golgi ribbon organization occurs, thus promoting vesicle transport to the leading edge for directional migration. At mitosis, centrosomes orchestrate mitotic spindle poles for segregating duplicated chromosomes equally;24 disorganized mitotic spindles result in misalignment of chromosomes thus leading to genomic instability.25
Cordycepin is an active anti-cancer component and has been used for treating several cancers. However, its role in human gestational choriocarcinoma has not been examined yet. Centrosome coordinates cell cycle progression, mitotic division, and directional migration. We thus investigate the effect of cordycepin on human gestational choriocarcinoma and uncovered whether the centrosome plays roles in contributing to the underlying molecular mechanism.
Methods
Materials
The following antibodies were obtained commercially: anti‐γ‐tubulin (T6557), anti‐cyclin A (C4710), and anti‐α‐tubulin (T9026) (Sigma, St. Louis, MO), anti‐ATR, anti‐ATR phospho‐Ser428, anti‐CDK2 (#2546), anti‐CDK2 phospho‐Thr160 (#2561), anti‐cleaved caspase‐3 (Asp175) (#9661), anti‐Chk2, anti‐Chk2 phospho‐Thr68, anti‐AKT, anti‐phospho‐AKT (Ser473), anti‐ERK1/2 (#4695), anti‐phospho‐ERK1/2 on Thr202/Tyr204 (#4370) and anti‐LC3A/B (#12,741) (Cell Signaling, Beverly, MA), anti‐Chk1 phospho‐Ser345 and anti‐H2AX phospho‐Ser139 (ab2893, Abcam, Cambridge, UK), anti‐Ku70 and anti‐ATM (Genetex, Trvine, CA), anti‐ATM phospho‐Ser1981 (Epitomics, Burlingame, CA), anti‐cyclin E (HE‐12, GTX23927), anti‐actin (GTX109639) (GeneTex, Irvine, CA), anti-FAK (EP695Y, ab40794), anti-E cadherin (HECD-1, ab1416), and anti-cyclin F antibody (ab203117) (Abcam, Cambridge, UK).
For drug treatment, the following chemicals are treated for 24 h before analysis. Ku55933 (ATM inhibitor, SML1109, 10 μM), cytochalasin D (C2618, 5 μg/mL), vanillin (V1104, 1 mM), U0126 (U120, 10 μM), and chloroquine (CQ, 50–63-5, 50 μM) were purchased from Sigma, St. Louis, MO. Bafilomycin-A1 (Baf.A1, BML-CM110, 10 nM) was purchased from Enzo, NY, USA.
Cell Culture
The human embryonic kidney HEK (ATCC, CRL-1573) cells and human gestational choriocarcinoma JAR cells (ATCC, HTB-144) were purchased from the ATCC. These cells were grown in Dulbecco’s modified Eagle medium (DMEM)-F12; human immortalized chorionic villi cells of first-trimester placenta (HTR-8/SVneo) were grown in Roswell Park Memorial Institute (RPMI)-1640 medium, medium supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere at 5% CO2. The mycoplasma contamination is examined regularly by immunofluorescence microscopy with DAPI staining according to the guidelines.
Western Blot Assay
For the preparation of cell extracts, cells were lysed by RIPA cell lysis buffer supplemented with cocktail protease inhibitors. The lysates were collected on ice for 10 min followed by centrifugation (15,000 g) at 4°C for 10 min. The supernatant was collected and quantified by Bradford protein quantity assay (Bio‐Rad, Hercules, CA). Then, the quantified cell extracts were mixed with an equal amount of two-fold sample buffer followed by heating at 100°C for 10 min. Then, all prepared samples were loaded onto and separated by SDS‐PAGE (150 V, 120 min) in a cold-room. Next, the separated samples in the SDS-PAGE gel were transferred to the PVDF membrane, which was rinsed in the transfer buffer, by wet-transfer caseate (25 V, 720 min) in the cold-room. After transfer, the PVDF membrane was washed extensively by TBST buffer and blocked in 3% BSA at room temperature for 1 h followed by incubating with primary antibodies at 4°C overnight. After incubation, the primary antibodies were washed out with TBST three times. Then, the membrane was incubated with HRP‐conjugated secondary antibody followed by washing with TBST three times. After extensive washing, the target signal was detected by chemiluminescent substrates.
Immunofluorescence Assay
For the immunofluorescence assay, cells were fixed with ice‐cold methanol at −20°C for 6 min, then washed with PBS three times. Cells were then blocked with 5% FBS for 1 h. Then, cells were incubated with antibodies for 24 h at 4°C followed by washing with PBS three times. After extensive washing, cells were co-incubated with fluorescein isothiocyanate‐conjugated and Cy3‐conjugated secondary antibodies (Invitrogen, Carlsbad, CA) with 4ʹ,6‐diamino‐2‐phenylindole (DAPI, 0.1 µg/mL) simultaneously in the dark for 1 h. Then, these cells were washed with PBS three times, and the coverslips were mounted in 50% glycerol on glass slides. The fluorescence signals were examined with an AxioImager Z1 fluorescence microscope (Zeiss, Switzerland).
Flow Cytometry
Trypsinized cells were re‐suspended with PBS containing 1 mM EDTA (PBS-E) followed by centrifugation at 1000 rpm for 5 min. After centrifugation, the cells were suspended with PBS‐E (0.5 mL) with ice‐cold 70% ethanol (4.5 mL) at 4°C at least for 12 h. The ethanol was washed out and cells were resuspended by PBS‐E followed by staining with propidium iodide (PI, SouthernBiotech, Birmingham, AL) for 1 h. The population of each DNA content of cells was counted by FACScan (Becton‐Dickinson, San Diego, CA) and further analyzed by Kaluza software (Beckman Coulter, Brea, CA).
EdU Incorporation Assay
EdU positive cells were stained by detecting fluorescence EdU signaling according to the manufacturer’s instruction (Invitrogen, Carlsbad, CA). An EdU incorporation assay was performed in asynchronized cells after JAR cells were treated with cordycepin for 24 h.
Microtubule Regrowth Assay
Microtubules were depolymerized by treating cells with nocodazole (5 μM) for 1 h. After depolymerization, cells were washed by PBS to remove the nocodazole and cultured in the fresh medium for 1 or 10 min for microtubule to regrow. Then, an immunofluorescence assay was performed for observing microtubule arrays.
Autophagic Flux Assay
An autophagic flux assay was performed according to the published method.26 Briefly, cells were treated with a lysosomal inhibitor to block the lysosomal degradation of LC3 II. Then, the conversion of LC3 I to II was evaluated by immunoblotting assay.
Statistical Analysis
Unpaired two-tailed t-tests were used for comparing the differences between the two groups. A p-value of less than 0.05 was considered as statistically significant. All experimental data are shown as the mean ± SD of three independent experiments, at least 100 cells in each individual group were counted.
Results
Cordycepin is an anti-cancer compound used for treating several cancers. To test its effect on choriocarcinoma, human gestational choriocarcinoma JAR cells were treated with cordycepin followed by counting cell numbers. Treatment of cells with cordycepin reduced the cell density dramatically in JAR cells (Figure 1A), and these results were quantified by counting cell numbers (Figure 1B). The half-maximal inhibitory concentration (IC50) values of cordycepin of JAR cells was about 150 μM. Thus, we treated cells with cordycepin at 150 μM concentration in the following experiments. Choriocarcinoma derives from chorionic villi cells, so to examine whether cordycepin affected the growth of normal cells, chorionic villi cells derived from human first-trimester placenta, HTR8 cell line, were used. Unlike JAR cells, treatment of cordycepin at different concentrations had no effect on HTR8 cell growth (Figure 1C), suggesting that cordycepin inhibited cell growth of choriocarcinoma but not normal chorionic villi cells.
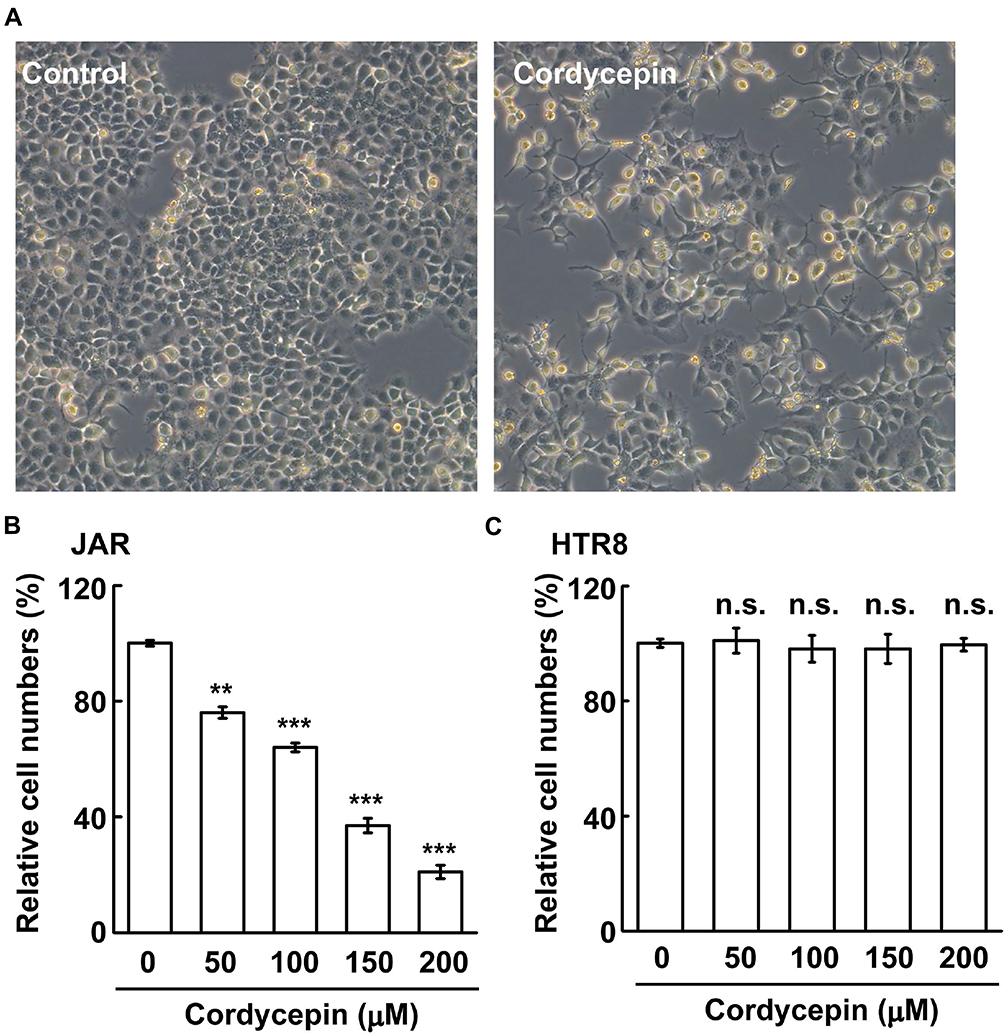 |
Figure 1 Cordycepin inhibits human gestational choriocarcinoma JAR cell growth. (A, B) Cordycepin inhibits JAR cell growth in a dose-dependent manner. (A) The bright‐field images are shown in the presence or absence of cordycepin treatment at a concentration of 150 μM. (B) Treatment of cordycepin at different concentrations for 24 h reduces growth of JAR cells by counting cell numbers (N=3). (C) Cordycepin has no effect on HTR8 cell growth (N=3). **P < 0.01; ***P < 0.001. Abbreviation: n.s., no significance. |
The effect of cordycepin on cell growth was further analyzed by flow cytometry. Upon cordycepin treatment, the population of cells in the subG1 phase increased (Figure 2A–B), suggesting that cordycepin might induce apoptosis. Indeed, several apoptotic bodies were observed in cordycepin-treated cells (Figure 2C) and the abundance of cleaved-caspase 3 was increased (Figure 2D and E). Thus, cordycepin induces cellular apoptosis. The population of cells at S phase was reduced upon cordycepin treatment (Figure 2A and B); we then checked the ability of cells to enter S phase by EdU incorporation assay. As shown in Figure 3A and B, the population of EdU positive cells was reduced dramatically when JAR cells were treated with cordycepin (Figure 3A and B), suggesting that cordycepin inhibited S phase entry. To further confirm this, the levels of cyclin A and cyclin E and the activation of CDK2 were examined by immunoblotting assay. Upon cordycepin treatment, the abundance of cyclin E was not affected (Figure 3C and D). However, the expression of cyclin A was reduced (Figure 3C and E); and the activation of CDK2 was also inhibited (Figure 3F and G). Collectively, cordycepin inhibits S phase entry by reducing cyclin A expression and CDK2 activation.
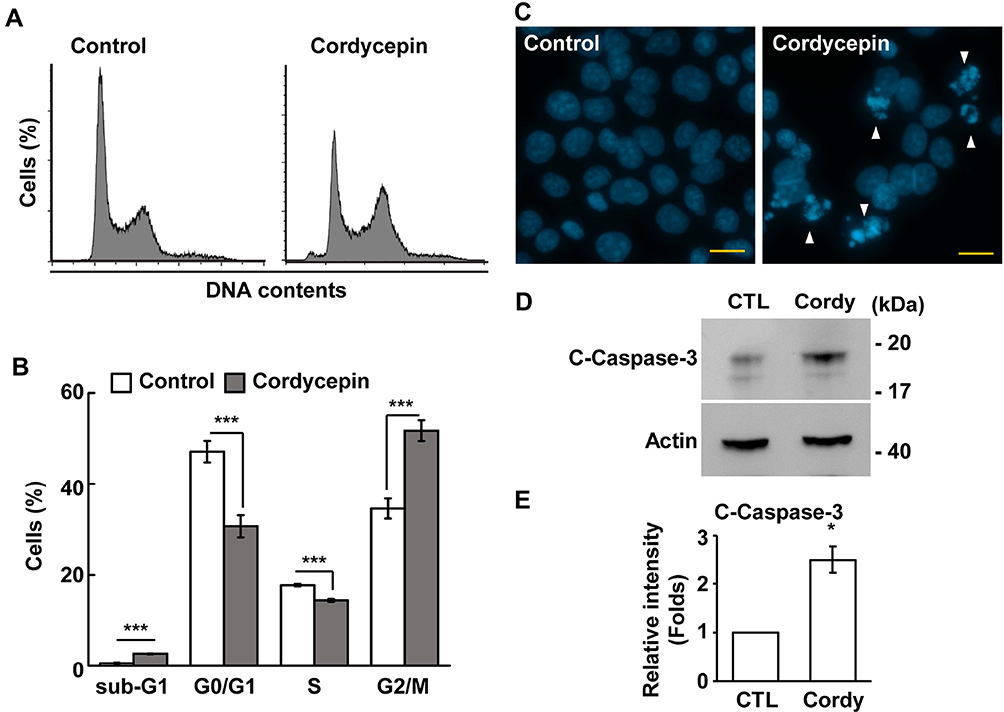 |
Figure 2 Cordycepin treatment leads to reduced S phase entry and apoptosis. (A, B) Cordycepin disturbs cell cycle progression. (A) Cell cycle profiles are analyzed by flow cytometry in the presence or absence of cordycepin treatment. (B) Quantification of % of cells at different cell cycle phases in (A) (N=3). ***P < 0.001. (C, D) The population of apoptotic cells is increase in cordycepin-treated JAR cells. (C) Apoptotic bodies (as shown in white arrowhead) are observed by DAPI staining in cordycepin-treated cells. (D) Cordycepin increases the expression of cleaved‐caspase 3 in JAR cells. Whole-cell extracts of cordycepin‐treated JAR cells are analyzed by Western blotting with antibodies against cleaved‐caspase 3 (c-caspase 3) and actin. (E) Quantification results of relative intensity (folds) of cleaved-caspase 3 in (D) *P < 0.05. |
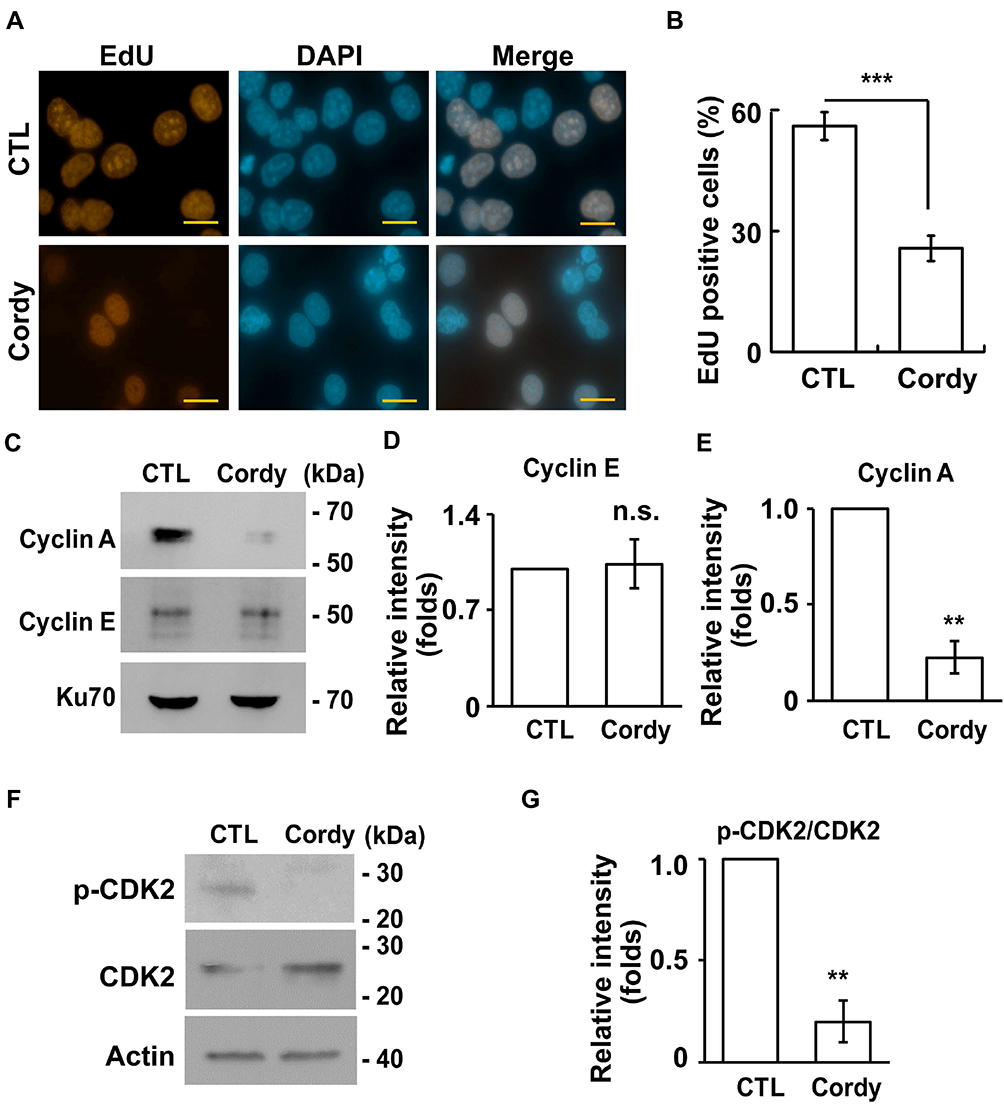 |
Figure 3 Cordycepin inhibits S phase entry. (A, B) Cordycepin treatment leads to reduced S phase entry. (A) EdU incorporation are reduced in cordycepin‐treated JAR cells. Immunostaining of EdU (red) and DAPI (blue) in scramble control (CTL) or cordycepin (Cordy)‐treated JAR cells. (B) Quantitation of EdU incorporation in (A) (N=3). These results are mean ± SD from three independent experiments; more than 1000 cells were counted in each individual group. ***P < 0.001. (C–E) Cordycepin reduces cyclin A expression. (C) Whole-cell extracts of cordycepin‐treated JAR cells are analyzed by immunoblotting assay with antibodies against cyclin A, cyclin E and Ku70 (loading control). (D, E) Quantification results of relative intensity of cyclin E (D) and cyclin A (E) in (C). **P<0.01. (F, G) Cordycepin reduces CDK2 activation. (F) Whole-cell extracts of cordycepin‐treated JAR cells are analyzed by immunoblotting assay with antibodies against phosphorylated CDK2 at Thr161 (p-CDK2), CDK2 and actin (loading control). (G) Quantification results of relative intensity of p-CDK2 to CDK2 in (F). **P<0.01. Abbreviation: n.s., no significance. |
Then, the G2/M transition was examined. The mitotic index was reduced in cordycepin-treated JAR cells, suggesting that cordycepin reduced M phase entry (Figure 4A). Next, the mitotic spindles were further examined. Normally, the mitotic spindle aligns the duplicated chromosomes in the middle of the cells (Figure 4B, upper panel). However, abnormal spindles with misaligned chromosomes were observed in cordycepin-treated JAR cells (Figure 4B, lower panel). The abnormal spindles might be caused by multiple mitotic spindle poles,26 so we further examined the spindle poles by γ-tubulin staining. Two mitotic spindle poles appeared at the opposite sites of the well-aligned chromosomes. However, multiple mitotic spindle poles with misaligned chromosomes were observed in cordycepin-treated cells (Figure 4C and D), suggesting cordycepin treatment induced multiple mitotic spindle poles. Defective mitotic segregation might lead to cytokinetic failure as shown by cells with dinuclei.27 The population of cells with dinuclei increased in cordycepin-treated JAR cells (Figure 4E and F). Thus, cordycepin treatment leads to disorganized mitotic apparatus.
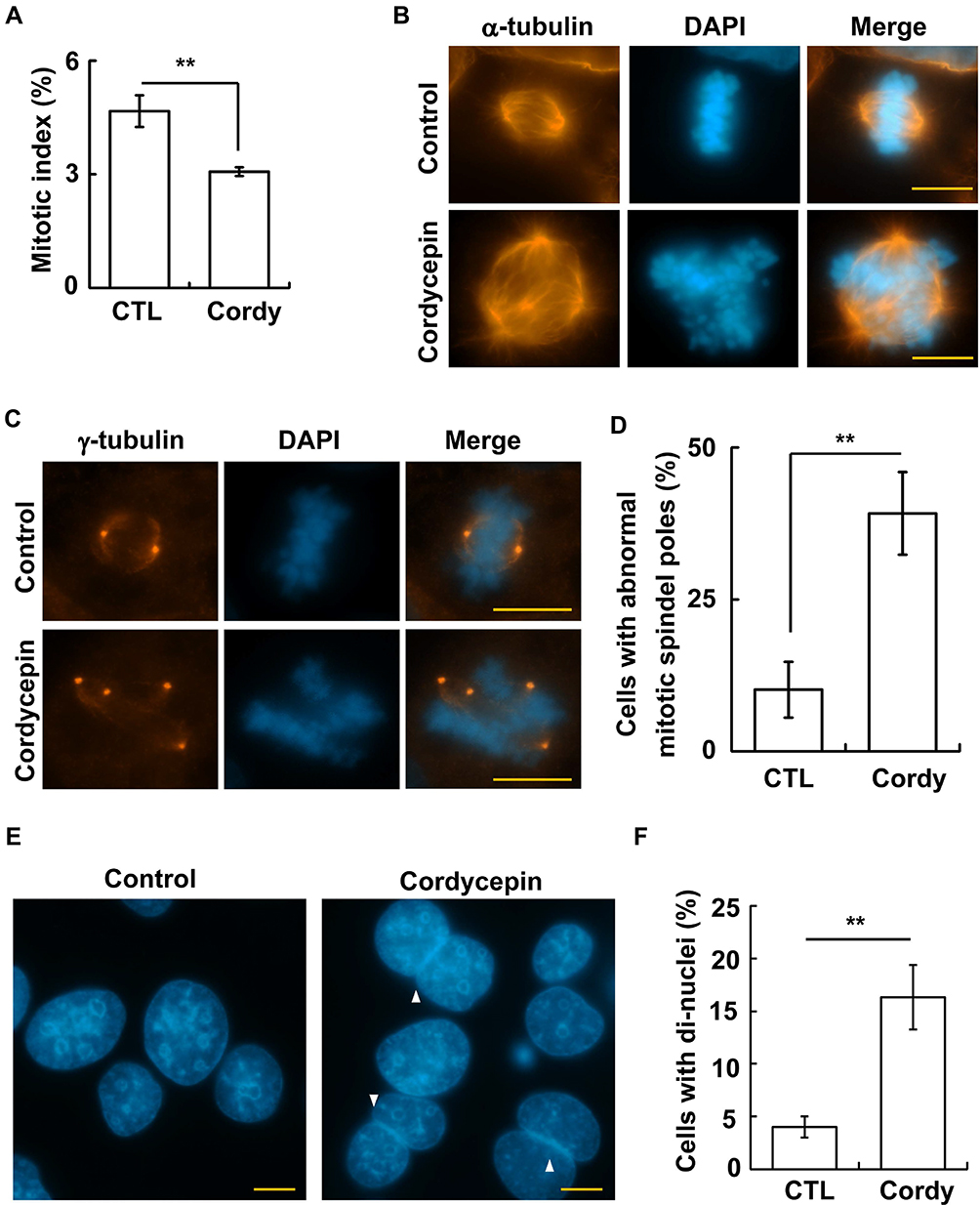 |
Figure 4 Cordycepin treatment leads to aberrant mitosis. (A) Cordycepin treatment reduces mitotic entry. Quantification of mitotic index in the absence (CTL) or presence (Cordy) of cordycepin (N=3). (B–D) Cordycepin treatment induces aberrant mitotic apparatus in JAR cells. (B, C) Aberrant mitotic spindles (B, staining of α-tubulin) and amplified mitotic spindle poles (C, staining of γ-tubulin) are observed in cordycepin-treated cells. Nuclei are stained with DAPI: blue. Scale bar: 10 µm. (D) Quantitation of results of cells with aberrant mitotic spindle poles in (C) (N=3). (E, F) Cordycepin treatment leads to cytokinetic failure. (E) DAPI staining shows cell with di-nuclei (arrowhead). (F) Quantification results of (E) (N=3). **P < 0.01. |
Multiple mitotic spindle poles might result from centrosome amplification during interphase;28 thus, centrosome copy numbers were examined upon cordycepin treatment. Normally, cells contain one (before duplication) or two (after duplication) centrosomes. In the presence of cordycepin, centrosome amplification (cells with more than two centrosomes) was observed (Figure 5A and B), suggesting that centrosome homeostasis was disrupted. Downregulation of cyclin F leads to centrosome amplification.29 Cordycepin treatment downregulated the level of cyclin F, supporting the suggestion that cordycepin disrupted centrosome homeostasis (Figure 5C and D). The centrosome is known as a microtubule organizing center; we thus examined the ability of microtubule nucleation in cordycepin-treated cells by using a microtubule regrowth assay.30 After the microtubule regrew for 1 min, microtubules nucleated around the centrosome in both control and cordycepin-treated cells (Figure 5E, 1 min). However, upon cordycepin treatment, the density of emanated microtubules was stronger when compared with that of in control cells. After the microtubule regrew for 10 min, well-organized microtubule arrays were observed in control cells (Figure 5E, 10 min). However, emanated microtubule arrays were bundled and did not spread out clearly in the cytoplasm of cordycepin-treated cells, suggesting that treatment of cordycepin disrupts microtubule-organizing arrays.
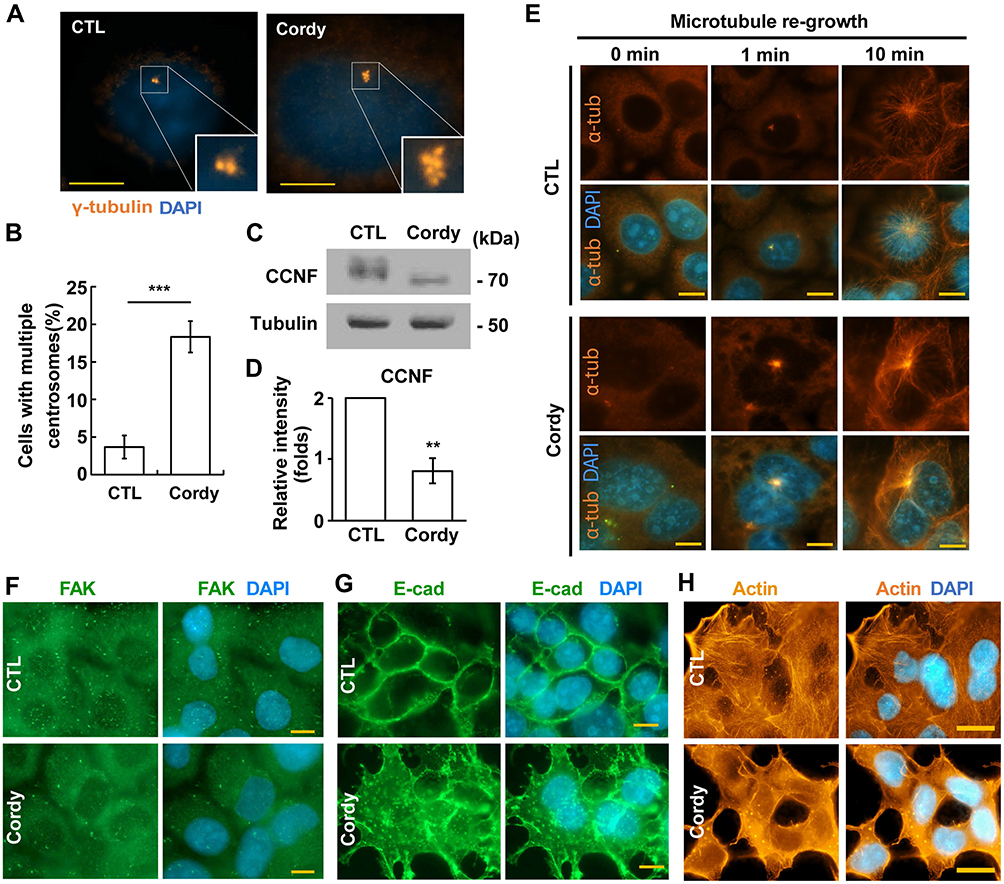 |
Figure 5 Cordycepin treatment disrupts centrosome homeostasis. (A–D) Cordycepin induces centrosome amplification by downregulating cyclin F. (A) Centrosome copy numbers are examined by immunostaining with antibody against γ-tubulin. (B) Quantification results of cells with multiple centrosomes (more than two centrosomes) in (A) (N=3). ***P < 0.001. (C) Cyclin F is downregulated by cordycepin treatment. Extracts of control or cordycepin-treated cells are analyzed by immunoblotting with antibodies against cyclin F (CCNF) and tubulin. (D) Quantification results of relative intensity of CCNF in (C) **P < 0.01. (E) Cordycepin disturbs microtubule organization. Microtubules are depolymerized followed by culturing in drug-free medium for microtubule to regrow for 0, 1, and 10 min in the absence (CTL, upper panel) or presence of cordycepin (Cordy, lower panel). Microtubules are shown by immunostaining with antibody against α-tubulin (α-tub). (F–H) Cordycepin disrupts cytoskeleton-related structures. Cytoskeleton-related structures including FAK (F), E-cadherin (G), and actin (H) are examined by immunostaining in the absence of presence of cordycepin. Nuclei are stained with DAPI: blue. Scale bar: 10 µm. Abbreviations: CTL, control; Cordy, cordycepin. |
Microtubule arrays maintain focal adhesion organization and alignment of actin stress fibers.31 We then examined the effect of cordycepin on focal adhesion organization and alignment of stress fibers. Puncta of focal adhesion kinase (FAK), the key regulator of adhesion,32 were clearly observed (Figure 5F, upper panels) and the E-cadherin was well organized in the peripheral part of the cell boundary (Figure 5G, upper panels). In addition, the actin stress fibers were well aligned (Figure 5H, upper panels). However, when JAR cells were treated with cordycepin, FAK puncta apparently reduced (Figure 5F, lower panels). In addition, reduced peripheral E-cadherin and scattered E-cadherin in the cytoplasm were also observed (Figure 5F and G, lower panels). Moreover, the actin stress fiber became blurry (Figure 5H, lower panels). These data suggest that focal adhesion organization and alignment of actin stress fibers are disrupted in cordycepin‐treated JAR cells.
Next, the underlying molecular signaling by which cordycepin induced centrosome amplification was examined. DNA damage responses induce centrosome amplification;33 we thus checked whether cordycepin induced DNA damage in JAR cells. Upon cordycepin treatment, level of γ-H2AX, a marker of DNA damage, increased, suggesting that cordycepin induced DNA damage (Figure 6A). Then, the DNA damage response was examined by checking members of the phosphatidylinositol 3-kinase-related kinase superfamily, including ATM (ataxia telangiectasia, mutated), ATR (ataxia telangiectasia, mutated, and Rad3-related) and DNA-PK (DNA-dependent protein kinase).34 Upon cordycepin treatment, levels of phosphorylated ATM and DNA-PK catalytic subunit (DNA-PKcs) were increased (Figure 6B–D). However, cordycepin had no effect on ATR activation (Figure 6B), suggesting that ATM and DNA-PK are activated by cordycepin treatment in JAR cells. Next, we tested whether ATM and DNA-PK activation participated in inducing centrosome amplification. Inhibition of ATM had no effect on cordycepin-induced centrosome amplification (Figure 6E). However, treatment of vanillin, the DNA-PK specific inhibitor, reduced cordycepin-induced centrosome amplification (Figure 6F), suggesting that cordycepin induces centrosome amplification via DNA-PK activation. Then, the downstream effectors were examined. Cordycepin had no effect on Chk1 activation but slightly inhibited Chk2 activation (Figure 6G and H). However, the level of phosphorylated ERK increased when JAR cells were treated with cordycepin (Figure 6I and J). Inhibition of ERK activation by treating cells with ERK specific inhibitor U0126 reduced cordycepin-induced centrosome amplification (Figure 6K), suggesting that activated ERK induces centrosome amplification upon cordycepin treatment. Collectively, activation of DNA-PK and ERK induces centrosome amplification in cordycepin-treated JAR cells.
 |
Figure 6 Cordycepin induces centrosome amplification via DNA-PK and ERK signaling. (A) Cordycepin induces DNA damage. DNA damage is shown by immunostaining with antibody against γ-H2AX. Nuclei are stained with DAPI: blue. Scale bar: 10 µm. (B–F) DNA-PK induces centrosome amplification upon cordycepin treatment. (B) Extracts of cordycepin-treated JAR cells are analyzed by immunoblotting with antibodies against phosphorylated ATM (p-ATM), ATM, phosphorylated ATR (p-ATR), ATR, phosphorylated DNA-PK catalytic subunit (p-PKcs), DNA-PKcs (PKcs), and Ku70 (as loading control). (C, D) Quantification results of relative intensity of p-ATM to ATM (C) and p-PKcs to PKcs (D) in (B). (E) Inhibition of ATM has no effect on cordycepin-induced centrosome amplification. Quantification results of % of cells with multiple centrosomes in the absence of presence of ATM inhibitor (ATMi) (N=3). (F) Inhibition of DNA-PK reduces centrosome amplification in cordycepin-treated cells. Quantification results of % of cells with multiple centrosomes in the absence of presence of DNA-PK inhibitor (Vanillin, Vani), (N=3). (G–K) Activated ERK induces centrosome amplification upon cordycepin treatment. (G) Extracts of cordycepin-treated JAR cells are analyzed by immunoblotting with antibodies against phosphorylated Chk2 (p-Chk2), Chk2, phosphorylated Chk1 (p-Chk1), Chk1, and tubulin (loading control). (H) Quantification results of relative intensity of p-Chk2 to Chk2 in (G). (I) Extracts of cordycepin-treated JAR cells are analyzed by immunoblotting with antibodies against phosphorylated ERK (p-ERK), ERK, and tubulin (loading control). (J) Quantification results of relative intensity of p-ERK to ERK in (I). (K) Inhibition of ERK reduces cordycepin-induced centrosome amplification. Quantification results of % of cells with multiple centrosomes in the absence of presence of ERK inhibitor (ERKi, U0126) (N=3). *P<0.05, **P < 0.01, ***P < 0.001. Abbreviation: n.s, no significance. |
DNA damage activates autophagy.26 Cordycepin treatment induced DNA damage (Figure 6A). Thus, we tested whether cordycepin treatment affected autophagy. LC3 signal was barely detectable by immunofluorescence staining in JAR cells. However, upon cordycepin treatment, the LC3 signal increased dramatically (Figure 7A), suggesting that cordycepin treatment affects autophagy. Accumulation of LC3 might result from accelerating autophagic flux or blocking autophagic degradation. Thus, autophagy was checked by examining the conversion of LC3 I to LC3 II. Compared with control cells, upon cordycepin treatment, LC3 I was slightly reduced and the LC3 II was slightly increased, suggesting cordycepin accelerates autophagy (Figure 7B and C). To further confirm this, an autophagic flux assay was performed. The LC3 II to LC3 I ratio was increased significantly in cordycepin-treated cells when compared with that of in control cells (Figure 7D and E). Thus, cordycepin facilitates autophagic flux. Then, the role of cordycepin-induced autophagy was examined. Autophagy might facilitate cell death or prevent apoptosis upon chemotherapeutic drug treatment.35 Thus, the effect of cordycepin-induced autophagy on JAR cell survival was examined. Cordycepin reduced JAR cell viability; however, when cells were co-treated with cordycepin and autophagy inhibitor chloroquine, viable cells increased, suggesting cordycepin-induced autophagy inhibits cell growth (Figure 7F).
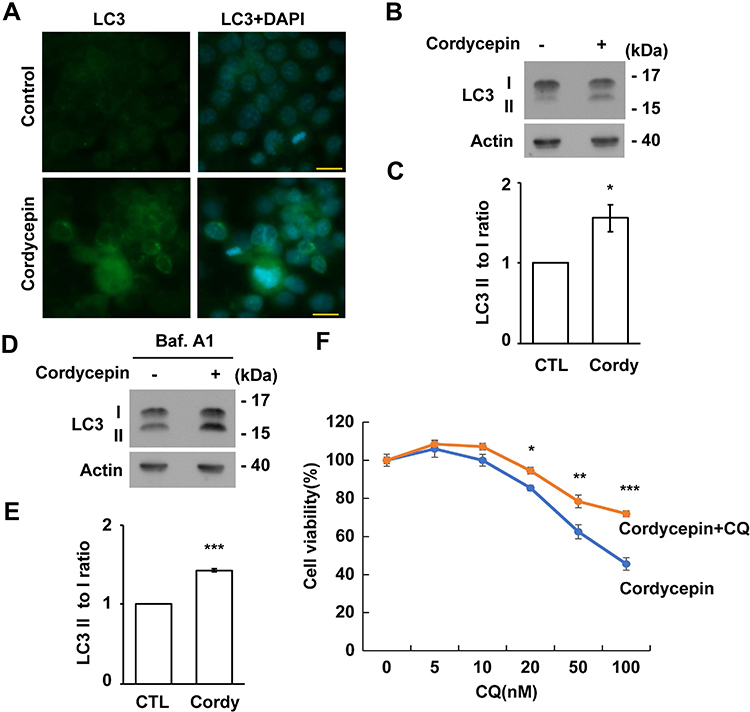 |
Figure 7 Cordycepin-induced autophagy reduces cell viability. (A–E) Cordycepin induces autophagy. (A) Autophagy is induced by cordycepin as shown by immunostaining with antibodies against LC3. Nuclei are stained with DAPI: blue. Scale bar: 10 µm. (B) Extracts of cordycepin-treated cells are analyzed with antibodies against LC3 and actin. (C) Quantification results of LC3 II to I ratio in (B). (D) Extracts of cordycepin-treated cells in the presence of bafilomycin A1 (Baf.A1) are analyzed with antibodies against LC3 and actin. (E) Quantification results of LC3 II to I ratio in (D). (F) Chloroquine (CQ) accelerates cell viability in cordycepin‐treated JAR cells. Quantitation results of relative cell viability of control or cordycepin‐treated cells in the presence of CQ at different concentrations. Each cordycepin‐treated sample is normalized to the cells without cordycepin treatment (defined as 100%) in either control or CQ‐treated cells independently (N=3). *P < 0.05; **P < 0.01; ***P < 0.001. |
Discussion
Cordycepin has been considered as an active compound in reducing cancer cell growth. The molecular mechanism by which cordycepin inhibits cancer cell growth including activation of apoptosis and cell cycle arrest and prevention of chemoresistance in several cancers, such as gastric, liver, kidney, bladder, and testicular cancer cells.5,7,11,20 In our study, we demonstrated that cordycepin induced apoptosis and blocked cell cycle progression in human gestational choriocarcinoma JAR cells but did not affect normal placental cell growth. Interestingly, centrosome amplification was also induced. This is the first study that shows the effect of cordycepin on the centrosome. The centrosome is known for its function in nucleating microtubule networks for maintaining cell polarity and shapes. Treatment with cordycepin induced centrosome amplification via DNA-PK and ERK signaling, thus leading to aberrant mitotic spindles and defective microtubule arrays (Figure 8). These events might eventually, at least in part, contribute to cell death.36 In addition, we also showed that cordycepin-activated autophagy reduced JAR cell viability. Thus, in addition to intrinsic nuclear signals, centrosome-mediated cytoplasmic events also contribute to the anti-cancer effect of cordycepin.
 |
Figure 8 A working model for the effect of cordycepin on human gestational choriocarcinoma. Upon cordycepin treatment, human gestational choriocarcinoma suffers from DNA damage, thus, activating DNA-PK/ERK signaling for inducing centrosome amplification. These amplified centrosomes lead to formation of aberrant mitotic apparatus during M phase and disrupt microtubule arrays. In addition, cordycepin also triggers autophagy to reduce cell viability. |
So far, no cordycepin clinical trials have been performed. When we compared the concentration of cordycepin we used with other studies, the concentration used in treating human gestational choriocarcinoma (150 μM) in our study is less than that used for treating other cancers (240 and 320 μM in lung cancer;1 135 and 270 μM in colorectal cancer37). Interestingly, by using an in vivo mice xenograft model, cordycepin inhibits esophageal cancer growth and prolongs survival by treating mice with cordycepin at 5 or 10 mg/kg concentration without causing toxicity effects.38 Meanwhile, n in vitro cancer cell model was also established and it was found that IC50 of cordycepin inhibited the human esophageal squamous carcinoma cell viability ranges from 240 to 320 μM (depending on different cell lines). The IC50 of cordycepin in treating human esophageal squamous carcinoma is higher than that used in treating human gestational choriocarcinoma. Importantly, the concentration of cordycepin they used in treating esophageal squamous carcinoma in a xenograft mice model prevented tumorigenesis efficiently without side effects. Thus, we speculate cordycepin at 5 or 10 mg/kg concentration might be suitable for treating gestation choriocarcinoma in a xenograft mice model. However, this hypothesis should be tested in the future.
DNA damage response induces centrosome amplification in cancers.33 In osteosarcoma, hydroxyurea treatment (prolonged replication stress) induces centrosome amplification via DNA-PK-Chk2 signaling;25 and irradiation-activated Chk1 facilities centrosome over-duplication.39 These data suggest different DNA damage stresses induce centrosome amplification via different signaling pathways. Here we showed that cordycepin-induced DNA damage triggers DNA-PK activation for centrosome amplification in JAR cells. The canonical downstream effectors of DNA-PK are Chk1 and Chk2;25,40 however, cordycepin treatment activated neither Chk1 nor Chk2. A previous study showed that, under DNA damage, ATM and DNA-PK activate NF-κB via inducing ERK signaling.41 Indeed, ERK activation was observed when JAR cells were treated with cordycepin, and inhibition of EKR activation alleviated cordycepin-induced centrosome amplification. Thus, our study uncovers a novel function of the DNA-PK-ERK cascade for mediating centrosome copy numbers.
Here we showed that cordycepin inhibited human gestational choriocarcinoma JAR cell growth. In addition, centrosome homeostasis was disrupted, thus leading to defective mitotic spindles, aberrant chromosome alignment, and disorganized microtubule arrays. Furthermore, autophagy is activated by cordycepin treatment and inhibition of autophagy by chloroquine prevented cordycepin-induced cell death. Thus, our study reveals a potential therapeutic effect of cordycepin and uncovered the underlying molecular mechanism in treating human gestational choriocarcinoma.
Abbreviations
ATM, ataxia telangiectasia-mutated; ATR, ataxia telangiectasia and Rad3-related; CCNF, cyclin F; Chk1, checkpoint kinase 1; Chk2, checkpoint kinase 2; DAPI, 4ʹ,6-diamidino-2-phenylindole; DNA-PK, DNA-dependent protein kinase; DNA-PKcs, DNA-dependent protein kinase, catalytic subunit; ERK, extracellular signal-regulated kinases; FAK, focal adhesion kinase.
Acknowledgments
- We are grateful for the support from the Core Research Laboratory, College of Medicine, National Cheng Kung University.
- This study was supported by grants from An Nan Hospital, China Medical University (ANHRF108-18) to Shih-Wei Tsai.
- This study was supported by grants from the Ministry of Science and Technology (MOST106-2320-B-006-056-MY3 and MOST109-2320-B-006-042-MY3) to Chia-Yih Wang.
Disclosure
The authors declare no competing financial interests in this work. Competing interests: Non-financial competing interests.
References
1. Frijstein MM, Lok CAR, Short D, et al. The results of treatment with high-dose chemotherapy and peripheral blood stem cell support for gestational trophoblastic neoplasia. Eur J Cancer. 2019;109:162–171. doi:10.1016/j.ejca.2018.12.033
2. Lee JB, Adrower C, Qin C, Fischer PM, de Moor CH, Gershkovich P. Development of cordycepin formulations for preclinical and clinical studies. AAPS PharmSciTech. 2017;8(8):3219–3226. doi:10.1208/s12249-017-0795-0
3. Ahn YJ, Park SJ, Lee SG, Shin SC, Choi DH. Cordycepin: selective growth inhibitor derived from liquid culture of Cordyceps militaris against Clostridium spp. J Agric Food Chem. 2000;48(7):2744–2748. doi:10.1021/jf990862n
4. Li Y, Li K, Mao L, et al. Cordycepin inhibits LPS-induced inflammatory and matrix degradation in the intervertebral disc. PeerJ. 2016;4:e1992. doi:10.7717/peerj.1992
5. Chen YC, Chen YH, Pan BS, Chang MM, Huang BM. Functional study of cordyceps sinensis and cordycepin in male reproduction: a review. J Food Drug Anal. 2017;25(1):197–205. doi:10.1016/j.jfda.2016.10.020
6. Leu SF, Chien CH, Tseng CY, Kuo YM, Huang BM. The in vivo effect of cordyceps sinensis mycelium on plasma corticosterone level in male mouse. Biol Pharm Bull. 2005;28(9):1722–1725. doi:10.1248/bpb.28.1722
7. Pan BS, Wang YK, Lai MS, Mu YF, Huang BM. Cordycepin induced MA-10 mouse Leydig tumor cell apoptosis by regulating p38 MAPKs and PI3K/AKT signaling pathways. Sci Rep. 2015;5:13372. doi:10.1038/srep13372
8. Zeng Y, Lian S, Li D, et al. Anti-hepatocarcinoma effect of cordycepin against NDEA-induced hepatocellular carcinomas via the PI3K/Akt/mTOR and Nrf2/HO-1/NF-kappaB pathway in mice. Biomed Pharmacother. 2017;95:1868–1875. doi:10.1016/j.biopha.2017.09.069
9. Hwang JH, Joo JC, Kim DJ, et al. Cordycepin promotes apoptosis by modulating the ERK-JNK signaling pathway via DUSP5 in renal cancer cells. Am J Cancer Res. 2016;6(8):1758–1771.
10. Wang XA, Xiang SS, Li HF, et al. Cordycepin induces S phase arrest and apoptosis in human gallbladder cancer cells. Molecules. 2014;19(8):11350–11365. doi:10.3390/molecules190811350
11. Liao Y, Ling J, Zhang G, et al. Cordycepin induces cell cycle arrest and apoptosis by inducing DNA damage and up-regulation of p53 in leukemia cells. Cell Cycle. 2015;14(5):761–771. doi:10.1080/15384101.2014.1000097
12. Tsai YJ, Lin LC, Tsai TH. Pharmacokinetics of adenosine and cordycepin, a bioactive constituent of cordyceps sinensis in rat. J Agric Food Chem. 2010;58(8):4638–4643. doi:10.1021/jf100269g
13. Kristensen AR, Schandorff S, Hoyer-Hansen M, et al. Ordered organelle degradation during starvation-induced autophagy. Mol Cell Proteomics. 2008;7(12):2419–2428. doi:10.1074/mcp.M800184-MCP200
14. Bae H, Guan JL. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol Cancer Res. 2011;9(9):1232–1241. doi:10.1158/1541-7786.MCR-11-0098
15. Lee SW, Kim H-K, Lee N-H, et al. The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells. Cancer Lett. 2015;360(2):195–204. doi:10.1016/j.canlet.2015.02.012
16. Sehgal AR, Konig H, Johnson DE, et al. You eat what you are: autophagy inhibition as a therapeutic strategy in leukemia. Leukemia. 2015;29(3):517–525. doi:10.1038/leu.2014.349
17. Xie BS, Zhao HC, Yao SK, et al. Autophagy inhibition enhances etoposide-induced cell death in human hepatoma G2 cells. Int J Mol Med. 2011;27(4):599–606. doi:10.3892/ijmm.2011.607
18. Park JM, Jung CH, Seo M, et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy. 2016;12(3):547–564. doi:10.1080/15548627.2016.1140293
19. Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol. 2008;445:77–88. doi:10.1007/978-1-59745-157-4_4
20. Li Y, Li R, Zhu S, et al. Cordycepin induces apoptosis and autophagy in human neuroblastoma SK-N-SH and BE(2)-M17 cells. Oncol Lett. 2015;9(6):2541–2547. doi:10.3892/ol.2015.3066
21. Lee HH, Kim SO, Kim GY, et al. Involvement of autophagy in cordycepin-induced apoptosis in human prostate carcinoma LNCaP cells. Environ Toxicol Pharmacol. 2014;38(1):239–250. doi:10.1016/j.etap.2014.06.003
22. Bettencourt-Dias M, Glover DM. Centrosome biogenesis and function: centrosomics brings new understanding. Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol. 2007;8(6):451–463. doi:10.1038/nrm2180
23. Schliwa M, Euteneuer U, Graf R, Ueda M. Centrosomes, microtubules and cell migration. Biochem Soc Symp. 1999;65:223–231.
24. Hinchcliffe EH, Sluder G. “It takes two to tango”: understanding how centrosome duplication is regulated throughout the cell cycle. Genes Dev. 2001;15(10):1167–1181. doi:10.1101/gad.894001
25. Wang CY, Huang EY, Huang SC, Chung BC. DNA-PK/Chk2 induces centrosome amplification during prolonged replication stress. Oncogene. 2015;34(10):1263–1269. doi:10.1038/onc.2014.74
26. Lien WC, Chen TY, Sheu SY, et al. 7-hydroxy-staurosporine, UCN-01, induces DNA damage response, and autophagy in human osteosarcoma U2-OS cells. J Cell Biochem. 2018;119(6):4729–4741. doi:10.1002/jcb.26652
27. Lai PY, Wang CY, Chen WY, et al. Steroidogenic factor 1 (NR5A1) resides in centrosomes and maintains genomic stability by controlling centrosome homeostasis. Cell Death Differ. 2011;18(12):1836–1844. doi:10.1038/cdd.2011.54
28. Chen TY, Syu JS, Lin TC, Cheng HL, Lu FL, Wang CY. Chloroquine alleviates etoposide-induced centrosome amplification by inhibiting CDK2 in adrenocortical tumor cells. Oncogenesis. 2015;4:e180. doi:10.1038/oncsis.2015.37
29. D’Angiolella V, Donato V, Vijayakumar S, et al. SCFCyclin F controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature. 2010;466(7302):138–142. doi:10.1038/nature09140
30. Wang CY, Hong YH, Syu JS, et al. LRWD1 regulates microtubule nucleation and proper cell cycle progression in the human testicular embryonic carcinoma cells. J Cell Biochem. 2018;119(1):314–326. doi:10.1002/jcb.26180
31. Chen TY, Lien WC, Cheng HL, Kuan TS, Sheu SY, Wang CY. Chloroquine inhibits human retina pigmented epithelial cell growth and microtubule nucleation by downregulating p150(glued). J Cell Physiol. 2019;234(7):10445–10457. doi:10.1002/jcp.27712
32. Schaller MD. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci. 2010;123(Pt 7):1007–1013. doi:10.1242/jcs.045112
33. Dodson H, Bourke E, Jeffers LJ, et al. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J. 2004;23(19):3864–3873. doi:10.1038/sj.emboj.7600393
34. Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol. 2001;13(2):225–231. doi:10.1016/s0955-0674(00)00201-5
35. Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306(5698):990–995. doi:10.1126/science.1099993
36. Ishikawa F, Ushida K, Mori K, Shibanuma M. Loss of anchorage primarily induces non-apoptotic cell death in a human mammary epithelial cell line under atypical focal adhesion kinase signaling. Cell Death Dis. 2015;6:e1619. doi:10.1038/cddis.2014.583
37. Li SZ, Ren JW, Fei J, Zhang XD, Du RL. Cordycepin induces baxdependent apoptosis in colorectal cancer cells. Mol Med Rep. 2019;19(2):):901–908. doi:10.3892/mmr.2018.9717
38. Xu JC, Zhou XP, Wang XA, et al. Cordycepin induces apoptosis and G2/M phase arrest through the ERK pathways in esophageal cancer cells. J Cancer. 2019;10(11):2415–2424. doi:10.7150/jca.32071
39. Bourke E, Dodson H, Merdes A, et al. DNA damage induces Chk1-dependent centrosome amplification. EMBO Rep. 2007;8(6):603–609. doi:10.1038/sj.embor.7400962
40. Lin YF, Shih HY, Shang Z, Matsunaga S, Chen BP. DNA-PKcs is required to maintain stability of Chk1 and claspin for optimal replication stress response. Nucleic Acids Res. 2014;42(7):4463–4473. doi:10.1093/nar/gku116
41. Panta GR, Kaur S, Cavin LG, et al. ATM and the catalytic subunit of DNA-dependent protein kinase activate NF-kappaB through a common MEK/extracellular signal-regulated kinase/p90(rsk) signaling pathway in response to distinct forms of DNA damage. Mol Cell Biol. 2004;24(5):1823–1835. doi:10.1128/mcb.24.5.1823-1835.2004
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.