")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Combined treatment with olmesartan medoxomil and amlodipine besylate attenuates atherosclerotic lesion progression in a model of advanced atherosclerosis
Authors Sievers P, Uhlmann L, Korkmaz-Icöz S, Fastner C, Bea F, Blessing E, Katus HA, Preusch MR
Received 21 March 2015
Accepted for publication 30 April 2015
Published 29 July 2015 Volume 2015:9 Pages 3935—3942
DOI https://doi.org/10.2147/DDDT.S85203
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Philipp Sievers,1 Lorenz Uhlmann,2 Sevil Korkmaz-Icöz,3 Christian Fastner,1 Florian Bea,1 Erwin Blessing,1 Hugo A Katus,1 Michael R Preusch1
1Department of Internal Medicine III, 2Institute of Medical Biometry and Informatics, 3Department of Cardiac Surgery, University of Heidelberg, Heidelberg, Germany
Introduction: Besides their blood pressure-lowering effects, olmesartan medoxomil and amlodipine besylate exhibit additional anti-inflammatory mechanisms in atherosclerosic disease. Most of the studies investigating the effects of atherosclerosis focused on early atherosclerotic lesions, whereas lesions in human disease, at the time when medical treatment is started, are already well established. Therefore, we set up a model of advanced atherosclerosis and investigated the effects of olmesartan medoxomil, amlodipine besylate, and the combination of both on atherosclerotic lesion size and lesion composition.
Materials and methods: Olmesartan medoxomil (1 mg/kg/day), amlodipine besylate (1.5 mg/kg/day), and the combination of both was added to chow and was fed to apolipoprotein E-deficient (ApoE-/-) mice at 25 weeks of age. Mice were sacrificed after 25 weeks of drug administration and perfused with formalin. Innominate arteries were dissected out and paraffin embedded. Serial sections were generated, and lesion sizes and their composition – such as minimal thickness of the fibrous cap, size of the necrotic core, and presence of calcification – were analyzed. Electrophoretic mobility shift assays were used to detect DNA-binding activity of the transcription factor nuclear factor-kappa B (NF-κB) in aortic tissue.
Results: Treatment with the combination of olmesartan medoxomil and amlodipine besylate led to a significant reduction in atherosclerotic lesion size in ApoE-/- mice (olmesartan medoxomil/amlodipine besylate: 122,277±6,795 µm2, number [n]=14; versus control: 177,502±10,814 µm2, n=9; P<0.001). Treatment with amlodipine besylate (n=5) alone did not reach significance. However, a trend toward a decrease in lesion size in the amlodipine besylate-treated animals could be observed. In the histological analysis of atherosclerotic lesion composition, significantly thicker fibrous caps were found in treatment with amlodipine besylate (amlodipine: 5.12±0.26 µm, n=6; versus control: 3.98±0.18 µm, n=10; P<0.01). Furthermore, all sections revealed morphological signs of calcification, but no difference could be detected. Treatment with the combination of olmesartan medoxomil and amlodipine besylate showed no effect on lesion composition. Electrophoretic mobility shift assays of nuclear extracts demonstrated reduced activity of the transcription factor NF-κB when treated with olmesartan medoxomil, amlodipine besylate, or their combination, as compared to controls.
Conclusion: Combined treatment with olmesartan medoxomil and amlodipine besylate attenuated atherosclerotic lesion progression, possibly due to anti-inflammatory mechanisms. Our data support the hypothesis that even in advanced atherosclerosis anti-inflammatory treatment, using angiotensin II type 1 receptor blockers and calcium channel antagonists of the dihydropyridine type can attenuate atherosclerotic lesion progression.
Keywords: advanced atherosclerosis, AT1 receptor blocker, calcium channel antagonist, inflammation, NF-κB, ApoE
Introduction
Atherosclerosis is a progressive disease of the arterial wall and a leading cause of death worldwide.1–3 In our current understanding of the pathophysiology of atherosclerosis, the concept of inflammation plays a pivotal role and provides a common link between risk factors and the cellular and molecular alterations.2,4 In this concept, atherosclerosis is seen as a lipid-driven inflammatory disease, characterized by the accumulation of macrophage-derived foam cells in the arterial wall and accompanied by a cascade of proinflammatory cytokines and chemokines.4,5 Vascular inflammation contributes to the initiation, progression, and even complications of atherosclerotic lesions. Many of the inflammatory genes involved in the pathogenesis of atherosclerosis are induced by nuclear factor-kappa B (NF-κB), which acts as an important factor during atherogenesis.6 With increasing recognition of the role of inflammation in atherosclerosis, anti-inflammatory treatment strategies have become more important and provide new therapeutic options.7–9 Current clinical strategies against atherosclerosis still focus on the attenuation of risk factors like hypertension and hyperlipidemia, or the prevention of thrombembolic complications, but they do not directly address the inflammatory mechanisms of atheroprogression.9 In addition to the effects on hypertension, the widely used antihypertensive drug classes of angiotensin II type 1 (AT1) receptor blockers and calcium channel antagonists have shown additional anti-inflammatory properties. Previous studies suggest that these antihypertensive drugs exhibit atheroprotective effects independent of lowering blood pressure, leading to a reduction of atherosclerotic lesion progression.10–14 Furthermore, coadministration of AT1 receptor blockers and calcium channel antagonists have shown antiatherogenic effects.15
The apolipoprotein E-deficient (ApoE−/−) mouse model is well established and frequently used to study mechanisms of atherosclerosis.14,16 The majority of these experimental studies using hyperlipidemic mice have focused on early atherosclerotic processes, and so far, there have been only limited data relating to the effects on complex advanced lesions as they occur in human disease. However, approximately two-thirds of cardiovascular events, like myocardial infarction and stroke, are caused by rupture of a vulnerable atherosclerotic plaque, which underlines the enormous relevance of advanced stages of atherosclerosis.17
Despite suggestive evidence of the beneficial effect of AT1 receptor blockers and calcium channel antagonists in early stages, the role of the drugs in advanced atherosclerosis remains vague due to the lack of experimental validation. Here, we investigated the effects of the AT1 receptor blocker olmesartan medoxomil and the calcium channel antagonist amlodipine besylate on atherosclerotic progression and vascular inflammation using an ApoE−/− mouse model of advanced atherosclerosis.
Materials and methods
Animals and treatment
Twenty-five-week-old female ApoE−/−-deficient mice (number [n]=63) on a C57BL/6 background (Charles River Laboratories International, Inc., Sulzfeld, Germany) exhibiting advanced atherosclerotic lesions within the innominate artery were kept within the animal care facility of the University of Heidelberg (Heidelberg, Germany). Mice were randomized into four groups and were fed a chow supplemented with olmesartan medoxomil (1 mg/kg/day; n=15), amlodipine besylate (1.5 mg/kg/day; n=15), or their combination (n=17). Sixteen control mice received regular chow. We chose the dose of olmesartan medoxomil and amlodipine besylate in accordance with a study of Suzuki et al13 and Yoshii et al14 in which the authors demonstrated significant antiatherosclerotic properties of a single treatment with olmesartan medoxomil or amlodipine besylate without affecting systolic blood pressure. All animal procedures were approved by the Federal Animal Care and Use Committee of the Regierungspräsidium Karlsruhe, Germany.
The housing and care of animals and all of the procedures performed in the study were conducted in accordance with the guidelines and regulations of the Animal Care Committee of the University of Heidelberg.
Animal sacrifice and preparation of tissues
There was no significant loss of animals during treatment (n=1). After 25 weeks on the study diet, mice were heavily sedated (n=62; ketamine/xylazine), blood was collected from the inferior vena cava, and the animals were sacrificed by exsanguination. The animals were perfused via the left ventricle with 10 mL of phosphate buffered saline. The thoracic and abdominal aorta was removed for subsequent electrophoretic mobility shift assays (EMSAs). Followed by another perfusion of the heart with 4% buffered formalin for paraffin sections, the entire heart and the innominate artery from each animal were dissected out. The innominate arteries were embedded in paraffin and serially sectioned (5 μm). Every fifth section was stained with a modified Movat’s pentachrome stain.18
Determination of plasma lipid concentration
Plasma concentrations of total cholesterol, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, and triglycerides were determined enzymatically in heparinized plasma.
Evaluation of lesion size and lesion composition
The cross-sectional area of each Movat’s pentachrome-stained section was determined using computer-assisted morphometry (ImageJ, National Institutes of Health, Bethesda, MD, USA) and reported as the mean plaque area per animal (data expressed in μm2). Furthermore, characteristic features of atherosclerotic plaque morphology and composition were evaluated. These included minimal thickness of the fibrous cap (data given in μm), size of the necrotic core (given as the ratio mean size of the necrotic core to the mean lesion area, also measured by morphometry), and the presence of calcification (detected morphologically).
Preparation of nuclear extracts
Aortas were separated from connective tissue and homogenized in 400 μL of hypotonic buffer (10 mmol/L HEPES, pH 7.9; 10 mmol/L KCl; 0.1 mmol/L EDTA; 0.1 mmol/L EGTA; 2 mmol/L dithiothreitol [DTT]) supplemented with proteinase and phosphatase inhibitors (5 μg/mL E64; 1 mmol/L NaF; 0.2 mmol/L Na3VO4; and 0.5 mg/L Pefabloc), incubated on ice for 15 minutes, and afterward supplemented with 25 μL of 10% NP-40. The nuclei were recovered by centrifugation (14,000 rpm, 1 minute, 4°C). The nuclear pellets were resuspended in 50 μL of buffer (20 mmol/L HEPES, pH 7.9; 0.4 mol/L NaCl; 1 mmol/L EDTA; 1 mmol/L EGTA; 2 mmol/L DTT supplemented with 5 μg/mL E64; 1 mmol/L NaF; 0.2 mmol/L Na3VO4; and 0.5 mg/mL Pefabloc). After centrifugation (14,000 rpm, 5 minutes, 4°C), the nuclear protein was collected and stored at −80°C until used.
Electrophoretic mobility shift assay
Protein concentrations were measured by the Bradford method. Nuclear extracts (10 μg of protein in each assay) were incubated with labeled oligonucleotide probes. The sequence of the oligonucleotide used in the present study was as follows: NF-κB, 5′-AGTTGAGGGGACTTTCCCAGGC-3′. The oligonucleotide (1.75 pmol/μL) was labeled with ([gamma]32P)-adenosine triphosphate using T4 polynucleotide kinase. Specific activities used in each assay were approximately 10,000 cpm. A 100-fold excess of unlabeled oligonucleotides was used for cold inhibition. Binding reactions were resolved on 4% native polyacrylamide gel and exposed to X-ray film for 12–24 hours. Gels were analyzed using densitometric analysis (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
All data were expressed as the mean ± standard error of the mean. Differences between means in body weight and plasma lipid profiles were tested with the two-tailed unpaired Student’s t-test. For the analysis, plaque morphometry (lesion size) and morphology (area of necrotic core, thickness of fibrous cap), as well as densitometric analysis of the EMSA groups were compared using the Mann–Whitney U-test, as the data could not be assumed to be normally distributed. P-values <0.05 were considered statistically significant. As this was an exploratory analysis, no adjustments for multiple testing were performed. Statistical analyses and graphs were performed using GraphPad Prism version 6.00 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Plasma lipid levels and body weight
At the time of sacrifice, animals showed no significant difference in body weight or lipid profile between the groups (Table 1).
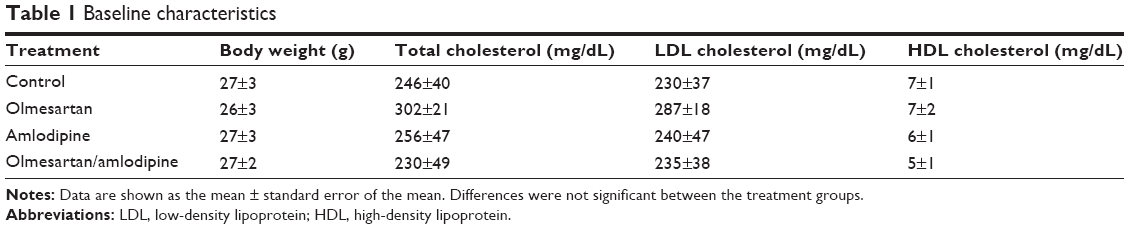 | Table 1 Baseline characteristics |
Mean lesion area within the innominate artery
Morphometric analysis of Movat’s pentachrome staining revealed a markedly significant reduction of atherosclerotic lesion size in the group receiving the combination of olmesartan medoxomil and amlodipine besylate after 25 weeks on the study diet. Mean lesion area averaged 122,277±6,795 μm2 (n=14) in the olmesartan medoxomil/amlodipine besylate group compared with 177,502±10,814 μm2 (n=9) in the control group (P<0.001; Figure 1).
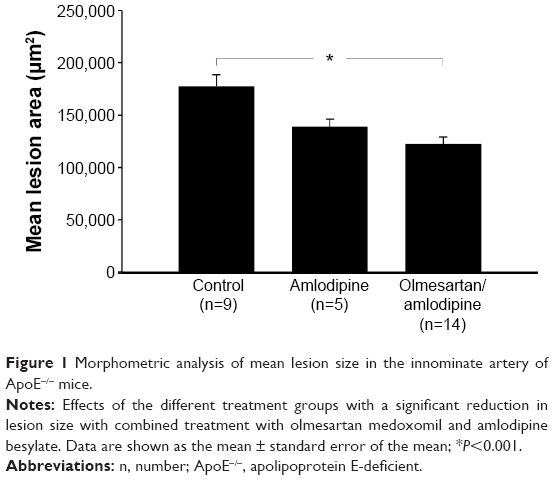 | Figure 1 Morphometric analysis of mean lesion size in the innominate artery of ApoE−/− mice. |
Treatment with amlodipine besylate (n=5) alone did not reach levels of significance (Figure 1). However, a trend toward a decrease in lesion size in the amlodipine besylate-treated animals could be observed (amlodipine: 138,885±7,118 μm2, n=5; versus control: 177,502±10,814 μm2, n=9; P=0.06). Due to the small number of high-quality slides in the olmesartan medoxomil group (n=2), we could not perform a statistical analysis.
Lesion composition
Evaluation of plaque morphology showed significantly thicker fibrous caps within the amlodipine besylate group compared to controls (amlodipine: 5.12±0.26 μm, n=6; versus control: 3.98±0.18 μm, n=10; P=0.006; Figure 2A). Furthermore, all sections revealed morphological signs of calcification, but no difference could be detected (Figures 2B and 3). Treatment with the combination of olmesartan medoxomil and amlodipine besylate showed no significant effect on lesion composition. Due to the small number of high-quality slides in the olmesartan medoxomil group (n=2), we could not perform statistical analyses.
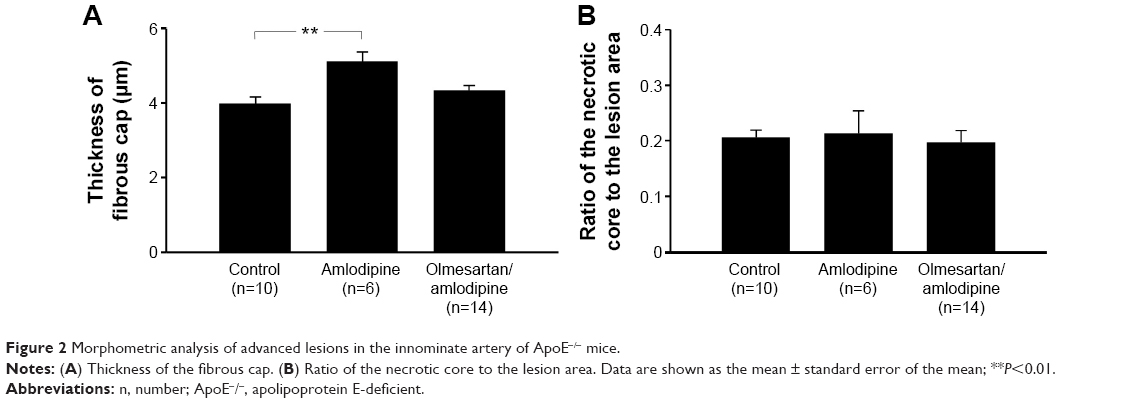 | Figure 2 Morphometric analysis of advanced lesions in the innominate artery of ApoE−/− mice. |
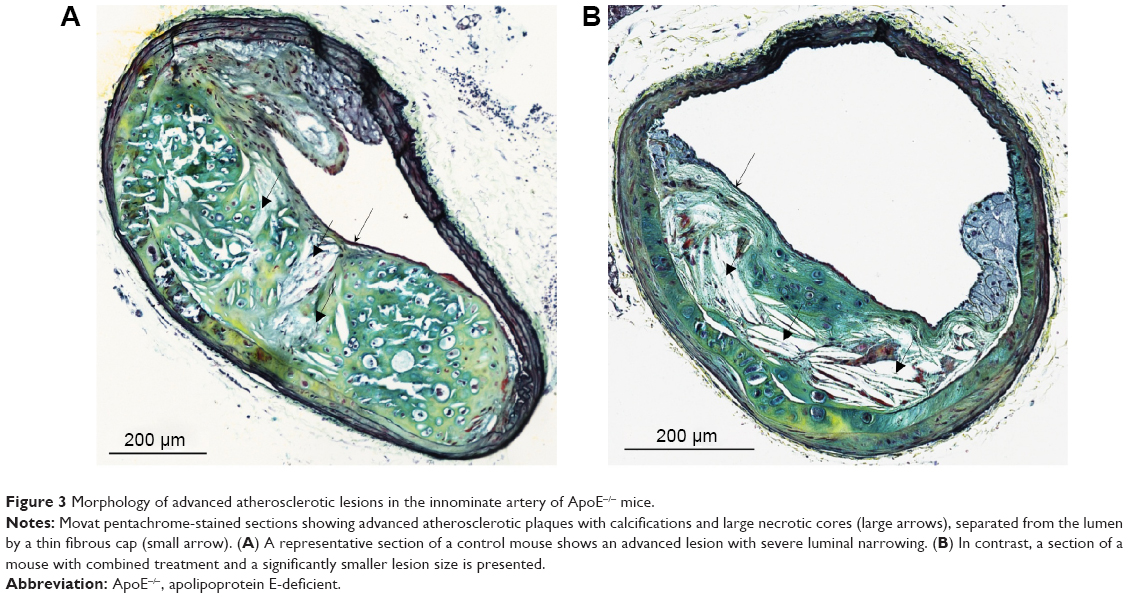 | Figure 3 Morphology of advanced atherosclerotic lesions in the innominate artery of ApoE−/− mice. |
EMSA
EMSAs of nuclear extracts (n=7/group) and subsequent densitometric evaluation showed a significant reduction of DNA-binding activity of the transcription factor NF-κB in the aortic tissue of mice treated with olmesartan medoxomil (P<0.05) or amlodipine besylate (P<0.05), as well as the combined treatment (P<0.05). There was a trend to a more intense downregulation of NF-κB in the combined treatment group compared to monotherapy with olmesartan medoxomil or amlodipine besylate, but this effect did not reach significance (Figure 4).
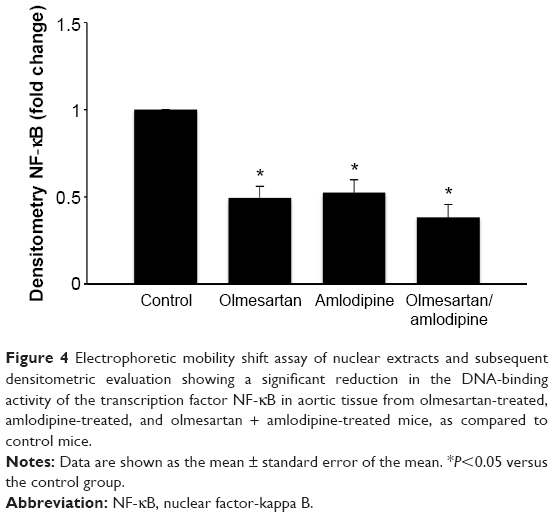 | Figure 4 Electrophoretic mobility shift assay of nuclear extracts and subsequent densitometric evaluation showing a significant reduction in the DNA-binding activity of the transcription factor NF-κB in aortic tissue from olmesartan-treated, amlodipine-treated, and olmesartan + amlodipine-treated mice, as compared to control mice. |
Discussion
The present study shows that combined treatment with olmesartan medoxomil and amlodipine besylate attenuates atherosclerotic progression in a mouse model of advanced atherosclerosis. Furthermore, EMSAs of nuclear extracts demonstrate that the combination of olmesartan medoxomil and amlodipine besylate leads to a significant downregulation of the transcription factor NF-κB in aortic tissue. However, in our present study, effects on plaque composition seem to be poor in very advanced stages of atherosclerosis.
Most of the pharmacological intervention studies using hyperlipidemic mouse models have focused their investigations on early atherosclerotic processes describing the effects and mechanisms of disease initiation and early progression.19 So far, there have been only very limited data concerning the effects on complex advanced lesions as they occur in human disease. However, clinical manifestations of atherosclerosis are caused by progressive arterial occlusion (stenosis) or plaque rupture and thrombosis,4,9 which typically occur in advanced stages. Advanced atherosclerotic lesions are characterized by the presence of a lipid-rich necrotic core, which is separated from the vessel lumen by a protective fibrous cap. Rupture of this fragile cap of an advanced atherosclerotic plaque is the main underlying cause of cardiovascular events like myocardial infarction and stroke.17,20 These conditions illustrate the enormous relevance of advanced and vulnerable stages. Due to the fact that early atherosclerotic lesions do not represent the point of interest for clinical investigations, as already established lesions do, our studies focused on the evaluation of effects on advanced atherosclerosis, which is more relevant in human disease.
Previous mouse models of atherosclerosis predominately concentrated on quantification of early lesions in the aortic sinus, and in the thoracic or abdominal aorta. The complex morphology of advanced lesions, such as thinning of the fibrous cap and plaque hemorrhage, is not addressed in these studies. In contrast, the model of advanced lesions in the innominate artery of hyperlipidemic ApoE−/− mice, as described by Rosenfeld et al,19 enables the assessment of features such as plaque destabilization, plaque hemorrhage, and plaque rupture. ApoE−/− mice develop advanced atherosclerotic lesions in the innominate arteries that have many of the morphological features of advanced atherosclerotic lesions in humans.19,21 Using older ApoE−/− mice and analyzing the plaque morphology in the innominate artery, we were able to explore such features of advanced atheroprogression.
The reduction of atherosclerotic progression in the early stages of atherosclerosis by AT1 receptor blockers and calcium channel antagonists has been documented in several hyperlipidemic mouse models.10–14 However, the impact on already established advanced atherosclerosis has not yet been sufficiently evaluated. Our data show that combined treatment with olmesartan medoxomil and amlodipine besylate leads to a significant reduction in lesion size in the innominate artery in comparison to controls, indicating a therapeutic effect even in advanced stages. This observation is consistent with studies that have shown a beneficial effect on lesion size in early stages using a combination of AT1 receptor blockers and calcium channel antagonists.13,15 To exclude a possible confounding effect of blood pressure and lipid levels on lesion progression, we chose the dose of olmesartan medoxomil and amlodipine besylate based on a study of Suzuki et al13 and Yoshii et al14 where the authors demonstrated significant antiatherosclerotic properties of olmesartan medoxomil and amlodipine besylate without affecting systolic blood pressure. Furthermore, we obtained blood samples to determine plasma lipid levels in ApoE−/− mice, which did not show significant differences in the lipid profile after 25 weeks on the diet, when the mice were 50 weeks old.
A trend toward a decrease in lesion size in the amlodipine besylate-treated animals could be observed. However, likely due to the small number, the data did not reach levels of significance. Statistical analyses of monotherapy with olmesartan medoxomil could not be performed, due to the small number of high-quality slides. Nevertheless, the reduction in lesion size with combined treatment suggests an additive effect of olmesartan medoxomil and amlodipine besylate, which underlines a possible influence of olmesartan medoxomil.
The histological analysis of atherosclerotic lesion composition widely revealed hypocellular, fibrotic, and calcified lesions covered by thin fibrous caps, which are typical features of advanced stages.22 However, we could not detect relevant beneficial effects on plaque composition in any of the groups, although significantly thicker fibrous caps were found following treatment with amlodipine besylate. The risk of plaque rupture and thrombosis depends more on plaque composition than on lesion size.23,24 Although the degree of luminal obstruction correlates with the morphometric stage of disease, the lesion size is not the major determinant leading to acute cardiovascular complications. Our data show that combined treatment with olmesartan medoxomil and amlodipine besylate significantly reduces lesion size, but we did not observe significant effects on plaque composition. These results strongly suggest an influence of AT1 receptor blockers and calcium channel antagonists on atheroprogression, even in advanced stages of atherosclerosis, and the decrease in lesion size may result in a reduction of flow-limiting stenoses and tissue ischemia. However, plaque composition, which is linked to atherosclerotic lesion destabilization and cardiovascular complications, was unaffected.
Chronic inflammation plays a fundamental role in the pathogenesis of atherosclerosis and is mediated by a cascade of proinflammatory cytokines and chemokines. Experimental data have shown a critical role of the NF-κB signaling pathway in the development of atherosclerosis. NF-κB induces the expression of several genes involved in the initiation and progression of vascular inflammation, including proinflammatory cytokines, adhesion molecules, and chemokines.6 Because of its role in regulating the transcriptional expression of different genes involved in atherogenesis, inhibition of NF-κB has become an attractive target for therapeutic intervention.25,26 The EMSAs of nuclear extracts demonstrated that combination of olmesartan medoxomil and amlodipine besylate, as well as amlodipine besylate and olmesartan medoxomil monotherapy, leads to a significant downregulation of the transcription factor NF-κB in aortic tissue when compared to controls. Furthermore, there was a trend toward a more intense downregulation in the combined treatment group compared to monotherapy, which supports our in vivo data. This observation provides a possible anti-inflammatory mechanism by which the combination of olmesartan medoxomil and amlodipine besylate could influence atheroprogression; it also underlines the antiatherosclerotic property of the drugs, independent of the blood pressure-lowering effect. These potential pleiotropic effects have been reported for both drugs. Several studies illustrated that angiotensin II activates the NF-κB, and that this activation can be inhibited by AT1 receptor blockers.27,28 Similar effects of the inhibition of NF-κB activation have been identified by interventions with dihydropyridine calcium channel antagonists.29 In our study, we could also observe a significant reduction of the DNA-binding activity of the transcription factor NF-κB in aortic tissue with olmesartan medoxomil or amlodipine besylate monotherapy. Despite the apparent inhibitory property of olmesartan medoxomil and amlodipine besylate on NF-κB, the underlying molecular mechanisms are widely unclear.
There are several limitations to our study. The advanced atherosclerotic lesions in ApoE−/− mice were highly calcified, which made tissue preparation susceptible to damage by microtome. The amount of high-quality slides for staining was therefore lower than expected. Furthermore, we focused on the more important quantitative evaluation of lesions and plaque composition (necrotic core and fibrous caps; hemorrhage and medial erosions are not sufficiently detectable when the artery/plaque is damaged). Due to the small number of high-quality slides in the olmesartan medoxomil group, we could not perform statistical analysis within this group. Although we chose the dose of olmesartan medoxomil and amlodipine besylate in accordance with studies that demonstrated antiatherosclerotic properties without affecting blood pressure,13,14 we cannot completely exclude that antiatherosclerotic effects might be related to a reduction in blood pressure. Moreover, most of the blood samples had to be used in order to detect lipid levels, which is important to rule out any confounding effects on atherosclerotic lesion development.
Conclusion
In conclusion, our study demonstrates that combined treatment with olmesartan medoxomil and amlodipine besylate attenuates atherosclerotic progression in an ApoE−/− mouse model of advanced atherosclerosis. Furthermore, the combination of olmesartan medoxomil and amlodipine besylate leads to a significant downregulation of the transcription factor NF-κB in aortic tissue, thereby providing a possible anti-inflammatory mechanism influencing atheroprogression. The underlying mechanisms by which olmesartan medoxomil and amlodipine besylate contribute to the inhibition of atheroprogression are still unclear, but the NF-κB pathway might be a convergence point for the two different classes of antihypertensive drugs. These findings suggest that combined treatment with olmesartan medoxomil and amlodipine besylate could be a potentially important drug combination in advanced atherosclerosis. Future studies will be needed to understand the underlying mechanism behind the atheroprotective effects of this drug combination, and even to evaluate its beneficial clinical effects.
Acknowledgments
We thank Annette Buttler for her excellent technical assistance. This work was supported by a research grant from Daiichi Sankyo Deutschland GmbH, Munich, Germany.
Disclosure
The authors report no conflicts of interest in this work.
References
Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–325. | ||
Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38(6):1092–1104. | ||
Silvestre-Roig C, de Winther MP, Weber C, Daemen MJ, Lutgens E, Soehnlein O. Atherosclerotic plaque destabilization: mechanisms, models, and therapeutic strategies. Circ Res. 2014;114(1):214–226. | ||
Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. 2010;74(2):213–220. | ||
Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19(9):1166–1172. | ||
de Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor kappaB signaling in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25(5):904–914. | ||
Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–212. | ||
Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(18):1685–1695. | ||
Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17(11):1410–1422. | ||
Cristofori P, Lanzoni A, Quartaroli M, et al. The calcium-channel blocker lacidipine reduces the development of atherosclerotic lesions in the apoE-deficient mouse. J Hypertens. 2000;18(10):1429–1436. | ||
Shimada K, Murayama T, Yokode M, Kita T, Fujita M, Kishimoto C. Olmesartan, a novel angiotensin II type 1 receptor antagonist, reduces severity of atherosclerosis in apolipoprotein E deficient mice associated with reducing superoxide production. Nutr Metab Cardiovasc Dis. 2011;21(9):672–678. | ||
Takaya T, Kawashima S, Shinohara M, et al. Angiotensin II type 1 receptor blocker telmisartan suppresses superoxide production and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. Atherosclerosis. 2006;186(2):402–410. | ||
Suzuki J, Iwai M, Li Z, et al. Effect of combination of calcium antagonist, azelnidipine, and AT1 receptor blocker, olmesartan, on atherosclerosis in apolipoprotein E-deficient mice. J Hypertens. 2005;23(7):1383–1389. | ||
Yoshii T, Iwai M, Li Z, et al. Regression of atherosclerosis by amlodipine via anti-inflammatory and anti-oxidative stress actions. Hypertens Res. 2006;29(6):457–466. | ||
Noda K, Hosoya M, Nakajima S, Ohashi J, Fukumoto Y, Shimokawa H. Anti-atherogenic effects of the combination therapy with olmesartan and azelnidipine in diabetic apolipoprotein E-deficient mice. Tohoku J Exp Med. 2012;228(4):305–315. | ||
Pendse AA, Arbones-Mainar JM, Johnson LA, Altenburg MK, Maeda N. Apolipoprotein E knock-out and knock-in mice: atherosclerosis, metabolic syndrome, and beyond. J Lipid Res. 2009;50 Suppl:S178–182. | ||
Charo IF, Taub R. Anti-inflammatory therapeutics for the treatment of atherosclerosis. Nat Rev Drug Discov. 2011;10(5):365–376. | ||
Movat HZ. Demonstration of all connective tissue elements in a single section; pentachrome stains. AMA Arch Pathol. 1955;60(3): 289–295. | ||
Rosenfeld ME, Polinsky P, Virmani R, Kauser K, Rubanyi G, Schwartz SM. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol. 2000;20(12):2587–2592. | ||
Johnson JL, Jackson CL. Atherosclerotic plaque rupture in the apolipoprotein E knockout mouse. Atherosclerosis. 2001;154(2): 399–406. | ||
Williams H, Johnson JL, Carson KG, Jackson CL. Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2002;22(5):788–792. | ||
Rosenfeld ME, Averill MM, Bennett BJ, Schwartz SM. Progression and disruption of advanced atherosclerotic plaques in murine models. Curr Drug Targets. 2008;9(3):210–216. | ||
Ylä-Herttuala S, Bentzon JF, Daemen M, et al. Stabilisation of atherosclerotic plaques. Position paper of the European Society of Cardiology (ESC) Working Group on atherosclerosis and vascular biology. Thromb Haemost. 2011;106(1):1–19. | ||
Moons AH, Levi M, Peters RJ. Tissue factor and coronary artery disease. Cardiovasc Res. 2002;53(2):313–325. | ||
Madonna R, De Caterina R. Relevance of new drug discovery to reduce NF-κB activation in cardiovascular disease. Vascul Pharmacol. 2012;57(1):41–47. | ||
Stellos K, Dimmeler S. Vascular microRNAs: from disease mechanisms to therapeutic targets. Circ Res. 2014;114(1):3–4. | ||
Blessing E, Preusch M, Kranzhöfer R, et al. Anti-atherosclerotic properties of telmisartan in advanced atherosclerotic lesions in apolipoprotein E deficient mice. Atherosclerosis. 2008;199(2):295–303. | ||
Esteban V, Lorenzo O, Rupérez M, et al. Angiotensin II, via AT1 and AT2 receptors and NF-kappaB pathway, regulates the inflammatory response in unilateral ureteral obstruction. J Am Soc Nephrol. 2004;15(6):1514–1529. | ||
Cominacini L, Garbin U, Fratta Pasini A, et al. Lacidipine inhibits the activation of the transcription factor NF-kappaB and the expression of adhesion molecules induced by pro-oxidant signals on endothelial cells. J Hypertens. 1997;15(12 Pt 2):1633–1640. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.