")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Characterization of preclinical in vitro and in vivo pharmacokinetics properties for KBP-7018, a new tyrosine kinase inhibitor candidate for treatment of idiopathic pulmonary fibrosis
Authors Huang Z, Li H, Zhang Q, Tan X, Lu F, Liu H, Li S
Received 17 February 2015
Accepted for publication 18 March 2015
Published 5 August 2015 Volume 2015:9 Pages 4319—4328
DOI https://doi.org/10.2147/DDDT.S83055
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Zhenhua Huang,1 Heran Li,1 Qian Zhang,2 Xiaojuan Tan,2 Fangzheng Lu,1 Hongzhuo Liu,1 Sanming Li1
1School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, 2KBP BioSciences Co. Ltd., Jinan, Shandong, People’s Republic of China
Abstract: KBP-7018 is a novel selective tyrosine kinase inhibitor with potential for the treatment of idiopathic pulmonary fibrosis. The objective of this study was to characterize the preclinical pharmacokinetics of KBP-7018 in vitro and in vivo, and then to assess the likelihood of developing KBP-7018 as a clinical candidate. The systemic clearance (CL) of KBP-7018 was relatively low in rodents and monkeys with a value of less than 30% of hepatic blood flow, while it was high in dogs. The steady-state volume of distribution (Vss) ranged from 1.51 L/kg to 4.65 L/kg across the species tested. The maximum concentration (Cmax) of KBP-7018 occurred at 0.25–6 hours after oral dosing, and the bioavailability was moderate (21%–68%). The human CL (~20% of hepatic blood flow) and Vss (1.6–5.3 L/kg) were predicted by allometric scaling method and together with the other modeling methods indicated low metabolism and acceptable half-time (4.8–19.3 hours) in vivo. Overall, the preclinical data make it amenable to further oral solid dosage from design for the upcoming Phase I trials in human.
Keywords: idiopathic pulmonary fibrosis, tyrosine kinase inhibitor, pharmacokinetics, KBP-7018
Introduction
Idiopathic pulmonary fibrosis (IPF) is a debilitating disease characterized by a progressive decline in lung function, poor survival, and limited therapeutic options.1 Its pathogenesis is thought to involve aberrant wound healing mediated by multiple signaling pathways, resulting in progressive lung injury and scarring. The course of the disease is difficult to predict, but it generally involves progressive deterioration, with a median survival time of 2.9 years after diagnosis.2
KBP-7018 is a potent intracellular inhibitor of tyrosine kinases that shows promising outcomes for the treatments of IPF. Its targets included platelet-derived growth factor receptors α and β (half maximal inhibitory concentration [IC50], 26 nM and 34 nM, respectively), c-kit receptor ([IC50], 10 nM), and RET receptor ([IC50], 7.6 nM) (unpublished data). Because signaling pathways activated by these tyrosine kinase receptors have been shown to be involved in lung fibrosis, inhibition of the receptors may slow the progression of IPF.3 In a mice model, such inhibition was shown to prevent the development of bleomycin-induced lung fibrosis when KBP-7018 was administrated before or during the fibrotic phase of the disease. The improved efficacy of KBP-7018 over nintedanib, a drug approved by Food and Drug Administration (FDA) for the treatment of IPF, was even observed (unpublished data).
The purpose of the current study was to characterize the in vitro pharmacokinetic (PK) profile of KBP-7018, including metabolic stability, permeability, plasma protein binding, and CYP inhibition, as well as its in vivo PKs, in anticipation of further preclinical research.
Experimental
Compounds, reagents, and solvents
KBP-7018 (the chemical structure is shown in Figure 1; the hydrochloride salt was used in all the in vitro and in vivo studies) and the structural analog KHP-2052, used as internal standards (also shown in Figure 1), were synthesized at KBP BioSciences Co., Ltd. (Jinan, Shandong, People’s Republic of China). Liver microsomes were purchased from Research Institute for Liver Disease (Shanghai, People’s Republic of China). Purified recombinant CYP450 isozymes, CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, were purchased from BD Biosciences (San Jose, CA, USA). For the blood to plasma concentration ratio study, freshly collected mouse, rat, dog, and monkey blood was obtained from in-house animals. Human blood and plasma were obtained from the volunteers. Standard substrates, inhibitors, and NADPH used for the CYP inhibition study were obtained from Sigma-Aldrich Co. (St Louis, MO, USA), BD Biosciences, and TRC (Toronto, Canada), respectively. Formic acid and methanol were purchased from Fluka (Buchs, Switzerland). Injection graded hydroxpropyl-β-cyclodextrin and 5% glucose were purchased from Xi’an Lide Biochemical Co., Ltd. (Xi’an, People’s Republic of China) and Cisen Pharmaceutical Co., Ltd. (Jining, People’s Republic of China), respectively. The water used for the preparation of all solutions was obtained from a Milli-Q apparatus (EMD Millipore, Billerica, MA, USA). All other reagents were of research or analytical grade.
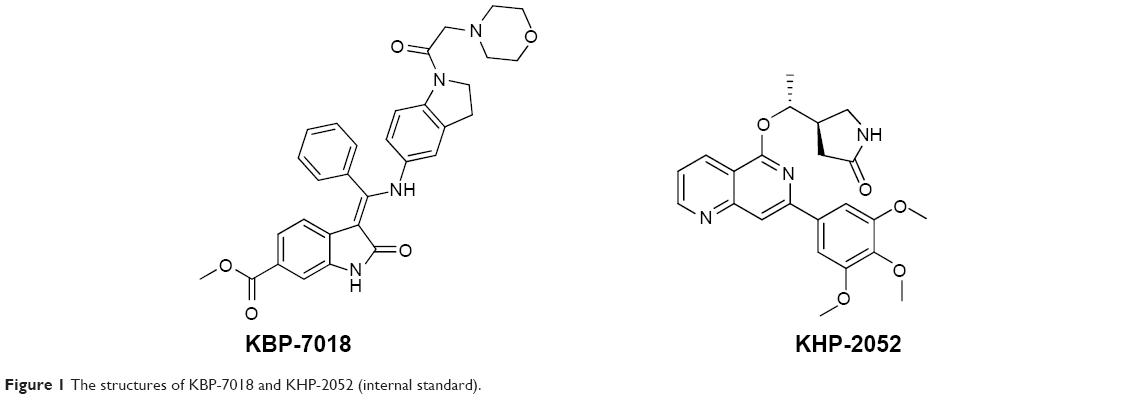 | Figure 1 The structures of KBP-7018 and KHP-2052 (internal standard). |
Animal
All the animal studies were done as per approved protocols by the Institutional Animal Care and Use Committee at KBP BioSciences Co., Ltd. Male CD-1 mice weighing 20–25 g, male Sprague-Dawley (SD) rats weighing 250–300 g (Beijing Vital River Lab, Beijing, People’s Republic of China), male beagle dogs weighing 7–10 kg (Marshall BioResource, Beijing, People’s Republic of China), and adult male cynomolgus monkeys weighing 4–6 kg (Shandong Hongli Medical Animal Experiment Research Co., Ltd., Jinan, People’s Republic of China) were used for in vivo studies. Animals were maintained under a 12-hour light/dark cycle with free access to food and water.
Stability in blood plasma
For blood–plasma stability, KBP-7018 was incubated at 1 μM in CD-1 mouse, SD rat, beagle dog, cynomolgus monkey, and human plasma samples for up to 4 hours at 37°C. At the desired time points, an aliquot from the incubation was quenched by adding 10 volumes of 100% methanol supplemented with internal standard. Following the last collection, the samples were centrifuged at 12,000 rpm for 5 minutes and the supernatants were transferred to a new plate containing an equal volume of water for analysis by liquid chromatography coupled to triple quadrupole mass spectrometry (LC-MS/MS).4
Microsomal stability
Microsomes from CD-1 mouse, SD rat, beagle dog, cynomolgus monkey, and human were used for assay. Incubation (1 mL) consisted of liver microsomes (1 mg/mL), NADPH (10 mM), and 100 mM phosphate buffer (pH 7.4). Following preincubation (3 minutes, 37°C), reactions were initiated by adding KBP-7018 (10 μM). Samples (70 μL) were withdrawn at 0 minute, 5 minutes, 10 minutes, 20 minutes, 30 minutes, and 60 minutes and quenched with 300 μL acetonitrile containing internal standard. Concomitant NADPH-free control incubations were made similarly with samples collected at 0 minute and 60 minutes. The samples were centrifuged for 3 minutes at 12,000 rpm to pellet proteins, and the supernatant was subjected to LC-MS/MS analysis. Log-linear plots of percentage compound remaining versus time were plotted, and the slope of the curve was calculated by linear regression of the log-linear curve. The half-life (t1/2) of metabolic stability was estimated using the first-order equation t1/2 0.693/Kel, where Kel (elimination rate constant) is the slope of the linear portion of the log-linear curve. The microsomal intrinsic clearance (CL’int) was estimated using the equation CL’int (0.693/t1/2) × (volume of incubation/milligram microsomal protein). The hepatic intrinsic clearance (CL’h,int) of KBP-7018 in various species was estimated from liver microsomal data using the following equation:5
|
|
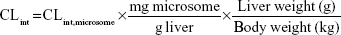
where 45 mg of microsomal protein per gram of liver tissue was applied to all species, and 87 g, 40 g, 30 g, 32 g, and 26 g of liver tissue per kilogram of body weight (BW) were used for mice, rats, dogs, monkeys, and humans, respectively. Then in vivo hepatic clearance of KBP-7018 was calculated by the well-stirred model6 by
|
|
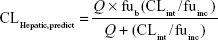
where Q is hepatic blood flow (90 mL/min/kg, 55 mL/min/kg, 31 mL/min/kg, 44 mL/min/kg, and 21 mL/min/kg in mice, rats, dogs, monkeys, and humans, respectively), fub and fuinc were the unbound fraction of protein binding in blood and microsomes, respectively, and CLint is the scaled intrinsic clearance determined from Equation 1. fuinc was calculated by substitution of clog P(3.81) to the following equation for liver microsomes:
|
|
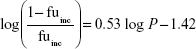
Cytochrome P450 inhibition
The potential of KBP-7018 to inhibit major human CYP enzymes was also evaluated.7 Briefly, the test compound, the substrate, purified isozyme, 2 μL of 100× regeneration system, 20 μL of 10× NADP+, 20 μL of 10× buffer, and deionized water were added to a final volume of 0.2 mL/well in a 96-well plate. CYP isozyme-specific probe substrates used were CYP1A2 (phenacetin, 10 μM), CYP2C9 (diclofenac, 5 μM), CYP2C19 (S-mephenytoin, 30 μM), CYP2D6 (dextromethorphan, 5 μM), and CYP3A4 (midazolam and testosterone for 2 μM and 50 μM, respectively). KBP stock solutions were spiked (2 μL) to achieve the final target inhibitor concentrations (0.05 μM, 0.3 μM, 1 μM, 3 μM, 10 μM, 30 μM, 80 μM). After incubation for 10 minutes, the reaction was stopped by the addition of stop solution. The samples (100 μL) were centrifuged at 4,000 rpm for 10 minutes, and the supernatants were transferred to a new plate containing an equal volume of water for analysis by LC-MS/MS. The IC50 values were estimated using Prism software.8
Plasma protein binding
A modified equilibrium dialysis method was applied in this study to determine the plasma protein binding of KBP-7018 in plasma.9 The stability in plasma was evaluated at 2 μM and 20 μM of KBP-7018 for each species after incubation at 37°C for 6 hours. Plasma protein binding studies were performed using a 96-well HT Dialysis equilibrium dialysis chamber apparatus (HTDialysis®, Gales Ferry, CT, USA). An aliquot (150 μL) of KBP-7018 spiked plasma was added to the donor side of each designated well. An equal volume of phosphate buffer was added to the receiver side. The plate was covered with adhesive sealing film to prevent evaporation and is placed in a water bath. The resulting plasma and buffer dialyzates were promptly recovered and analyzed by LC-MS/MS after sample preparation as described above. The binding degree of KBP-7018 in equilibrium dialysis experiments is expressed according to the following equation:
|
|
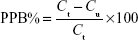
where PPB is plasma protein binding, Ct is the total concentration of drug in the plasma solution at equilibrium, and Cu is the free drug concentration at the end of the dialysis.
Blood–plasma partition
KBP-7018 (200 μM) was added to whole blood to obtain final concentrations of 1 μM and incubated at 37°C for 15 minutes. Blood and plasma were separated from blood samples and analyzed by LC-MS/MS. The blood–plasma partitioning (BPP) was determined by dividing the total initial concentration in blood by that found in plasma separated from blood samples.9
Caco-2 permeability
Human colon carcinoma (Caco-2) cells were grown in Dulbecco’s Modified Eagle’s Medium, supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. The cells were seeded at a density of 1×105 cells/cm2 in 96-well Transwell® inserts and cultured at 37°C in a humidified incubator with a 5% CO2 atmosphere. In all, 22 days post-seeding, permeability of KBP-7018 was determined in apical to basolateral (A–B) and basolateral to apical (B–A) directions. Following preincubation in Hank’s balanced salt solution (HBSS)–2-[4-2-(Hydroxyethyl)-1-piperazinyl]ethane sulfonic acid (HEPES) buffer in an orbital shaker (37°C, 5% CO2, 30 minutes), lucifer yellow ratio values were measured and only those inserts with values ≤1% were considered for assay. The buffer was removed and KBP-7018 spiked buffer (1% final Dimethyl Sulphoxide [DMSO] concentration) was added to each donor compartment in triplicate. Blank buffer containing 1% DMSO was added to the receiver compartment. Samples were withdrawn from the receiver chamber and the donor chamber at 0 minute and 120 minutes, respectively, and fresh buffer was added in the same amount from the basolateral or apical side. The samples were stored at -20°C until KBP-7018 analysis by LC-MS/MS. Lucifer yellow ratio values were measured after completion of assay to ensure monolayer integrity. At the end of the experiment, cells were washed with cold buffer and lysed with acetonitrile to assess cell accumulation and estimate the recovery. Apparent permeability (Papp), efflux ratio (Papp(B-A)/Papp(A-B)), cell accumulation, and recovery were calculated. Digoxin (substrate for P-glycoprotein [P-gp]) was run as positive control.10,11
In vivo PK
The intravenous (IV) solution vehicle for CD-1 mice and beagle dogs comprised 30% (v/v) polyethylene glycol 400 (PEG-400) and 70% (v/v) glucose injection (adjusted to pH 4). The IV solution vehicle for SD rats and cynomolgus monkeys comprised 20% and 10% hydroxypropyl-β-cyclodextrin in 5% glucose, respectively. Oral solution in CD-1 mice, SD rats, and cynomolgus monkeys used the same vehicles as IV administration. Suspension formulations comprising 0.5% (w/v) methyl cellulose was used for oral administration to beagle dogs. The IV dose was 2 mg/kg for SD rats, beagle dogs, and cynomolgus monkeys and 10 mg/kg for CD-1 mice. The oral dose was 50 mg/kg for CD-1 mice and beagle dogs, 10 mg/kg for SD rats, and 5 mg/kg for cynomolgus monkeys.
For PK studies, a sparse sampling design was used in mice (n=3 per time point). Serial blood sampling was used for rats (parallel groups, n=3), dogs (crossover group, n=3, 10-day washout period), and monkeys (crossover group, n=3, 10-day washout period). In mice, approximately, 100 μL of blood samples were collected at 0.083 hour, 0.25 hour, 0.5 hour, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, and 24 hours post-dose. In rats, dogs, and monkeys, blood samples were collected at 0.083 hour (only IV), 0.166 hour (only PO [oral administration]), 0.25 hour, 0.5 hour, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 24 hours, and 30 hours (only for monkeys) post-administration. The plasma generated from the blood samples were placed in clean, tightly capped, pre-labeled heparinized plastic vials and frozen at approximately -80°C to await bioanalysis. In each case, a 20 μL of plasma sample was mixed with 200 μL of internal standard working solution. The samples were then vortexed for 3 minutes and subsequently centrifuged at 12,000 rpm for 5 minutes. A 100 μL of liquid supernatant samples were collected and diluted by 100 μL of water for ultrafast liquid chromatography-tandem mass spectrometry (UFLC-MS/MS) analysis.
UFLC-MS/MS analysis
Quantitative analysis of the plasma samples at each time point was performed by UFLC-MS/MS (HPLC system: Shimadzu UFLC). Chromatographic separations were accomplished on a Waters XBridge-C18 (50 mm ×2.1 mm, 5 μm, USA) column using two mobile phases, 0.1% formic acid in methanol (v/v) and 0.1% formic acid in water (v/v) with a flow rate of 0.50 mL/min. The column temperature was maintained at 35°C. The linear gradient was delivered as follows: 0–0.4 minutes, hold at 5% organic; 0.4–0.8 minutes, from 5% to 98% organic; hold for 1 minute; return to initial conditions at 1.9 minutes; and total run time 3 minutes.
Analysis was conducted on API 4000 Qtrap mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) operated with electrospray ionization interface in positive ionization mode. The working parameters of the mass spectrometer were the following: 5,500 V of ion spray voltage, 550°C of source temperature, 55 psi of ion source gas (gas 1), and 55 psi of ion source gas (gas 2). Quantification was performed in multiple-reaction monitoring mode with specific ion transition of protonated precursor ion to product ion at m/z 539.2→410.3 for analyte and at m/z 424.3→313.3 for internal standard. The results were valid since the relative standard deviation of replicates was below 15%.
Plasma concentration data processing
PK analysis was performed on plasma concentration–time data obtained after IV or oral administration. Peak plasma concentration (Cmax) and time of Cmax (Tmax) were determined directly from the individual observed concentration–time data. PK parameters (AUC0–T last and t1/2) were derived by using a model-independent approach [noncompartmental analysis] according to a uniform weighting scheme (WinNonlin version 6.3, Pharsight Corporation, Cary, NC, USA). AUC0–T last was determined by using the linear up–log down trapezoidal rule. The area was extrapolated to infinity (AUC0-∞) using the rate constant of the terminal elimination phase (λz), which was determined from the slope of the terminal log-linear portion of the concentration–time curve by using a minimum of three measureable time points after Cmax was achieved. In addition, t1/2 was calculated by dividing λz into the natural logarithm of 2. Clearance (CL) and volume of distribution (V) were calculated by using the following equations:
|
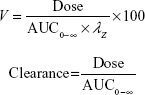
|
|
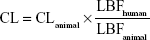
Prediction of human clearance and volume of distribution
Prior to the investigational new drug filing, the PKs in preclinical species were used to predict human PKs for the first-in-human clinical study. In the present study, allometric scaling method,12,13 liver blood flow method,6 and estimation based on monkey data alone14 were used for the prediction of human CL of KBP-7018. The allometric scaling employed in the present study is universal equation based on a log–log relationship with a given PK parameter × brain weight (BrW) and animal BW. Since monkey provided the most accurate prediction of both clearance and volume of distribution, predicting human PK profiles on the basis of monkey data was the most accurate of the various approaches considered.14 In this evaluation, CL in monkeys can be expressed as a fraction of liver blood flow, and the human CL was estimated using the following equation:
|
|
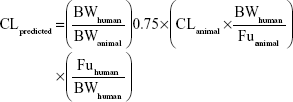
Or directly prediction was performed by single species scaling from monkey PK studies using the following equation:
|
|
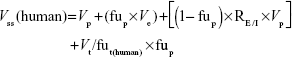
where BW is the body weight and Fu is the fraction unbound in plasma.
Besides allometric scaling method, the Oie-Tozer approach,15 which was found as the top method for predicting human Vss, was also used for predicting human Vss by using the following equation:
where Vp (l/kg) =0.0313, 0.0313, 0.515, 0.0448, and 0.0436 for mouse, rat, dog, monkey, and human, respectively. Ve (l/kg) =0.265, 0.265, 0.216, 0.208, and 0.151 for mouse, rat, dog, monkey, and human, respectively. RE/I =1.4. Vt/fut(human) is estimated by allometry of Vt/fut(animal).
Result
Stability in plasma and liver microsomes in vitro
When KBP-7018 (1 μM) was incubated for 4 hours with plasma from human, cynomolgus monkey, beagle dog, SD rat, and CD-1 mouse, the remaining ratios were 109%, 104%, 105%, 67%, and 85% of the original amount, respectively. It was concluded that KBP-7018 was stable in the plasma from all of the above species except SD rat (Table 1). Table 1 also summarized the in vitro metabolism of KBP-7018, as measured by the disappearance of the molecules, in incubations with NADPH-fortified liver microsomes from various preclinical species and human. The rate of disappearance of KBP-7018 was significantly greater in microsomes prepared from monkeys (0.44 mL/min/mg) compared with that observed with mouse, rat, dog, or human microsomes (0.04–0.12 mL/min/mg). As observed in Table 1, the predicted CLHepatic,predict values of KBP-7018 for mice, rats, dogs, monkeys, and humans were 7.2 mL/min/kg, 5.6 mL/min/kg, 6.4 mL/min/kg, 18.4 mL/min/kg, and 4.2 mL/min/kg, respectively. The in vivo blood clearance for the monkey (10.2 mL/min/kg) was predicted well using microsomal data (within twofold). However, the predicted plasma clearance by using the microsomal was over estimating as compared with in vivo data.
 | Table 1 The stability of KBP-7018 in plasma and microsomes (n=2) |
CYP inhibition
The ability of KBP-7018 and nintedanib to inhibit the major human cytochrome P450 isozymes, including 1A2, 2C9, 2C19, 2D6, and 3A4, was compared (Table 2). For KBP-7018, no inhibition of 1A2, 2C19, 2D6, and 3A4 (midazolam and nifedipine as probe substrate) was observed. There was weak inhibition of 2C9 (IC50 35.4 μM) and strong inhibition of 3A4 (testosterone as probe substrate, IC50 0.087 μM). Nintedanib did not show inhibition of 3A4 (midazolam as substrate), but limited inhibition of 2C9, 2C19, and 2D6 (IC50 value ≥33 μM). However, inhibition of 1A2 and 3A4 mediated by testosterone was observed (IC50, 17.5 μM and 2.3 μM, respectively). Our results indicate that KBP-7018 and nintedanib showed strong inhibition of 3A4 (testosterone as probe substrate); thus, clinical interaction between KBP-7018 and these CYPs would be significant.
 | Table 2 Comparative P450 inhibition potential of KBP-7018 and nintedanib (IC50, μM) |
Plasma protein binding and BPP
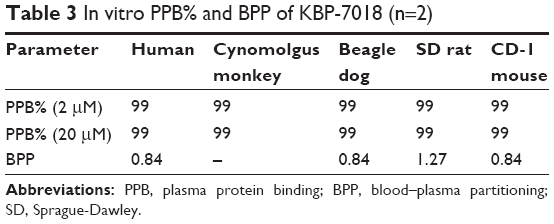
The extent of KBP-7018 binding to human and animal plasma proteins was determined using equilibrium dialysis method at drug concentrations of 2 μM and 20 μM, respectively. As shown in Table 3, KBP-7018 showed very high protein binding to human, cynomolgus monkey, beagle dog, SD rat, and CD-1 mouse plasma (99%). Blood–plasma partition of KBP-7018 in human, beagle dog, SD rat, and CD-1 mouse plasma was also assessed in vitro by LC-MS/MS method at drug concentrations of 1 μM. As shown in Table 3, the results of in vitro blood–plasma partition ratio was approximately 0.8, indicating that KBP-7018 did not partition much into red blood cells in humans, dogs, and mice except rats (~1.3 in rats).
 | Table 3 In vitro PPB% and BPP of KBP-7018 (n=2) |
Caco-2 cell permeability
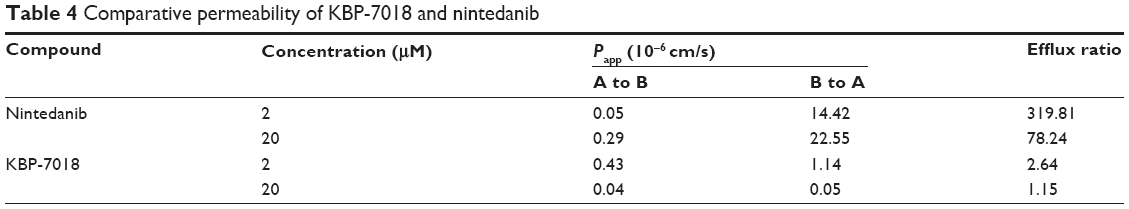
To predict intestinal absorption, we performed an in vitro cell permeability experiment using a Caco-2 cell monolayer transport system. The bidirectional permeability of KBP-7018 was measured at 2 μM and 20 μM. As shown in Table 4, KBP-7018 exhibited low Papp values in the absorptive direction (A to B), with values of 0.43×10−6 cm/s and 0.04×10−6 cm/s, respectively, at concentrations of 2 μM and 20 μM. On the other hand, the Papp values in the B to A direction were greater than those in the A to B direction (efflux ratio of ~2.6 and ~1.1 for 2 μM and 20 μM, respectively), suggesting that KBP-7018 may be a substrate of an active efflux transporter, for example, P-gp. However, nintedanib has higher efflux ratio (~319 and ~78 for 2 μM and 20 μM, respectively), appeared to be P-gp substrates.
 | Table 4 Comparative permeability of KBP-7018 and nintedanib |
In vivo PKs of KBP-7018 in monkeys after IV and oral administration
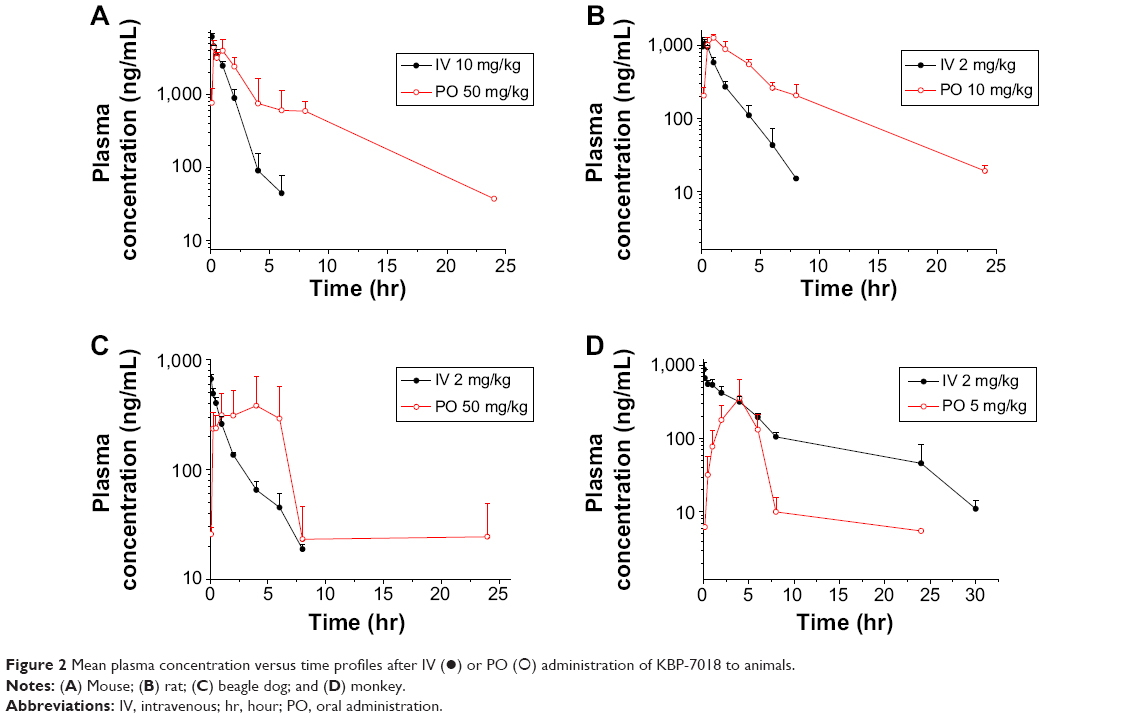
The PK profiles of KBP-7018 in mice, rats, dogs, and monkeys after a single IV dose and oral dosing are shown in Figure 2. The noncompartmental analysis PK parameters of KBP-7018 in mice, rats, and monkeys are summarized in Table 5.
 | Figure 2 Mean plasma concentration versus time profiles after IV (•) or PO (○) administration of KBP-7018 to animals. |
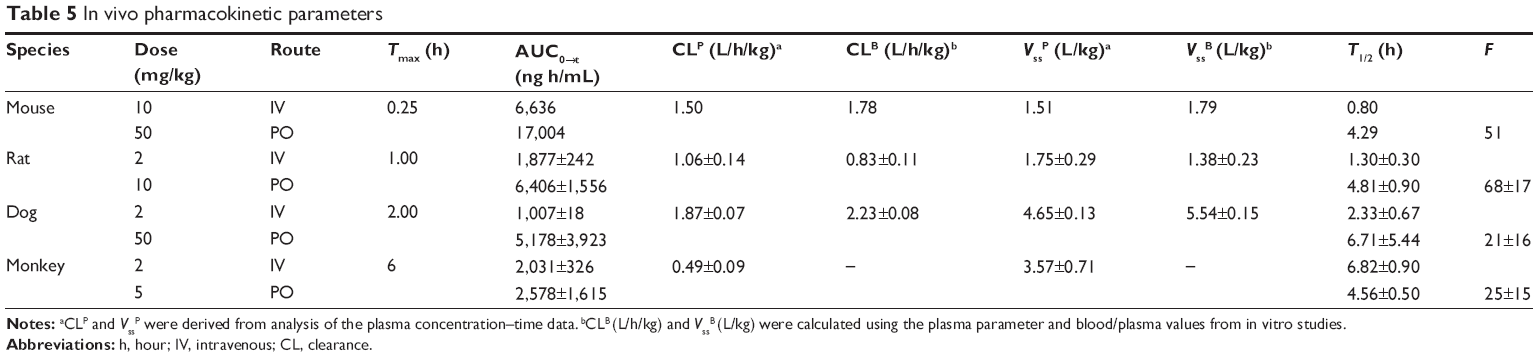 | Table 5 In vivo pharmacokinetic parameters |
Following IV administration of KBP-7018 to the mouse (Figure 2A), mean plasma concentrations declined exponentially with t1/2 of 0.8 hour. Plasma clearance was 1.5 L/h/kg, and the volume of distribution at steady state was 1.51 L/kg. Using a blood to plasma concentration ratio of 0.8 (Table 3), the blood CL was estimated to be 1.78 L/h/kg or approximately 30% of liver blood flow in the mouse. The blood Vss estimated from the plasma Vss, and the blood to plasma ratio, was 1.79 L/kg. The value is ~2.5 times of body water in the mouse, suggesting there was substantial penetration of KBP-7018 into tissues. Oral bioavailability of a solution formulation in mouse was moderate (~50%, Table 5). Following oral administration, maximum plasma concentration was attained at 15 minutes, indicating rapid absorption; thereafter, mean plasma concentrations declined exponentially with a terminal half-life of 4.29 hours. The PKs in rat (Figure 2B) were similarly profiled as found in mouse after IV and PO administration. In rats, t1/2 was 1.3 hours and 4.8 hours following IV and PO administration, respectively. Blood CL and Vss were derived from the plasma parameter (Table 5) and in vitro BPP data as described for the mouse. Blood clearance in the rat was estimated to be 0.83 L/h/kg or approximately 30% of liver blood flow. Blood Vss was 1.38 L/kg, 1.2 times of total body water in this species. Oral bioavailability was high (68%).
In the dog, KBP-7018 displayed exponential disposition with first-order kinetics after a single IV dose (Figure 2C). The plasma CL was high (1.87 L/h/kg), with high Vss (4.65 L/kg, estimated blood Vss 7.5 times of total blood water) and a mean t1/2 of 2.3 hours. Mean blood clearance was 2.23 L/h/kg or approximately 100% of liver blood flow in the dogs. After a single oral dose, the mean terminal t1/2 was 6.7 hours and oral bioavailability was low (21%).
In the case of monkeys (Figure 2D), the concentration of KBP-7018 declined exponentially following IV infusion, with mean plasma t1/2 of 6.8 hours. The lower plasma CL (0.49 L/h/kg) and higher Vss (3.57 L/kg) were observed, indicating a preferential partitioning into tissues. After a single oral dose, the exposure of KBP-7018 was much low in cynomolgus monkey. The mean dose-normalized Cmax after oral dosing reached only 73±54 ng/mL at a Tmax of 6.0 hours post-administration. The average elimination half-life after PO dosing was 4.6 hours and similar to the half-life observed after IV dosing. The bioavailability after oral dosing was 25%.
Allometry
The PK parameters of KBP-7018 were also predicted in humans using allometric scaling. The allometric relationships for CL and Vss and predicted values in humans for a 70-kg individual are shown in Figure 3. The relationship between BrW × CL and BW (Figure 3A) and between BrW × Vss and BW showed good linearity (Figure 3B). The mean allometric exponent and coefficient for CL were 0.8123 and 0.0091, respectively, and the corresponding values for Vss were 1.1009 and 0.0228, respectively. The allometric scaling method gave estimates of 0.19 (L/h/kg) for plasma clearance and 1.6 (L/kg) for volume of distribution for a 70 kg human. These human clearance and volume estimates gave an estimated blood half-life (t1/2=0.693 Vss/CL) for KBP-7018 in humans of approximately 5.9 hours.
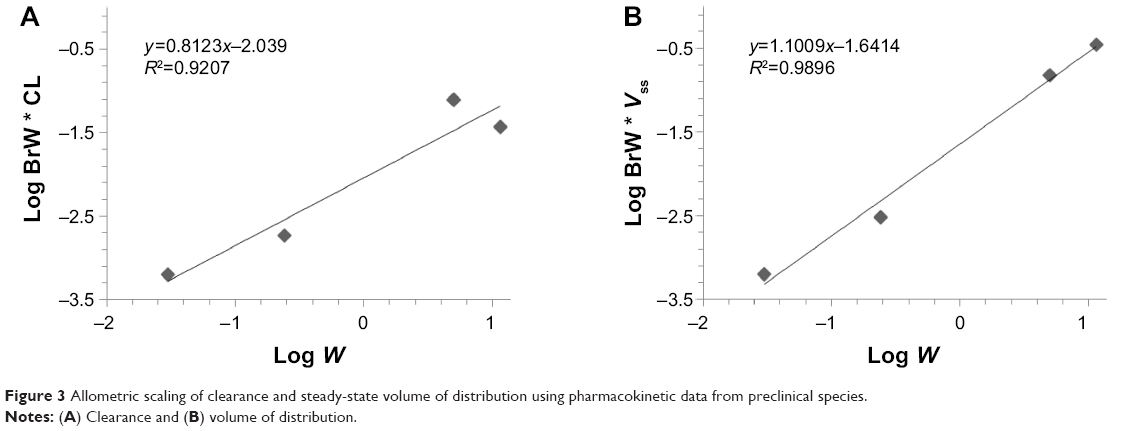 | Figure 3 Allometric scaling of clearance and steady-state volume of distribution using pharmacokinetic data from preclinical species. |
The predicted human CL and Vss values for KBP-7018 by different methods also are summarized in Table 6. The predicted human Vss from Oie-Tozer approach was 5.53 L/kg, much higher than the total body water (0.7 L/kg). The prediction of human plasma CL was 0.23 L/h/kg obtained by both liver blood flow methods and estimation by monkeys data, agreed with the value given by allometric scaling (within 1.5-fold). The human blood CL was then calculated as the value of 0.28 L/h/kg, accounting for approximately 20% of the human hepatic blood flow rate (1.24 L/h/kg). The prediction suggested that KBP-7018 is likely slow clearance compound in humans, which was in consistence with the prediction from microsomal data in vitro.
 | Table 6 Predicted plasma CL and Vss values using different methods |
Discussion
Abnormal tyrosine kinase activities disturb the physiological cell homeostasis and eventually lead to cancer, vascular disease, and fibrosis. With regard to fibrosis, different tyrosine kinases were found to play key roles in disease progression and then identified as potential targets for anti-fibrotic therapies. This involved both receptor tyrosine kinases (eg, PDGF receptor, VEGF receptor) as well as non-receptor tyrosine kinases (eg, c-Abl, c-Kit).16 Imatinib (targeted PDGF, c-Abl and c-Kit receptor) was first investigated whether it is available in IPF. Disappointingly, patients treated with imatinib did not show significant improvement in lung function and survival during the follow-up period in experimental models of IPF. This prompted a double-blind randomized, placebo-controlled trial.17 Besides, unexpectedly high rates of adverse events, including edema, fatigue, nausea, muscle weakness, and so on, finally led to the withdrawal of imatinib in many patients.18 Later, the multi-tyrosine kinase inhibitor nintedanib has recently drawn great interest in the treatment of IPF. However, Richeldi and colleagues also failed to achieve the primary outcomes but only improvements in secondary outcome measures.19 Also encouraged by its well tolerance, nintedanib was just approved by FDA for its promising improvement in life quality of patients.
KBP-7018 is a recently identified potent new compound for the treatment of IPF, which is currently under preclinical development. Compared with nintedanib, the approved kinase inhibitor in IPF, KBP-7018, showed better selectivity in the inhibition of kinases than nintedanib at 1,000 nM and excellent activity from 10 mg/kg/day to 100 mg/kg/day in a bleomycin-induced pulmonary fibrosis model in C57 mice both in a prophylactic and a treatment study (unpublished data). As such, KBP-7018 represents a major breakthrough for an unmet medical need. The studies described here show that KBP-7018 possesses feasible preclinical in vitro and in vivo PK properties in a variety of models.
The rate of metabolism of KBP-7018 determined in liver microsomes was used to predict the in vivo clearance using physiologically based scaling factor. There was a good correlation between predicted and observed clearance for the monkeys (within twofold), whereas there was a moderate correlation in the rats (within threefold) and poor correlation in the mice and dogs (fivefold). The predicted clearance value scaling from microsomal data in preliminary animals underpredicted in vivo clearance. Although the inclusion of both plasma and microsomal unbound fraction has been suggested as a generally acceptable approach, previous reports pointed out, for lipophilic basic molecules, the inclusion of both correction factors (plasma and microsomal) results in underprediction of clearance.20,21
Experiments were also conducted in an effort to identify which liver microsomal enzymes were involved in the metabolism of KBP-7018. When incubated with purified cytochromes, P450 enzymes, it appeared that the CYP3A4 was predominantly involved in the overall metabolism of KBP-7018. As suggested, KBP-7018 could be at risk for drug–drug interactions if co administered with potent inhibitors of CYP3A4 such as azole antifungals or antimicrobials such as erythromycin. Clinical drug–drug interaction studies should have to be conducted to determine the magnitude of this potential interaction.
The absorption of orally administered KBP-7018 was variable in terms of both onset and rate (Cmax and Tmax) with an incomplete bioavailability (21%–68%) observed in all preclinical species. The time to achieve peak concentrations of KBP-7018 ranged from 0.25 hour to 6 hours after oral dosing (Table 5). KBP-7018 distributed slightly better into plasma than red blood cells in all species except rats. Plasma protein binding was extensive (99%) in all species and was found to be independent of concentration in the range of 2–20 μM across these species.
The utility of allometric scaling, together with the other modeling methods, can make the prediction of PK parameters in humans more informative and accurate. It was found that the predicted values for CL and Vss showed no large differences from the different prediction methods used in the current study. Based on the prediction results, the human CL in blood (~0.25 L/h/kg) only accounted for approximately 20% of the hepatic blood flow rate, similar to the clearance ratio observed in rodents and monkeys. Because of a larger Vss scaled from Oie-Tozer approach, the half-life when allometric scaling was used to predict clearance was longer (19.3 hours) than the value scaled from lower Vss and a little rapid clearance (4.8 hours), suggesting potent exposure after administration. It makes it amenable to further oral solid dosage from design for the upcoming Phase I trials in human.
Conclusion
KBP-7018, a candidate for treatment of IPF with good PK properties, was identified and developed. In general, KBP-7018 showed acceptable half-life and oral bioavailability in preclinical species. The predicted PK parameters in human indicated low metabolism and acceptable half-time in vivo. In addition, KBP-7018 has better pharmacodynamic properties than nintedanib, the well-characterized approved drug in the treatment of IPF to date. The excellent efficacy and receptor selectivity, combining with its acceptable PK properties, make it a valuable compound for further upcoming Phase I trials in human.
Disclosure
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this paper.
References
Hostettler KE, Zhong J, Papakonstantinou E, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15(1):157. | ||
Kondoh Y, Taniguchi H, Ogura T, et al. Disease progression in idiopathic pulmonary fibrosis without pulmonary function impairment. Respirology. 2013;18(5):820–826. | ||
Coward WR, Saini G, Jenkins G. The pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2010;4(6):367–388. | ||
Chaudhary NI, Roth GJ, Hilberg F, et al. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29(5):976–985. | ||
Liu F, Zhuang X, Yang C, et al. Characterization of preclinical in vitro and in vivo ADME properties and prediction of human PK using a physiologically based pharmacokinetic model for YQA-14, a new dopamine D3 receptor antagonist candidate for treatment of drug addiction. Biopharm Drug Dispos. 2014;35(5):296–307. | ||
Tamaki S, Komura H, Kogayu M, Yamada S. Comparative assessment of empirical and physiological approaches on predicting human clearances. J Pharm Sci. 2011;100(3):1147–1155. | ||
Li J, Dake C, Bi H, Jin J, Huang M. Effects of flavonoids derived from Taxus yunnanensis on p-glycoprotein and cytochrome P450 3A4. Asian J Pharm Sci. 2013;8(3):168–173. | ||
Mosure KW, Knipe JO, Browning M, et al. Preclinical pharmacokinetics and in vitro metabolism of asunaprevir (BMS-650032), a potent hepatitis C virus NS3 protease inhibitor. J Pharm Sci. 2015. doi:10.1002/jps.24356. | ||
Davis CB, Bambal R, Moorthy GS, et al. Comparative preclinical drug metabolism and pharmacokinetic evaluation of novel 4-aminoquinoline anti-malarials. J Pharm Sci. 2009;98(1):362–377. | ||
Oltra-Noguera D, Mangas-Sanjuan V, Centelles-Sanguesa A, et al. Variability of permeability estimation from different protocols of subculture and transport experiments in cell monolayers. J Pharmacol Toxicol Methods. 2015;71:21–32. | ||
Deshmukh DD, Nagilla R, Ravis WR, Betageri GV. Effect of dodecylmaltoside (DDM) on uptake of BCS III compounds, tiludronate and cromolyn, in Caco-2 cells and rat intestine model. Drug Deliv. 2010;17(3):145–151. | ||
Mahmood I. Interspecies scaling of biliary excreted drugs: a comparison of several methods. J Pharm Sci. 2005;94(4):883–892. | ||
Mahmood I, Sahajwalla C. Interspecies scaling of biliary excreted drugs. J Pharm Sci. 2002;91(8):1908–1914. | ||
Ward KW, Smith BR. A comprehensive quantitative and qualitative evaluation of extrapolation of intravenous pharmacokinetic parameters from rat, dog, and monkey to humans. II. Volume of distribution and mean residence time. Drug Metab Dispos. 2004;32(6):612–619. | ||
Jones RD, Jones HM, Rowland M, et al. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 2: comparative assessment of prediction methods of human volume of distribution. J Pharm Sci. 2011;100(10):4074–4089. | ||
Beyer C, Distler JH. Tyrosine kinase signaling in fibrotic disorders: translation of basic research to human disease. Biochim Biophys Acta. 2013;1832(7):897–904. | ||
Daniels CE, Lasky JA, Limper AH, et al; Imatinib-IPF Study Investigators. Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am J Respir Crit Care Med. 2010; 181(6):604–610. | ||
Ottmann OG, Druker BJ, Sawyers CL, et al. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood. 2002;100(6):1965–1971. | ||
Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011; 365(12):1079–1087. | ||
Obach RS, Baxter JG, Liston TE, et al. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther. 1997;283(1):46–58. | ||
De Buck SS, Sinha VK, Fenu LA, Nijsen MJ, Mackie CE, Gilissen RA. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab Dispos. 2007;35(10):1766–1780. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.