")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 15
Budd–Chiari Syndrome as an Initial Presentation of Systemic Lupus Erythematosus Associated with Antiphospholipid Syndrome: A Case Report with Review of the Literature
Received 12 June 2023
Accepted for publication 11 August 2023
Published 16 August 2023 Volume 2023:15 Pages 139—143
DOI https://doi.org/10.2147/OARRR.S425535
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Gashaw Solela,1 Merga Daba2
1Department of Internal Medicine, Yekatit 12 Hospital Medical College, Addis Ababa, Ethiopia; 2Department of Internal Medicine, College of Health Sciences, Addis Ababa University, Addis Ababa, Ethiopia
Correspondence: Gashaw Solela, Department of Internal Medicine, Yekatit 12 Hospital Medical College, Addis Ababa, Ethiopia, Email [email protected]
Introduction: Budd–Chiari syndrome (BCS) is a rare disorder characterized by hepatic outflow obstruction. It can be classified as primary or secondary BCS. Common causes of BCS include myeloproliferative diseases, infections, malignancies, and systemic autoimmune illnesses. Systemic lupus erythematosus (SLE) can be complicated with BCS. However, only a few case reports have described the uncommon occurrence of BCS as a primary presentation of SLE.
Case Presentation: We report the case of a 32-year-old female patient who presented with progressive abdominal distension of four months. On the abdominal CT scan, the left and middle hepatic veins were not visualized; the right hepatic vein and intrahepatic IVC had luminal narrowing; and there was caudate lobe enlargement suggestive of Budd–Chiari syndrome (BCS). Six months after the diagnosis of BCS, the patient developed other clinical features suggestive of systemic lupus erythematosus (SLE) and was finally diagnosed with SLE.
Conclusion: Acquired or inherited thrombotic conditions are the most common underlying causes of Budd–Chiari syndrome. Systemic lupus erythematosus (SLE) is the most common cause of secondary APS and most patients present with Budd–Chiari syndrome as a manifestation of APS after the diagnosis of SLE. In rare cases, such as the current case, Budd–Chiari syndrome can present even before the diagnosis of SLE. Hence, we would like to emphasize that Budd–Chiari syndrome can be an initial presentation of SLE.
Keywords: Budd–Chiari syndrome, systemic lupus erythematosus, antiphospholipid syndrome
Introduction
Budd-Chiari syndrome (BCS) is a rare disorder characterized by hepatic venous outflow blockage, regardless of the severity or mechanism of obstruction, with the exception of functional obstruction caused by congestive heart failure.1 It can be classified as primary or secondary BCS. Primary BCS is caused by primary venous lesions, including thrombosis, phlebitis, or fibrosis, whereas secondary BCS occurs when the initial lesion is outside the hepatic venous system.1,2
In a case series, the most common underlying causes for BCS were myeloproliferative disorders and other conditions, including inherited or acquired thrombophilia, oral contraceptive use, pregnancy within three months of diagnosis, connective tissue diseases, inflammatory bowel disease, Behçet’s disease, and sarcoidosis.3
Hepatic involvement in patients with systemic lupus erythematosus (SLE) may be due to a wide range of factors, including drug-induced damage, steatosis, viral hepatitis, vascular thrombosis, overlap with autoimmune hepatitis, or the SLE itself.4 Hepatic complications attributable to antiphospholipid antibodies include Budd–Chiari syndrome, hepatic veno-occlusive disease, nodular regenerative hyperplasia, liver infarction, and transient elevation of hepatic enzymes resulting from multiple fibrin thrombi.5
It is rare to see Budd–Chiari syndrome as an initial presentation of SLE and we report a 32-year-old female patient who was initially diagnosed with Budd–Chiari syndrome, and 6 months later started to develop other clinical features of SLE.
Case Presentation
A 32-year-old female patient presented with progressive abdominal distension of four months. She had also right upper quadrant abdominal pain, low-grade intermittent fever, and night sweating of the same duration, along with a weight loss of 6 kg over a 4-month period. For these complaints, she visited a nearby hospital, and with the impression of peritoneal tuberculosis, she was started on antituberculosis drugs, but there was no improvement. The patient had no history of oral contraceptive use or recurrent pregnancy loss. The patient tested negative for HIV. The patient had no history of diabetes mellitus, hypertension, alcohol consumption, or cigarette smoking.
Upon physical examination, she was well looking, with a blood pressure of 100/75 mmHg, pulse rate of 80 beats per minute, respiratory rate of 18 breaths per minute, and body temperature of 37.1 °C. She had pink conjunctivae and non-icteric sclerae. Abdominal examination revealed a grossly distended abdomen that moved with respiration, with positive shifting dullness and fluid thrill. No abdominal tenderness or palpable organomegaly was noted.
She had a complete blood count of 4800 cells/µL, hemoglobin of 14.6 g/dL, and platelet count of 227,000 cells/µL. Liver enzyme, serum albumin, and bilirubin levels were within normal ranges. Tests for hepatitis B and C viral markers yielded negative results. Ascitic fluid analysis showed a white blood cell count of 250 cells/µL, protein of 4.1 g/dL, glucose of 106 g/dL, negative gram stain and negative AFB. The serum ascitic albumin level gradient (SAAG) was 1.3. Cytology of the ascitic fluid was significant for lymphocytosis, but no malignant cells were observed.
Abdominal ultrasonography revealed a liver size of 16 cm with mild coarse echo texture, an enlarged caudate lobe, nonvisualized hepatic veins, and free fluid collections within the peritoneum. On the abdominal CT scan, the left and middle hepatic veins were not visualized; the right hepatic vein and intrahepatic IVC had luminal narrowing, and there was caudate lobe enlargement (Figures 1 and 2). Upper gastrointestinal endoscopy revealed grade 1 esophageal varices.
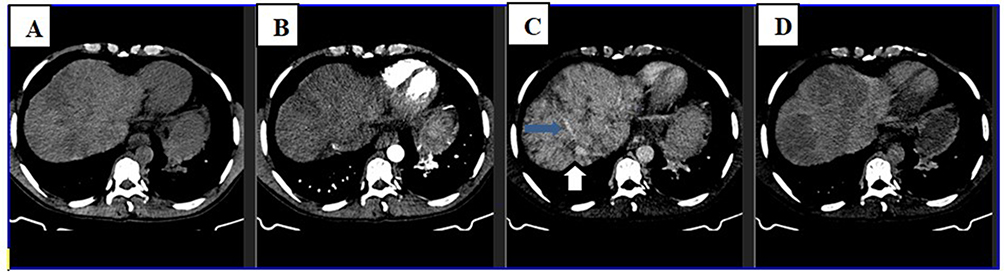 |
Figure 1 Axial section of triphasic abdominal CT scan at the level of upper liver shows heterogeneous liver parenchyma on precontrast image (A) that shows caudate lobe and central parenchymal enhancement on arterial and portal phases (B and C). The periphery of the liver enhances with central low density on delayed image (D). The middle and left hepatic veins are not visualized, while right hepatic vein and intrahepatic IVC have luminal narrowing (blue and white arrows on C respectively). |
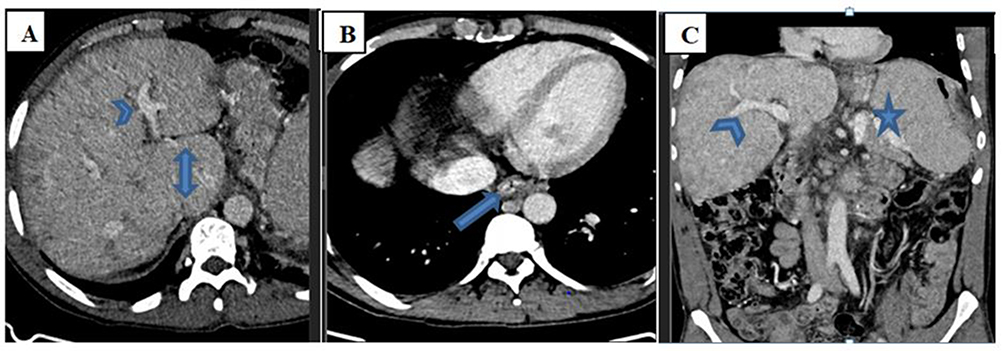 |
Figure 2 Axial (A and B) and coronal (C) portal venous phase abdominal CT scan at different levels show caudate lobe enlargement (double arrow on A), splenomegaly (star on C) and esophageal varices (arrow on B). The portal veins are patent (arrowheads on A and C). |
After the diagnosis of Budd–Chiari syndrome (BCS) was confirmed by abdominal computed tomography (CT), the patient was started on anticoagulation therapy with unfractionated heparin overlapped with warfarin followed by warfarin alone. The patient was also started on diuretics (furosemide with spironolactone) and beta blocker (propranolol 20mg PO BID). During follow-up, she showed marked symptomatic improvement with a decrease in the amount of ascites. However, after 6 months of BCS diagnosis, while on follow-up, she started to develop bilateral knee joint pain with swelling, alopecia, and oral lesions. After these manifestations developed, the patient was further investigated for systemic lupus erythematosus (SLE) and antiphospholipid (APS) and laboratory tests revealed positive antinuclear antibodies (ANA) with titer of 1:320, positive anti-double-stranded DNA (dsDNA) antibodies with level of 40 IU/mL (reference range: <30 IU/mL), and positive antiphospholipid antibodies (anticardiolipin antibodies and beta 2 glycoprotein 1 antibodies).
The patient was finally diagnosed with Budd–Chiari syndrome secondary to SLE associated with APS and was initiated on chloroquine phosphate 250mg PO 5 times per week and steroid (prednisolone), while warfarin, diuretics and propranolol were continued. The patient was counseled on the need of advanced therapies like transjugular intrahepatic portosystemic shunting (TIPS) placement, surgical shunting or liver transplantation if any worsening of the symptoms of BCS despite medical therapies.
Discussion
Patients with Budd-Chiari syndrome (BCS) often present with abdominal pain and/or distension, hepatomegaly, and gastrointestinal bleeding. Fewer common presenting features include jaundice, fever, lower extremity edema, and hepatic encephalopathy.6 Most of these common clinical manifestations were present in the current case, though the liver size was borderline and there was no jaundice.
As reported in a case series of 163 patients with BCS, acquired or inherited thrombotic conditions like myeloproliferative disorders are the most common underlying causes of BCS. Other risk factors identified are the use of oral contraceptives, pregnancy, connective tissue diseases, inflammatory bowel disease, Behcet's disease, and sarcoidosis.3 Unlike the common underlying conditions of BCS reported in this case series, our patient had systemic lupus erythematous (SLE) associated with antiphospholipid syndrome (APS) as a rare cause of BCS.
Thrombosis is a hallmark of APS, with venous thrombosis being more common than arterial thrombosis. While the most common site of thrombosis is the deep veins of the lower extremities, other sites include the inferior vena cava and pelvic, renal, pulmonary, hepatic, portal, subclavian, and cerebral veins.7
A prospective study of 1000 patients with antiphospholipid syndrome (APS) found that 53.1% had primary APS, and the most common associated disorder (36.2%) was systemic lupus erythematosus.8 A cross-sectional study found that arterial thrombosis, venous thrombosis, and fetal loss were more frequent in patients with APS and SLE than in those with primary APS.9 Similar to the findings of this study, our patient had clinical and laboratory features of SLE and APS and presented with BCS (hepatic vein thrombosis), although evidences of arterial thrombosis and fetal loss were lacking.
There are only a few case reports of APS with Budd-Chiari syndrome, and most of these patients were diagnosed with APS after a diagnosis of SLE had been made. Contrary to the common temporal association, our patient was initially diagnosed with Budd–Chiari syndrome, and several months later, she developed the clinical features of SLE. Such presentations are rare, with the exception of a few case reports in the literature.10–12
According to the Asian Pacific Association for the Study of the Liver (APASL) consensus guidelines, anticoagulants are recommended as first-line therapy, with vitamin K antagonists preferred over direct-acting anticoagulants.13 Hepatic venous revascularization with angioplasty and stenting is recommended as the treatment of choice for patients with short-length obstructive lesions, while transjugular intrahepatic portosystemic shunting (TIPS) is recommended in patients who lack improvement in symptoms of portal hypertension or liver function.13 Consistent with other guidelines, surgical shunting and liver transplantation are recommended when other modalities are not effective.13 Patients with BCS treated with such a stepwise management strategy had good long-term outcomes.14,15 Based on the APASL consensus guidelines, our patient was put on anticoagulation (warfarin) and the other supportive therapies, while the other options of management like TIPS and surgical shunting were planned if any worsening of the symptoms of BCS despite medical therapies.
Conclusion
Acquired or inherited thrombotic conditions are the most common underlying causes of Budd–Chiari syndrome. Systemic lupus erythematosus (SLE) is the most common cause of secondary APS and most patients present with Budd–Chiari syndrome as a manifestation of APS after the diagnosis of SLE. In rare cases, such as the current case, Budd–Chiari syndrome can present even before the diagnosis of SLE. Hence, we would like to emphasize that Budd–Chiari syndrome can be an initial presentation of SLE.
Ethical Approval
Institutional approval was not required for the publication of the patient’s case details.
Consent
Informed written consent was obtained from the patient for the publication of her case details including the history, physical findings, laboratory reports and images.
Funding
No funding organization was involved in this case report.
Disclosure
The authors declared no conflicts of interest in this work.
References
1. Ludwig J, Hashimoto E, McGill DB, van Heerden JA. Classification of hepatic venous outflow obstruction: ambiguous terminology of the Budd-Chiari syndrome. Mayo Clin Proc. 1990;65:51–55. doi:10.1016/S0025-6196(12)62109-0
2. Valla DC. Budd-Chiari syndrome and veno-occlusive disease/sinusoidal obstruction syndrome. Gut. 2008;57:1469. doi:10.1136/gut.2007.133637
3. Darwish Murad S, Plessier A, Hernandez-Guerra M, et al. EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med. 2009;151(3):167–175. PMID: 19652186. doi:10.7326/0003-4819-151-3-200908040-00004
4. Grover S, Rastogi A, Singh J, Rajbongshi A, Bihari C. Spectrum of histomorphologic findings in liver in patients with SLE: a review. Hepat Res Treat. 2014;2014:562979. PMID: 25136456; PMCID: PMC4130189. doi:10.1155/2014/562979
5. Hallegua DS, Venuturupalli S. Gastrointestinal and hepatic manifestations. In: Wallace DJ, Hahn BH, editors. Dubois’ Lupus Erythematosus and Related Syndromes. Vol. 8. Philadelphia: Elsevier Saunders; 2013:420.
6. Darwish Murad S, Plessier A, Hernandez-Guerra M, et al. Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med. 2009;151:167. doi:10.7326/0003-4819-151-3-200908040-00004
7. Zuily S, Regnault V, Guillemin F, et al. Superficial vein thrombosis, thrombin generation and activated protein C resistance as predictors of thromboembolic events in lupus and antiphospholipid patients. A prospective cohort study. Thromb Res. 2013;132:e1. doi:10.1016/j.thromres.2013.04.012
8. Cervera R, Serrano R, Pons-Estel GJ, et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis. 2015;74:1011. doi:10.1136/annrheumdis-2013-204838
9. Danowski A, de Azevedo MN, de Souza Papi JA, Petri M. Determinants of risk for venous and arterial thrombosis in primary antiphospholipid syndrome and in antiphospholipid syndrome with systemic lupus erythematosus. J Rheumatol. 2009;36(6):1195–1199. PMID: 19447935. doi:10.3899/jrheum.081194
10. Espinosa G, Font J, García-Pagan JC, et al. Budd-Chiari syndrome secondary to antiphospholipid syndrome: clinical and immunologic characteristics of 43 patients. Medicine. 2001;80(6):345–354. doi:10.1097/00005792-200111000-00001
11. Ilkgül O, Içöz G, Dayangaç M, Tokat Y, Ozütemiz O. A case of antiphospholipid antibody syndrome with Budd-Chiari and colonic ulcers complicated with gastrointestinal hemorrhage. Turk. J Gastroenterol Off J Turk Soc Gastroenterol. 2004;15(2):115–116.
12. Seijo S, Plessier A, Hoekstra J, et al. Good long-term outcome of Budd-Chiari syndrome with a step-wise management. Hepatology. 2013;57:1962–1968. doi:10.1002/hep.26306
13. Shukla A, Shreshtha A, Mukund A, et al. Budd-Chiari syndrome: consensus guidance of the Asian Pacific Association for the study of the liver (APASL). Hepatol Int. 2021;15:531–567. doi:10.1007/s12072-021-10189-4
14. Pandiaraja J, Sathyaseelan A. Budd- Chiari syndrome as an initial manifestation of systemic lupus erythematosus. J Clin Diagn Res. 2016;10(4):OD01–2. PMID: 27190864; PMCID: PMC4866162. doi:10.7860/JCDR/2016/16623.7532
15. Rhee J, Demetris AJ, Abu Elmagd K, Rabinovitz M. Suprahepatic Budd Chiari syndrome treated with thrombectomy and cavoplasty. Dig Dis Sci. 2003;48(8):1637–1641. doi:10.1023/A:1024740512762
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.