")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Bardoxolone methyl induces apoptosis and autophagy and inhibits epithelial-to-mesenchymal transition and stemness in esophageal squamous cancer cells
Authors Wang Y, Yang Y, Zhao R, Pan S, Zhe H, He Z, Duan W, Zhang X, Yang T, Qiu J, Zhou S
Received 30 August 2014
Accepted for publication 7 October 2014
Published 17 February 2015 Volume 2015:9 Pages 993—1026
DOI https://doi.org/10.2147/DDDT.S73493
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Min Li
Yan-Yang Wang,1,2 Yin-Xue Yang,3 Ren Zhao,1 Shu-Ting Pan,2,4 Hong Zhe,1 Zhi-Xu He,5 Wei Duan,6 Xueji Zhang,7 Tianxin Yang,8 Jia-Xuan Qiu,4 Shu-Feng Zhou2,5
1Department of Radiation Oncology, General Hospital of Ningxia Medical University, Yinchuan, People’s Republic of China; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA; 3Department of Colorectal Surgery, General Hospital of Ningxia Medical University, Yinchuan, People’s Republic of China; 4Department of Oral and Maxillofacial Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, 5Guizhou Provincial Key Laboratory for Regenerative Medicine, Stem Cell and Tissue Engineering Research Center and Sino-US Joint Laboratory for Medical Sciences, Guiyang Medical University, Guiyang, People’s Republic of China; 6School of Medicine, Deakin University, Waurn Ponds, VIC, Australia; 7Research Center for Bioengineering and Sensing Technology, University of Science and Technology Beijing, Beijing, People’s Republic of China; 8Department of Internal Medicine, University of Utah and Salt Lake Veterans Affairs Medical Center, Salt Lake City, UT, USA
Abstract: Natural and synthetic triterpenoids have been shown to kill cancer cells via multiple mechanisms. The therapeutic effect and underlying mechanism of the synthetic triterpenoid bardoxolone methyl (C-28 methyl ester of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid; CDDO-Me) on esophageal cancer are unclear. Herein, we aimed to investigate the anticancer effects and underlying mechanisms of CDDO-Me in human esophageal squamous cell carcinoma (ESCC) cells. Our study showed that CDDO-Me suppressed the proliferation and arrested cells in G2/M phase, and induced apoptosis in human ESCC Ec109 and KYSE70 cells. The G2/M arrest was accompanied with upregulated p21Waf1/Cip1 and p53 expression. CDDO-Me significantly decreased B-cell lymphoma-extra large (Bcl-xl), B-cell lymphoma 2 (Bcl-2), cleaved caspase-9, and cleaved poly ADP ribose polymerase (PARP) levels but increased the expression level of Bcl-2-associated X (Bax). Furthermore, CDDO-Me induced autophagy in both Ec109 and KYSE70 cells via suppression of the phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) signaling pathway. There were interactions between the autophagic and apoptotic pathways in Ec109 and KYSE70 cells subject to CDDO-Me treatment. CDDO-Me also scavenged reactive oxygen species through activation of the nuclear factor (erythroid-derived 2)-related factor 2 (Nrf2) pathway in Ec109 and KYSE70 cells. CDDO-Me inhibited cell invasion, epithelial–mesenchymal transition, and stemness in Ec109 and KYSE70 cells. CDDO-Me significantly downregulated E-cadherin but upregulated Snail, Slug, and zinc finger E-box-binding homeobox 1 (TCF-8/ZEB1) in Ec109 and KYSE70 cells. CDDO-Me significantly decreased the expression of octamer-4, sex determining region Y-box 2 (Sox-2), Nanog, and B lymphoma Mo-MLV insertion region 1 homolog (Bmi-1), all markers of cancer cell stemness, in Ec109 and KYSE70 cells. Taken together, these results indicate that CDDO-Me is a promising anticancer agent against ESCC. Further studies are warranted to explore the molecular targets, efficacy and safety of CDDO-Me in the treatment of ESCC.
Keywords: CDDO-Me, esophageal squamous cell carcinoma, cell cycle, apoptosis, autophagy, EMT, stemness, Akt, mTOR
Introduction
Esophageal cancer is the eighth most frequent cancer, and is the sixth leading cause of cancer-related death worldwide. It is a growing health concern that is expected to increase in incidence by 140% by 2025.1 There were an estimated 482,300 new esophageal cancer cases and 406,800 deaths globally in 2008.2 Incidence rates vary internationally by nearly 16-fold, with the highest rates observed in Southern and Eastern Africa and Eastern Asia, and the lowest rates in Western and Middle Africa and Central America in both males and females.2 According to the National Cancer Institute, it was estimated that there were 18,170 new esophageal cancer cases and 15,450 deaths from esophageal cancer in the USA in 2014.3 The age-adjusted incidence of esophageal cancer in the USA is 4.4 per 100,000 men and women per year based on 2007–2011 case numbers. This disease is three to four times more common among men than among women. In People’s Republic of China, the estimated age-adjusted incidence rate of esophageal cancer in 2008 was 16.7 per 100,000 population.4 About half of all cases globally occur in People’s Republic of China. Esophageal cancer is the 19th most common cancer in Europe, with around 45,900 new cases diagnosed in 2012 (1% of the total). In 2011, there were 8,332 new cases of esophageal cancer in the UK, 5,582 (67%) in men and 2,750 (33%) in women. The two main subtypes of esophageal cancer are squamous cell carcinoma, which is more common in the developing countries, and adenocarcinoma, which is more common in the developed countries. The risk factors of esophageal squamous cell carcinoma (ESCC) include tobacco, alcohol, very hot drinks, and a poor diet, while the most common causes of the adenocarcinoma type are smoking tobacco, obesity, and acid reflux.5,6 Squamous cell carcinoma used to be the predominant histological type of esophageal cancer (ESCC), but now, adenocarcinoma has become the primary type of esophageal cancer in the USA, accounting for 80% of cases.6,7 Surgical resection is considered the mainstay of curative treatment for ESCC, though it can only be performed in a small subgroup of patients. Most patients diagnosed with advanced disease or recurrence within 5 years after surgery will receive chemotherapeutic treatment. However, the response rate of traditional chemotherapy drugs, such as fluorouracil and cisplatin, is low (15%–20%) with remarkable side effects.5,6,8 Radiotherapy is given before, during, or after chemotherapy or surgery. Five-year survival rates for esophageal cancer are around 13%–18%.8 Thus, there is an urgent need to find more effective and safer therapeutic agents for esophageal cancer.
Naturally occurring triterpenoids like oleanolic acid and ursolic acid only show weak anti-inflammatory and anticarcinogenic activities.9 To increase their pharmacological efficacy, oleanolic acid has been modified to synthesize a series of new derivatives. The synthetic triterpenoids, such as 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO) and its derivatives including CDDO methyl ester (CDDO-Me; also called RTA 402, NSC 713200, and TP-155; its chemical structure is shown in Figure 1A), the methyl amide of CDDO, the ethyl amide of CDDO, and CDDO-imidazolide, are compounds originally developed for the prevention and treatment of inflammation and cancer.9,10 These synthetic triterpenoids are potent inhibitors of the de novo synthesis of inflammatory enzymes such as inducible nitric oxide synthase and inducible cyclooxygenase 2.10 CDDO-Me has improved the renal function in patients with chronic kidney disease associated with type 2 diabetes in a Phase II study.11 However, a higher rate of cardiovascular events including heart failure, hospitalizations, and deaths with CDDO-Me than with placebo prompted termination of a Phase III trial in October 2012.12 CDDO-Me is under clinical investigation for the treatment of pulmonary hypertension in the USA and chronic kidney disease associated with type 2 diabetes in Japan. Synthetic triterpenoids represent a novel group of small molecules currently being evaluated as anticancer agents. CDDO-Me (also known as bardoxolone methyl and RTA402) is more potent than CDDO in anticancer and cancer-preventive activities and in the activation of Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1/nuclear factor (erythroid-derived 2)-related factor 2/antioxidant response element (Keap1/Nrf2/ARE) pathway, which is involved in cytoprotection in the presence of excessive electrophiles or oxidative stress.9,13,14 The cytotoxic and antitumor effects of CDDO-Me have been reported in a variety of cancer cells in vitro and xenograft tumor-bearing nude mice.15–19 Cancer cells tested for CDDO-Me included prostate cancer, breast cancer, lung cancer, pancreatic cancer, osteosarcoma, and leukemia cell lines.18,19 CDDO-Me has also shown promising activities against several types of cancers in clinical trials. Nagaraj et al20 explored the efficacy and safety of CDDO-Me in 19 patients with pancreatic adenocarcinoma. Patients were treated intravenously with gemcitabine (1,000 mg/m2) weekly on days 1, 8, and 15. CDDO-Me was administered orally once daily for 21 days. Nine patients received a dose of 150 mg/day; two patients, 200 mg/day; six patients, 250 mg/day; and two patients, 300 mg/day. CDDO-Me abrogated the immune-suppressive effect of myeloid-derived suppressor cells and improved immune responses in cancer patients. CDDO-Me was well tolerated. Hong et al21 investigated the efficacy and safety of CDDO-Me in 47 patients with advanced solid tumors and lymphoma in a Phase I study. The maximum tolerated dose was established as 900 mg/day. A complete tumor response occurred in a mantle cell lymphoma patient, and a partial response was observed in an anaplastic thyroid carcinoma patient.21 NAD(P)H:quinone oxidoreductase-1 (NQO1) mRNA levels were increased in peripheral blood mononuclear cells, while nuclear factor-κB (NF-κB) and cyclin D1 levels were decreased in tumor biopsies. CDDO-Me was tolerated well.
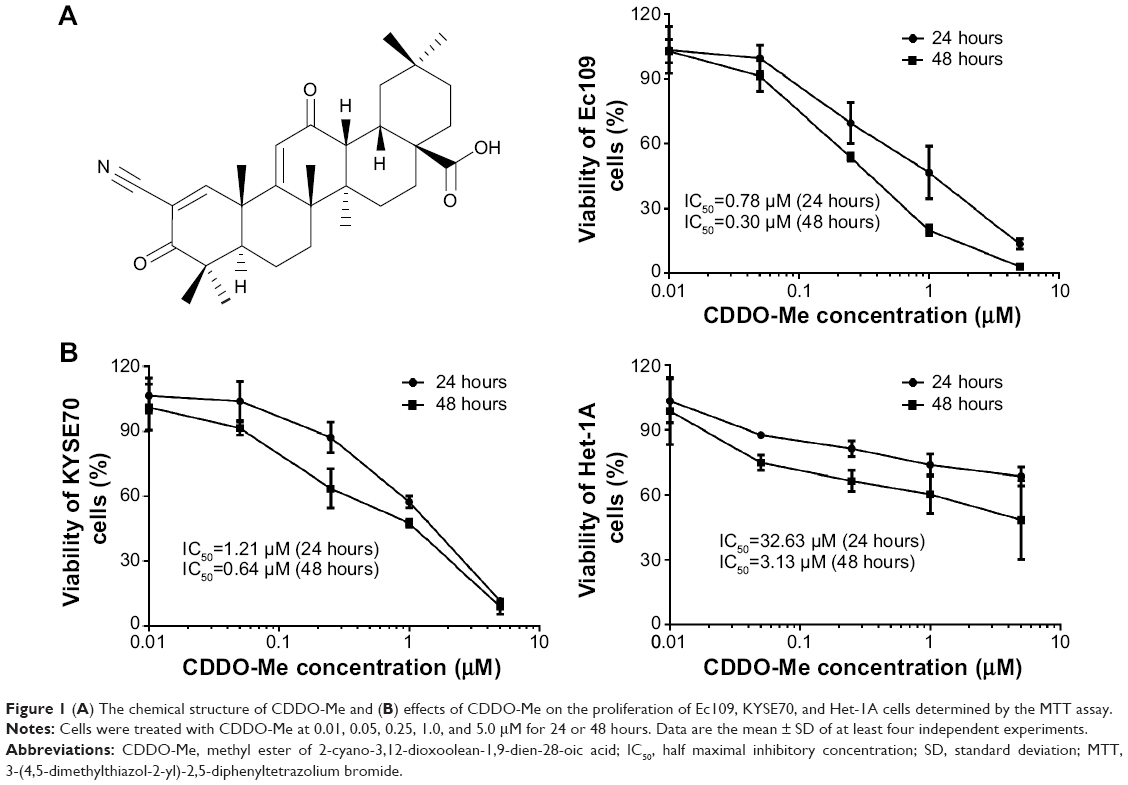 | Figure 1 (A) The chemical structure of CDDO-Me and (B) effects of CDDO-Me on the proliferation of Ec109, KYSE70, and Het-1A cells determined by the MTT assay. |
CDDO-Me acts as an activator of the Nrf2 pathway and an inhibitor of the NF-κB pathway. It contains α,β-unsaturated carbonyl groups on rings A and C that can generate reversible adducts with the thiol groups of Cys residues in target proteins such as Keap1 and the inhibitor of κB kinase.9,13,14,18 At low nanomolar concentrations, CDDO-Me protects the cells against oxidative stress via inhibition of reactive oxygen species (ROS) generation, while CDDO-Me at low micromolar concentrations induces apoptosis by increasing ROS and decreasing intracellular glutathione levels.9,13,14 The mechanisms for the anticancer activities of CDDO-Me are not fully understood. CDDO-Me has been shown to inhibit the proliferation of cancer cells, arrest cancer cells in G2/M phase, and induce apoptosis.22–24 Preclinical studies have suggested that the mitochondrial pathway, Keap1/Nrf2/ARE pathway, phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) pathway, Janus-activated kinase-1/signal transducer and activator of transcription-3 pathway, Notch 1 signaling pathway, and many other signaling pathways are involved in the anticancer effect of CDDO-Me. However, the therapeutic effects of CDDO-Me on ESCC and the underlying mechanisms have not been reported. In this study, we evaluated the therapeutic potential and the underlying mechanisms of CDDO-Me in human ESCC Ec109 and KYSE70 cells.
Materials and methods
Chemicals and reagents
CDDO-Me (also called RTA 402, NSC 713200, and TP-155) was obtained from Cayman Chemical Inc. (Ann Arbor, MI, USA) with a purity of more than 98%. The PI3K inhibitor wortmannin, 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), and the specific p38 mitogen-activated protein kinase (p38 MAPK) inhibitor SB202190 [4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole] were bought from Invitrogen Inc. (Carlsbad, CA, USA). The protein kinase B (Akt) inhibitor MK-2206 and the mTOR inhibitor rapamycin were purchased from Selleck Chemicals Inc. (Houston, TX, USA). The 5′-AMP-activated protein kinase (AMPK) inhibitor Compound C (dorsomorphin; 6-[4-(2-piperidin-1-ylethoxy)phenyl]-3-pyridin-4-ylpyrazolo[1,5-a]pyrimidine) was obtained from Abcam Inc. (Cambridge, MA, USA). Trypsin, heat-inactivated fetal bovine serum (FBS), and penicillin/streptomycin antibiotic mixture were from Mediatech Inc. (Manassas, VA, USA). The Roswell Park Memorial Institute medium (RPMI)-1640 medium was obtained from Coring Cellgro Inc. (Herndon, VA, USA). The bronchial epithelial cell growth medium (BEGM™) BulletKit™ was bought from Lonza Group Ltd. Co. (Walkersville, MD, USA). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), and Dulbecco’s phosphate-buffered saline (PBS) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). The BD Pharmingen™ annexinV:phycoerythrin (PE) apoptosis detection kit was purchased from BD Biosciences Inc. (San Jose, CA, USA). Cyto-ID® Green Autophagy Detection Kit was obtained from Enzo Life Sciences Inc. (Farmingdale, NY, USA). QCM™ ECMatrix 96-well Cell Invasion Assay kit and polyvinylidene difluoride (PVDF) membrane were purchased from EMD Millipore Inc. (Bedford, MA, USA). Western blot substrate was obtained from Thermo Scientific Inc. (Hudson, NH, USA). Primary antibodies against human p21Waf1/Cip1, p53, cyclin B1, cyclin-dependent kinase 1 (CDK1/Cdc2), the p53 upregulated modulator of apoptosis (PUMA), B-cell lymphoma 2 (Bcl-2), Bcl-2-like protein 4/Bcl-2-associated X protein (Bax), Bcl-extra large (Bcl-xl), cytochrome c, cleaved caspase-3, cleaved caspase-9, poly ADP ribose polymerase (PARP), p38, phosphorylated (p-) p38 at Thr180/Tyr182, AMPK, p-AMPK at Thr 172, Akt, p-Akt at Ser473, mTOR, p-mTOR at Ser2448, PI3K, p-PI3K/p85 at Tyr458, phosphatase and tensin homolog (PTEN), beclin1, microtubule-associated protein 1A/1B-light chain 3 (LC3)-I, LC3-II, N-cadherin, E-cadherin, zona occludens protein 1 (ZO-1), β-catenin, Snail, Slug, zinc finger E-box-binding homeobox 1 (TCF-8/ZEB1), vimentin, claudin 1, octamer-4 (Oct-4), B lymphoma Mo-MLV insertion region 1 homolog (Bmi-1), Nanog, or sex determining region Y-box 2 (Sox-2) were all purchased from Cell Signaling Technology Inc. (Beverly, MA, USA). The antibodies against human Nrf2, heme oxygenase-1 (HO-1), NQO1, glutathione S-transferase (GST), histone H3, and β-actin were purchased from Santa Cruz Inc. (Santa Cruz, CA, USA).
Cell lines, cell culture, and CDDO-Me treatment
The human ESCC cell line Ec109 was obtained from AddexBio Inc. (San Diego, CA, USA), and KYSE70 cells were purchased from Sigma-Aldrich Co. The normal human esophageal epithelial cell line Het-1A was bought from American Type Culture Collection (Manassas, VA, USA). The Het-1A cell line was derived from human esophageal autopsy tissue by transfection with plasmid pRSV-T consisting of the RSV-LTR promoter and the sequence encoding the simian virus 40 large T-antigen. The Ec109 and KYSE70 cells were cultured in RPMI-1640 medium with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% heat-inactivated FBS, and Het-1A was cultured in BEGM™ BulletKit™. All cells were maintained at 37°C in a saturated humidity atmosphere containing 95% air and 5% CO2. For experimental purposes, cells in exponential growth phase (approximately 70%–80% confluency) were used. The stock solution of CDDO-Me was prepared and stored at −20°C. The working solution of CDDO-Me was freshly diluted to the desired concentrations with culture medium. The final concentration of DMSO was at 0.05% (v/v).
Cell viability assay
The cell viability analysis was performed by MTT assay as described previously.25 Briefly, Ec109, KYSE70, and Het-1A cells were seeded in a 96-well plate at a density of 8,000 cells each well and cultured overnight for attachment. The next day, the cells were treated with varying concentrations of CDDO-Me (0–5 μM) for 24 and 48 hours. Following drug treatment, the cells were incubated with 5 mg/mL MTT (10 μL/well) for 4 hours at 37°C. The reaction was terminated by the addition of 100 μL DMSO. The absorbance was measured using a Synergy™ H4 Hybrid microplate reader (BioTek Inc., Winooski, VT, USA) at a wavelength of 450 nm. The absorbance values were normalized by assigning the value of the control line in the medium without drug to 1.0 and the value of the no-cell control to 0. The half inhibitory concentration (IC50) was defined as the CDDO-Me concentration required for decreasing the cells to 50% of the control value. Experiments were performed in at least triplicate.
Cell cycle analysis using flow cytometry
Flow cytometry was used to analyze the distribution of the cell cycle. The Ec109 and KYSE70 cells were incubated with various concentrations (0.25, 0.5, and 1.0 μM) of CDDO-Me for 24 hours. In a separate experiment, Ec109 and KYSE70 cells were treated with 0.5 μM CDDO-Me for 4, 8, 12, 24, 48, and 72 hours. After incubation, cells were harvested and fixed with 70% ethanol overnight at 4°C. Fixed cells were washed with PBS. Cells were then incubated with 1 U/mL of RNase A and 5 μg/mL of propidium iodide for 30 minutes at room temperature in the dark. Samples were analyzed for their DNA content by using an FACS Calibur flow cytometry with CellQuest™ software (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA). The number of cells in G0/G1, G2/M, and S phases was determined as a percentage of the total number of cells.
Quantification of cellular apoptosis using flow cytometry
Annexin V is a member of a calcium and phospholipid binding family of proteins with vascular anticoagulant activity. Annexin V is a 35-36 kDa Ca2+ dependent phospholipid-binding protein that has a high affinity for phosphatidylserine, and binds to cells with exposed phosphatidylserine. Annexin V can be conjugated to the fluorochrome PE, retaining its high affinity for phosphatidylserine and thus serving as a sensitive probe for flow cytometric analysis of apoptotic cells. Induction of apoptosis was assessed by the binding of annexin V to phosphatidylserine, which is externalized to the outer leaflet of the plasma membrane early during induction of apoptosis following the manufacturer’s protocols. In short, Ec109 and KYSE70 cells were treated with CDDO-Me at various concentrations (0.25, 0.5, and 1.0 μM) for 24 hours or with 0.5 μM CDDO-Me for different time intervals (4, 8, 12, 24, and 48 hours) and were resuspended in the binding buffer provided in the BD Pharmingen™ annexinV:PE apoptosis detection kit (BD Biosciences). Cells were mixed with 5 μL of annexin V:PE reagent and 5 μL of 7-amino-actinomycin D (7-AAD) and incubated for 15 minutes at room temperature in the dark. The double stained cells were analyzed by flow cytometer (Becton Dickinson Immunocytometry Systems) within 1 hour. By use of this detecting strategy, annexin V-negative/7-AAD-negative cells were defined as viable, annexin V-positive/7-AAD-negative cells as early apoptosis, annexin V-negative/7-AAD-positive cells as death, and annexin V-positive/7-AAD-positive cells as late apoptosis or necrosis.
Quantification of cellular autophagy using flow cytometry
Cellular autophagy was assessed using Cyto-ID® Green Autophagy Detection Kit (Enzo Life Sciences Inc.) by flow cytometry (LSRFortessa; BD Biosciences Inc.) according to the manufacturer’s instructions. The Cyto-ID® fluorescent reagents specifically detect the autophagic vacuoles formed during autophagy. In brief, Ec109 and KYSE70 cells were treated with CDDO-Me at various concentrations (0.25, 0.5, and 1.0 μM) for 24 hours. In separate experiment, Ec109 and KYSE70 cells were treated with 0.5 μM CDDO-Me for 4, 8, 12, 24, and 48 hours. At the end of the treatment, sample was washed by resuspending the cell pellet in 1× assay buffer (Enzo Life Sciences Inc.) and cells were collected by centrifugation as described previously.25 Cells were then resuspended in 250 μL phenol red-free culture medium (Invitrogen) containing 5% FBS, and 250 μL of the diluted Cyto-ID® Green stain solution was added to each sample and mixed well. Samples were then incubated at 37°C for 30 minutes in dark and analyzed after wash with 1× assay buffer in the green channel (FL1) of an FACS Calibur flow cytometer (BD Biosciences). The flow cytometer collected 10,000 events. The percentage of cells with Cyto-ID® staining was used to represent the relative formation or accumulation of autophagosomes. Results are expressed as the percentage of Cyto-ID®-positive cells of total live cells.
Quantification of cellular autophagy using confocal fluorescence microscopy
The autophagic level of Ec109 and KYSE70 cells was detected with Cyto-ID® Green Autophagy Detection Kit using confocal fluorescence microscopy. In brief, cells were plated on eight-well chamber slides, which were pre-coated with collagen I (BD Biosciences) at 40% confluency. After incubation with control or various concentrations of CDDO-Me (0.25, 0.5, and 1.0 μM), the cells were washed twice with 1× assay buffer and stained with Cyto-ID® detection agent according to the manufacturer’s instructions. The live cell imaging was performed on a Leica TCS SP2 laser scanning confocal microscopy (Leica Microsystems, Wetzlar, Germany) equipped with a live cell chamber device to maintain 37°C in 5% CO2 condition during experiments. A standard FITC filter set for imaging the autophagic signal at wavelength of 405 nm/488 nm was used for the detection. The intensity of cell fluorescence was calculated for the corrected total cell fluorescence using the program ImageJ (National Institute of Health, Bethesda, MD, USA).
Western blotting analysis
Nuclear and cytoplasmic extracts for Western blotting assay were obtained by using NE-PER® Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific Inc., Rockford, IL, USA) following the manufacturer’s instructions. Total protein (cytoplasm plus nucleus) was obtained from cell lysates using the radioimmunoprecipitation assay buffer containing a protease inhibitor cocktail. Protein levels were determined using the bicinchoninic acid assay kit. The samples (30 μg) were separated by 7%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis using a Mini-Protean® TGX™ Gel (BioRad Inc., Hercules, CA, USA) after boiling at 95°C for 5 minutes. The separated protein bands were then transferred to PVDF membranes. The membrane was incubated in a blocking buffer containing 5% nonfat dry milk in Tris-buffered saline and Tween 20 (TBST) buffer for 1 hour at room temperature followed by overnight incubation at 4°C with the indicated primary antibody. Then, the membranes were washed with TBST and incubated with the appropriate secondary antibody (1:5,000) for 1 hour at room temperature. Visualization was performed using an enhanced chemiluminescence kit (BioRad Inc.) with electro-chemiluminescence substrate. Protein level was normalized to the matching densitometric value of the internal control.
Simultaneous determination of apoptosis and autophagy using flow cytometry
To explore the potential cross talk between CDDO-Me-induced apoptosis and autophagy, we further determined the two modes of programmed cell death simultaneously after the combined treatment of different kinase inhibitors of autophagic pathway and CDDO-Me. In brief, the p38 MAP kinase inhibitor SB202190 (20 μM), the PI3K inhibitor wortmannin (10 μM), the Akt inhibitor MK-2206 (2.5 μM), the mTOR inhibitor rapamycin (0.5 μM), or the AMP-kinase inhibitor Compound C (10 μM) were used alone or in combination with 0.5 μM CDDO-Me to treat the cells for 24 hours. All these inhibitors were dissolved in DMSO at a final concentration of 0.05% (v/v). At the end of the treatment, the cells were trypsinized and centrifuged at 1,000× g for 5 minutes to pellet the cells. The cells were divided into two equal volume samples, and washed with PBS. The apoptosis was detected using the annexinV:PE apoptosis detection kit and the autophagy was determined using Cyto-ID® Green Autophagy Detection Kit by flow cytometry (LSRFortessa™; BD Biosciences Inc.) according to the manufacturer’s instructions at the same time. The flow cytometer collected 10,000 events.
Intracellular ROS measurement
Intracellular ROS levels were assessed using CM-H2DCFDA ROS detection kit (Invitrogen Inc.) according to the manufacturer’s protocol. CM-H2DCFDA is cleaved by intracellular esterases to produce an impermeable, nonfluorescent active form, which further reacts with ROS to form a fluorescent product. In short, Ec109 and KYSE70 cells were seeded in a 96-well plate at a density of 1×104 cells/well. Cells were treated with CDDO-Me at different concentrations or evaluated for different time intervals. After treatment, the cells were washed with PBS and then stained with CM-H2DCFDA (5 μM) for 30 minutes at 37°C, and analyzed for ROS levels using a Synergy™ H4 Hybrid microplate reader (BioTek Inc.) set at 485 nm and 530 nm excitation and emission wavelengths, respectively. After reading of ROS, cultures were then treated with Janus green, and cell counts were determined with the plate reader set to an absorbance of 610 nm, and ROS intensities were then corrected accordingly.
Cell invasion assay
Cell invasion assay was performed using a QCM™ ECMatrix Cell Invasion kit (EMD Millipore Inc.; Cat #: ECM555). In brief, 1×105 cells in FBS-free RPMI were added to the top chamber, and 10% FBS in RPMI was added to the bottom chamber as a chemoattractant. The plate was incubated for 24 hours at 37°C in a 5% CO2 incubator. At the end of the incubation period, the invasive cells were dissociated from the membrane with the cell detachment solution for 30 minutes at 37°C. Next, 50 μL of lysis buffer/CyQuant® GR Dye Solution (1:75) was added to each well and incubated for 15 minutes at room temperature. Finally, 150 μL of the mixture was transferred to a new 96-well plate, and the fluorescence value was detected with a Synergy™ H4 Hybrid microplate reader (BioTek Inc.) using 480 nm/520 nm filter set.
Statistical analysis
All statistical calculations were performed using Prism 5.0 (GraphPad Software Inc., San Diego, CA, USA). Experimental values were presented as the mean ± standard deviation (SD) of the mean. Comparisons of multiple groups were evaluated by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison procedure. A P-value of <0.05 was considered statistically significant.
Results
CDDO-Me inhibits the proliferation of Ec109 and KYSE70 cells
To test the effect of CDDO-Me on the proliferation of human ESCC cells, 8,000 Ec109 and KYSE70 cells were plated in 96-well plates overnight and then treated with CDDO-Me at concentrations of 0.01–5 μM for 24 and 48 hours. The normal human esophageal epithelial Het-1A cells were also treated with the same concentrations of CDDO-Me as human ESCC cell lines. At the end of the treatment, the viability of cultures was determined by the MTT assay. As shown in Figure 1B, treatment with CDDO-Me resulted in proliferative inhibition of ESCC and normal human esophageal epithelial cells in a concentration- and time-dependent manner. When cells were treated with CDDO-Me at concentrations from 0.01 to 5 μM for 24 and 48 hours, the percentage of cellular viability of Ec109 was 102.8%–13.5% and 102.9%–3.2%, respectively; the percentage of cellular viability of KYSE70 was 106.7%–11.6% and 101.2%–8.9%, respectively; and the percentage of cellular viability of Het-1A was 103.5%–68.6% and 98.8%–48.4%, respectively. The IC50 of CDDO-Me in Ec109 cells was 0.78 and 0.30 μM for 24- and 48-hour drug treatment, respectively (Figure 1B), and was 1.21 and 0.64 μM for 24- and 48-hour drug treatment in KYSE70 cells, respectively (Figure 1B). Moreover, the IC50 of CDDO-Me in Het-1A cells was 32.63 and 3.13 μM when cells were incubated for 24 and 48 hours, respectively (Figure 1B). These results suggest that CDDO-Me suppresses the growth of both human ESCC cells in a concentration- and time-dependent manner. CDDO-Me shows a significantly higher antiproliferative activity in ESCC cells than in normal human esophageal epithelial cells. When the drug exposure time is increased, the sensitivity of normal human esophageal epithelial cells to CDDO-Me was greatly enhanced.
CDDO-Me induces G2/M arrest in Ec109 and KYSE70 cells
To identify the mechanisms behind the cell growth inhibition, we determined the effect of CDDO-Me on cell cycle progression. Ec109 and KYSE70 cells were treated with various concentrations of CDDO-Me (0.25, 0.5, and 1.0 μM) and analyzed using flow cytometry. As shown in Figure 2A, both Ec109 and KYSE70 cells were arrested in G2/M phase after incubation with CDDO-Me for 24 hours, especially at the high concentration. The percentage of cells arrested in G2/M phase was increased by 16.3% in Ec109 cells (P<0.001 by one-way ANOVA) and 12.2% in KYSE70 cells (P<0.01 by one-way ANOVA) at 1.0 μM of CDDO-Me in comparison to the control cells. Exposure of human ESCC cells to CDDO-Me significantly reduced the number of cells in G1 phase compared to the control cells treated with the control vehicle only. The number of cells in G1 phase was significantly reduced by 16.2% (P<0.001 by one-way ANOVA) and 14.3% (P<0.001 by one-way ANOVA) in Ec109 and KYSE70 cells, respectively, when treated with CDDO-Me at 1.0 μM. In addition, treatment of Ec109 and KYSE70 cells with CDDO-Me at 0.5 μM significantly decreased the number of cells in S phase. The percentage of cells in S phase was decreased by 1.6% (P<0.05 by one-way ANOVA) and 6.6% (P<0.001 by one-way ANOVA), respectively, in Ec109 and KYSE70 cells. A lower or higher concentration of CDDO-Me (0.25 and 1.0 μM) did not significantly affect the number of both Ec109 and KYSE70 cells in S phase (P>0.05 by one-way ANOVA).
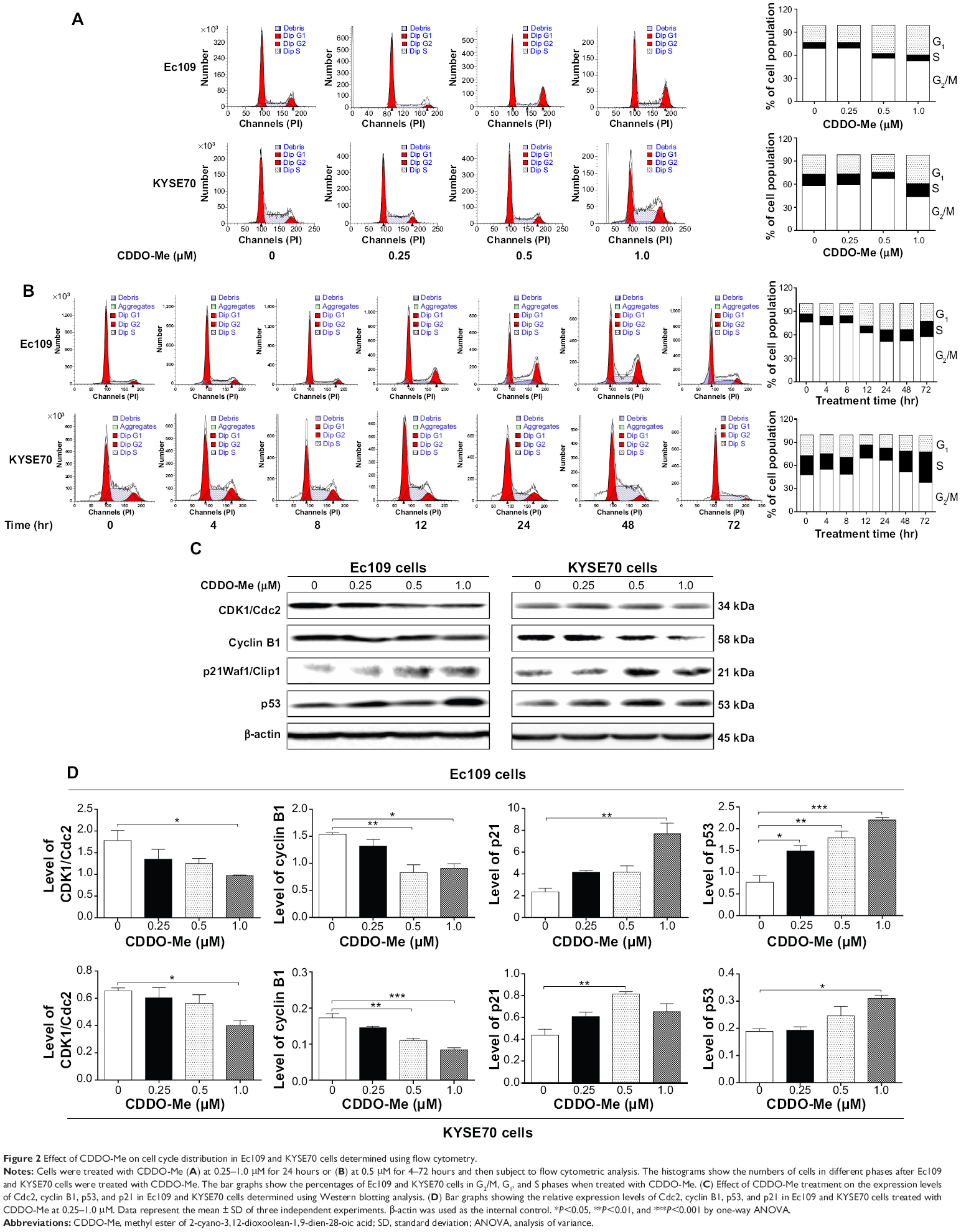 | Figure 2 Effect of CDDO-Me on cell cycle distribution in Ec109 and KYSE70 cells determined using flow cytometry. |
Next, we evaluated the cell cycle-arresting effect of CDDO-Me at 0.5 μM on both Ec109 and KYSE70 cells with different incubation times in a separate experiment. Treatment of Ec109 cells with 0.5 μM CDDO-Me for 4–72 hours significantly increased the percentage of cells in G2/M phase from 13.1% at basal level to 22.9% (P<0.01 by one-way ANOVA; Figure 2B). The number of Ec109 cells in G1 phase significantly decreased from 76.0% at basal level to 57.5% after 4–72-hour incubation with 0.5 μM CDDO-Me (P<0.01 by one-way ANOVA; Figure 2B). However, there was no significant difference observed in the number of cells in S phase in the time course evaluation. For KYSE70 cells, CDDO-Me exhibited a differential effect on the cell cycle progression. Compared to the control cells, the percentage of cells in G1 phase was increased from 48.1% at basal level to 51.7% after 4–48-hour treatment with CDDO-Me and declined to 38.1% after 72-hour treatment with CDDO-Me, while 0.5 μM CDDO-Me treatment reduced the percentage of cells in S phase from 24.9% at basal level to 15.9% after 4–24-hour incubation and the percentage of cells raised to 39.8% after 48–72-hour incubation with CDDO-Me. There was a significant decrease in the percentage of KYSE70 cells in G2/M phase after treatment with CDDO-Me for 12 hours (P<0.01 by one-way ANOVA; Figure 2B).
To delineate the molecular mechanism of CDDO-Me-mediated G2/M arrest, we determined its effect on the key regulators of cell cycle including Cdc2 and cyclin B1 for G2-to-M phase transition26 using Western blotting assay. A significant decrease in the Cdc2 and cyclin B1 expression level was observed in human ESCC cells treated with CDDO-Me. In Ec109 cells, treatment with CDDO-Me at 0.25, 0.5, and 1.0 μM for 24 hours significantly decreased the level of Cdc2 by 24.2%, 30.3%, and 44.9% (P<0.05 by one-way ANOVA; Figure 2C and D), and suppressed the expression of cyclin B1 by 14.4%, 46.1%, and 41.6%, respectively, compared to the control cells (P<0.05 or 0.01 by one-way ANOVA; Figure 2C and D). In KYSE70 cells, in comparison to the control cells, the expression level of Cdc2 was decreased 1.6-fold when treated with 1.0 μM CDDO-Me for 24 hours. In addition, there was a 42.1% and 55.7% reduction in the expression level of cyclin B1 in KYSE70 cells treated with CDDO-Me at 0.5 and 1.0 μM for 24 hours, respectively. Taken together, these results demonstrate that CDDO-Me arrests human ESCC cells in G2/M phase in a concentration- and time-dependent manner, with the involvement of Cdc2 and cyclin B1.
CDDO-Me upregulates p21Waf1/Cip1 and p53 expression in Ec109 and KYSE70 cells
To further investigate the molecular mechanism of CDDO-Me-mediated G2/M arrest, we determined the expression levels of p21Waf1/Cip1 and p53 in human ESCC cells by Western blot analysis. p21Waf1/Cip1 is a potent CDK inhibitor that binds to and inhibits the activity of cyclin–CDK2, cyclin–CDK1, and cyclin–CDK4/6 complexes, and thus functions as a regulator of cell cycle progression.27 The expression of p21Waf1/Cip1 is tightly controlled by the tumor suppressor protein p53, through which this protein mediates the p53-dependent cell cycle arrest in response to a variety of stress stimuli.28 Our results showed that there was a substantial increase in the p21Waf1/Cip1 expression level in Ec109 cells exposed to CDDO-Me. After incubation with 1.0 μM CDDO-Me, the expression level of p21Waf1/Cip1 was increased 3.3-fold compared to control cells (P<0.01 by one-way ANOVA; Figure 2C and D). In addition, there was a marked increase in the expression of p53 in a concentration-dependent manner in Ec109 cells. Treatment of Ec109 cells with CDDO-Me at 0.25, 0.5, and 1.0 μM for 24 hours significantly increased the level of p53 by 94.7%, 134.2%, and 188.2%, respectively (P<0.05, 0.01 or 0.001 by one-way ANOVA; Figure 2C and D).
Similarly, there was also a significant increase in the expression of p21Waf1/Cip1 in KYSE70 cells treated with CDDM-Me. Compared to the control cells, there was a 2.1-fold increase in the expression of p21Waf1/Cip1 in KYSE70 cells treated with CDDO-Me at 0.5 μM (P<0.01 by one-way ANOVA; Figure 2C and D). Similarly, the expression of p53 was also significantly increased in KYSE70 cells treated with CDDO-Me. CDDO-Me at 1 μM increased the expression level of p53 in KYSE70 cells by 64.9%, compared to the control cells (P<0.05 by one-way ANOVA; Figure 2C and D). These results indicate that CDDO-Me increases the level of p21Waf1/Cip1 via upregulating p53, contributing to its cell cycle-arresting effect in human ESCC cells.
CDDO-Me induces the apoptosis of Ec109 and KYSE70 cells via activation of mitochondrial pathway
To elucidate whether CDDO-Me decreases cell survival through the induction of apoptosis in human ESCC cells, we examined the effects of CDDO-Me on apoptosis in Ec109 and KYSE70 cells using flow cytometry. The number of apoptotic cells was first quantified by the binding of annexin V:PE to Ec109 and KYSE70 cells treated with CDDO-Me using flow cytometric analysis. As shown in Figure 3A, a small percentage of untreated Ec109 and KYSE70 cells bound annexin V:PE (5.3% and 5.6%, respectively). The percentage of annexin V:PE-binding Ec109 and KYSE70 cells markedly increased following treatment with CDDO-Me at concentrations of 0.25, 0.5, and 1 μM for 24 hours (Ec109: 6.7%, 10.6%, and 26.0%; KYSE70: 15.4%, 20.2%, and 25.2%, respectively).
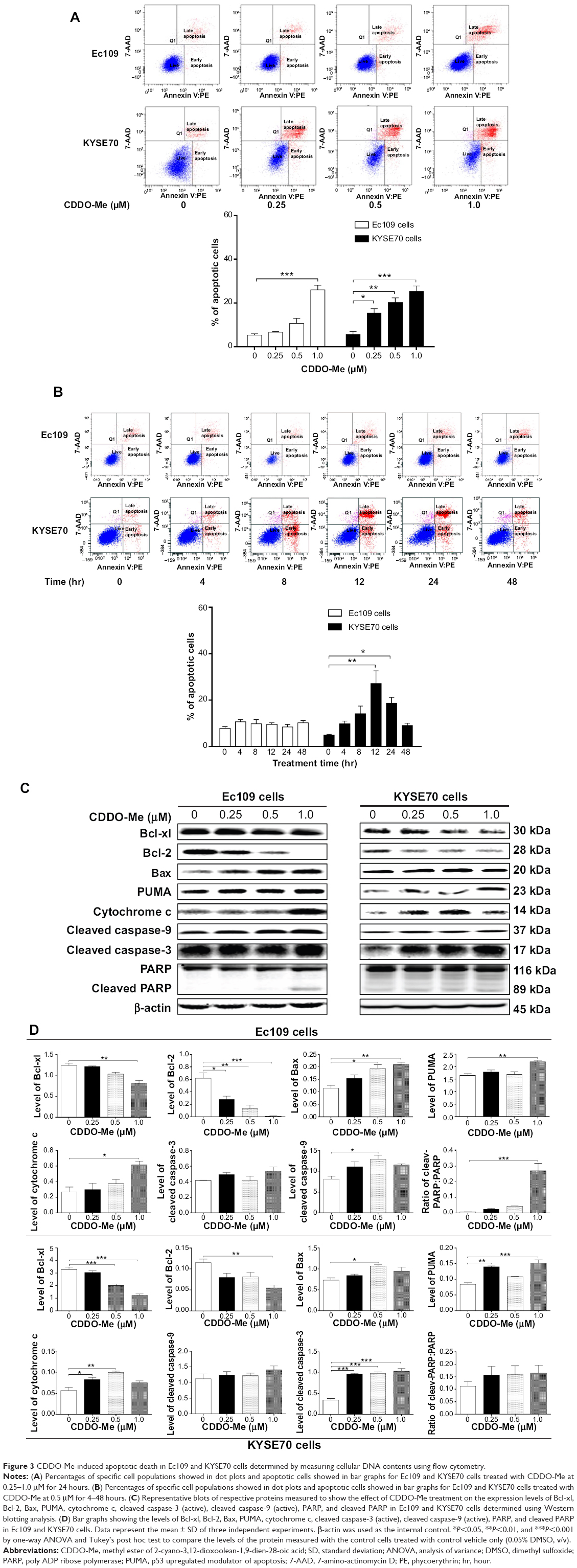 | Figure 3 CDDO-Me-induced apoptotic death in Ec109 and KYSE70 cells determined by measuring cellular DNA contents using flow cytometry. |
Next, we tested the apoptotic effect of 0.5 μM CDDO-Me on Ec109 and KYSE70 cells with different incubation time intervals by flow cytometric analysis. The results showed that 0.5 μM CDDO-Me did not trigger the apoptotic effect in Ec109 cells. However, in KYSE70 cells, a time-dependent apoptotic effect was demonstrated in the analysis. The percentage of the apoptotic cells was increased from 4.9% at the basal level to 18.7% after 24-hour incubation with 0.5 μM CDDO-Me (Figure 3B).
Bcl-2, Bcl-xl, Bax, and PUMA are major proteins involved in the apoptosis-regulating pathways.29 To investigate the mechanisms underlying CDDO-Me-induced apoptosis, we examined the expression of these key proteins upon CDDO-Me treatment by Western blotting assay. Ec109 and KYSE70 cells were exposed to different concentrations of CDDO-Me for 24 hours and cellular lysates were obtained for analysis. The results showed that CDDO-Me consistently decreased the level of Bcl-xl and Bcl-2 and increased the level of Bax and PUMA. Incubation of Ec109 cells with CDDO-Me at 1.0 μM significantly decreased Bcl-xl and Bcl-2 expression level by 34.7% and 98.3%, respectively, compared to the control cells (P<0.05, 0.01, or 0.001 by one-way ANOVA; Figure 3C and D), but significantly increased the level of Bax and PUMA by 90.9% and 33.5% (P<0.05 or 0.01 by one-way ANOVA; Figure 3C and D), respectively. Similarly, incubation of KYSE70 cells with CDDO-Me at 1.0 μM significantly decreased Bcl-xl and Bcl-2 expression level by 62.6% and 54.5% (P<0.01 or 0.001 by one-way ANOVA; Figure 3C and D), respectively, compared to the control cells, but significantly increased the level of PUMA by 46.7% compared to the control cells (P<0.01 or 0.001 by one-way ANOVA; Figure 3C and D). There was an increase in the expression level of Bax by CDDO-Me treatment at 0.5 μM in KYSE70 cells, whereas no such change was observed at 1.0 μM CDDO-Me.
In addition, the activation of caspases upon CDDO-Me treatment in human ESCC cells was also observed in the present study. We detected the presence of cleaved caspase-9, caspase-3, and PARP by Western blotting assay with specific antibodies, and found that their activation was significantly triggered upon CDDO-Me treatment for 24 hours. In Ec109 cells, the increase fold was 1.6 for cleaved caspase-9 and 26.0 for cleaved PARP. The cleaved caspase-3 level did not change under CDDO-Me treatment during the period of monitoring. For KYSE70 cells, the increase fold was 3.0 for cleaved caspase-3, but the levels of cleaved caspase-9 and cleaved PARP were not affected by CDDO-Me.
Cytochrome c is a component of the electron transport chain in mitochondria and is involved in the initiation of apoptosis.30 The release of cytochrome c was evaluated in both Ec109 and KYSE70 cells in the current study. Treatment of Ec109 and KYSE70 cells with CDDO-Me for 24 hours significantly increased the release of cytochrome c. Taken together, CDDO-Me decreases cell survival through the induction of apoptosis via activation of mitochondrial pathway in human ESCC cells.
CDDO-Me induces autophagy in Ec109 and KYSE70 cells via inhibition of PI3K/mTOR pathway
To check if CDDO-Me induced autophagy in human ESCC cells and the underlying mechanisms, we evaluated the autophagic cell death in Ec109 and KYSE70 cells exposed to various concentrations of CDDO-Me using flow cytometric analysis. As shown in Figure 4A and B, a small percentage of autophagic cells were detected in untreated Ec109 and KYSE70 cells (1.1% and 2.7%, respectively). The percentage of autophagic cells in Ec109 and KYSE70 cells markedly increased following treatment with CDDO-Me at concentrations of 0.25, 0.5, and 1 μM for 24 hours (Ec109 cells: 5.3%, 6.1%, and 11.3%; and KYSE70 cells: 3.0%, 3.7%, and 6.8%, respectively).
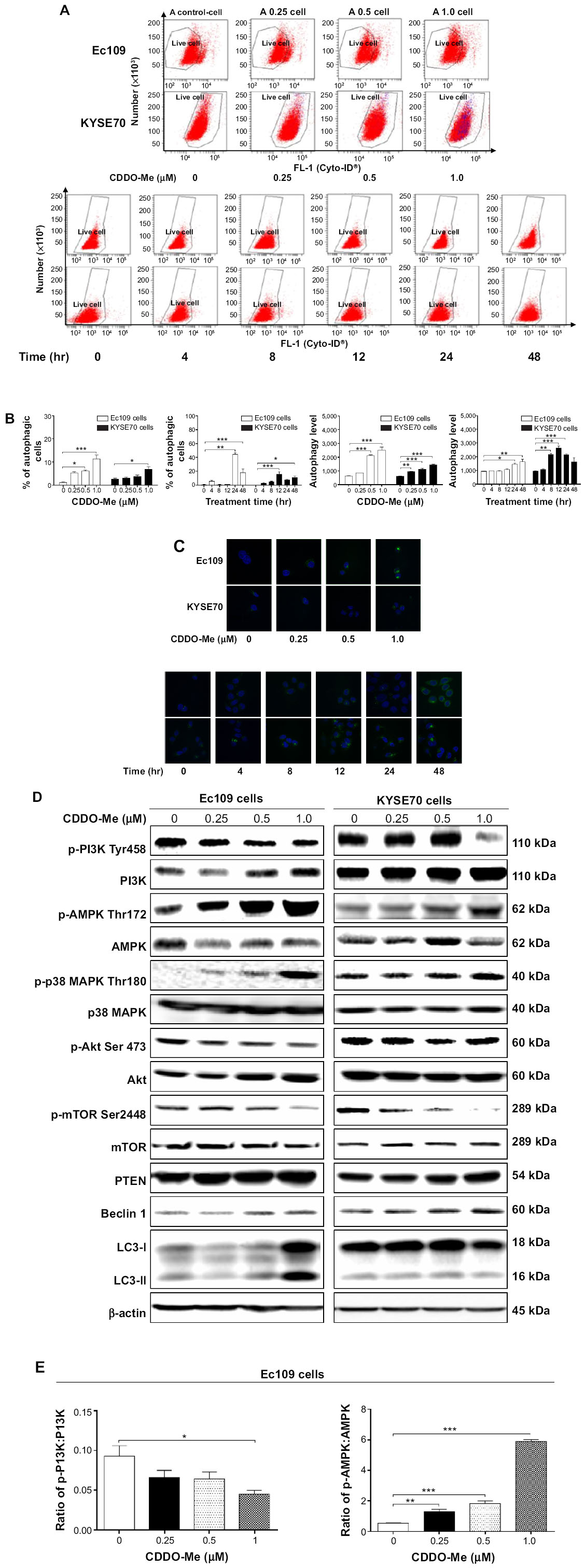
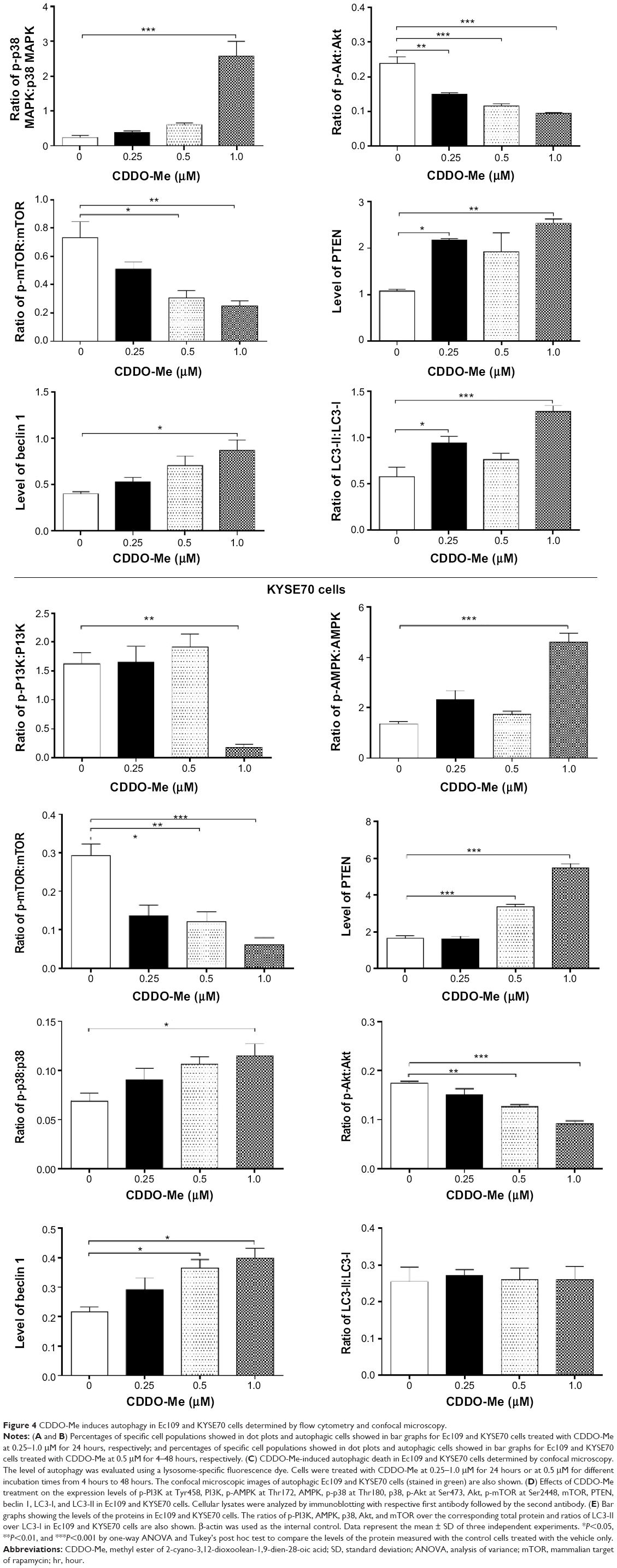 | Figure 4 CDDO-Me induces autophagy in Ec109 and KYSE70 cells determined by flow cytometry and confocal microscopy. |
Next, we tested the autophagic effect of 0.5 μM CDDO-Me on Ec109 and KYSE70 cells with different incubation time intervals by flow cytometric analysis. A time-dependent autophagic effect was demonstrated in both cell lines. The percentage of the autophagic cells was increased from 0.7% at the basal level to 44.2% after 24-hour incubation with CDDO-Me in Ec109 cells (Figure 4A and B). In KYSE70 cells, the number of the autophagic cells was increased 36.3-fold with 48-hour treatment of 0.5 μM CDDO-Me.
LC3 is one of the autophagy-related proteins participating in the process of autophagosome formation, and is frequently used as an autophagosome marker.31 To further confirm the autophagic effect of CDDO-Me on human ESCC cells, we examined the changes of LC3 after incubation with various concentrations of CDDO-Me using confocal microscopy. We demonstrated that the population of LC3-positive cells increased after incubation of human ESCC cells with CDDO-Me. Figure 4C clearly shows that after 24-hour incubation with CDDO-Me, the cells had obvious increases in green punctate structures distributed in perinuclear regions and focally throughout the cytoplasm in Ec109 and KYSE70 cells, whereas these structures were almost not observed in control cells. The intensity of fluorescence, which indicated the autophagic death of cells, was increased 3.3- and 3.9-fold when treated with CDDO-Me at 0.25 and 0.5 μM for 24 hours, respectively, in Ec109 cells. In KYSE70 cells, the intensity of fluorescence was increased by 53.0%, 80.4%, and 132.8% when treated with 0.25, 0.5, and 1.0 μM CDDO-Me, respectively. In addition, Cyto-ID® Green reagent staining also showed that the relative fluorescence intensity of cells was increased in a time-dependent manner, indicating the occurrence of autophagy (Figure 4C). These results are consistent with the autophagic effect of CDDO-Me on human ESCC cells, which are detected by flow cytometric analysis in the same study.
As mTOR is a well-known regulator of autophagy,32 the involvement of mTOR signaling pathway in the initiation of autophagy in CDDO-Me-treated cells was further investigated using Western blotting analysis. CDDO-Me treatment led to a marked decrease in p-mTOR at Ser2448 in both Ec109 and KYSE70 cells. There was no significant change in the expression level of total mTOR in both cell lines when treated with CDDO-Me for 24 hours. Thus, a decreased ratio of p-mTOR/mTOR was observed in both cell lines when treated with different concentrations of CDDO-Me. In Ec109 cells, the ratio of p-mTOR/mTOR was decreased from 0.7 at basal level to 0.5, 0.3, and 0.2 when treated with 0.25, 0.5, and 1 μM of CDDO-Me, respectively (P<0.05 or 0.01 by one-way ANOVA; Figure 4D and E). In KYSE70 cells, the ratio of p-mTOR/mTOR was decreased from 0.3 at basal level to 0.13, 0.12, and 0.06 when treated with 0.25, 0.5, and 1 μM of CDDO-Me, respectively (P<0.05, 0.01 or 0.001 by one-way ANOVA; Figure 4D and E).
To further explore the mechanism of CDDO-Me-induced autophagy in human ESCC cells, several key proteins of upstream signaling pathways of mTOR, such as PI3K, Akt, AMPK, p38 MAPK, and PTEN, were measured after CDDO-Me treatment by Western blotting assay. CDDO-Me treatment significantly decreased the levels of p-PI3K at Tyr458 and p-Akt at Ser473 but increased the levels of p-AMPK at Thr172, p-p38 MAPK at Thr180, and PTEN in a dose-dependent manner in both Ec109 and KYSE70 cells.
Additionally, the expression level of beclin 1 and LC3, two autophagy-related proteins, was also determined using Western blotting assay. Beclin 1 is an essential autophagic protein that contributes to the initial vesicle nucleation and formation of autophagosome.33 Figure 4D clearly shows that both CDDO-Me-treated cell types had a notable increase in beclin 1 expression levels compared to control cells. Treatment of Ec109 cells with CDDO-Me at 0.25, 0.5, and 1.0 μM significantly increased the level of beclin 1 by 30.0%, 77.5%, and 117.5%, respectively, compared to the control cells (P<0.05 by one-way ANOVA; Figure 4D and E). In KYSE70 cells, the level of beclin 1 was increased by 38.0%, 71.4%, and 90.4% at 0.25, 0.5, and 1.0 μM CDDO-Me, respectively, compared to the control cells (P<0.05 by one-way ANOVA; Figure 4D and E).
The conversion of cytosolic LC3-I to membrane-bound LC3-II indicates autophagic activity.34,35 When autophagy is induced, LC3-I is conjugated to phosphatidylethanolamine to form LC3-II, the 16-kDa form of the LC3 protein that is specific for the membranes of autophagosomes.34,35 Therefore, the conversion of LC3-I to LC3-II is correlated with increased autophagic flux. In Figure 4D, the conversion of LC3-I to LC3-II was remarkably increased in Ec109 cells after 24-hour incubation with CDDO-Me. CDDO-Me significantly increased the ratio of LC3-II over LC3-I from 0.57 (basal) to 0.76–1.28 (P<0.05 or 0.001 by one-way ANOVA; Figure 4D and E). However, the ratio of LC3-II/LC3-I did not change significantly in KYSE70 cells. These data together demonstrate that CDDO-Me induces autophagy in human ESCC cells through suppression of mTOR signaling pathway.
There is a crosstalk between CDDO-Me-induced apoptotic and autophagic pathways in Ec109 and KYSE70 cells
Considering the paradoxical role of autophagy in promoting cell death or survival, we further treated human ESCC cells with several kinase inhibitors of autophagic pathway, including p38 MAPK, PI3K, Akt, AMPK, and mTOR inhibitor, either alone or in combination with CDDO-Me to determine the crosstalk between CDDO-Me-induced apoptosis and autophagy. The apoptosis and autophagy were simultaneously determined by flow cytometry. The results demonstrated that CDDO-Me predominantly induced apoptosis, with autophagy being the minor pathway in programmed cell death of Ec109 and KYSE70 cells. In Ec109 cells, the CDDO-Me-induced apoptosis was 6.7%, 10.7%, and 26.0%, and autophagy was 5.3%, 6.1%, and 11.3% at 0.25, 0.5, and 1.0 μM of CDDO-Me, respectively. In KYSE70 cells, CDDO-Me-induced apoptosis was 15.3%, 20.2%, and 25.2%, and autophagy was 3.0%, 3.7%, and 6.7% at 0.25, 0.5, and 1.0 μM of CDDO-Me, respectively.
Next, we examined the effect of inhibition of the key kinases of the autophagic pathway on basal and CDDO-Me-induced apoptosis in Ec109 and KYSE70 cells. Under the basal condition, treatment of ESCC cells with 20 μM SB202190 or 10 μM Compound C significantly enhanced both autophagy and apoptosis in human ESCC cells (Figure 5A–C). In Ec109 cells, the percentage of autophagic cells was increased 6.6- and 13.8-fold after the incubation with 20 μM SB202190 and 10 μM Compound C, respectively. The percentage of apoptotic cells was increased 4.0-fold after 24-hour incubation with 20 μM SB202190. In KYSE70 cells, the number of autophagic cells were increased by 1,664.8% when treated with 20 μM SB202190 for 24 hours and apoptotic cells were increased by 292.9% when treated with 10 μM Compound C for 24 hours. In addition, 2.5 μM MK-2206 also enhanced the autophagy of KYSE70 cells. However, 10 μM wortmannin and 0.5 μM rapamycin had no impact on the basal level of autophagy and apoptosis in human ESCC cells (Figure 5A–C).
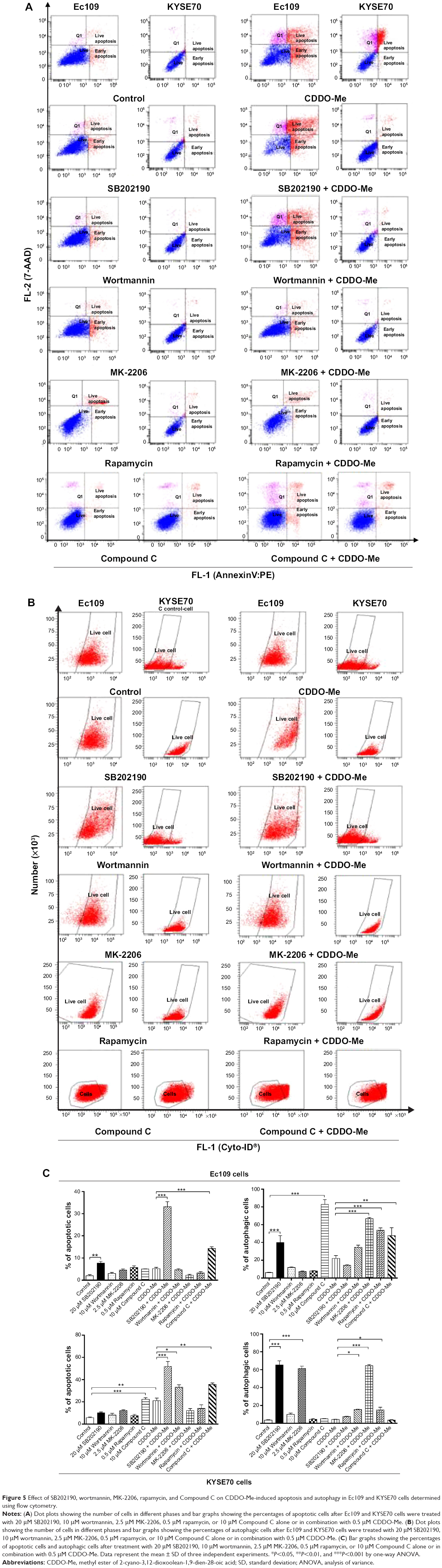 | Figure 5 Effect of SB202190, wortmannin, MK-2206, rapamycin, and Compound C on CDDO-Me-induced apoptosis and autophagy in Ec109 and KYSE70 cells determined using flow cytometry. |
In the combination experiment, 10 μM wortmannin or 10 μM Compound C significantly enhanced the CDDO-Me-induced autophagy and apoptosis. 0.5 μM rapamycin or 2.5 μM MK-2206 significantly enhanced the CDDO-Me-induced autophagy, while incubation with 20 μM SB202190 significantly enhanced CDDO-Me-induced apoptosis. In Ec109 cells, the percentage of CDDO-Me-induced autophagic cells was increased 2.2-, 2.4-, and 3.0-fold when incubated with 10 μM Compound C, 0.5 μM rapamycin, or 2.5 μM MK-2206, respectively (Figure 5A–C). The percentage of CDDO-Me-induced apoptotic cells was increased 2.8-and 6.4-fold after 24-hour incubation with 10 μM Compound C and 20 μM SB202190, respectively. In KYSE70 cells, the number of CDDO-Me-induced autophagic cells was increased by 254.2%, 260.4%, and 1,397.6% when treated with 0.5 μM rapamycin, 10 μM wortmannin, or 2.5 μM MK-2206 for 24 hours, respectively, and CDDO-Me-induced apoptotic cells were increased by 58.1%, 69.5%, and 146.7% when treated with 10 μM wortmannin, 10 μM Compound C, or 20 μM SB202190 for 24 hours, respectively (Figure 5A–C).
These results demonstrate that there are interactions between the autophagic and apoptotic pathways in Ec109 and KYSE70 cells subject to CDDO-Me treatment. PI3K and AMPK play an important role in the interplay for their dual modulating functions in both CDDO-Me-induced autophagy and apoptosis.
CDDO-Me inhibits the generation of intracellular ROS and activates the Nrf2 pathway in Ec109 and KYSE70 cells
To further explore the mechanism of CDDO-Me-induced cell proliferation inhibition and programmed cell death including both apoptosis and autophagy, we determined the effect of CDDO-Me on intracellular ROS level in Ec109 and KYSE70 cells treated with 0.1, 0.25, 0.5, and 1.0 μM CDDO-Me for 24 hours. The intracellular level of ROS was significantly decreased by CDDO-Me in both cell lines. In Ec109 cells, treatment with CDDO-Me at 0.1 and 1.0 μM for 24 hours reduced the intracellular level of ROS by 21.1% and 36.8%, respectively, compared to the control cells (P<0.01 or 0.001 by one-way ANOVA; Figure 6A). Similarly, incubation of KYSE70 cells with CDDO-Me at 0.1, 0.25, and 0.5 μM for 24 hours significantly decreased the intracellular level of ROS by 16.8%, 18.3%, and 20.7%, respectively, compared to the control cells (P<0.01 by one-way ANOVA; Figure 6A).
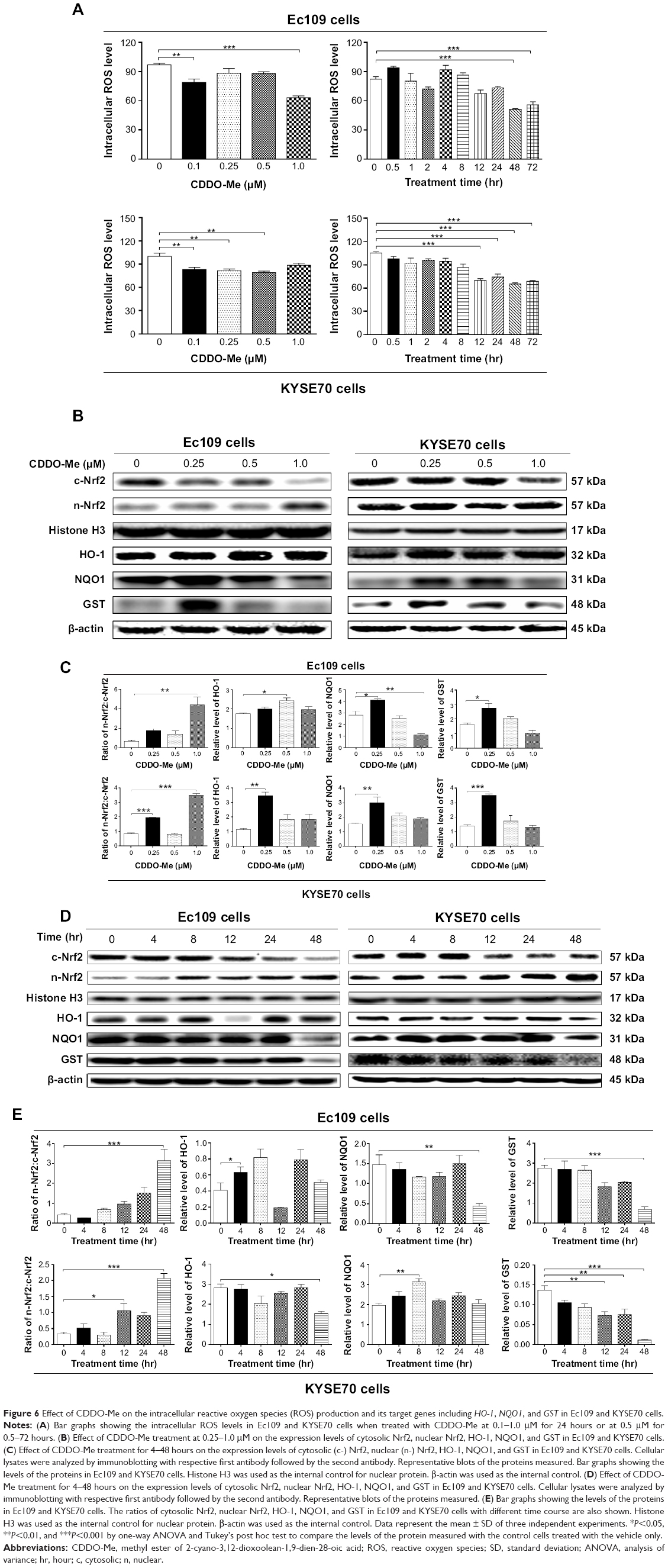 | Figure 6 Effect of CDDO-Me on the intracellular reactive oxygen species (ROS) production and its target genes including HO-1, NQO1, and GST in Ec109 and KYSE70 cells. |
In addition, the intracellular ROS levels in Ec109 and KYSE70 cells treated with 0.5 μM CDDO-Me at different time intervals were also determined in this study. In Ec109 cells, the intracellular ROS level was decreased by 48.9% and 44.3% when incubated with 0.5 μM CDDO-Me for 48 and 72 hours, respectively (P<0.01 by one-way ANOVA; Figure 6A). In KYSE70 cells, the intracellular ROS level was decreased from 31.2% to 40.3% when treated with 0.5 μM CDDO-Me for 12–72 hours (P<0.01 by one-way ANOVA; Figure 6A).
Nrf2 is a nuclear factor that controls the expression and coordinated induction of a battery of genes that encode detoxifying enzymes, drug transporters, anti-apoptotic proteins, and proteasomes.36 Under basal conditions, Nrf2 is retained in the cytoplasm by Keap1 and cullin 3 with a half-life of only 20 minutes, which is degraded rapidly by ubiquitination. Under oxidative stress, critical cysteine residues in Keap1 are disrupted, and Nrf2 will build up and then translocate to the nucleus where it forms a heterodimer with a small Maf protein and binds to a DNA promoter and initiates transcription of antioxidative genes.36 To further investigate the molecular mechanisms of the ROS-scavenging effects of CDDO-Me in human ESCC cells, we examined if CDDO-Me activated the Nrf2 pathway. Nuclear proteins were isolated and analyzed by Western blotting assay using anti-Nrf2 antibody. The expression levels of the target genes of Nrf2 including HO-1, NQO1, and GST were also analyzed using Western blotting assay. Cytosolic (c)-Nrf2, nuclear (n)-Nrf2, HO-1, NQO1, and GST expression levels were increased in both Ec109 and KYSE70 cells treated with a low concentration of CDDO-Me as compared to the control cells. In Ec109 cells, the HO-1 expression level was elevated 1.4-fold when treated with 0.5 μM CDDO-Me for 24 hours (P<0.05 by one-way ANOVA; Figure 6B & C). The NQO1 and GST levels were elevated 1.5-fold (P<0.05 or 0.01 by one-way ANOVA; Figure 6B & C) and 1.7-fold (P<0.05 by one-way ANOVA; Figure 6B & C), respectively, when treated with 0.25 μM CDDO-Me for 24 hours. In KYSE70 cells treated with 0.25 μM CDDO-Me, the ratio of n-Nrf2/c-Nrf2, HO-1, NQO1, and GST levels was increased by 135.4%, 200.8%, 96.7%, and 152.5%, respectively (P<0.05, 0.01, or 0.001 by one-way ANOVA; Figure 6B & C).
In addition, c-Nrf2, n-Nrf2, HO-1, NQO1, and GST expression levels were also examined with the treatment of 0.5 μM CDDO-Me at different incubation time intervals. The accumulation of Nrf2 in nucleus was detected after 48-hour incubation with CDDO-Me in both cell lines. However, the time-dependent activation of Nrf2 target genes was not observed. The dramatically reduced expression level of HO-1, NQO1, and GST was observed in both Ec109 and KYSE70 cells after 48-hour incubation with 0.5 μM CDDO-Me (Figure 6D & E). The reason for this is unknown. These enzymes may also be regulated by other genes such as nuclear receptors, which might be modulated by CDDO-Me.
CDDO-Me suppresses Ec109 and KYSE70 cell invasion in vitro
To determine the effect of CDDO-Me on the invasive potential of human ESCC cells, the QCM™ 96-well cell invasion assay was performed. The invasive potential of cancer cells in this assay was determined by the ability of cells to invade ECM layer, the major components of the basement membrane. As demonstrated in Figure 7A, both Ec109 and KYSE70 cells had a tendency of higher invasive phenotype at the base level. The ratio of the fluorescence value for Ec109 cells cultured in media with FBS over that without FBS was 2.7, while this ratio for KYSE70 cells was 3.3. The invasive capacity of both cell lines was reduced by CDDO-Me. After incubation with 0.5 μM CDDO-Me for 24 hours, the ratio of the fluorescence value for Ec109 cells cultured in media with FBS over that without FBS was reduced to 1.7, while this ratio for KYSE70 cells was reduced to 1.5. These results suggest that CDDO-Me attenuates the invasive capacity of human ESCC cells in vitro.
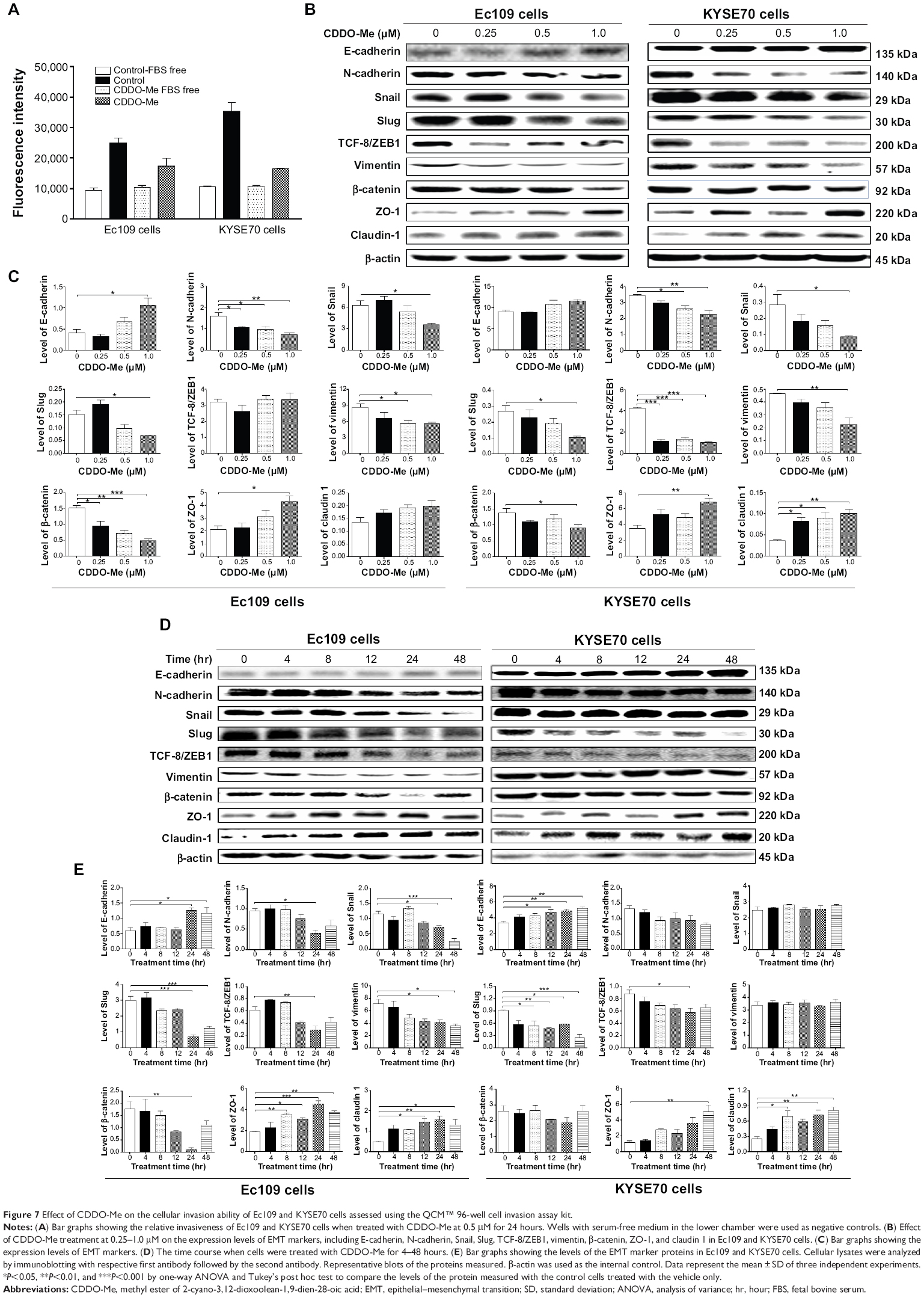 | Figure 7 Effect of CDDO-Me on the cellular invasion ability of Ec109 and KYSE70 cells assessed using the QCM™ 96-well cell invasion assay kit. |
CDDO-Me inhibits the EMT phenotype of Ec109 and KYSE70 cells
Epithelial–mesenchymal transition (EMT) is integral in development, wound healing, and stem cell behavior, and contributes pathologically to fibrosis and cancer progression.37,38 It is a fine-tuned process regulated by a number of proteins that are strategically distributed in the nuclear and cytosolic compartments of cancer cells.39 To further examine the mechanisms of the therapeutic effect of CDDO-Me on human ESCC cells, we evaluated the change of EMT-associated markers in Ec109 and KYSE70 cells exposed to CDDO-Me using Western blotting assay. The most critical and well-studied EMT molecule is E-cadherin.37 The E-cadherin was upregulated by the treatment with CDDO-Me; however, the statistical significance was achieved only in Ec109 cells. The E-cadherin expression level was increased 2.6-fold when treated with 1.0 μM CDDO-Me for 24 hours, compared to the control cells (P<0.05 by one-way ANOVA; Figure 7B and C).
Snail, Slug, and TCF-8/ZEB1 are the critical members of the family of transcription factors that repress E-cadherin, leading to upregulation of mesenchymal genes.37 These three markers were downregulated after the incubation with various concentrations of CDDO-Me for 24 hours in both Ec109 and KYSE70 cells. In Ec109 cells, the Snail and Slug expression levels were reduced by 44.4% and 53.3% (P<0.05 by one-way ANOVA; Figure 7B and C), respectively, when treated with 1.0 μM CDDO-Me for 24 hours, compared to the control cells. The TCF-8/ZEB1 expression level was not impacted by CDDO-Me. In KYSE70 cells, the Snail, Slug, and TCF-8/ZEB1 expression levels were reduced by 71.4%, 61.5%, and 76.2% (P<0.05 or 0.001 by one-way ANOVA; Figure 7B and C), respectively, when treated with 1.0 μM CDDO-Me for 24 hours, compared to the control cells. N-cadherin and vimentin are important mesenchymal markers,37 which were also evaluated in this study. The N-cadherin and vimentin levels in Ec109 cells were decreased 2.1-fold and 1.6-fold (P<0.05 or 0.01 by one-way ANOVA; Figure 7B and C), respectively, when treated with 1.0 μM CDDO-Me for 24 hours, compared to the control cells. In KYSE70 cells, the expression levels of N-cadherin and vimentin were decreased by 1.5- and 2.1-fold (P<0.05 or 0.01 by one-way ANOVA; Figure 7B and C), respectively.
In addition, the expression levels of the tight junction proteins ZO-1 and claudin 1 were also determined in Ec109 and KYSE70 cells exposed to CDDO-Me. In Ec109 cells, both ZO-1 and claudin 1 were upregulated when treated with CDDO-Me for 24 hours; however, only the elevated level of ZO-1 achieved statistical significance. Treatment with 1.0 μM CDDO-Me significantly increased the level of ZO-1 by 107.2% (P<0.05 by one-way ANOVA; Figure 7B and C). In KYSE70 cells, 1.0 μM of CDDO-Me significantly increased the level of ZO-1 and claudin 1 by 95.7% and 233.3% (P<0.05 or 0.01 by one-way ANOVA; Figure 7B and C), respectively.
β-Catenin is a transcription factor in the Wnt signaling pathway and is involved in the regulation of cell adhesion, and it is typically more abundant in epithelial-like cells.40 β-Catenin binds to cadherin receptor intracellular cytoplasmic tail domains, acting as an integral component of a protein complex in adherens junctions that help cells maintain epithelial layers.40 The expression level of β-catenin was downregulated by 68.2% and 30.8% (P<0.05, 0.01, or 0.001 by one-way ANOVA; Figure 7B and C) in Ec109 and KYSE70 cells, respectively, when treated with 1.0 μM CDDO-Me for 24 hours, compared to the control cells.
In a separate experiment, we evaluated the impact of different incubation times with 0.5 μM CDDO-Me on the expression of EMT-related markers. The change in the expression of these EMT-related markers showed the same trend as the concentration-dependent effect of CDDO-Me in Ec109 and KYSE70 cells. The most significant change of these markers occurred in the longer incubation time interval of CDDO-Me. In Ec109 cells, E-cadherin, ZO-1, and claudin 1 were upregulated by 95.0%, 101.6%, and 63.4%, respectively, while Snail, Slug, and vimentin were downregulated by 78.3%, 59.2%, and 50.9%, respectively, after 48-hour incubation with 0.5 μM CDDO-Me, compared to the control cells (Figure 7D and E). In KYSE70 cells, there was 53.2%, 326.1%, and 221.0% increase in E-cadherin, ZO-1, and claudin 1, respectively, whereas there was a 73.6% reduction in Slug after 48-hour incubation with 0.5 μM CDDO-Me (Figure 7D and E). These results suggested that CDDO-Me inhibits EMT in human ESCC cells.
CDDO-Me attenuates the stemness of Ec109 and KYSE70 cells
There is increasing evidence that diverse tumors are hierarchically organized and sustained by a distinct subpopulation of cancer stem cells.41–43 On the basis of the cancer stem cell hypothesis, a tumor may be sustained by a subset of cancer cells with stem-cell-like features that have the ability for self-renewal and pluripotency. Because stemness is a characteristic hallmark of malignant disease, which has an impact on the efficiency of cancer therapy, chemotherapeutic agents that block the stemness have been an important focus in the field of cancer chemotherapy.41,42 Oct-4, a member of the POU family, is considered to be an important cancer stem cell marker and essential transcription factor during embryogenesis.44 Sox-2 is a member of the Sox (SRY-related HMG box) gene family that encodes transcription factors with a single HMG DNA-binding domain, which is expressed in many types of cancer.45 Nanog is believed to function in conjunction with other factors such as Oct-4 and Sox-2 to form an embryonic stem cell identity.46 Bmi-1 is a transcriptional repressor that belongs to the polycomb-group family of proteins that are involved in hematopoiesis, regulation of proliferation, and senescence.47 It is a potent repressor of retinoblastoma and the p53 pathway with an important role in tumorigenesis. To further explore the mechanisms of therapeutic effect of CDDO-Me on human ESCC cells, several selected stemness markers, including Oct-4, Sox-2, Nanog, and Bmi-1, were analyzed using Western blotting assay. The results showed that CDDO-Me significantly decreased the expression of Oct-4, Sox-2, Nanog, and Bmi-1 in Ec109 and KYSE70 cells. Incubation of Ec109 cells with CDDO-Me at 1.0 μM significantly decreased Oct-4, Sox-2, Nanog, and Bmi-1 expression levels by 44.3%, 53.4%, 26.8%, and 44.2%, respectively, compared to the control cells (Figure 8A and B). In KYSE70 cells, there was 3.1-fold, 1.7-fold, and 2.1-fold decrease in Oct-4, Nanog, and Bmi-1, respectively, when treated with 1.0 μM CDDO-Me for 24 hours (Figure 8A and B).
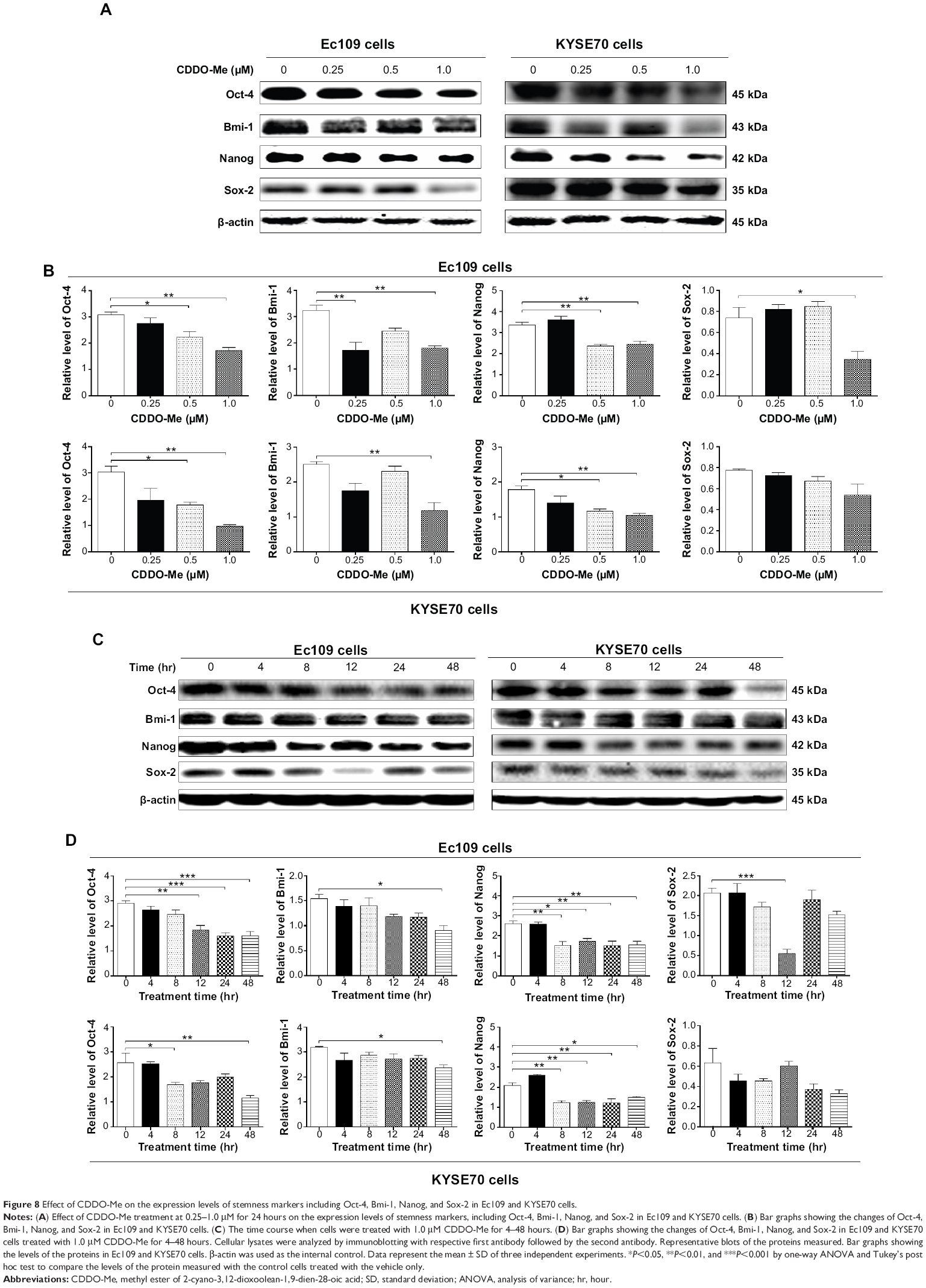 | Figure 8 Effect of CDDO-Me on the expression levels of stemness markers including Oct-4, Bmi-1, Nanog, and Sox-2 in Ec109 and KYSE70 cells. |
In a separate experiment, we evaluated the effects of different incubation times with 0.5 μM CDDO-Me on the stemness of human ESCC cells. In Ec109 cells, Oct-4, Nanog, and Bmi-1 were downregulated by 44.8%, 38.0%, and 41.6%, respectively, after 48-hour incubation with 0.5 μM CDDO-Me, compared to the control cells (Figure 8C and D). The decreased expression level of Sox-2 was observed only after 12-hour incubation of 0.5 μM CDDO-Me. Sox-2 was decreased by 73.3% when compared to the basal level. In KYSE70 cells, all four stemness makers were suppressed with the 48-hour treatment with CDDO-Me; however, the decreased level of Sox-2 did not achieve statistical significance. Oct-4, Nanog, and Bmi-1 were downregulated 2.2-, 1.4-, and 1.4-fold, respectively, after 48-hour incubation with 0.5 μM CDDO-Me, compared to the control cells (Figure 8C and D). These results indicate that CDDO-Me suppresses the stemness of human ESCC cells.
Discussion
There is an increased interest in the use of CDDO-Me as a new anticancer agent. However, the effect of CDDO-Me on ESCC remains unclear. Furthermore, the effects of CDDO-Me on autophagy, EMT, and stemness in solid cancer cells have not been reported. In this study, we found that CDDO-Me effectively suppressed the proliferation and induced G2/M cell cycle arrest and apoptosis in human ESCC cells. We also found that CDDO-Me scavenged ROS level through activation of the Nrf2 pathway. For the first time, our study has demonstrated that CDDO-Me induced autophagy and suppressed EMT and stemness, contributing to the anticancer activity of CDDO-Me. To the best of our knowledge, this is the first study that has confirmed the autophagy-inducing ability of CDDO-Me in solid cancer cells.
A number of studies have shown that CDDO-Me displayed a remarkable concentration-dependent inhibitory effect on the proliferation and growth of many cancer cell lines.19,48,49 The present study showed that CDDO-Me significantly inhibited the growth of Ec109 and KYSE70 cells. However, CDDO-Me did not show remarkable inhibition on the proliferation of the normal human epithelial Het-1A cells compared to the human ESCC cells. Difference in the proliferation inhibitory effect between human ESCC and normal human epithelial cells is essential for the clinical usage of CDDO-Me when a low sensitivity to the drug in normal tissues is needed.
Cell cycle control is one of the major regulatory mechanisms of cell growth, and uncontrolled cell division or propagation of damaged DNA can contribute to genomic instability and tumorigenesis.50 Many anticancer agents have been reported to arrest the cell cycle at a specific checkpoint and thereby induce the cytotoxicity effect.51 CDDO-Me induced G0/G1 arrest in the breast cancer-associated gene 1 (BRCA1)-deficient cells, G2/M arrest in 4T1 breast cancer cells, and severe depletion in the S phase in neuroblastoma cells.52–54 In the current study, we observed a G2/M arrest in a concentration- and time-dependent manner in human ESCC cells following treatment with CDDO-Me. The transition from G2 to M phase of cell cycle is regulated in part by the mitosis-promoting factor, which consists of cyclin B1 and the activated CDK1 (Cdc2).50 Cdc2 binds to cyclin B1 and forms a Cdc2–cyclin complex, which is suppressed by the phosphorylation of Tyr15 of Cdc2. Moreover, the inhibition of cyclin–CDK complex might be amplified by simultaneous upregulation of the p21Waf1/Cip1 protein, a CDK inhibitor.55 Activated p53 itself can inhibit cell cycle progression by regulation of p21Waf1/Cip1.56 In the subsequent experiment, we found that CDDO-Me treatment decreased the expression of cyclin B1 and Cdc2, and increased the levels of p21Waf1/Cip1 and p53 in both Ec109 and KYSE70 cells. These results provided an explanation for the G2/M-arresting effect of CDDO-Me on Ec109 and KYSE70 cells.
Next, we evaluated the apoptotic effect of CDDO-Me on human ESCC cell using flow cytometry analysis. Consistent with the pro-apoptotic effects in other cancer cell lines reported in previous studies,57–59 our data showed that CDDO-Me induced concentration- and dose-dependent apoptosis in Ec109 and KYSE70 cells. Apoptosis is regulated by two major pathways, direct activation of caspase-8 by a signaling complex that includes a cell surface receptor and activation of caspase-8 via direct signaling transduction, and the convergence of the signaling at the mitochondrion, such as those mediated by the Bcl-2 family of proteins.60,61 The proteins of the Bcl-2 family are the key regulators of apoptosis, exerting both pro- and anti-apoptotic effects.62 Anti-apoptotic members of Bcl-2 (Bcl-xl and Bcl-2) can be inhibited by posttranslational modification and/or by increased expression of PUMA, which is an essential regulator of p53-mediated cell apoptosis.63 The possible molecular mechanisms responsible for CDDO-Me-induced apoptosis were explored in the present study. The decreased expression level of the pro-survival proteins Bcl-2 and Bcl-xl and increased expression level of the pro-apoptotic molecule Bax and its regulator PUMA were observed. Caspases are proteins that are highly conserved, cysteine-dependent aspartate-specific proteases that play a central role in apoptosis initiation and execution. There are two types of caspases: initiator caspases including caspases 2, 8, 9, 10, 11, and 12 and effector caspases including caspases 3, 6, and 7.64–66 Caspase activation is generally considered to be a key mark of apoptosis, playing an important role in the induction and regulation of apoptosis.64,66 PARP can be activated in cells experiencing stress and/or DNA damage, depleting the ATP of a cell and eventually inducing apoptosis and autophagy.67 Our study clearly showed that after 24-hour treatment with CDDO-Me, the expression of cleaved caspase-3 and -9 and PARP was significantly increased. These data indicate that CDDO-Me induces the apoptosis of human ESCC cells through mitochondrial membrane dysfunction.
Autophagic cell death that is different from both apoptosis and necrosis has independent morphological and biochemical features.34,35,68 So it is considered a second type of programmed cell death (sometimes referred to “type II cell death”).68 Accumulating evidence has demonstrated that various anticancer therapies induce autophagy in different cancer cell types.69,70 These findings have shed light from different directions on the potential of modulating autophagy as a novel therapeutic strategy for malignant diseases. The current study showed that a proportion of human ESCC cells underwent autophagic cell death in a dose- and time-dependent manner following CDDO-Me treatment, which was confirmed by both flow cytometric and confocal microscopy analysis. CDDO-Me enhanced the conversion of LC3 from cytosolic form (LC3-I) to the proteolytic and lipidated form (LC3-II) in Ec109 cells. Additionally, beclin 1 is an essential modifier of the autophagic process and is commonly used to monitor autophagy activity combined with other biochemical factors.33 The upregulation of beclin 1 by CDDO-Me was observed in our study.
The kinase mTOR is a critical regulator of autophagy induction, with activated mTOR (Akt and MAPK signaling) suppressing autophagy and negative regulation of mTOR (AMPK and p53 signaling) promoting it.34,71–73 Class III PI3K complex, containing hVps34, beclin 1 (a mammalian homolog of yeast Atg6), p150 (a mammalian homolog of yeast Vps15), and Atg14-like protein (Atg14L or Barkor) or ultraviolet irradiation resistance-associated gene (UVRAG), is required for the induction of autophagy.33 The Atg genes control the autophagosome formation through Atg12-Atg5 and LC3-II (Atg8-II) complexes.31 Akt regulates multiple biological processes including cell survival, proliferation, growth, and glycogen metabolism. Activated Akt triggers activation of the lipid kinase PI3K, which generates PIP3 at the plasma membrane.74 Akt binds PIP3 through its pleckstrin homology domain, resulting in translocation of Akt to the membrane. Akt directly phosphorylates and activates mTOR, as well as inhibits the mTOR inhibitor proteins PRAS40 and tuberin. Akt promotes cell survival directly by its ability to phosphorylate and inactivate several pro-apoptotic targets, including Bad, Bim, Bax, and the forkhead (FoxO1/3a) transcription factors.74 PTEN, a lipid phosphatase that catalyzes the dephosphorylation of PIP3, is a major negative regulator of Akt signaling.75 Loss of PTEN function has been implicated in many human cancers. p38 MAPKs (α, β, γ, and δ) are members of the MAPK family that are activated by a variety of environmental stresses and inflammatory cytokines.76 As with other MAPK cascades, the membrane-proximal component is an MAPK kinase kinase (MAPKKK), typically an MAP kinase kinase kinase (MEKK) or a mixed lineage kinase. As the inhibition of mTOR pathway is the best-known mechanism through which autophagy is induced, many other kinases or proteins which modulated this pathway and contributed to the regulation of autophagic cell death were also evaluated in this study. The results showed that CDDO-Me suppressed the phosphorylation of PI3K and Akt but elevated AMPK, p-p38 MAPK, and PTEN expression levels in both Ec109 and KYSE70 cells. This will trigger the initiation of autophagy in ESCC cells when exposed to CDDO-Me.
Despite marked differences between apoptosis and autophagy, recent studies have suggested that there is a cross talk between the autophagic and apoptotic pathways.77–80 Autophagy can be activated in response to anticancer therapy by apoptosis-inducing agents that limit drug efficacy, and blockage of autophagy can facilitate apoptosis. This illustrates that the apoptotic and autophagic response machineries share common pathways that either link or polarize the cellular responses. One example is the interaction between anti-apoptotic protein Bcl-2 and autophagy-related protein beclin 1. Bcl-2 antagonizes Bax and Bak, preventing apoptosis, whereas the interaction between Bcl-2 and beclin 1 inhibits autophagy.81 Therefore, the Bcl-2 level is of great significance for apoptosis and autophagy. As indicated previously, the mTOR has been confirmed as a key negative regulator of autophagy in mammalian cells.32 In the present study, we focused on the modulation of mTOR signaling pathway and evaluated the interactions between CDDO-Me-induced autophagy and apoptosis using flow cytometric analysis. p38 MAPK inhibitor SB202190, AMPK inhibitor Compound C, Akt inhibitor MK-2206, PI3K inhibitor wortmannin, and mTOR inhibitor rapamycin were used in the analysis. We found that PI3K and AMPK participated in both the CDDO-Me-induced apoptosis and autophagy. AMPK plays a key role as a master regulator of cellular energy homeostasis.82 The kinase is activated in response to stresses that deplete cellular ATP supplies such as low glucose, hypoxia, ischemia, and heat shock. It exists as a heterotrimeric complex composed of a catalytic α subunit and regulatory β and γ subunits.82 Binding of AMP to the γ subunit allosterically activates the complex, making it a more attractive substrate for phosphorylation at Thr172 in the activation loop of the α subunit by its major upstream AMPK kinase.
ROS are generated intracellularly as byproducts of normal aerobic metabolism or as second messengers in various signal transduction pathways or in response to environmental stress.83 Increased generation of ROS and an altered redox status have long been observed in cancer cells, and recent studies suggest that modulating the unique redox regulatory mechanisms of cancer cells might be an effective strategy to eliminate these cells.84 Previous studies have found that CDDO-Me inhibited the proliferation of cancer cells via the ROS generation.59,85,86 In contrast, the results of the current study showed that CDDO-Me attenuated the ROS level in human ESCC cells. To elucidate the underlying mechanism, the Nrf2 pathway87 that plays a critical role in the antioxidative modulation was evaluated in this study. CDDO-Me has been used as an Nrf2 activator in treating the chronic kidney disease and showed promise results.11 However, the role of Nrf2 pathway activation for CDDO-Me was not well documented in solid cancer before. Subsequently, we found that CDDO-Me induced the nuclear translocation of Nrf2 and activated the downstream target genes, such as HO-1, NQO1, and GST, especially at the lower concentration. However, for the cell signaling pathway hijack effect of cancer, the contribution of Nrf2 pathway activation in the scavenging of ROS was less important. As indicated in the “Results” section, the reduction level of ROS in Ec109 and KYSE70 cells was moderate after the treatment with CDDO-Me.
It is well known that cancer metastasis occurs in many steps including vessel formation, cell attachment, adhesion, invasion, migration, and proliferation, and these events are regulated by extremely complex mechanisms.42 Cell invasion is one of the essential steps of cancer metastasis.88 In this study, the cell invasion suppression effect of CDDO-Me on human ESCC cells was analyzed. Our findings indicated that following treatment with 0.5 μM CDDO-Me for 24 hours, the invasive ability of both Ec109 and KYSE70 cells was remarkably reduced. Accumulating evidence has shown that EMT plays an important role in cancer invasion. To further check the underlying mechanism of the suppression of cancer cell invasion by CDDO-Me, the EMT process was evaluated in the present study. EMT is such process which allows epithelial cells to change their phenotype, acquire mesenchymal properties, and increase their capability to migrate.37,39,89 It is a fine-tuned process regulated by a number of proteins that are strategically distributed in the nuclear and cytosolic compartments of cancer cells. A number of different parallel signaling pathways interact in the development of EMT.39 A key feature of EMT is decreased expression of E-cadherin, a transmembrane cell adhesion molecule necessary for maintenance of intercellular contacts and cellular polarity in epithelial tissue, and increased expression of one or more of the mesenchymal markers vimentin, fibronectin, and N-cadherin.90 The transcription factors Snail, Slug, Twist, and ZEB1 play a central role in the induction of EMT and downregulation of E-cadherin. In particular, Snail has been reported to interact with nuclear β-catenin, promoting activation of Wnt target genes and further driving EMT.91 Moreover, tight junction proteins ZO-1 and claudin 1 also have a contribution role in the EMT process.37 Interestingly, here we showed that incubation of CDDO-Me led to a reversion of EMT progress, characterized by decreased expression of transcription factors Snail, Slug, and ZEB1, and mesenchymal markers vimentin and N-cadherin, and increased expression of epithelial markers E-cadherin, and tight junction proteins ZO-1 and claudin 1, which was in agreement with the cell invasion assay of human ESCC cell lines in vitro. These findings suggest a potential role of CDDO-Me in suppression of metastasis of ESCC.
A growing body of research demonstrated that, similar to normal tissue, some cancers have a hierarchical organization where tumorigenic cancer stem cells differentiate into non-tumorigenic progenies.41,43 Experimental and clinical oncogenomics data provide powerful evidence in support of a cancer stem-cell hypothesis and suggest that gene expression signatures associated with the stemness state of a cell (defined as phenotypes of self-renewal, asymmetric division, and pluripotency) might perform critical role in cancer therapy resistance.92,93 Oct-4, one of the stem-related transcription factors, together with Sox-2 and Nanog, regulates tumor proliferation and self-renewal and maintains stem pluripotency of cancer cell.44,46,94 Bmi-1, a member of the polycomb-group proteins, was identified as one of the key regulatory proteins of “stemness” functions in both normal and cancer stem cells.47,95 In the current study, these four stemness markers were selected for the evaluation of the inhibitory ability of CDDO-Me in cancer stemness. The results showed that CDDO-Me suppressed the expression level of these stemness markers, especially in the higher concentration and longer incubation time. Since stemness reflects the self-renewal ability of cancer cells, CDDO-Me may inhibit the growth of ESCC via suppression of self-renewal.
Conclusion
In conclusion, we first investigated the anticancer effects of CDDO-Me on human ESCC cells. We demonstrated that CDDO-Me induced cell cycle arrest, apoptosis, and autophagy, and inhibited ROS level, cell invasion, EMT, and stemness in ESCC cells. Induction of apoptosis by CDDO-Me is via activation of the mitochondrial pathway, while the induction of autophagy is via the inhibition of the PI3K/Akt/mTOR pathway. CDDO-Me might be a potent and promising agent to combat esophageal cancer. Further mechanistic studies are needed to elucidate the target network of CDDO-Me in ESCC treatment.
Acknowledgments
The authors appreciate the financial support from the Startup Fund of the College of Pharmacy, University of South Florida, Tampa, FL 33612, USA. They also appreciate the technical assistance for this study from Dr Zhi-Wei Zhou, PhD, College of Pharmacy, University of South Florida, Tampa, FL 33612, USA; and Dr Jin-Ping Li, MD, General Hospital, Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China.
Disclosure
The authors report no conflicts of interest in this work.
References
Simard EP, Ward EM, Siegel R, Jemal A. Cancers with increasing incidence trends in the United States: 1999 through 2008. CA Cancer J Clin. 2012;62:118–128. | ||
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Lin Y, Totsuka Y, He Y, et al. Epidemiology of esophageal cancer in Japan and China. J Epidemiol. 2013;23(4):233–242. | ||
Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349(23):2241–2252. | ||
Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381(9864):400–412. | ||
Eslick GD. Epidemiology of esophageal cancer. Gastroenterol Clin North Am. 2009;38(1):17–25,vii. | ||
Kim T, Grobmyer SR, Smith R, et al. Esophageal cancer – the five year survivors. J Surg Oncol. 2011;103(2):179–183. | ||
Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7(5):357–369. | ||
Honda T, Rounds BV, Bore L, et al. Synthetic oleanane and ursane triterpenoids with modified rings A and C: a series of highly active inhibitors of nitric oxide production in mouse macrophages. J Med Chem. 2000;43(22):4233–4246. | ||
Pergola PE, Raskin P, Toto RD, et al; BEAM Study Investigators. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365(4):327–336. | ||
de Zeeuw D, Akizawa T, Audhya P, et al; BEACON Trial Investigators. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013;369(26):2492–2503. | ||
Petronelli A, Pannitteri G, Testa U. Triterpenoids as new promising anticancer drugs. Anticancer Drug. 2009;20(10):880–892. | ||
Shanmugam MK, Dai XY, Kumar AP, Tan BKH, Sethi G, Bishayee A. Oleanolic acid and its synthetic derivatives for the prevention and therapy of cancer: preclinical and clinical evidence. Cancer Lett. 2014;346(2):206–216. | ||
Jutooru I, Chadalapaka G, Abdelrahim M, et al. Methyl 2-cyano-3,12-dioxooleana-1,9-dien-28-oate decreases specificity protein transcription factors and inhibits pancreatic tumor growth: role of microRNA-27a. Mol Pharmacol. 2010;78(2):226–236. | ||
Liby K, Risingsong R, Royce DB, et al. Triterpenoids CDDO-methyl ester or CDDO-ethyl amide and rexinoids LG100268 or NRX194204 for prevention and treatment of lung cancer in mice. Cancer Prev Res (Phila). 2009;2(12):1050–1058. | ||
Liby K, Risingsong R, Royce DB, et al. Prevention and treatment of experimental estrogen receptor-negative mammary carcinogenesis by the synthetic triterpenoid CDDO-methyl ester and the rexinoid LG100268. Clin Cancer Res. 2008;14(14):4556–4563. | ||
Wang YY, Yang YX, Zhe H, He ZX, Zhou SF. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties. Drug Des Devel Ther. 2014;8:2075–2088. | ||
Wang YY, Zhe H, Zhao R. Preclinical evidences toward the use of triterpenoid CDDO-Me for solid cancer prevention and treatment. Mol Cancer. 2014;13:30. | ||
Nagaraj S, Youn JI, Weber H, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. 2010;16(6):1812–1823. | ||
Hong DS, Kurzrock R, Supko JG, et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2012;18(12):3396–3406. | ||
Hyer ML, Shi R, Krajewska M, et al. Apoptotic activity and mechanism of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic-acid and related synthetic triterpenoids in prostate cancer. Cancer Res. 2008;68(8):2927–2933. | ||
Ikeda T, Sporn M, Honda T, Gribble GW, Kufe D. The novel triterpenoid CDDO and its derivatives induce apoptosis by disruption of intracellular redox balance. Cancer Res. 2003;63(17):5551–5558. | ||
Konopleva M, Tsao T, Estrov Z, et al. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous leukemia. Cancer Res. 2004;64(21):7927–7935. | ||
Li YC, He SM, He ZX, et al. Plumbagin induces apoptotic and autophagic cell death through inhibition of the PI3K/Akt/mTOR pathway in human non-small cell lung cancer cells. Cancer Lett. 2014;344(2):239–259. | ||
Hu X, Moscinski LC. Cdc2: a monopotent or pluripotent CDK? Cell Prolif. 2011;44(3):205–211. | ||
Yoon MK, Mitrea DM, Ou L, Kriwacki RW. Cell cycle regulation by the intrinsically disordered proteins p21 and p27. Biochem Soc Trans. 2012;40(5):981–988. | ||
Okorokov AL. p53 in a crosstalk between DNA repair and cell cycle checkpoints. Cell Cycle. 2003;2(3):233–235. | ||
Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev. 2011;21(1):12–20. | ||
Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–489. | ||
Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130(1):165–178. | ||
Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–1295. | ||
Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. | ||
Hurley JH, Schulman BA. Atomistic autophagy: the structures of cellular self-digestion. Cell. 2014;157(2):300–311. | ||
Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75. | ||
Niture SK, Khatri R, Jaiswal AK. Regulation of Nrf2 - an update. Free Radic Biol Med. 2014;66:36–44. | ||
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. | ||
Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16(6):488–494. | ||
De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. | ||
Mosimann C, Hausmann G, Basler K. β-Catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol. 2009;10(4):276–286. | ||
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768. | ||
Goding CR, Pei D, Lu X. Cancer: pathological nuclear reprogramming? Nat Rev Cancer. 2014;14(8):568–573. | ||
Rossi DJ, Jamieson CH, Weissman IL. Stems cells and the pathways to aging and cancer. Cell. 2008;132(4):681–696. | ||
Lee J, Kim HK, Rho JY, Han YM, Kim J. The human OCT-4 isoforms differ in their ability to confer self-renewal. J Biol Chem. 2006;281(44):33554–33565. | ||
Weina K, Utikal J. SOX2 and cancer: current research and its implications in the clinic. Clin Trans Med. 2014;3:19. | ||
Chambers I, Colby D, Robertson M, et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113(5):643–655. | ||
Grinstein E, Mahotka C. Stem cell divisions controlled by the proto-oncogene BMI-1. J Stem Cells. 2009;4(3):141–146. | ||
Suh N, Wang Y, Honda T, et al. A novel synthetic oleanane triterpenoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, with potent differentiating, antiproliferative, and anti-inflammatory activity. Cancer Res. 1999;59(2):336–341. | ||
Ryu K, Susa M, Choy E, et al. Oleanane triterpenoid CDDO-Me induces apoptosis in multidrug resistant osteosarcoma cells through inhibition of Stat3 pathway. BMC Cancer. 2010;10:187. | ||
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166. | ||
Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–294. | ||
Alabran JL, Cheuk A, Liby K, et al. Human neuroblastoma cells rapidly enter cell cycle arrest and apoptosis following exposure to C-28 derivatives of the synthetic triterpenoid CDDO. Cancer Biol Ther. 2008;7(5):709–717. | ||
Kim EH, Deng C, Sporn MB, et al. CDDO-methyl ester delays breast cancer development in BRCA1-mutated mice. Cancer Prev Res (Phila). 2012;5(1):89–97. | ||
Ling X, Konopleva M, Zeng Z, et al. The novel triterpenoid C-28 methyl ester of 2-cyano-3,12-dioxoolen-1,9-dien-28-oic acid inhibits metastatic murine breast tumor growth through inactivation of STAT3 signaling. Cancer Res. 2007;67(9):4210–4218. | ||
Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–414. | ||
Aylon Y, Oren M. Living with p53, dying of p53. Cell. 2007;130(4):597–600. | ||
Shishodia S, Sethi G, Konopleva M, Andreeff M, Aggarwal BB. A synthetic triterpenoid, CDDO-Me, inhibits IκBα kinase and enhances apoptosis induced by TNF and chemotherapeutic agents through down-regulation of expression of nuclear factor κB-regulated gene products in human leukemic cells. Clin Cancer Res. 2006;12(6):1828–1838. | ||
Konopleva M, Tsao T, Ruvolo P, et al. Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood. 2002;99(1):326–335. | ||
Deeb D, Gao X, Jiang H, et al. Oleanane triterpenoid CDDO-Me inhibits growth and induces apoptosis in prostate cancer cells through a ROS-dependent mechanism. Biochem Pharmacol. 2010;79(3):350–360. | ||
Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4(2):139–163. | ||
Khosravi-Far R, Esposti MD. Death receptor signals to mitochondria. Cancer Biol Ther. 2004;3(11):1051–1057. | ||
Hardwick JM, Soane L. Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5(2):a008722. | ||
Yu J, Zhang L. PUMA, a potent killer with or without. Oncogene. 2008;27(suppl 1:S71–S83):53. | ||
McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. | ||
Parrish AB, Freel CD, Kornbluth S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol. 2013;5(6):a008672. | ||
Poreba M, Strozyk A, Salvesen GS, Drag M. Caspase substrates and inhibitors. Cold Spring Harb Perspect Biol. 2013;5(8):a008680. | ||
Aredia F, Scovassi AI. Poly(ADP-ribose): a signaling molecule in different paradigms of cell death. Biochem Pharmacol. 2014;92(1):157–163. | ||
Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004–1010. | ||
Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res. 2014;74(3):647–651. | ||
Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol. 2014;85(6):830–838. | ||
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. | ||
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. | ||
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. | ||
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. | ||
Rossi DJ, Weissman IL. Pten, tumorigenesis, and stem cell self-renewal. Cell. 2006;125(2):229–231. | ||
Peti W, Page R. Molecular basis of MAP kinase regulation. Protein Sci. 2013;22(12):1698–1710. | ||
Giansanti V, Torriglia A, Scovassi AI. Conversation between apoptosis and autophagy: “Is it your turn or mine?”. Apoptosis. 2011;16(4):321–333. | ||
Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. | ||
Booth LA, Tavallai S, Hamed HA, Cruickshanks N, Dent P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal. 2014;26(3):549–555. | ||
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833(12):3448–3459. | ||
Maiuri MC, Le Toumelin G, Criollo A, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in beclin-1. EMBO J. 2007;26(10):2527–2539. | ||
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–1023. | ||
Shaughnessy DT, McAllister K, Worth L, et al. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect. 2014;122(12):1271–1278. | ||
Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. | ||
Deeb D, Gao X, Liu YB, Gautam SC. Inhibition of cell proliferation and induction of apoptosis by CDDO-Me in pancreatic cancer cells is ROS-dependent. J Exp Ther Oncol. 2012;10(1):51–64. | ||
Gao X, Liu Y, Deeb D, et al. ROS mediate proapoptotic and antisurvival activity of oleanane triterpenoid CDDO-Me in ovarian cancer cells. Anticancer Res. 2013;33(1):215–221. | ||
Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12(8):564–571. | ||
Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–458. | ||
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | ||
Bednarz-Knoll N, Alix-Panabieres C, Pantel K. Plasticity of disseminating cancer cells in patients with epithelial malignancies. Cancer Metastasis Rev. 2012;31(3–4):673–687. | ||
Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays. 2001;23(10):912–923. | ||
Maugeri-Sacca M, Vigneri P, De Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res. 2011;17(15):4942–4947. | ||
Zhao Y, Bao Q, Schwarz B, et al. Stem cell-like side populations in esophageal cancer: a source of chemotherapy resistance and metastases. Stem Cells Dev. 2014;23(2):180–192. | ||
Boiani M, Scholer HR. Regulatory networks in embryo-derived pluripotent stem cells. Nat Rev Mol Cell Biol. 2005;6(11):872–884. | ||
Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397(6715):164–168. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.