")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Atrial natriuretic peptide modified oleate adenosine prodrug lipid nanocarriers for the treatment of myocardial infarction: in vitro and in vivo evaluation
Received 27 February 2018
Accepted for publication 10 April 2018
Published 11 June 2018 Volume 2018:12 Pages 1697—1706
DOI https://doi.org/10.2147/DDDT.S166749
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Georgios Panos
Jianjun Yu,1,* Wei Li,2,* Dongmei Yu3
1Department of Emergency, Shandong Jining No 1 People’s Hospital, Jining 272011, Shandong, People’s Republic of China; 2Department of Outpatient, Shandong Jining No 1 People’s Hospital, Jining 272011, Shandong, People’s Republic of China; 3Department of Public Health, Shandong Jining No 1 People’s Hospital, Jining 272011, Shandong, People’s Republic of China
*These authors contributed equally to this work
Purpose: Myocardial infarction is a major cause of mortality and heart failure worldwide. One of the most effective methods of this injury is direct delivery of cardioprotective drugs to ischemia–reperfusion (IR) myocardium. The aim of the present study was to fabricate an adenosine (Ade) prodrug-based, atrial natriuretic peptide (ANP)-modified nanosystem for the treatment of myocardial infarction.
Materials and methods: Oleate adenosine prodrug (Ade-OA) and ANP-distearoylphosphatidylethanolamine-polyethylene glycol were synthesized. ANP-modified Ade-loaded lipid nanocarriers (ANP Ade/LNCs) were then self-assembled by using solvent evaporation method. In vitro drug release in the presence of plasma was evaluated. In vivo inhibition effect on infarct size, tissue distribution, and pharmacokinetics were investigated in rats with ischemic myocardium after intravenous injection.
Results: In vivo inhibition effect on infarct size, tissue distribution, and pharmacokinetics studies in acute myocardial infarction (AMI) rats showed that ANP Ade/LNCs exhibited better efficiency than non-modified Ade/LNCs and free Ade in all respects.
Conclusion: These results indicated that the ANP Ade/LNCs can be used as a promising system for the treatment of cardiovascular diseases.
Keywords: myocardial infarction, atrial natriuretic peptide, lipid nanoparticles, adenosine, prodrug
Introduction
Myocardial infarction is a major cause of mortality and heart failure worldwide.1 Compared with the United States or Europe, the incidence of acute myocardial infarction (AMI) increases dramatically in China, which is ~2.5 million AMI patients at present and estimated to be 23 million in 2030.2 Therefore, significant clinical effort has been devoted to preventing AMI and reduce the infarction size. Currently, early reperfusion therapies, including thrombolysis and percutaneous coronary intervention, are standard strategies in patients with ST-segment elevation AMI for the reduction of infarction size.3,4 However, it is well recognized that reperfusion therapy would induce myocardial ischemia–reperfusion (IR) injury, which further induces calcium overload, impaired glycemic control, increased oxidative stress, and inflammatory response.5,6 One of the most effective methods of this injury is direct delivery of cardioprotective drugs to IR myocardium.
Adenosine (Ade) is a ubiquitous endogenous nucleoside that regulates many physiological functions particularly in the heart and brain.7 It has most of the IR injury mechanisms, restoring calcium homeostasis, reducing reactive oxygen species (ROS) synthesis, inhibiting the inflammatory response, promoting the preservation of microvascular flow and stabilizing cellular membranes, reducing free radical formation, and promoting angiogenesis following vascular injury.6,8–11 However, its systemic administration would bring side effects and limited pharmacological efficiency, and it’s extremely short plasma half-life has hampered its clinical application.12–14
Lipid prodrug-based nanosystems are receiving considerable attentions.15 This strategy provides important benefits: 1) an increase of drug chemical stability in vivo; 2) a sustained drug release; and 3) a reduced toxicity before the metabolization occurs.16 Lipid covalently conjugated to the drugs contains fatty acids, cholesterol derivatives, phospholipids, and so on.17 In this study, oleic acid (OA) was chosen as the lipid to conjugate to Ade, so as to achieve sustained release. By combining the lipid prodrug strategy with the use of lipid nanoparticulate systems as drug carriers, oleate adenosine prodrug (Ade-OA)-loaded lipid nanocarriers (LNCs) were developed.
Lipid nanoparticulate systems can be harnessed to deliver therapeutic agents precisely to the infarcted heart by passive targeting.18–20 Compared with passive targeting, active targeting due to the specific interaction of the tissue-recognition ligand and receptor may deliver more drugs to the injured tissue than the intact tissue.21 Atrial natriuretic peptide (ANP), a member of the natriuretic peptide family, exerts its cardioprotective functions not only as a circulating hormone but also as a local autocrine and/or paracrine factor.22 Research studies22,23 have shown that ANP inhibits IR injury and reduces infarct size. Moreover, ANP could be bound to specific natriuretic peptide receptors (NPRs), which are expressed predominantly in the endocardium of the ischemic heart.23
In this study, ANP-modified and Ade-OA (oleic acid) prodrug-loaded LNCs were fabricated. Ade and OA were conjugated to obtain an amphiphilic prodrug (Ade-OA). ANP was conjugated to distearoylphosphatidylethanolamine-polyethylene glycol (DSPE-PEG) by a coupling reaction to get ANP-PEG-DSPE. LNCs were then self-assembled by using the solvent evaporation method. Particle size and morphological analyses revealed uniform spherical shape of NLC. In vitro drug release in the presence of plasma was evaluated. In vivo inhibition effect on infarct size, tissue distribution, and pharmacokinetics were investigated in rats with ischemic myocardium after intravenous injection.
Materials and methods
Materials
Ade (≥99%, A9251), ANP rat (amino acid sequence: Ser-Leu-Arg-Arg-Ser-Ser-Cys-Phe-Gly-Gly-Arg-Ile-Asp-Arg-Ile-Gly-Ala-Gln-Ser-Gly-Leu-Gly-Cys-Asn-Ser-Phe-Arg-Tyr, ≥97%, HPLC, A1663), OA (≥99%, GC, O1008), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, 39391), dimethyl sulfoxide (DMSO, D 2650), Dulbecco’s Modified Eagle’s Medium (DMEM, D5796), fetal bovine serum (FBS, F9665), and 3-[4,5-dimethylthiazol-2yl]-2,5diphenyltetrazolium (MTT, M2128) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). DSPE-PEG (MW~5 kDa)-COOH (DSPE-PEG5000-COOH, PS2-ODE-5K) and DSPE-PEG-FITC (PS2-DEFT-5K) were obtained from Shanghai Pengsheng Biological Co., Ltd. (Shanghai, China). Injectable soyabean lecithin (ISL, TW-SL-IJ01) was provided by Shanghai Taiwei Pharmaceutical Co., Ltd. (Shanghai, China).
Synthesis and characterization of Ade-OA prodrug and ANP-PEG-DSPE ligand
Ade-OA prodrug was synthesized by forming an amide linkage between Ade-NH2 and OA-COOH (Figure 1A).23 OA-COOH was dissolved in DMSO and activated by adding EDC·HCl (1.5 equivalents) and NHS (1.5 equivalents) for 2 h at room temperature with stirring (400 rpm). One equivalent of Ade-NH2 was added to OA-COOH and stirred (400 rpm) for 24 h in the dark at room temperature. Then it was dialyzed against water for 24 h and lyophilized to obtain Ade-OA prodrug.
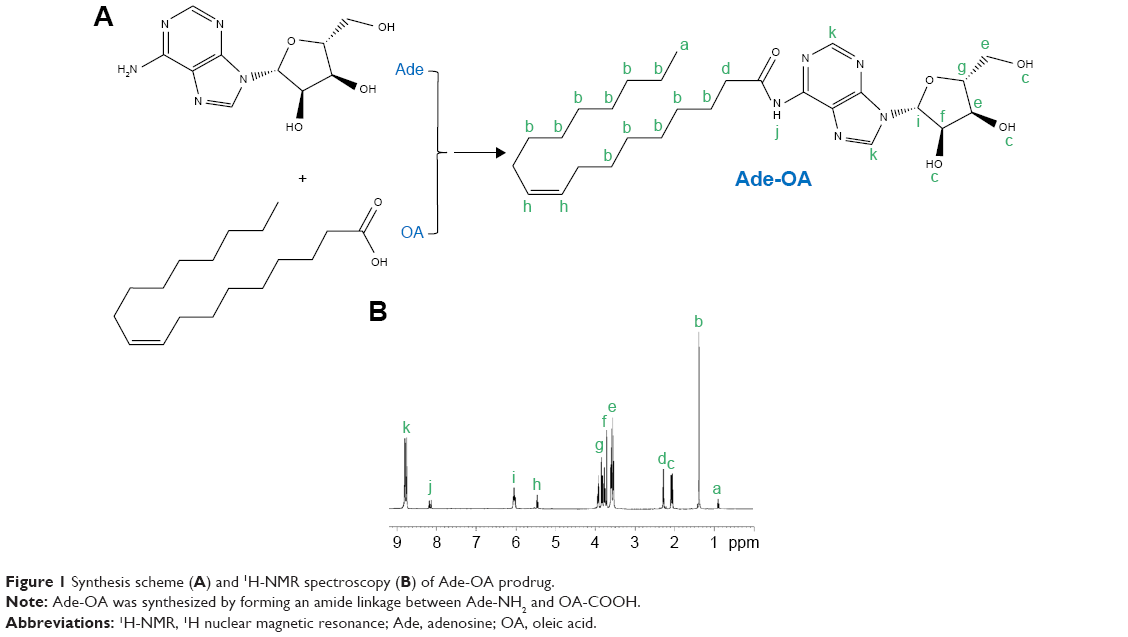 | Figure 1 Synthesis scheme (A) and 1H-NMR spectroscopy (B) of Ade-OA prodrug. |
ANP-PEG-DSPE ligand was synthesized by conjugating APN-NH2 to DSPE-PEG5000-COOH (DSPE-PEG) by amide bond (Figure 2A).22 The free carboxyl groups of DSPE-PEG were again activated by EDC·HCl (1.5 equivalents) and NHS (1.5 equivalents) for 2 h at room temperature with stirring (400 rpm). ANP was dissolved in phosphate-buffered saline (PBS, pH <7.2) and incubated with DSPE-PEG for 24 h in the dark with stirring (400 rpm) at room temperature, and then it was dialyzed against water for 24 h and lyophilized to obtain ANP-PEG-DSPE.
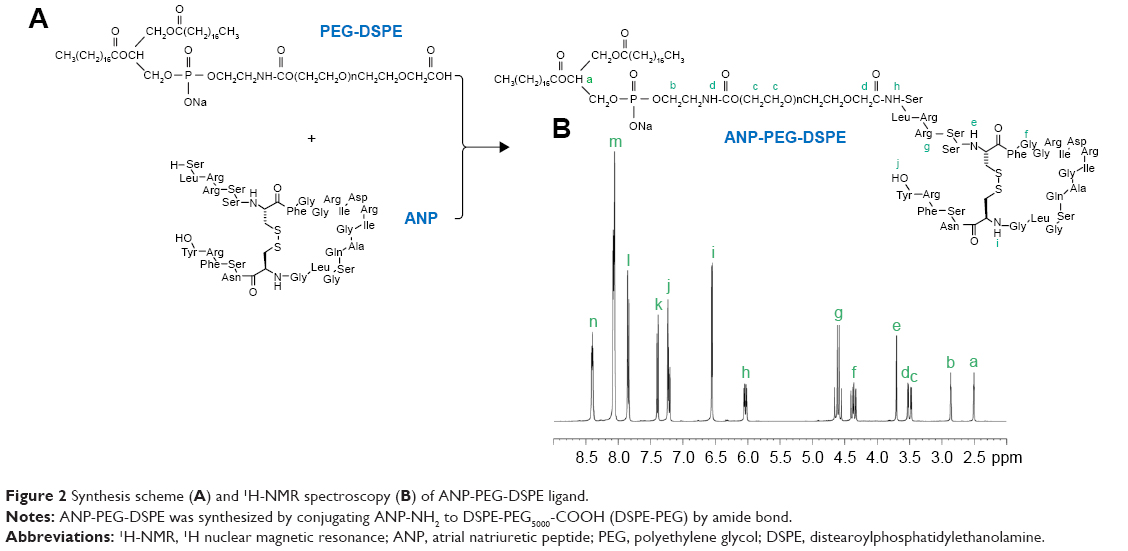 | Figure 2 Synthesis scheme (A) and 1H-NMR spectroscopy (B) of ANP-PEG-DSPE ligand. |
The chemical structure of Ade-OA and ANP-PEG-DSPE was determined by using 1H nuclear magnetic resonance (1H-NMR) analysis by using DMSO-d6 as solvent.
Preparation and characterization of LNCs
LNCs were then self-assembled by using the solvent evaporation method.24 Briefly, Ade-OA (50 mg) and ISL; (200 mg) were dissolved in acetone (5 mL) (1). ANP-PEG-DSPE (100 mg) was dissolved in acetone–water solution (5 mL, 1:1, v/v) (2). 0.05% poloxamer 188 was dissolved in Milli-Q® water (10 mL) (3). Then solutions (1) and (2) were simultaneously and separately added dropwise into solution (3). The mixture was stirred at 600 rpm at room temperature until complete evaporation of the acetone. Redundant stabilizers were removed by centrifugation at 1,000× g and 4°C for 30 min. The pellet was resuspended in Milli-Q water, vortexed and washed three times, filtered through a 0.45 μm membrane, and adjusted to pH 7.4. The obtained ANP-PEG-DSPE-modified Ade-OA-loaded LNCs (ANP Ade/LNCs, Figure 3) were stored at 2–8°C. ANP-PEG-DSPE-modified, no Ade-loaded blank LNCs (ANP LNCs) were prepared by the same method, using OA instead of Ade-OA. Ade-loaded LNCs without ANP modification (Ade/LNCs) were prepared by the same method, using PEG-DSPE instead of ANP-PEG-DSPE.
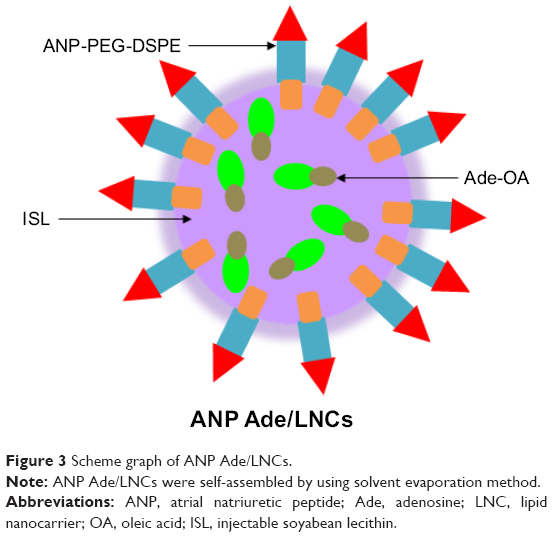 | Figure 3 Scheme graph of ANP Ade/LNCs. |
Particle size, polydispersity index (PDI), and ζ-potential of each sample were measured at room temperature by Zeta Sizer Nano ZS apparatus (Malvern Instruments, Malvern, UK).25 Samples were prepared in disposable capillary cells without dilution. The measurements were performed under conditions of low ionic strength where the surface charge of the particles can be measured accurately. The average particle size was reflected in volume mean diameter.
The Ade encapsulated in LNCs was separated from the LNCs by dissolving in aqueous HCl (2 M), sonicated for 30 min and stirred for 3 h.7 The solution was then centrifuged at 2,000× g for 30 min, the supernatant was collected, and the concentration of Ade was measured at 280 nm by using UV-vis spectrophotometer (UV-1700; SHIMADZU, Kyoto, Japan). The entrapment efficiency (EE) was calculated as (encapsulated Ade in LNCs/mass of the total Ade added) × 100%. The loading capacity (LC) was evaluated by the formula (encapsulated Ade in LNCs/mass of LNCs) × 100%.
In vitro Ade release of LNCs
In vitro release of Ade from the LNCs was studied by dialysis method in a pH 7.4 medium containing 10% FBS.26 Briefly, suspensions of Ade/LNCs and ANP Ade/LNCs were placed inside a dialysis bag (cutoff 12,000 Da) and stirred at 37°C for 72 h. At predetermined time points, 100 μL samples were withdrawn and replaced with fresh medium intervals. The Ade concentration in samples was analyzed by the same method mentioned in the “Preparation and characterization of LNCs” section.
Cells
The H9c2 cells (rat cardiomyoblasts) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in DMEM (Sigma-Aldrich Co., USA) supplemented with 10% fetal FBS (Fisher Chemicals, Fairlawn, NJ, USA) and maintained in a humidified incubator at 37°C and 5% CO2.
Cellular uptake of LNCs
Fluorescently tagged LNCs were prepared by using DSPE-PEG-FITC instead of DSPE-PEG. H9c2 cells were replaced in 96-well plates with fresh media after 24 h of incubation and treated with fluorescently tagged Ade/LNCs, ANP Ade/LNCs, and Ade/LNCs. After 4, 24, and 48 h of incubation, the cells were harvested and washed in cold PBS for three times and determined the fluorescence intensity of the cells by flow cytometer (BD Biosciences, San Jose, CA, USA) equipped with a 488 nm argon laser for excitation.27
Cytotoxicity of LNCs
Cytotoxicity of LNCs was determined by using the MTT assay.28 Briefly, H9c2 cells were seeded in 96-well plates at a density of 1×104 cells per well with fresh media and incubated for 24 h prior to drug treatment. Subsequently, cells were treated separately with Ade/LNCs, ANP Ade/LNCs, ANP LNCs, free Ade, along with 0.9% saline control and incubated for 72 h. 20 μL PBS containing 5 mg/mL MTT reagent was then added to each well, incubated for an additional 4 h at 37°C. 200 μL of DMSO was used to dissolve the formed formazan crystals, and absorbance was read at 570 nm. Cell viability and half-maximal inhibitory concentration (IC50) was then calculated for each sample.
Animals and AMI model induction
Sprague-Dawley rats (220–240 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and fed regular chow, and water was available ad libitum. All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved the Medical Ethics Committee of Jining Medical University (no. JNMC201712.2-001). AMI rats were induced as follows:29 rats were anesthetized with a combination of ketamine (40 mg/kg) and xylazine (10 mg/kg), incubated, and mechanically ventilated. The chest was opened by left thoracotomy, the left anterior descending was ligated for 45 min with an intramural 5-0 silk suture, followed by persistent reperfusion. Then chest was closed, and rats could recover.
In vivo effects of LNCs on infarct size
AMI rats were randomly divided into six groups (n=8) that received an intravenous injection of 1 mL of Ade/LNCs (1), ANP Ade/LNCs (2), ANP LNCs (3), free Ade (4), respectively, along with AMI group (AMI rats received physiological saline (5), and sham-operated group (identical surgery except for the coronary artery ligation and injected with physiological saline (6). The hearts were excised and cut into 2 mm thick slices parallel to the atrioventricular groove.30 The slices were immersed in a 1% solution of 2,3,5-triphenyltetrazolium chloride at 37°C (pH 7.4) for 15 min and photographed again for the identification of infarct area. The area of AMI was calculated by using ImageJ (National Institutes of Health, Bethesda, MD, USA) software for analyzing the stained or unstained areas of the heart tissue. The size of the infarct area was evaluated as a percentage of whole size of the left ventricle.
In vivo tissue distribution of LNCs
After intravenous injection, rats of Ade/LNCs (1), ANP Ade/LNCs (2), and free Ade (4) were sacrificed at predefined times (1 and 24 h postadministration).31 Various tissues (heart, kidney, liver, brain, and lung) were immediately harvested, kept in saline solution to remove the blood and contents, blotted on filter paper, and then weighed for wet weight. After homogenization using a threefold volume of 0.1 M phosphate buffer (pH 7.4), tissue samples were stored at −20°C for further analysis. Concentrations of Ade in rat plasma were analyzed by the same method mentioned in the “Preparation and characterization of LNCs” section.
In vivo pharmacokinetics of LNCs
After intravenous injection, blood samples of Ade/LNCs (1), ANP Ade/LNCs (2), and free Ade (4) were collected into heparinized centrifuge tubes just before intravenous administration (0 h) and after at 0.25, 0.5, 1, 2, 4, 6, 8, 12, 16, 24, 36, and 48 h.32 We then centrifuged the blood sample at 13,000× g for 3 min and isolated the plasma. All samples were stored at −20°C until later analysis. Concentrations of Ade in rat plasma were analyzed by the same method mentioned in the “Preparation and characterization of LNCs” section.
Statistical analysis
The statistical analyses were performed with computer software SPSS 20.0 (IBM Corp, Armonk, NY, USA). Data distribution was tested with Kolmogorov-Smirnov test. Unpaired t-test was used to compare 2 independent groups and statistical significances in pharmacokinetics were evaluated using one-way analysis of variance (ANOVA) followed by Tukey’s test. The levels of the significant differences were set at probabilities of *P<0.05, **P<0.01, and ***P<0.001.
Results
Characterization of Ade-OA and ANP-PEG-DSPE
1H-NMR spectrograms of Ade-OA and ANP-PEG-DSPE in DMSO-d6 are presented with point to point of each proton peak according to the structure. Ade-OA (Figure 1B): δ 0.97 (a, −CH3, OA); 1.35 (b, −CH2−, OA); 2.06 (c, −OH, Ade); 2.31 (d, −CH2−CO−N−, amide linkage); 3.51–3.93 (e, f, g, −CH−CH2−, Ade); 5.45 (h, −CH=CH−, OA); 6.04 (i, −N−CH−O−, Ade); 8.17 (j, −CO−NH−, amide linkage); 8.79 (k, −N−CH=N−, Ade). ANP-PEG-DSPE (Figure 2B): δ 2.49 (a, −CO−O−CH−, DSPE); 2.86 (b, −P−O−CH2−, DSPE); 3.44 (c, −CH2−CH2−O−, PEG); 3.52 (d, −CO−NH−, amide linkage between PEG and DSPE); 6.03 (h, −CO−NH−, amide linkage between ANP and PEG); 4.37, 6.53–8.39 (e, i–n, chemical shifts belong to ANP). The chemical shifts of the amide linkages, Ade, OA, PEG, and DSPE could determine the formation of Ade-OA and ANP-PEG-DSPE.
Characterization of LNCs
The particle size, PDI, ζ-potential, EE, and LC of LNCs were analyzed and are summarized in Table 1. The size of the ANP Ade/LNCs and Ade/LNCs was 168 and 122 nm, respectively. The PDI of the LNCs was <0.2. The ζ-potential of ANP Ade/LNCs was 30 mV; however, the ζ-potential of Ade/LNCs was 16 mV. The EE of Ade-loaded LNCs was >80%. The LC of Ade/LNCs and ANP Ade/LNCs was 10.2 and 7.1%, respectively.
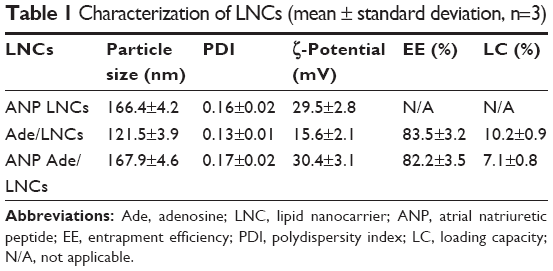 | Table 1 Characterization of LNCs (mean ± standard deviation, n=3) |
In vitro Ade release of LNCs
The in vitro Ade release profiles (Figure 4) demonstrate that both ANP Ade/LNCs and Ade/LNCs show sustained release behavior. The results illustrate that the release of Ade from ANP Ade/LNCs was slower than Ade/LNCs. Over 80% of drug release was achieved at 24 and 48 h, respectively.
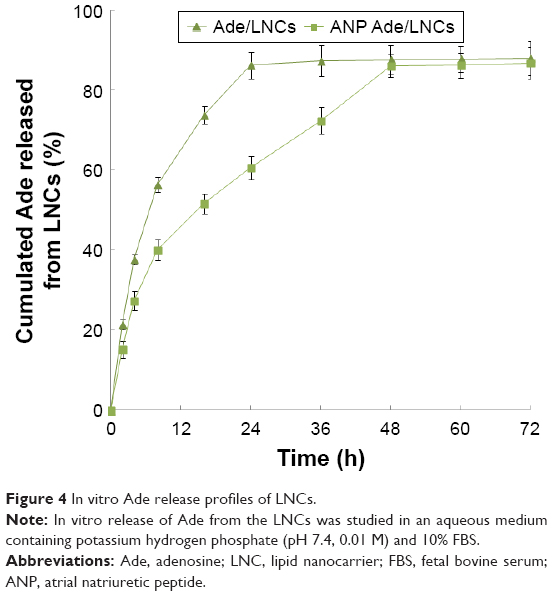 | Figure 4 In vitro Ade release profiles of LNCs. |
Cellular uptake
Flow cytometry analysis (Figure 5) showed that ANP Ade/LNCs had higher cell uptake compared with Ade/LNCs at 24 (P=0.0309) and 48 h (P<0.01). ANP LNCs also had higher cell uptake compared with Ade/LNCs at 24 (P=0.0293) and 48 h (P<0.01). Higher cellular uptake efficiency was observed in the studied groups along with the time (from 4 to 48 h). There are no significant differences in cell uptake efficiency between ANP Ade/LNCs and ANP LNCs group at all the time points tested (P>0.05).
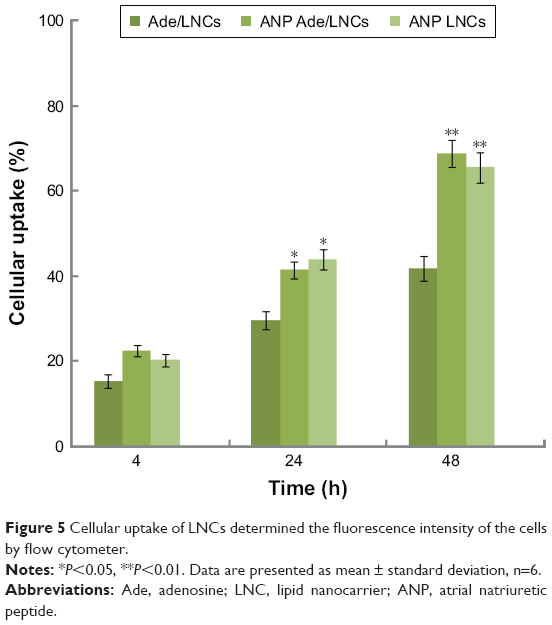 | Figure 5 Cellular uptake of LNCs determined the fluorescence intensity of the cells by flow cytometer. |
In vitro cytotoxicity
Cell viability (Figure 6) of the LNCs groups over the concentration range from 1 to 200 μM was over 80% compared with the controls. These results mean than neither Ade nor LNCs would introduce toxicity to the H9c2 cells when used at concentrations below 200 μM.
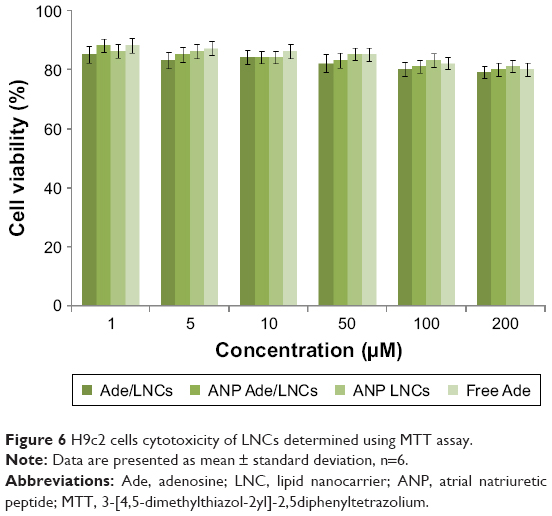 | Figure 6 H9c2 cells cytotoxicity of LNCs determined using MTT assay. |
In vivo effects on infarct size
In vivo experiment (Figure 7) showed that the infarct size was significantly inhibited in all the Ade-containing groups than the AMI group (58%). Compared with AMI group, the infarct size was obviously decreased in the free Ade group (47%) (P=0.0426), Ade/LNCs group (39%) (P<0.01), and ANP Ade/LNCs group (27%) (P<0.001). ANP Ade/LNCs group exhibited the strongest efficiency in the inhibition of infarct size.
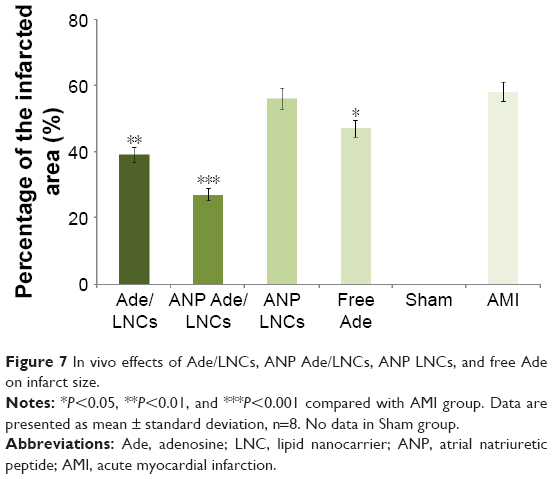 | Figure 7 In vivo effects of Ade/LNCs, ANP Ade/LNCs, ANP LNCs, and free Ade on infarct size. |
In vivo tissue distribution
Tissue distribution of Ade/LNCs, ANP Ade/LNCs, and free Ade are different at 1 (Figure 8A) and 24 h (Figure 8B) post administrations in vivo. At 1 h, Ade/LNC (P=0.0227) and ANP Ade/LNC (P=0.0246) distributions in the heart were higher than free Ade. At 24 h, ANP Ade/LNC distribution in the heart was significantly higher compared with free Ade (P<0.001). Ade/LNCs also showed more obvious heart distribution at 24 h in comparison with free Ade (P<0.01).
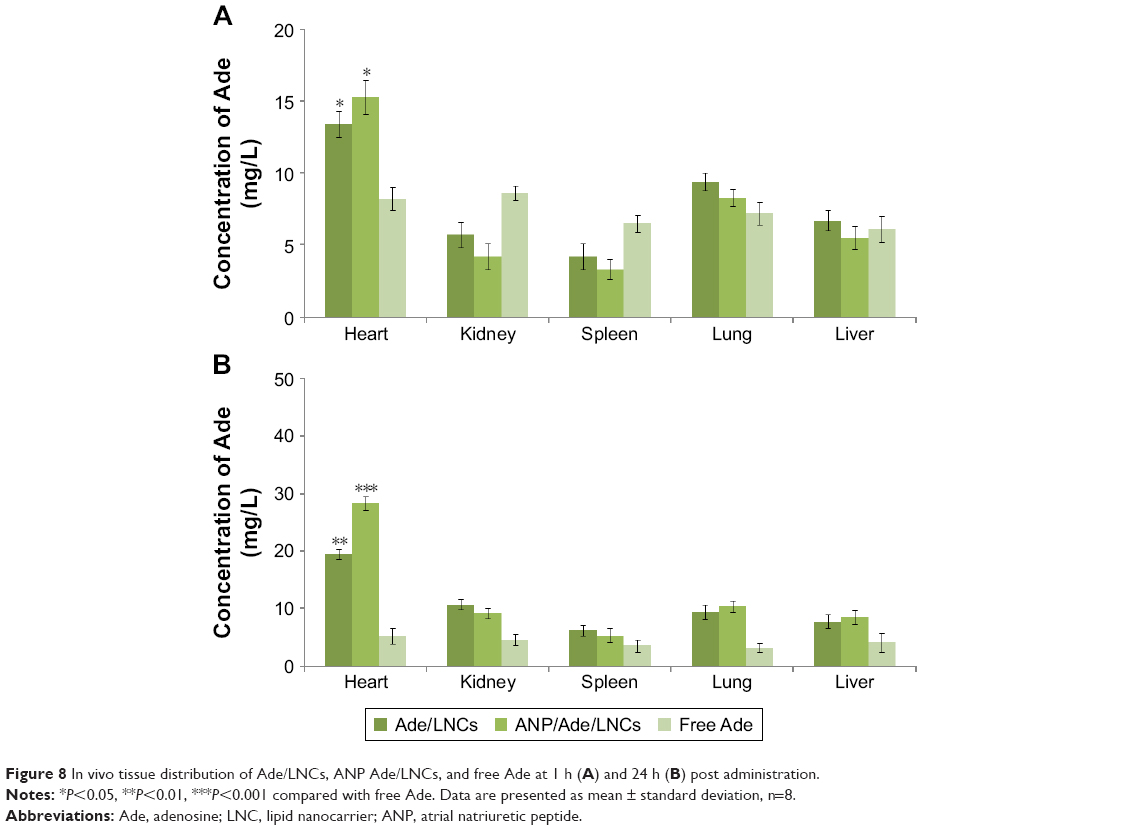 | Figure 8 In vivo tissue distribution of Ade/LNCs, ANP Ade/LNCs, and free Ade at 1 h (A) and 24 h (B) post administration. |
In vivo pharmacokinetics
The plasma drug concentration–time profile (Figure 9) illustrated that the free Ade was rapidly cleared from the circulation in the first few hours, while Ade/LNCs and ANP Ade/LNCs exhibited a longer plasma circulation time. The half-lives of Ade/LNCs (2.0±0.4 h) and ANP Ade/LNCs (3.4±0.7 h) were longer than that of the free Ade (0.8±0.1 h) (Table 2). ANP Ade/LNCs exhibited the maximum area under the curve (AUC) of 138.5 mg/L·h. The plasma clearance (CL) and volume of distribution (V) of ANP Ade/LNCs were significantly higher than those of Ade/LNCs and free Ade.
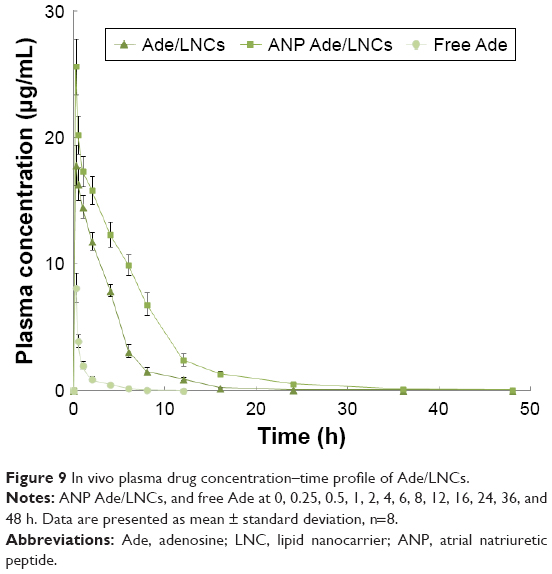 | Figure 9 In vivo plasma drug concentration–time profile of Ade/LNCs. |
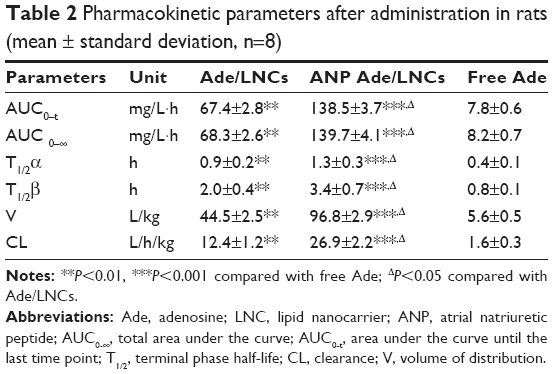 | Table 2 Pharmacokinetic parameters after administration in rats (mean ± standard deviation, n=8) |
Discussion
Lipid nanoparticulate systems can be harnessed to deliver therapeutic agents precisely to the infarcted heart. In this study, we would like to make a comparison of the lipid prodrug to cell-membrane-based nanoparticles (NPs). Cell-membrane-coated nanocarriers have been explored for their beneficial biocompatibility and modifications through lipid-insertion or chemical conjugation.33,34 Cell-membrane-coated surface engineering approach provides a platform to functionalize nanocarriers without sacrificing the natural function of cell membranes. In contrast, lipid prodrug-based nanocarriers provide an increase of drug chemical stability in vivo and a reduced toxicity before the metabolization occurs. The aim of the present study was to fabricate an Ade prodrug-based, ANP-modified nanosystem for the treatment of myocardial infarction. First, Ade-OA prodrug and ANP-PEG-DSPE were synthesized. ANP-PEG-DSPE is an amphiphilic compound constructed by a lipophilic DSPE head and a hydrophilic ANP-PEG tail. The amphiphilic nature of ANP-PEG-DSPE makes it possible to self-assemble to LNCs with a lipid inner core and a hydrophilic shell. The hydrophobic Ade-OA could also be assembled to the lipid core during the preparation process. Larger size of ANP Ade/LNCs than that of Ade/LNCs could be explained by ANP modification on the LNC surface that increased the diameter of the nanocarriers. This could also be taken as an evidence that ANP was successfully modified on the outer layer of the system.35 Particle size is a key effect that can influence the in vivo distribution of carriers. The great advantages of nano-sized carriers include prolonged blood circulation time and improved bioavailability.36 PDI exhibits the size distribution of NPs. PDI <0.2 could be considered as a uniform system. The positive charge of LNCs could exploit the negative charge present at the cell surface, increase the residence time of the carriers, and promote cell penetration, thus the internalization process would be facilitated.37
The in vitro Ade release from ANP Ade/LNCs was slower than Ade/LNCs. This difference may be due to the ANP modification on the surface of LNCs hindered the drug release.38 The reason behind this sustained release behavior is attributed to slow degradation of the nanomaterials, and drugs were diffused from the matrix in a sustained manner. Moreover, the PEG shell on the outside of carriers also protects the drugs and let them have longer release time. In addition, in the presence of serum, the release of drugs was not affected, indicating the stability of LNCs.
Cellular uptake research could provide some circumstantial evidence to display the advantages of the nanocarriers to enter the cells.39 Cell uptake efficiency of ANP-modified LNCs was significantly higher compared with Ade/LNCs. This could be attributed to the ability of ANP modification that promotes the LNCs pass through the cells and accumulate in the cells, and also the PEG could prolong blood circulation time, enhance the solubility of hydrophobic drugs and improve cellular uptake.
The cytotoxicity of any nanoparticulate systems is dependent on several material-related and biological factors, such as cell type and exposure conditions.40 For example, the particle size, hydrophobicity, and surface charge can be crucial for the production of ROS and create potential sites of interaction with receptors, leading to variable degrees of cytotoxicity. In the present study, we have evaluated the cytocompatibility of cell viability of the LNCs over the concentration range from 1 to 200 μM on H9c2 cells that were to elucidate the best non-toxic concentration to be used later in the in vivo studies. In all cases, it was possible to observe that there is no obvious cytotoxicity of the LNCs in the studied concentrations. So, the concentration below 200 μM can be the safe range for further studies.
In vivo infarct therapy effect was evaluated by measuring the inhibition of the infarct size. Infarct size is conceived as one of critical indices for evaluating the cardiac damage in the generation of ischemic heart disease.41 To establish the AMI rat model for the evaluation of in vivo infarct therapy effect, coronary artery ligation and isoprenaline injection are the two methods that are commonly used.42 Between these two methods, coronary artery ligation is more clinically relevant for it can be used to imitate the clinical status of patients. In this study, compared with the AMI group, the infarct size was obviously decreased in the free Ade group, Ade/LNC group, and ANP Ade/LNC group. ANP Ade/LNC group exhibited the strongest efficiency in the inhibition of infarct size. This may attribute to ANP that could bind to specific NPRs, which are expressed predominantly in the endocardium of the ischemic heart. Moreover, ANP itself could inhibit IR injury and reduces infarct size. In vivo blood circulation half-lives were calculated based on two-compartment model of pharmacokinetics via PKSolver.43,44 In vivo tissue distribution and pharmacokinetics data showed long-circulating characteristics of LNCs than that of free drug, which proved that LNCs elongate the circulation time of drugs in serum and have great potential to accumulate in the myocardial infarct area. ANP-modified LNCs are candidates to target the infarct myocardium in a receptor-dependent manner.45 The aim of the ANP modification is to deliver more ingredient to the infarct zone. ANP Ade/LNCs exhibited higher heart concentration in comparison with Ade/LNCs in AMI rats which could be due to the targeted ability of ANP used for the modification. ANP Ade/LNCs exhibited higher AUC in comparison with Ade/LNCs and free Ade. This phenomenon could be due to the sustained release of Ade due to the long circulation effect of the PEG.46 It was reported that PEG could improve the surface hydrophilicity of nanocarriers and prevented the absorption of lipoproteins and opsonins effectively. Therefore, the conformational clouds of PEG over the nanocarriers could avoid the recognition of the reticuloendothelial system and prolong LNC circulation time in vivo.
Conclusion
In this research, Ade-OA prodrug and ANP-PEG-DSPE ligand were synthesized. ANP-PEG-DSPE-modified Ade-loaded LNCs were prepared as a novel drug delivery carrier due to the targeting ability and therapeutic efficiency to AMI. In vivo inhibition effect on infarct size, tissue distribution, and pharmacokinetics studies in AMI rats showed that ANP Ade/LNCs exhibited better efficiency than non-modified Ade/LNCs and free Ade in all respects. These results indicated that the ANP Ade/LNCs can be used as a promising system for the treatment of cardiovascular diseases.
Disclosure
The authors report no conflicts of interest in this work.
References
Benjamin EJ, Blaha MJ, Chiuve SE, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2017 update: a report from the American Heart Association. Circulation. 2017;135(10):e146–e603. | ||
Wang L, Zhou Y, Qian C, Wang Y. Clinical characteristics and improvement of the guideline-based management of acute myocardial infarction in China: a national retrospective analysis. Oncotarget. 2017;8(28):46540–46548. | ||
Writing Committee Members; Yancy CW, Jessup M, Bozkurt B, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;128(16):e240–e327. | ||
Lundy DJ, Chen KH, Toh EK, Hsieh PC. Distribution of systemically administered nanoparticles reveals a size-dependent effect immediately following cardiac ischaemia-reperfusion injury. Sci Rep. 2016;6:25613. | ||
Nakano Y, Matoba T, Tokutome M, et al. Nanoparticle-mediated delivery of irbesartan induces cardioprotection from myocardial ischemia-reperfusion injury by antagonizing monocyte-mediated inflammation. Sci Rep. 2016;6:29601. | ||
Kassimis G, Davlouros P, Patel N, et al. Adenosine as an adjunct therapy in ST elevation myocardial infarction patients: myth or truth? Cardiovasc Drugs Ther. 2015;29(5):481–493. | ||
Kazemzadeh-Narbat M, Reid M, Brooks MS, Ghanem A. Chitosan nanoparticles as adenosine carriers. J Microencapsul. 2015;32(5):460–466. | ||
Forman MB, Stone GW, Jackson EK. Role of adenosine as adjunctive therapy in acute myocardial infarction. Cardiovasc Drug Rev. 2006;24(2):116–147. | ||
Layland J, Carrick D, Lee M, Oldroyd K, Berry C. Adenosine: physiology, pharmacology, and clinical applications. JACC Cardiovasc Interv. 2014;7(6):581–591. | ||
Ernens I, Bousquenaud M, Lenoir B, Devaux Y, Wagner DR. Adenosine stimulates angiogenesis by up-regulating production of thrombospondin-1 by macrophages. J Leukoc Biol. 2015;97(1):9–18. | ||
Chin KY, Qin C, May L, Ritchie RH, Woodman OL. New pharmacological approaches to the prevention of myocardial ischemia–reperfusion injury. Curr Drug Targets. 2017;18(15):1689–1711. | ||
Möser GH, Schrader J, Deussen A. Turnover of adenosine in plasma of human and dog blood. Am J Physiol. 1989;256(4 Pt 1):C799–C806. | ||
Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets – what are the challenges? Nat Rev Drug Discov. 2013;12(4):265–286. | ||
Gaudin A, Lepetre-Mouelhi S, Mougin J, et al. Pharmacokinetics, biodistribution and metabolism of squalenoyl adenosine nanoparticles in mice using dual radio-labeling and radio-HPLC analysis. J Control Release. 2015;212:50–58. | ||
Mura S, Bui DT, Couvreur P, Nicolas J. Lipid prodrug nanocarriers in cancer therapy. J Control Release. 2015;208:25–41. | ||
Rautio J, Kumpulainen H, Heimbach T, et al. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7(3):255–270. | ||
Shao M, Yang W, Han G. Protective effects on myocardial infarction model: delivery of schisandrin B using matrix metalloproteinase-sensitive peptide-modified, PEGylated lipid nanoparticles. Int J Nanomedicine. 2017;12:7121–7130. | ||
Dong Z, Guo J, Xing X, Zhang X, Du Y, Lu Q. RGD modified and PEGylated lipid nanoparticles loaded with puerarin: formulation, characterization and protective effects on acute myocardial ischemia model. Biomed Pharmacother. 2017;89:297–304. | ||
Zhang S, Wang J, Pan J. Baicalin-loaded PEGylated lipid nanoparticles: characterization, pharmacokinetics, and protective effects on acute myocardial ischemia in rats. Drug Deliv. 2016;23(9):3696–3703. | ||
Cheraghi M, Negahdari B, Daraee H, Eatemadi A. Heart targeted nanoliposomal/nanoparticles drug delivery: an updated review. Biomed Pharmacother. 2017;86:316–323. | ||
Kasama S, Furuya M, Toyama T, Ichikawa S, Kurabayashi M. Effect of atrial natriuretic peptide on left ventricular remodelling in patients with acute myocardial infarction. Eur Heart J. 2008;29(12):1485–1494. | ||
Ferreira MPA, Ranjan S, Kinnunen S, et al. Drug-loaded multifunctional nanoparticles targeted to the endocardial layer of the injured heart modulate hypertrophic signaling. Small. Epub 2017 Jul 1. | ||
Gaudin A, Yemisci M, Eroglu H, et al. Squalenoyl adenosine nanoparticles provide neuroprotection after stroke and spinal cord injury. Nat Nanotechnol. 2014;9(12):1054–1062. | ||
Wang W, Xi M, Duan X, Wang Y, Kong F. Delivery of baicalein and paclitaxel using self-assembled nanoparticles: synergistic antitumor effect in vitro and in vivo. Int J Nanomedicine. 2015;10:3737–3750. | ||
Zhang B, Zhang Y, Yu D. Lung cancer gene therapy: transferrin and hyaluronic acid dual ligand-decorated novel lipid carriers for targeted gene delivery. Oncol Rep. 2017;37(2):937–944. | ||
Ghassami E, Varshosaz J, Jahanian-Najafabadi A, Minaiyan M, Rajabi P, Hayati E. Pharmacokinetics and in vitro/in vivo antitumor efficacy of aptamer-targeted Ecoflex(®) nanoparticles for docetaxel delivery in ovarian cancer. Int J Nanomedicine. 2018;13:493–504. | ||
Liu C, Liu F, Feng L, Li M, Zhang J, Zhang N. The targeted co-delivery of DNA and doxorubicin to tumor cells via multifunctional PEI-PEG based nanoparticles. Biomaterials. 2013;34(10):2547–2564. | ||
Zhang R, Ru Y, Gao Y, Li J, Mao S. Layer-by-layer nanoparticles co-loading gemcitabine and platinum (IV) prodrugs for synergistic combination therapy of lung cancer. Drug Des Devel Ther. 2017;11:2631–2642. | ||
Qiu J, Cai G, Liu X, Ma D. α(v)β(3) Integrin receptor specific peptide modified, salvianolic acid B and panax notoginsenoside loaded nanomedicine for the combination therapy of acute myocardial ischemia. Biomed Pharmacother. 2017;96:1418–1426. | ||
Galagudza M, Korolev D, Postnov V, et al. Passive targeting of ischemic-reperfused myocardium with adenosine-loaded silica nanoparticles. Int J Nanomedicine. 2012;7:1671–1678. | ||
Lin X, Wang ZJ, Wang S, et al. Comparison of tissue distribution of a PEGylated Radix Ophiopogonis polysaccharide in mice with normal and ischemic myocardium. Eur J Pharm Biopharm. 2011;79(3):621–626. | ||
Luo CF, Yuan M, Chen MS, et al. Pharmacokinetics, tissue distribution and relative bioavailability of puerarin solid lipid nanoparticles following oral administration. Int J Pharm. 2011;410(1–2):138–144. | ||
Fang RH, Hu CM, Chen KN, et al. Lipid-insertion enables targeting functionalization of erythrocyte membrane-cloaked nanoparticles. Nanoscale. 2013;5(19):8884–8888. | ||
Zhou H, Fan Z, Lemons PK, Cheng H. A facile approach to functionalize cell membrane-coated nanoparticles. Theranostics. 2016;6(7):1012–1022. | ||
Li S, Wang XP. In vitro and in vivo evaluation of novel NGR-modified liposomes containing brucine. Int J Nanomedicine. 2017;12:5797–5804. | ||
De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. | ||
Harush-Frenkel O, Rozentur E, Benita S, Altschuler Y. Surface charge of nanoparticles determines their endocytic and transcytotic pathway in polarized MDCK cells. Biomacromolecules. 2008;9(2):435–443. | ||
Fang YP, Hu PY, Huang YB. Diminishing the side effect of mitomycin C by using pH-sensitive liposomes: in vitro characterization and in vivo pharmacokinetics. Drug Des Devel Ther. 2018;12:159–169. | ||
Ji P, Yu T, Liu Y, et al. Naringenin-loaded solid lipid nanoparticles: preparation, controlled delivery, cellular uptake, and pulmonary pharmacokinetics. Drug Des Devel Ther. 2016;10:911–925. | ||
Ferreira MP, Ranjan S, Correia AM, et al. In vitro and in vivo assessment of heart-homing porous silicon nanoparticles. Biomaterials. 2016;94:93–104. | ||
Pascual-Gil S, Simón-Yarza T, Garbayo E, Prosper F, Blanco-Prieto MJ. Tracking the in vivo release of bioactive NRG from PLGA and PEG-PLGA microparticles in infarcted hearts. J Control Release. 2015;220(Pt A):388–396. | ||
Yao C, Shi X, Lin X, Shen L, Xu D, Feng Y. Increased cardiac distribution of mono-PEGylated Radix Ophiopogonis polysaccharide in both myocardial infarction and ischemia/reperfusion rats. Int J Nanomedicine. 2015;10:409–418. | ||
Zhou H, Fan Z, Deng J, et al. Hyaluronidase embedded in nanocarrier PEG shell for enhanced tumor penetration and highly efficient antitumor efficacy. Nano Lett. 2016;16(5):3268–3277. | ||
Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99(3):306–314. | ||
Nguyen J, Sievers R, Motion JP, Kivimäe S, Fang Q, Lee RJ. Delivery of lipid micelles into infarcted myocardium using a lipid-linked matrix metalloproteinase targeting peptide. Mol Pharm. 2015;12(4):1150–1157. | ||
Pawar H, Surapaneni SK, Tikoo K, et al. Folic acid functionalized long-circulating co-encapsulated docetaxel and curcumin solid lipid nanoparticles: in vitro evaluation, pharmacokinetic and biodistribution in rats. Drug Deliv. 2016;23:1453–1468. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.