")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Antiproliferation effect of imatinib mesylate on MCF7, T-47D tumorigenic and MCF 10A nontumorigenic breast cell lines via PDGFR-β, PDGF-BB, c-Kit and SCF genes
Authors Kadivar A, Kamalidehghan B, Akbari Javar H, Karimi B, Sedghi R, MI Noordin
Received 7 October 2016
Accepted for publication 7 December 2016
Published 21 February 2017 Volume 2017:11 Pages 469—481
DOI https://doi.org/10.2147/DDDT.S124102
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Ali Kadivar,1 Behnam Kamalidehghan,1 Hamid Akbari Javar,2 Benyamin Karimi,3 Reihaneh Sedghi,4 Mohamed Ibrahim Noordin1
1Department of Pharmacy, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia; 2Department of Pharmaceutics, Faculty of Pharmacy, Tehran University of Medical Sciences (TUMS), Tehran, Iran; 3Department of Pharmacology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia; 4Faculty of Medicine, Shiraz University of Medical Sciences (SUMS), Shiraz, Iran
Abstract: Recent cancer molecular therapies are targeting main functional molecules to control applicable process of cancer cells. Attractive targets are established by receptor tyrosine kinases, such as platelet-derived growth factor receptors (PDGFRs) and c-Kit as mostly irregular signaling, which is due to either over expression or mutation that is associated with tumorigenesis and cell proliferation. Imatinib mesylate is a selective inhibitor of receptor tyrosine kinase, including PDGFR-β and c-Kit. In this research, we studied how imatinib mesylate would exert effect on MCF7 and T-47D breast cancer and MCF 10A epithelial cell lines, the gene and protein expression of PDGFR-β, c-Kit and their relevant ligands platelet-derived growth factor (PDGF)-BB and stem cell factor (SCF). The MTS assay was conducted in therapeutic relevant concentration of 2–10 µM for 96, 120 and 144 h treatment. In addition, apoptosis induction and cytostatic activity of imatinib mesylate were investigated with the terminal deoxynucleotidyl transferase dUTP nick end labeling TUNEL and cell cycle assays, respectively, in a time-dependent manner. Comparative real-time PCR and Western blot analysis were conducted to evaluate the expression and regulation of imatinib target genes and proteins. Our finding revealed that imatinib mesylate antiproliferation effect, apoptosis induction and cytostatic activity were significantly higher in breast cancer cell lines compared to MCF 10A. This effect might be due to the expression of PDGFR-β, PDGF-BB, c-Kit and SCF, which was expressed by all examined cell lines, except the T-47D cell line which was not expressed c-Kit. However, examined gene and proteins expressed more in cancer cell lines. Therefore, imatinib mesylate was more effective on them. It is concluded that imatinib has at least two potential targets in both examined breast cancer cell lines and can be a promising drug for targeted therapy to treat breast cancer.
Keywords: Gleevec, breast cancer, normal breast cell line, tyrosine kinase receptor, protein expression, comparative real-time PCR, cell cycle analysis, cell cycle arrest, cytostatic activity
Introduction
Uncontrolled proliferation of tumor cells as a result of abnormal activation of numerous receptor tyrosine kinases (RTKs) and respected ligands is pertinent to the events of human cancer development. RTKs establish a receptor family that control main cellular events, such as cell migration, differentiation, apoptosis and proliferation. The RTK family comprises growth factor receptors, such as the receptor of stem cell factor (SCF), c-Kit, platelet-derived growth factor receptor (PDGFR), the insulin growth factor receptor and epidermal growth factor receptor.1–4
Growth factors facilitate their signal initiating the activation of cell surface transmembrane receptors to intracellular effectors that regulate major cancer cells functions. PDGF can facilitate differentiating, migrating and proliferating roles in developed and developing cells. The PDGF family members (PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC and PDGF-DD) attach to structurally linked RTKs, PDGFR-β and PDGFR-α. PDGFRs and their ligands are co-expressed creating paracrine and autocrine loops in vitro causing several cellular changes.4–7 Abnormal activity of PDGFRs and their ligands have also been observed in breast, melanomas, lung, gastric, ovarian and prostatic cancers.8
The c-Kit proto-oncogene encrypts c-Kit receptor, which belongs to the same superfamily of PDGFRs, a transmembrane glycoprotein with a tyrosine kinase activity in the intracellular domain. SCF (c-Kit natural ligand) is a hematopoietic growth factor that triggers the receptor-promoting autophosphorylation and dimerization at certain tyrosine remains. c-Kit signals main cellular functions, such as cell chemotaxis, survival, differentiation, proliferation and adhesion. Moreover, it promotes apoptosis and improves the invasive potential stimulating several signal transduction pathways, such as the STAT3, MAP kinase cascade, PLC and PI3-K. Abnormal expression of SCF and/or c-Kit has been observed in human malignant cells from prostate, colorectal carcinoma, neural, lung and breast.9,10
Breast cancer is among three most prevalent cancers and the most common neoplasm malignant in women worldwide with constantly increasing incidence in recent years.11 Surgery is contemplated as the primary step of early-stage breast cancer management; many cases are managed and treated only through surgery. The breast cancer surgery through negative margins aims to do full resection of the main tumor to reduce the possibility of local recurrences as well as pathologic staging of tumor and the axillary lymph nodes and to investigate necessary prognostic information.12 Adjuvant management of breast cancer cures micrometastatic disease (breast cancer cells that have run away from the regional lymph nodes and breast but still do not have a proven detectable metastasis). This treatment includes systemic and radiation therapies (comprising a range of chemotherapeutic, hormonal and biologic agents).13
Breast cancer cells need high activation and/or expression of RTKs to proliferate, attack and metastasize.14 Recently, researchers have tried to find new molecular targets to improve in patients’ response and survival with fewer side effects. Therefore, new treatments that control the activity of receptors, such as RTKs, were integrated with treatment protocols.15–17
In the last decade, one of the interesting pathways for cancer therapy has been the kinase cascade pathway, which initiates with growth factors, such as PDGFs and RTKs, such as c-Kit and PDGFR.18
Due to the importance of PDGFRs and c-Kit in the tumorigenesis progress, they are targets for molecular cures. Many strategies have been developed to hinder aberrant tyrosine kinase activity, including small molecule inhibitors, antigenic stimulation and antisense oligonucleotides.19,20 As an antineoplastic small molecule, imatinib mesylate hinders specific protein tyrosine kinases interfere in oncogenesis, such as Brc-Abl that is the principal source of chronic myelogenous leukemia, PDGF and c-Kit receptors that control main cellular events in many solid tumors by preventing signaling of RTKs.21–24 Blocking the intracellular ATP-binding site inhibits their activation and the following signal transduction. Imatinib administration reduces autocrine receptor stimulation and the unusual stimulation of mutated kinases seen among some carcinoma cells. It also prevents the progression of dermatofibrosarcoma protuberans and promotes apoptosis in leukemia and neuroblastoma.4,25 There is evidence that imatinib incubation prevents PDGFRs/c-kit and cell growth in some types of cancers. Furthermore, imatinib hinders the proliferation and invasiveness of a panel of human epithelial breast cancer cells with different invasion potentials.25–28
It is also used for the treatment of chronic gastrointestinal stromal tumors and myelogenous leukemia and has exhibited promising in vivo/in vitro results in small cell lung and colon cancer cell lines, where unusual c-Kit expression is among the primary causes of tumor progression.29–31
It is essential to study the effects of imatinib on SCF/c-Kit- and PDGF-BB/PDGFR-β-mediated cellular effect, including apoptosis induction, cell proliferation suppression and their gene/protein modulation in breast cancer cells.23
Two breast cancer cell lines, namely T-47D and MCF7, were compared with a nontumorigenic breast epithelial cell line MCF 10A in this study. T-47D and MCF7 are human tumorigenic epithelial mammary gland cell lines, which are derived from the pleural effusion metastatic site of ductal carcinoma and adenocarcinoma from 54- to 65-year-old females, respectively. MCF 10A is a nontumorigenic immortal epithelial mammary gland cell line, derived from fibrocystic 36-year-old female.
Materials and methods
Materials
Osvah Pharmaceutical Company (Tehran, Iran) provided the Imatinib mesylate. The cell culture medium RPMI 1640 was supplied from the Malaysia Sigma-Aldrich (Kuala Lumpur, Malaysia), fetal bovine serum (FBS) from GIBCO-BRL (Bio-Diagnostics Sdn. Bhd., Petaling Jaya, Selangor, Malaysia) and MEGM kit media from Lonza/Clonetics Corporation (Basel, Switzerland). All other substances were employed at analytical grade, and deionized water was processed by reverse osmosis.
Cell culture
One breast adenocarcinoma cell line MCF7 (American Type Culture Collection [ATCC®], Manassas, Virginia, USA HTB-22) and one breast ductal carcinoma cell line T-47D (ATCC HTB-133) were studied and compared with a fibrocystic epithelial breast cell MCF 10A (ATCC CRL-10317) as nontumorigenic cell line. Cell lines were gifted from Institut Biosains, UPM (Serdang, Malaysia) followed the ATCC. MCF7 and T-47D cancer cell lines were grown with 5% CO2 at 37°C in RPMI 1640 medium containing 5% FBS and MCF 10A were grown in MEGM kit media without gentamycin/amphotericin B mix plus 100 ng/mL cholera toxin.
Cell treatment
Cell lines in culture condition were harvested from 75 cm2 flasks, rinsed with the phosphate-buffered saline (PBS) and moved to 96-well plates containing full medium at a seeding density of 1,000 cells/well for MCF7, 1,500 cells/well for T-47D and 2,000 cells/well for MCF 10A.
All the treatment groups were cultured in triplicates. Twenty-four hours later, the cells were incubated in the above-mentioned culture medium containing 2, 3, 4, 5, 6, 7, 8, 9 and 10 μM imatinib mesylate for 96, 120 and 144 h. Every 48 h, the medium with or without imatinib mesylate was completely replaced.23
Furthermore, for positive control, the cells were exposed to hydrogen peroxide 1 mM to achieve minimal viability, which caused full cell death, verified by the viability staining of trypan blue. For negative control or maximum viability, the untreated cells with intact medium and no drug were used.
In vitro proliferation assay
Following the cell treatment procedure, which was mentioned, the Cell Titer Aqueous One Solution assay (Promega Corporation, Madison, USA) was conducted according to the kit provided procedure due to the investigation of cell proliferation inhibition. Then, 50% mortality of viable cells, that is, half minimal inhibitory concentration (IC50) of imatinib, was calculated using Eq (1); thus, the IC50 was used in the advance experiments. The Student’s t-test was used for statistical analysis and P-value of <0.05 was considered as significant:
|
|
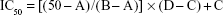
A represents the first point on the antiproliferation effect % vs imatinib mesylate concentration curve, which is <50%. B is the first point on the antiproliferation effect % vs. Imatinib mesylate concentration curve, which is ≥50%. C signifies the concentration of imatinib mesylate that provides A% of inhibition, and D is the concentration of imatinib mesylate that results in B% inhibition.32
TUNEL assay
The Cell Death Detection enzyme-linked immunosorbent assay (ELISA) plus kit (Roche Molecular Biochemicals, Mannheim, Germany) was employed to evaluate the apoptosis hallmark (oligonucleosomal DNA fragmentation), which set up the meticulous mode of apoptotic cells induced by the imatinib mesylate. Cell lines seeded in a 24-well plate with the seeding density of 6,500 cells per well for T-47D, 4,500 cells per well for MCF7 and 8,500 cell per well for MCF 10A.
After 24 h of cells attachment, the full cell culture medium with each cell corresponding IC50 concentration of imatinib mesylate was replaced with normal medium. Nontreated control group was cultured with just normal complete media. Besides, the kit provided the positive control. The culture medium with/without imatinib mesylate was replaced every 48 h. After 144 h, the kit manufacturer’s sample preparation protocol was followed, and ELISA reader was used to obtain spectrophotometric data at 405 nm.
Cell cycle assay
MCF7, T-47D and MCF 10A cell lines in the exponential phase of growth were treated with imatinib mesylate for 72 h, following the untreated group for each individual cell line. After the incubation, treated and untreated cell lines were harvested and washed twice with ice-cold PBS, then cells were fixed using 80% cold ethanol (700 μL) at 4°C, overnight. Later on, cells were washed with cold PBS and suspended in ice-cold PBS (500 μL). Cells were stained with 200 μL propidium iodide (PI) 50 μg/mL in the presence of 50 μL RNase A 100 μg/mL for 40 min at room temperature. To specify the PI-binding site to just the DNA content of experimental cell lines not the RNA content, RNase A was used to degrade the remaining RNA content. Cell cycle distribution was analyzed using a flow cytometer (BD FACSCanto II; BD Biosciences, San Jose, CA, USA) and a ModFit LT v3.0 software (Verity Software House, Inc., Topsham, ME, USA).
Imatinib mesylate treatment for gene expression study
Potential changes in PDGFR-β, c-Kit and their corresponding ligands PDGF-BB and SCF gene expression in the cell lines cured with imatinib were evaluated. To examine the gene expression, cell lines were cultured in six-well plates containing RPMI 1640 with 5% FBS. After 24 h of cells adhesion, the cell lines were cultured in normal medium in a way that each cell line relevant to IC50 of imatinib mesylate was treated for 144 h. The medium with/without imatinib was totally changed every 48 h. Afterward, the total RNA was extracted and gene expression study was conducted, which will be explained later.
Receptor expression
Expression profile of c-Kit, PDGFR-β and their ligands SCF and PDGF-BB were determined at the correlated transcriptional level in each individual cell line. The RNeasy mini kit (Qiagen, Kuala Lumpur, Malaysia) was used to extract total RNA, and RNase-Free DNase (Qiagen) ruled out the genomic DNA. The extracted total RNA was stored at −70°C.
Relative quantification of RNA transcriptional levels, which is encoding PDGFR-β, c-Kit, PDGF-BB and SCF genes, was investigated by the QuantiTect Primer Assay kit (Qiagen) applying reverse transcription polymerase chain reaction (RT-PCR) SYBR Green detection following One-Step RT-PCR Standard Protocol using the total extracted RNA with β-actin is used as the reference gene for gene expression analysis. Extracted RNA (0.1 μg) from the untreated and treated breast cancer cell lines with their relevant IC50 concentration of imatinib mesylate were applied to one-step RT-PCR amplifications in 50 μL reactions, including the QuantiTect Primer Assay (which includes primer pairs as shown in Figure 1), QuantiTect SYBR Green RT-PCR Master Mix and QuantiTect RT Mix.
 | Figure 1 QuantiTect Primer Assay specification. |
Applied biosystems StepOne Plus RT-PCR machine was employed for the RT-PCR amplifications following the subsequent protocol. Transcriptase was activated at 50°C for 30 min for reverse transcription, then for 15 min at 95°C to stimulate HotStar Taq DNA polymerase, inactivate transcriptase and denature resultant cDNA template. Next, 35 cycles of three-step cycling, including denaturation, were conducted at 94°C for 15 s, annealing at 55°C for 30 s and extension of 30 s at 72°C. Finally, amplification was completed with 10 min of final extension at 72°C.
Each test was repeated three times in duplicate tubes. To monitor the reagents used in the assay for probable contamination, control tubes without template RNA were incorporated into duplicates together with the appropriate primer sets in each test. The PCR amplifications were also conducted as negative control without template RNA. The amplification cycle was determined using the StepOne Software v2.3, in which product buildup was beyond the threshold (Ct). RT-PCR Ct values were analyzed using the standard REST 2009 software, which is a standalone software tool developed by Qiagen and M. Pfaffl (Technical University of Munich) for gene expression data analysis.33 To confirm the reliability of the results in each experiment, the mean Ct of each sample for a given genes in each experiment (repeated four times) was normalized to the mean Ct value of the reference gene in the untreated and treated cell lines, which was employed as control (β-actin).
The untreated relative genes expression was calculated according to housekeeping gene using the following equation:
|
|
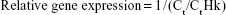
where Ct is the average of interested gene’s Ct values which were reported from four repeated samples in untreated cell lines and CtHk is the average Ct value of endogenous control β-actin.
Western blotting assay
To evaluate the presence of imatinib mesylate and its effect on protein expression of c-Kit and PDGFR-β and their correlated ligands PDGF-BB and SCF, cells were seeded in 12-well plates and were exposed to 0 and their own identified corresponding IC50 of imatinib mesylate for 144 h. Cell lysis buffer (120 mM NaCl, 50 mM Tris-HCl pH 8.0, 1 mM phenylmethane sulfonyl fluoride 0.5% NP-40) was used to extract the total cell proteins and 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis was used to separate 40 μg of extracted protein. Then, it was moved to a polyvinylidenedifluoride membrane (Bio-Rad), blocked at room temperature with 5% skimmed milk in the TBS-Tween buffer 7 (1.5 M NaCl, 0.1% Tween20, 0.12 M Tris-base) for 1 h and was incubated with the relevant primary antibodies at 4°C overnight. Next, it was incubated with alkaline phosphatase (AP)-conjugated secondary antibody for 30 min at room temperature. The primary antibodies, PDGFR-β, c-Kit, β-actin, PDGF-BB and SCF were acquired from Santa Cruz Biotechnology Inc. Then, the PVDF membranes were incubated for 45 min with AP-conjugated antimouse (1:5,000) or AP antirabbit (1:5,000) secondary antibodies, and then were washed twice with the TBST. The blots were developed on membranes by the colorimetric detection, BCIP/NBT, to quantify the target protein bands.
Statistical analysis
To test the significance of data and association between the treated and the untreated control groups, paired Student’s t-test analysis was performed (P>0.05). The REST 2009 was used to conduct the statistical analysis. It is the stand-alone software tool established by M Pfaffl (Technical University of Munich) and Qiagen for analysis of comparative-quantitative gene expression data33 and Microsoft Office Excel 2013.
Results
Antiproliferation effect of imatinib on breast cancer vs normal cell lines
In order to examine the suppression effect of imatinib mesylate on proliferation and growth of breast cancer cell lines, cell proliferation assay (MTS) was conducted in a dosage- and time-dependent manner. MCF 10A epithelial breast cell line and two examined breast cancer cell lines were treated for 96, 120 and 144 h with concentrations of 2–10 μM with the bovine serum-containing medium for MCF7 and T-47D and full media for MCF 10A. As shown in Figure 2, imatinib mesylate established a significant dose-dependent effect in both MCF7 and T-47-D breast cancer cell lines, but did not exert any remarkable effect on MCF 10A epithelial breast cell line. The breast cancer cell lines growth inhibition may well be associated with the Weigel et al and Malavaki et al findings.23,25 Imatinib mesylate established significant dose-dependent effect in both cancerous cell lines but did not exert any remarkable effect on MCF 10A epithelial breast cell line. Moreover in both breast cancer cell lines, no significant effect was detected by time-depended treatment in 48 and 96 h; however, longer treatment time caused more antiproliferation effect. Therefore, it was demonstrated that imatinib mesylate cancer cell treatment with at least 48 h of difference caused a significant time-dependent antiproliferation effect. The average IC50 of 6.33 μM for MCF7 and 5.14 μM for T-47D were determined on the viability of cell lines in 144 h of exposure (Figure 3).
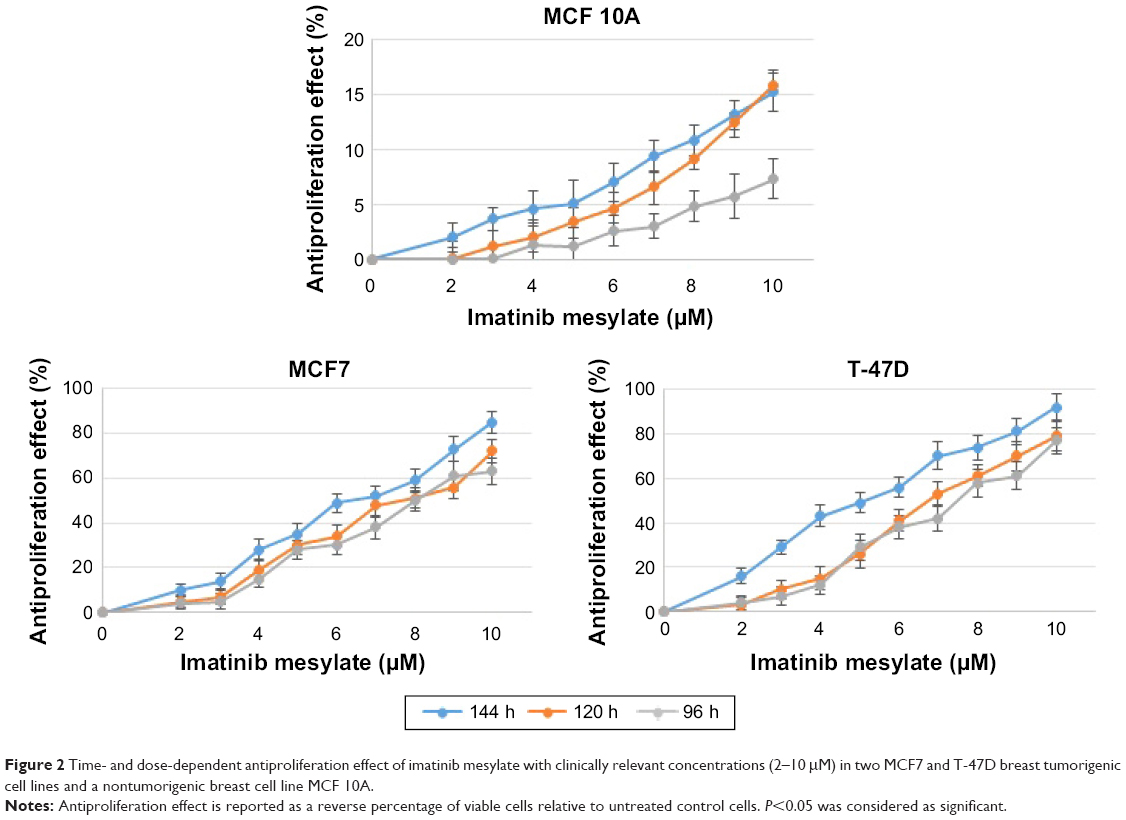 | Figure 2 Time- and dose-dependent antiproliferation effect of imatinib mesylate with clinically relevant concentrations (2–10 μM) in two MCF7 and T-47D breast tumorigenic cell lines and a nontumorigenic breast cell line MCF 10A. |
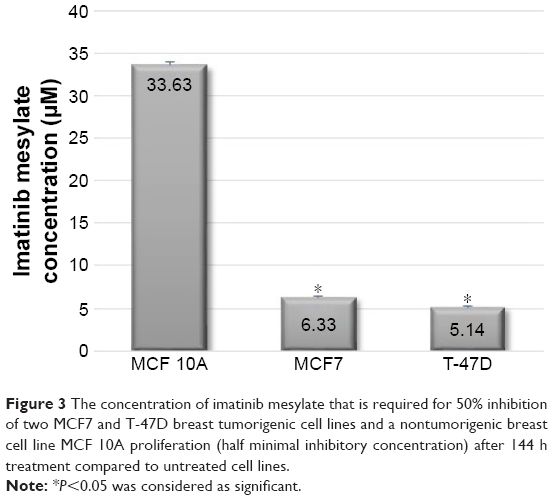 | Figure 3 The concentration of imatinib mesylate that is required for 50% inhibition of two MCF7 and T-47D breast tumorigenic cell lines and a nontumorigenic breast cell line MCF 10A proliferation (half minimal inhibitory concentration) after 144 h treatment compared to untreated cell lines. |
The results revealed that 144 h of imatinib mesylate treatment with the relevant medical concentration (2–10 μM) exerted the highest and most significant toxicity effect toward the two examined breast cancer cell lines. Hence, 144 h of exposure was selected and used for the rest of the study. However, MCF 10A treatment showed low cytotoxicity or insignificant effect even after 144 h of treatment.
Imatinib induces apoptosis only in cancer cell lines
The imatinib mesylate apoptosis induction on breast cancer cell lines was determined using the quantitative TUNEL assay, which detects DNA fragmentation in the late stage of apoptosis process. The results revealed that imatinib treatment with each cell line–specific IC50 concentration after 96 h led to a significantly greater apoptotic cell numbers compared to the untreated cells in both studied cancer cell lines. But in MCF 10A, even after 144 h, still no remarkable apoptosis induction was detected. In this study, we chose the longest imatinib mesylate exposure time (144 h) for further study to let the suppression effect be accomplished in the exposed cell line.
Figure 4 shows that MCF7 apoptosis index was 35.74% after 144 h of treatment, which was higher than T-47D cancer cell line with 30.58% apoptosis index. This indicates that MCF7 was more sensitive to imatinib mesylate apoptosis induction. Meanwhile, MCF 10A with apoptosis index of 5.2% was considered as a low sensitive cell line to imatinib mesylate apoptosis induction. In this experiment, induction of apoptosis was confirmed with positive control by the Cell Death Detection ELISA plus kit (data not shown).
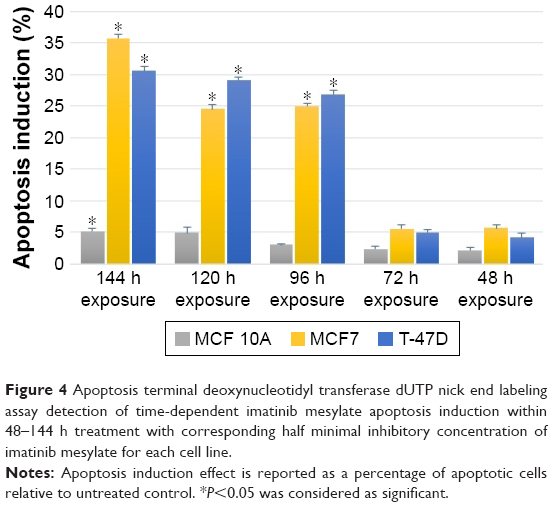 | Figure 4 Apoptosis terminal deoxynucleotidyl transferase dUTP nick end labeling assay detection of time-dependent imatinib mesylate apoptosis induction within 48–144 h treatment with corresponding half minimal inhibitory concentration of imatinib mesylate for each cell line. |
Imatinib induces cell cycle arrest at G2/M phase
As we reported, imatinib mesylate significantly inhibits breast cancer cell growth after 96 h of treatment; however, 72 h of imatinib mesylate antiproliferation effect was previously confirmed by Malavaki et al and Weigel et al23,25 for a panel of breast cancer cell lines. Moreover, apoptosis induction study revealed that after 72 h of treatment, imatinib was not able to induce apoptosis significantly nevertheless 96 h of treatment cause significant apoptosis induction in cancer cell lines but not in normal cells. Therefore, to investigate the effect of imatinib mesylate on cell cycle phase distribution (G0/G1 gap, S synthesis and G2/M mitosis) of MCF7, T-47D and MCF 10A cell lines, cell cycle analysis was conducted (Figure 5). Similar to the results of antiproliferation assay, the flow cytometric cell cycle analysis in both breast cancer cell lines revealed that imatinib inhibits cell proliferation exhibiting a significant cytostatic increase of the G2/M phase with a significant decrease at the S phase. Cell cycle and apoptosis induction results revealed that after 72 h of treatment cell growth suppression was only due to cell cycle arrest, but after 144 h treatment apoptosis induced with cell cycle arrest. However, surprisingly, imatinib mesylate was not significantly exert neither apoptosis nor cytostatic activity in MCF 10A normal mammary cell line, which can prove that imatinib mesylate targeted just cancer cell lines and not the normal cells.
 | Figure 5 The effect of imatinib mesylate on cell cycle distribution of MCF7, T-47D tumorigenic and MCF 10A nontumorigenic breast cell lines. |
PDGFR-β, c-Kit and their corresponding ligands gene expression in breast cancer vs normal cell lines
The expression and associated transcriptional level of genes encoding PDGFR-β/PDGF-BB and c-Kit/SCF in three breast cell lines were investigated using RT-PCR, and the comparative expression patterns of genes were normalized with β-actin housekeeping gene as an internal reference.
Comparative reverse transcription PCR analyses revealed variable relative expression level of PDGFR-β under culture condition in both cancer and normal cell lines, which was significantly more in cancer cell lines compared to MCF 10A normal mammary cell line. Similar to the corresponding receptor, PDGF-BB was expressed in all cell lines, which was significantly less in MCF 10A compared to the tumorigenic cell lines. The c-Kit expression was detectable only in MCF7 and MCF 10A, although there was no significant difference between them. The SCF was expressed in all three cell lines with almost the same expression pattern (Figure 6). These findings as well as previous studies by Roussidis et al and Weigel et al10,34 revealed that both examined breast cancer cell lines express at least one possible target receptor for imatinib treatment. Protein expression was then confirmed by Western blotting.
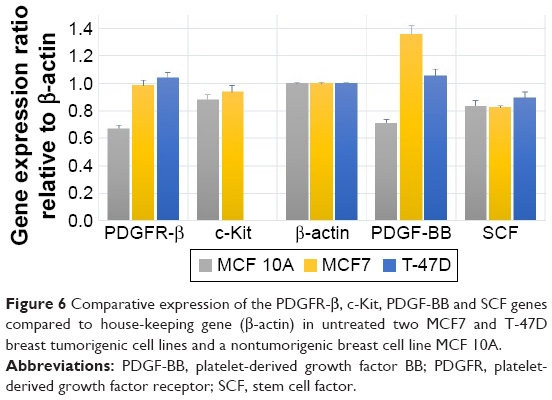 | Figure 6 Comparative expression of the PDGFR-β, c-Kit, PDGF-BB and SCF genes compared to house-keeping gene (β-actin) in untreated two MCF7 and T-47D breast tumorigenic cell lines and a nontumorigenic breast cell line MCF 10A. |
Imatinib suppresses PDGFR-β/PDGF-BB and c-Kit gene expression mostly in breast cancer cell lines
The comparative quantification approach was used for gene expression analysis, which was compared to the untreated control according to the REST 2009 software (Figure 7).
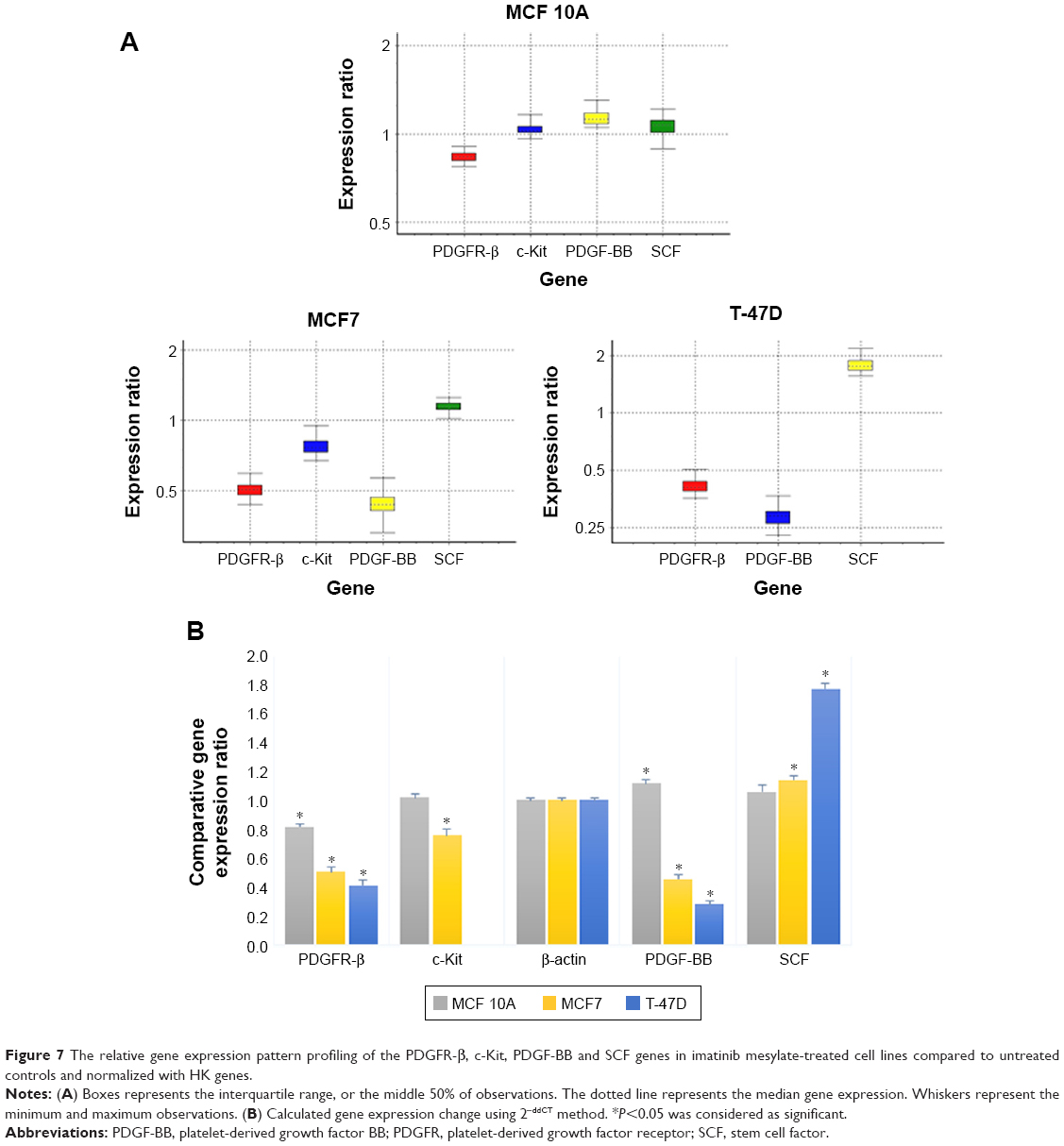 | Figure 7 The relative gene expression pattern profiling of the PDGFR-β, c-Kit, PDGF-BB and SCF genes in imatinib mesylate-treated cell lines compared to untreated controls and normalized with HK genes. |
As reported in Table 1, the expression level of PDGFR-β under its corresponding IC50 of imatinib mesylate treatment was significantly downregulated in all three cell lines (P<0.05), from which T-47D had the highest downregulation and MCF 10A exhibited less change in expression level. Similar to the corresponding receptor, the PDGF-BB results were revealed in two cancer cell lines. It was downregulated in the treated group compared to the untreated group by a mean factor of 0.46 in MCF7 cell line and a mean factor of 0.31 in T-47D cell line. However, the PDGF-BB was upregulated in the nontumorigenic MCF 10A by mean factor of 1.13. Moreover, the relative gene expression level of c-Kit was significantly decreased in MCF7, and no significant change was seen in MCF 10A. Its corresponding ligand SCF showed insignificant change in MCF 10A and significant upregulation in MCF7 and T-47D cell line with a mean factor of 1.13 and 1.78, respectively.
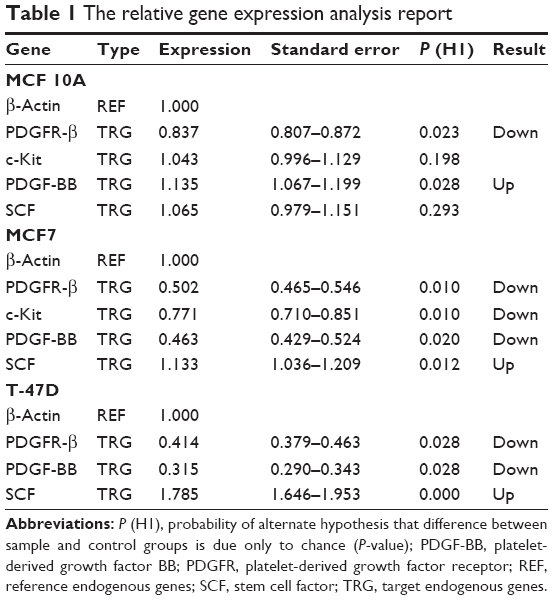 | Table 1 The relative gene expression analysis report |
PDGFR-β/PDGF-BB and c-Kit protein expression confirms the imatinib gene expression suppression in cancer cell lines
To evaluate the presence and regulation of considered genes under the imatinib mesylate treatment, a gene expression study and Western blotting were conducted. For the gene expression study, posttranscriptional regulation using Western blotting was relevant to genes transcription regulation as described.
The Western blot images were quantified three times using the ImageJ 1.47v software (Wayne Rasband National Institutes of Health, Bethesda, MD, USA). Also, the mean factors were considered as protein expression factors. Figure 8 shows relative protein expression ratio in the untreated MCF7, T-47D and MCF 10A cell lines. As demonstrated in Figure 9, the Western blot analysis showed that PDGFR-β, PDGF-BB and SCF proteins were expressed in all cell lines, whereas c-Kit protein was not expressed in T-47D cell line.
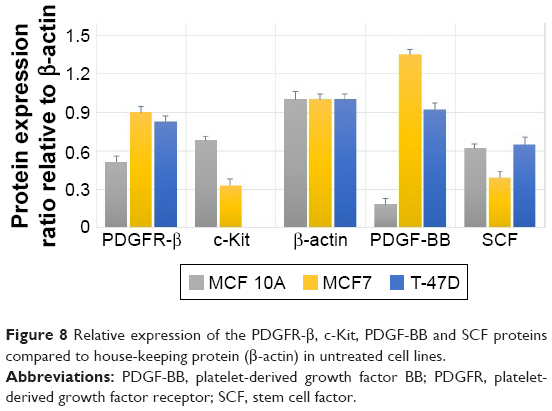 | Figure 8 Relative expression of the PDGFR-β, c-Kit, PDGF-BB and SCF proteins compared to house-keeping protein (β-actin) in untreated cell lines. |
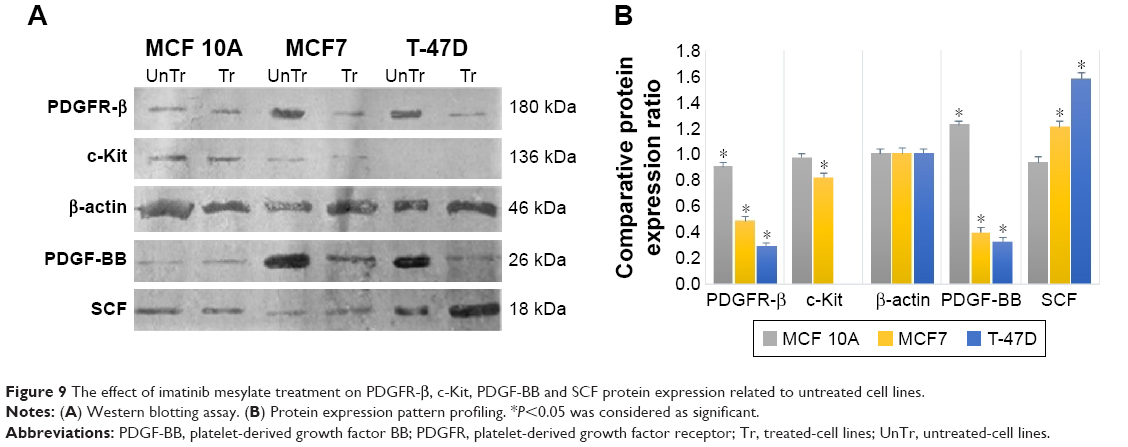 | Figure 9 The effect of imatinib mesylate treatment on PDGFR-β, c-Kit, PDGF-BB and SCF protein expression related to untreated cell lines. |
The relative protein expression results in the untreated cell lines revealed (Figure 8) that PDGF-β protein expression was almost the same in both breast carcinoma cell lines, whereas MCF 10A epithelial cell line expression was significantly lower. PDGF-BB has the highest protein expression in MCF7, whereas MCF 10A shows the lowest. The c-Kit protein was not expressed in T-47D; however, its expression was approximately two folds higher in MCF 10A compared to MCF7. The SCF expression was almost the same in MCF 10A and T-47D, whereas MCF7 expression was lower.
The Western blot comparative-quantitative analysis (Figure 9) revealed that PDGFR-β protein expression was significantly (P<0.05) downregulated under the imatinib mesylate treatment with cell lines corresponding IC50 compared to their untreated control in the same culture condition with β-actin as the internal control with a mean factor of 0.90, 0.48 and 0.29 in MCF10A, MCF7 and T-47D, respectively. It was demonstrated that the PDGF-BB expression level was significantly upregulated in MCF 10A with mean factor of 1.22, whereas downregulated in MCF7 and T-47D with a mean factor of 0.39 and 0.33, respectively. For c-Kit and SCF, the results did not show any significant change in MCF 10A, but the c-Kit protein expression was downregulated in MCF7 with a mean factor of 0.82. However, SCF expression did not significantly change in MCF 10A cell line, although it was upregulated significantly in MCF7 and T-47D cancer cell lines with a mean factor of 1.21 and 1.57, respectively.
Discussion
RTKs, such as PDGFRs and c-Kit, are essential for the development of malignancies and their progression.4,35 Most research studies on c-Kit detection have used immunohistochemistry through an antibody. Therefore, in the antibody-binding site, cancer-progressive posttranslational change may cause a remarkable receptor loss.36,37 It may be inferred that further research is necessary to understand the biological importance and the expression of c-Kit in breast cancer, because one detection technique might not be sufficient to show the expression of protein molecules, particularly at the presence of low quantities of c-Kit. Especially, cytoplasmic localization of the receptors endorses the assumption that the cytoplasm may contain a truncated receptor, in the same way as the mutated EGF receptors that do not contain parts of the extracellular portion and experience high risk transformations. Alternatively, a number of studies have exhibited the PDGFRs importance and expression.10 Therefore, we investigated the gene and protein expression of PDGFR-β/PDGF-BB and c-Kit/SCF in two breast carcinoma cell lines compared to a nontumorigenic mammary epithelial cell line using RT-PCR and Western blotting, both of which are gold standard tests to detect even small amount of considered genes and proteins in transcriptional and posttranscriptional levels.
An anticell proliferation study proved that imatinib mesylate has effect on breast cancer cell lines with a clinically relevant drug concentration range (2–10 μM).23 According to our findings, T-47D did not express the c-Kit but had a higher suppression effect in the PDGFR-β/PDGF-BB expression and was more sensitive to the imatinib with lower IC50 compared to MCF7 that expressed both imatinib mesylate target PDGFR-β and c-Kit, but had lower downregulation effect. However, imatinib treatment was not remarkably effective in the normal breast cell line MCF 10A, which can be proved by the inconsiderable suppression effect of imatinib for PDGFR-β and c-Kit expressions. Furthermore, the detected IC50 concentrations from 5 to 7 μM were obtained in vivo from the human serum.38,39 Therefore, it certifies the cell proliferation inhibitory effect of imatinib mesylate on experimented breast cancer cell lines. The reason might be the suppression of PDGFR-β and c-Kit expression and their importance in progression of cancer and cancer therapy.
Apoptosis induction study under the imatinib mesylate treatment revealed that both examined cancer cell lines experienced apoptosis, but the apoptosis induction was significant after 96 h of treatment. It can be attributed to the mechanism of action of imatinib mesylate as a RTK inhibitor, not as a toxic chemotherapeutic agent. Therefore, it prolongs the initial effect of imatinib mesylate on the internal signal transactions. The incubation period differs for other cell entities. For instance, neuroblastoma cells experience apoptosis 48 h after incubation, whereas osteosarcoma cells exhibit apoptosis 5 days later. Although these cancer cell types and some others have shown the proapoptotic effect, imatinib does not cause apoptosis at all times. For example, ovarian carcinoma cells stop in G1/G0 point of cell cycle. Tumor reduction in this case might be the result of apoptosis in vascular endothelial cells that causes an inadequate oxygen supply to the tumor.25,40,41 Moreover, Uziel et al42 also reported cell cycle arrest of Ewing sarcoma cells in G2/M phase of the cell cycle. Our finding further demonstrated a significant cell cycle arrest of the G2/M phase with a significant reduction at the S phase just in breast cancer cell lines with imatinib treatment, whereas imatinib antiproliferation effect was significantly noted as confirmed with Malavaki et al study.25 However, imatinib treatment did not cause any considerable apoptosis induction or cell cycle arrest in the normal mammary cell line MCF 10A, even after 144 h, which supports the results of proliferation study. Therefore, these findings can illustrate the targeted inhibitory effect of imatinib in breast cancer cells compared with the breast normal cell lines.
Breast carcinomas are known to express PDGF, and some to express c-Kit.23,43 Our studies revealed that PDGFR-β and PDGF-BB were remarkably expressed in both carcinoma cell lines. However, nontumorigenic mammary gland cell line, MCF 10A, did the same, but in a lower expression level. The c-Kit and SCF were expressed together in MCF7 and MCF 10A, which can confirm the autocrine loop cell signaling of PDGFR-β/PDGF-BB and c-Kit/SCF in the examined breast cell lines.7 However, expression of SCF occurred in T-47D without its corresponding ligand c-Kit, which can be relevant to the additional paracrine and/or endocrine loop of this cytokine.
Both PDGFR-β and c-Kit receptors were found effective therapeutic molecular targets in some cancers, and the selective inhibitor was observed to be against imatinib mesylate. Consequently, we were encouraged to further study the effect of imatinib mesylate in apoptosis induction and growth control of cancer cell lines in breast than nontumorigenic mammary gland cell line through PDGFR-β and c-Kit in addition to their relevant ligands PDGF-BB and SCF expression suppression.10,35
The PDGF and the RTK c-Kit are molecular targets for imatinib mesylate.21 In this study, we assured that MCF7 and T-47D express PDGFR-β, PDGF-BB and SCF, but c-Kit was expressed only in MCF7. Moreover, nontumorigenic mammary gland cell line MCF 10A expresses PDGFR-β, PDGF-BB, c-Kit and SCF. Therefore, to evaluate the effect of imatinib mesylate on the PDGFR-β/PDGF-BB and c-Kit/SCF expression modulation, we studied the transcriptional and posttranscriptional regulation of these genes. The comparison between the gene and protein expression studies are demonstrated in Figures 6 and 8, which revealed that the comparative protein expression confirmed the gene expression study. However, the difference between the protein expression level pattern and gene expression level testifies the influence of posttranscriptional regulation in the expression of the examined genes.
Our investigation demonstrated that with relevant IC50 concentration of imatinib mesylate treatment, PDGFR-β and PDGF-BB transcriptional levels are almost same as the post-transcriptional level, which were significantly downregulated in MCF7 and T-47D cell lines. Therefore, it can prove the effect of imatinib mesylate, not only in suppression of PDGFR-β expression but also in the downstream regulation of PDGF-BB expression as its corresponding ligand. Moreover, the imatinib mesylate inhibited c-Kit expression in MCF7, which can approve that both c-Kit and PDGFR-β are appropriate targets. However, in T-47D cell line, the results revealed that SCF expression is upregulated in the absence of its corresponding receptor c-Kit which, according to our finding, is not expressed in this cell line. This upregulation can be due to the endocrine signaling of this cytokine when its corresponding receptor was inhibited with imatinib mesylate treatment in other tumorigenic mammary cells. MCF 10A as a nontumorigenic mammary gland cell line control showed a minor downregulation of PDGFR-β, and an inconsiderable effect on other examined genes expression under the imatinib mesylate treatment.
Conclusion
It was revealed that imatinib mesylate is an anti-cancer targeted therapeutic agent, which can target and suppress receptor tyrosine kinases especially PDGFR-β and c-Kit pathways in breast cancer cell lines by influencing their expression down regulation. Moreover, imatinib suppressed breast cancer cell lines proliferation through apoptosis induction associated with cell cycle arrest in G2/M phase. However, its effect on breast nontumorigenic cell line was negligible. This makes it apparent to envisage the efficacy and to check the possible therapeutic effect of imatinib in patients with breast cancer and reduce the therapeutic adverse effects. Therefore, imatinib mesylate can be employed in future to target breast cancer cure.
Acknowledgments
The authors thank the University of Malaya through the Institute of Research Management & Services (IPPP) Research Grant (grant no PV047/2011A) for providing financial support to this study. They also thank the Osvah Pharmaceutical Company, Tehran, Iran, for donation of imatinib mesylate, the Department of Pharmaceutics, Faculty of Pharmacy, Tehran University of Medical Sciences and the Professor Alborzi Clinical Microbiology Research Center (PACMRC) for providing and supporting the technical equipment.
Disclosure
The authors report no conflicts of interest in this work.
References
Buchdunger E, Cioffi CL, Law N, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295(1):139–145. | ||
Porter AC, Vaillancourt RR. Tyrosine kinase receptor-activated signal transduction pathways which lead to oncogenesis. Oncogene. 1998;17(11):1343–1352. | ||
Jeffers M, Schmidt L, Nakaigawa N, et al. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci U S A.1997;94(21):11445–11450. | ||
Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315(3):971–979. | ||
Najy AJ, Won JJ, Movilla LS, Kim HRC. Differential tumorigenic potential and matriptase activation between PDGF B versus PDGF D in prostate cancer. Mol Cancer Res. 2012;10(8):1087–1097. | ||
Heldin C-H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal. 2013;11(1):1. | ||
de Jong JS, van Diest PJ, van der Valk P, Baak JP. Expression of growth factors, growth inhibiting factors, and their receptors in invasive breast cancer. I: an inventory in search of autocrine and paracrine loops. J Pathol. 1998;184(1):44–52. | ||
Tsimberidou A-M, Kurzrock R, Anderson KC. The role of angiogenesis in cancer. Target Ther Transl Cancer Res. 2015;5:64. | ||
Siemens H, Jackstadt R, Kaller M, Hermeking H. Repression of c-Kit by p53 is mediated by miR-34 and is associated with reduced chemoresistance, migration and stemness. Oncotarget. 2013;4(9):1399–1415. | ||
Roussidis AE, Theocharis AD, Tzanakakis GN, Karamanos NK. The importance of c-Kit and PDGF receptors as potential targets for molecular therapy in breast cancer. Curr Med Chem. 2007;14(7):735–743. | ||
International Agency for Research on Cancer. World cancer report 2014. In: Stewart BW, Wild CP, editors. World Cancer Report. Lyon: International Agency for Research on Cancer; 2015. | ||
Goldhirsch A, Winer EP, Coates A, et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen international expert consensus on the primary therapy of early breast cancer 2013. Annals of Oncology. 2013;24(9):2206–2223. | ||
Grantzau T, Mellemkjær L, Overgaard J. Second primary cancers after adjuvant radiotherapy in early breast cancer patients: a national population based study under the Danish Breast Cancer Cooperative Group (DBCG). Radiother Oncol. 2013;106(1):42–49. | ||
Biscardi JS, Ishizawar RC, Silva CM, Parsons SJ. Tyrosine kinase signalling in breast cancer: epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000;2(3):203. | ||
Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor–resistant disease. J Clin Oncol. 2013;31(8):1070–1080. | ||
Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the study of lung cancer/association of molecular pathologists guideline. J Clin Oncol. 2014;32(32):3673–3679. | ||
Kadivar A, Kamalidehghan B, Javar HA, et al. Formulation and in vitro, in vivo evaluation of effervescent floating sustained-release imatinib mesylate tablet. PloS One. 2015;10(6):e0126874. | ||
Boikos SA, Pappo AS, Killian JK, et al. Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors: a report from the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA oncology. 2016;2(7):922–928. | ||
Helena AY, Sima CS, Huang J, et al. Local therapy with continued EGFR tyrosine kinase inhibitor therapy as a treatment strategy in EGFR-mutant advanced lung cancers that have developed acquired resistance to EGFR tyrosine kinase inhibitors. J Thorac Oncol. 2013;8(3):346–351. | ||
Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature Reviews Cancer. 2009;9(1):28–39. | ||
Manley P, Cowan-Jacob S, Buchdunger E, et al. Imatinib: a selective tyrosine kinase inhibitor. Eur J Cancer. 2002;38(Suppl 5):S19–S27. | ||
Wilczynski SP, Chen YY, Chen W, Howell SB, Shively JE, Alberts DS. Expression and mutational analysis of tyrosine kinase receptors c-kit, PDGFRα, and PDGFRβ in ovarian cancers. Hum Pathol. 2005;36(3):242–249. | ||
Weigel MT, Meinhold-Heerlein I, Bauerschlag DO, et al. Combination of imatinib and vinorelbine enhances cell growth inhibition in breast cancer cells via PDGFR β signalling. Cancer Lett. 2009;273(1):70–79. | ||
Radford IR. Imatinib. Novartis. Curr Opin Investig Drugs. 2002;3(3):492–499. | ||
Malavaki CJ, Roussidis AE, Gialeli C, et al. Imatinib as a key inhibitor of the platelet-derived growth factor receptor mediated expression of cell surface heparan sulfate proteoglycans and functional properties of breast cancer cells. FEBS J. 2013;280(10):2477–2489. | ||
Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344(14):1052–1056. | ||
Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626–632. | ||
Srinivasan D, Plattner R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006;66(11):5648–5655. | ||
Demetri GD, Von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–480. | ||
Johnson BE. Imatinib for small cell lung cancer, aiming for a target in vivo. Clin Cancer Res. 2004;10(10):3235–3236. | ||
Lopes LF, Bacchi CE. Imatinib treatment for gastrointestinal stromal tumour (GIST). J Cell Mol Med. 2010;14(1–2):42–50. | ||
Mahata S, Maru S, Shukla S, et al. Anticancer property of bryophyllum pinnata (Lam.) oken. leaf on human cervical cancer cells. BMC Complement Altern Med. 2012;12(1):1–11. | ||
Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30(9):e36. | ||
Weigel MT, Dahmke L, Schem C, et al. In vitro effects of imatinib mesylate on radiosensitivity and chemosensitivity of breast cancer cells. BMC Cancer. 2010;10(1):412. | ||
Gross DJ, Munter G, Bitan M, et al. The role of imatinib mesylate (Glivec) for treatment of patients with malignant endocrine tumors positive for c-kit or PDGF-R. Endocr Relat Cancer. 2006;13(2):535–540. | ||
de Silva MC, Reid R. Gastrointestinal stromal tumors (GIST): C-kit mutations, CD117 expression, differential diagnosis and targeted cancer therapy with Imatinib. Pathol Oncol Res. 2003;9(1):13–19. | ||
Edris B, Willingham SB, Weiskopf K, et al. Anti-KIT monoclonal antibody inhibits imatinib-resistant gastrointestinal stromal tumor growth. Proc Natl Acad Sci U S A. 2013;110(9):3501–3506. | ||
Wolf A, Couttet P, Dong M, et al. Imatinib does not induce cardiotoxicity at clinically relevant concentrations in preclinical studies. Leuk Res. 2010;34(9):1180–1188. | ||
Peng B, Lloyd P, Schran H. Clinical pharmacokinetics of imatinib. Clin Pharmacokinet. 2005;44(9):879–894. | ||
Jacquel A, Herrant M, Legros L, et al. Imatinib induces mitochondria-dependent apoptosis of the Bcr-Abl-positive K562 cell line and its differentiation toward the erythroid lineage. FASEB J. 2003;17(14):2160–2162. | ||
Matei D, Chang DD, Jeng M-H. Imatinib mesylate (Gleevec) inhibits ovarian cancer cell growth through a mechanism dependent on platelet-derived growth factor receptor α and Akt inactivation. Clinical Cancer Res. 2004;10(2):681–690. | ||
Uziel O, Fenig E, Nordenberg J, et al. Imatinib mesylate (Gleevec) downregulates telomerase activity and inhibits proliferation in telomerase-expressing cell lines. Br J Cancer. 2005;92(10):1881–1891. | ||
Ulivi P, Zoli W, Medri L, et al. c-kit and SCF expression in normal and tumor breast tissue. Breast Cancer Res Treat. 2004;83(1):33–42. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.