")
Back to Journals » Drug Design, Development and Therapy » Volume 18
A Window for Enhanced Oral Delivery of Therapeutics via Lipid Nanoparticles
Authors Amekyeh H , Sabra R, Billa N
Received 12 September 2023
Accepted for publication 25 November 2023
Published 29 February 2024 Volume 2024:18 Pages 613—630
DOI https://doi.org/10.2147/DDDT.S439975
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Hilda Amekyeh,1 Rayan Sabra,2 Nashiru Billa3
1Department of Pharmaceutics, School of Pharmacy, University of Health and Allied Sciences, Ho, Ghana; 2Department of Pharmaceutical Sciences, University of Connecticut, Storrs, CT, USA; 3College of Pharmacy, Qatar University, Doha, Qatar
Correspondence: Nashiru Billa, Department of Pharmaceutical Sciences, College of Pharmacy, QU Health, Qatar University, P.O Box 2713, Doha, Qatar, Tel +974 4403-5642, Email [email protected]
Abstract: Oral administration of dosage forms is convenient and beneficial in several respects. Lipid nanoparticulate dosage forms have emerged as a useful carrier system in deploying low solubility drugs systemically, particularly class II, III, and IV drugs of the Biopharmaceutics Classification System. Like other nanoparticulate delivery systems, their low size-to-volume ratio facilitates uptake by phagocytosis. Lipid nanoparticles also provide scope for high drug loading and extended-release capability, ensuring diminished systemic side effects and improved pharmacokinetics. However, rapid gastrointestinal (GI) clearance of particulate delivery systems impedes efficient uptake across the mucosa. Mucoadhesion of dosage forms to the GI mucosa results in longer transit times due to interactions between the former and mucus. Delayed transit times facilitate transfer of the dosage form across the mucosa. In this regard, a balance between mucoadhesion and mucopenetration guarantees optimal systemic transfer. Furthermore, the interplay between GI anatomy and physiology is key to ensuring efficient systemic uptake. This review captures salient anatomical and physiological features of the GI tract and how these can be exploited for maximal systemic delivery of lipid nanoparticles. Materials used to impart mucoadhesion and examples of successful mucoadhesive lipid nanoformulations are highlighted in this review.
Keywords: gastrointestinal, lipid nanoparticle, mucin, mucoadhesion, mucopenetration
Graphical Abstract:
Introduction
Orally administered dosage forms have a natural predisposition to presenting an active pharmaceutical ingredient (API) systemically, following absorption from the gastrointestinal (GI) tract (GIT). Additionally, oral administration of medicines provides an avenue for treating localized GI diseases and disorders, such as ulcers, infections, inflammation, and cancer. These disorders may be GIT site-specific, requiring drug delivery systems to deploy APIs to the relevant sites.1 However, the vast majority of therapeutics administered orally are destined for systemic delivery. Oral delivery of medicines is more acceptable to patients and devoid of the constraints associated with parenteral administration, such as requiring the need for trained personnel for administration or the likelihood of sepsis.2 Submicron particles, particularly nanoparticulate delivery systems, have emerged as a frontier in the systemic delivery of APIs via the GIT.3,4 These particles are taken up and transported intracellularly through enterocytes, whilst larger particles are mainly absorbed by Peyer’s patch microfold (M) cells.5 Nanoparticle (NP) uptake by Peyer’s patches has been exploited as a means to improve the systemic bioavailability of poorly soluble drugs.6–8 Furthermore, oral administration of nanoencapsulated APIs potentially reduces the systemic side effects of the APIs.9 For example, reduction of the nephrotoxic side effects akin to parenteral delivery of amphotericin B can be achieved when the latter is administered orally in NPs, albeit, with lower bioavailability.10 Improved systemic bioavailability of poorly soluble APIs following oral administration in nanocarriers is well recognized.7,8 In this regard, the rate and extent of particle uptake (phagocytosis) is inversely related to the size of the particle administered.11
Aside from particle size, NP surface characteristics such as charge also play a very crucial role in GI absorption, whereby particles with positive surface charge are better absorbed compared to negatively charged surfaces. This has been attributed to the negatively charged sialic acid moieties in mucin.12 Systemic delivery of orally administered APIs in nanoparticulate dosage forms is also dependent on the predisposition of both the API and the delivery system in the GIT enzymatic milieu13 since certain drugs such as peptides are degraded by digestive enzymes. In fact, the degradation of nanoparticulate carrier systems has been used as a trigger for activating drug release from some dosage forms in the GIT.14 The mucosal layer presents a barrier to the systemic delivery of drugs from the GIT,15 whereby it acts as a pre-absorptive barrier next to the epithelia, and is arguably one of the most intensely studied areas in epithelial nanoparticulate drug delivery.16 The literature is rich with scientific reports on tenable nanoparticulate delivery systems developed to surmount the anatomical or physiological constraints within the GIT, prior to systemic absorption of APIs.
This review presents some of the key anatomical and physiological features of the GIT that are relevant in nanoparticulate uptake. The review also presents recent innovations employed to improve the systemic delivery of APIs encapsulated in nanoparticulate drug delivery systems. We also expound relevant physicochemical constraints of APIs that dictate the propensity for absorption across the GIT, and how nanoformulation technology is evolving towards ensuring efficient delivery. The review pays special attention to lipid NPs as a frontier in the systemic delivery of therapeutics, harnessing mucoadhesion within the GI epithelia and drainage into the lymphatics.
Oral Delivery of Dosage Forms
Oral drug delivery is essentially drug administration through the mouth. This route of drug administration is undoubtedly the most popular and preferred drug delivery route. It is estimated that approximately 50% of all medicines are administered by mouth.17 Dosage forms that are administered or used orally include tablets, capsules, powders, granules, drops, syrups, emulsions, solutions, suspensions, gels, and films, among others.
In general, drug absorption following oral administration is influenced by biological factors, which are mainly based on the anatomy and physiology of the gastrointestinal (GI) tract (GIT), and technical factors, which chiefly stem from physicochemical characteristics of active pharmaceutical ingredients (APIs) and the formulation strategy with respect to drug delivery system.18 Drug absorption following oral administration is favored by the large surface area for absorption in the GIT, particularly in the small intestine, and the residence or transit time of the administered formulation in the GIT. Other specific conditions in the GIT that can affect oral drug absorption include pH, mucus thickness, and the diversity and population of microbes in the different regions of the GIT.19–21
Basic Anatomy/Physiology of the GIT
The GIT is a hollow and muscular tube with varying diameters that stretches from the mouth to the anus. The main anatomical areas of the GIT are the esophagus, the stomach, the small intestine, and the large intestine (colon). The inner surface of the GIT is very rough, which helps to increase the surface area available for drug absorption. The GIT wall is made up of four principal histological layers, namely, the serosa, muscularis externa, submucosa, and mucosa. The GI epithelium is covered with a protective and mechanical mucus barrier.17 It is important to recognize the key features of the GIT as these have a strong influence on drug absorption following. The esophagus is approximately 25 cm in length,22 normally folded in a relaxed state but expands during swallowing.23 The pH of the contents of the esophageal lumen is usually between 5 and 6.17 On the other hand, the pH of the stomach is about 1−3.5.17,24 The stomach has a protective role of reducing the risk of toxic agents reaching the small intestine. It also majorly acts as a temporary reservoir for ingested materials and delivers them into the duodenum.17 The small intestine is the major site for oral drug absorption; however, the rate at which gastric emptying occurs may affect the onset of drug absorption.17 Furthermore, lipolysis starts in the stomach and direct absorption of short and medium chain fatty acids also occurs through the gastric mucosa.25,26 The small intestine has a pH of 6−7.5; it starts from the pylorus and ends at the ileocecal valve.17,27 It has numerous villi and microvilli.28 Peyer patches are also mostly found in the ileum.29 Among the three main parts of the small intestine, the jejunum and ileum have a higher absorption capability compared to the duodenum.30,31 Lipid digestion begins in the stomach and usually starts with the breakdown of triglycerides; however, lipolysis mostly occurs in the small intestine. Bile salts emulsify triglycerides via micelle formation to increase access of triglycerides to lipase.17,32 The large intestine has a pH of approximately 7–7.5,17,33 and its main functions are absorption of water and electrolytes and storage and compaction of luminal contents for controlled elimination of feces.17,33 Approximately 50% of the dose of a rectally administered drug bypasses the portal circulation, which may be beneficial and can be exploited if a drug has a low oral bioavailability.34 The importance of lymphatic drainage of the rectum with respect to drug absorption has not been fully established; however, it is possible that it contributes to the systemic absorption of highly lipophilic drugs.35
Advantages of Oral Drug Delivery
The oral route remains popular for drug delivery due to its unrivalled benefits such as being pain-free, non-invasive, and convenient.17,36 The oral route also allows for easy administration of intact solid dosage forms and is associated with better patient compliance. The viscous mucosal layer and large surface area (>300 m2) of the GIT allow for the adhesion of oral dosage forms to the GI lumen, which can enhance drug absorption21,37 and protect against shear stresses induced by flowing GI juices.38 However, it is not useful during emergency situations and is also associated with enzymatic, chemical, or physical barriers to absorption and the possibility of liver first pass.17,39 Several drugs have low GI permeability imposed by absorptive epithelial cells and mucus-secreting goblet cells.40 Paracellular drug uptake is impeded by intercellular tight junctions,41 and after traversing the GI epithelial layer, drugs must also traverse the endothelium before systemic entry.42 The propensity of drugs traversing the epithelia is a function of their lipid solubility.43–45
Improving the Oral Absorption of Poorly Soluble and/or Absorbable Drugs
The biopharmaceutical classification system (BCS) classifies drugs based on solubility and absorption propensity,46 which are also useful in the provision of biowaiver status in bioequivalence studies.47,48 A large number of drugs delivered orally are absorbed into the blood via transcellular passive transport. It follows that the intrinsic lipophilicity of an API will influence its own transport across enterocytes, such that lipophilic drugs tend to permeate favorably across the GI membrane.49 The intrinsic lipophilicity of a drug can be improved via modification of its chemical structure; however, this may modify its biological activity. GI absorption of low permeability drugs (ie, class III and IV drugs) can be improved through ion-pairing with penetration/permeation enhancers, and via encapsulation in lipid carriers such as liposomes, coarse emulsions, microemulsions, self-emulsifying drug delivery systems, self-microemulsifying drug delivery systems, and lipid NPs.44 Some of these systems may be used in conjunction with permeation enhancers. Unfortunately, the use of permeation enhancers may permanently affect the physical integrity of the GI membrane.50,51
Other techniques employed in improving the bioavailability of class II, III, and IV type drugs include pH alteration of the drug’s microenvironment, formation of eutectic mixtures, evaporative precipitation into aqueous solution, micronization, molecular encapsulation with cyclodextrins, nanonization, precipitation, selective adsorption on insoluble carriers, solvent deposition, spray-freezing into liquid, supercritical fluid recrystallization, preparation of solid dispersions, solid solutions, as well as the use of anhydrates, solvates, metastable polymorphs, amorphs, precipitation inhibitors, salt forms, and surfactants.49,52–54 Review of the aforementioned techniques for improving drug solubility and GI permeability points to nanoformulation technology as the spearhead in this pursuit. Importantly, lipid-based nanoformulations stand out because of their added ability to drain into the lymphatic system.
Nanoparticles as a Drug Delivery System
Drug delivery devices made of NPs normally range in sizes between 10 and 1000 nm, but typically in the 100’s range. These typically include micelles, liposomes, nanogels, dendrimers, and lipid, gold polymeric, and silica NPs, to name a few.55 They have the lowest size-to-volume ratio among the various drug delivery systems, which promotes a higher propensity for uptake across epithelia and a higher adsorption capacity for surface loading compared to conventional dosage forms. Drug cargoes are usually adsorbed, encapsulated, entrapped, or dissolved within a suitable matrix carrier system of the NP.56 Another superior feature of NPs is their capability of shielding drugs from degradation, whilst improving cellular uptake. Appropriate polymers may be incorporated into the formulation of NPs to impart mucoadhesion, with a resultant increase in transit times in the GIT.57,58 Crucially, NPs provide a wider scope for imparting modified drug release and functionalization of appropriate moieties that serve to target the NP to specific tissues.57,58 This is particularly important in cancer therapy, where minimization of side effects should be a prime focus. In this review, we are focusing on lipid NPs and present in the subsequent sections, salient features of the same that can be exploited for maximal therapeutic effect.
Lipid NPs
Lipid NPs can be fabricated as solid lipid nanoparticles (SLNs) or nanostructured lipid carriers (NLCs) (Figure 1), depending on the composition of the lipid matrix, which acts as the carrier/vehicle in the formulations. There are several advantages that lipid NPs have over other NPs. Firstly, they are relatively more stable and less expensive to formulate, especially compared to liposomes. It has been indicated that the stability of lipid NPs is largely dependent on the physicochemical stability of their solid lipids as a result of susceptibility to degradation and coalescence.59 Consequently, solid lipids must be carefully selected for lipid NP manufacture following adequate characterization. Another advantage of lipid NPs is that they can be up-scaled and formulated to improve the pharmacokinetic profile of encapsulated APIs. They can be used to deliver both hydrophilic and lipophilic APIs, and generally manifest lower toxicity as they can be fabricated using biocompatible lipids.60,61 Techniques used for the manufacture of lipid NPs on an industrial scale include high-pressure homogenization (hot/cold), membrane contactor technique, microemulsion cooling, emulsification-solvent evaporation, and coacervation. Other methods for the fabrication of lipid NPs include ultrasonication, high shear homogenization, microemulsion, phase inversion temperature, double emulsion, emulsification solvent diffusion, solvent injection, and supercritical fluid techniques.59,60,62
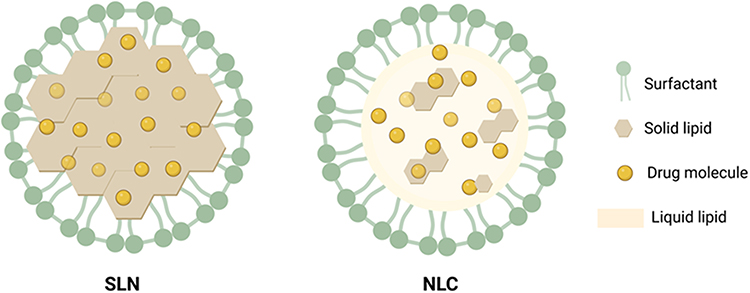 |
Figure 1 Schematic representation of an SLN and an NLC. Created with Biorender.com. |
SLNs are prepared with solid lipids such as highly purified triglycerides, complex glyceride mixtures, fatty acids, fats, and waxes, and stabilized with surfactants or co-surfactant solutions.63 SLNs have good physical stability, tolerability, and offer a good degree of protection to encapsulated drug cargoes.58 Additionally, drug release from the matrix can be controlled to achieve slow/sustained release profiles.64 One of the disadvantages of SLNs is that drug loading may be low, depending on drug solubility in the lipid matrix as well as the structure and polymorphic state of the lipid(s), which can result in lipid crystallization and drug expulsion.55,64,65 However, sufficiently high encapsulation efficiencies (>60%) have been obtained for drugs with different solubility characteristics using a theobroma oil and beeswax matrix,6 which indicates that a suitable solid lipid mix may be the key to achieving a good encapsulation efficiency for a given API. Another challenge that may be encountered in the development of SLNs is degradation of the loaded active drug as a result of high pressure applied during production. Fortunately, it has been indicated that the application of a back pressure can help curb this.59
NLCs, on the other hand, are lipid-based NPs that were developed to overcome some of the shortcomings of conventional SLNs. Unlike SLNs, NLCs are fabricated with both solid and liquid lipids, which results in an NLC lipid matrix with improved nanostructure that can support higher drug loads and prevent drug expulsion. There are three types of NLCs. The imperfect type NLCs, with general imperfections in their structure, result in the creation of domains for drug loading.66 In the “multiple” type NLCs, the API is dissolved in the liquid lipid and protected from degradation by the surrounding solid lipid. It is formed when the solubility of the drug in the liquid lipid is higher than that in the solid lipid and has been proposed to be similar to water-in-oil-in-water emulsions.61,67 In the “formless” or amorphous NLCs, drug crystallization is checked so that drug expulsion does not occur.66
Oral Absorption of Lipid NPs and Relevance to GIT Physiology
Following oral administration of lipid NPs, they transit through the GIT and are absorbed either as intact particles if they are able to withstand the effects of GI juices, or in their degraded form. Prior to uptake by epithelial cells, they interact with the mucus layer of the GIT. The characteristic smaller size and larger surface area of lipid NPs facilitate their uptake across the mucus layer and the epithelial monolayer.68 Degradation products of SLNs such as glycerides and fatty acids can enhance intestinal absorption of encapsulated drugs via micelle formation in the intestinal milieu. The lipid NPs and the degradation products can both traverse the enterocyte membrane via passive diffusion, facilitated diffusion, or active transport.68,69
Paracellular transport within the GIT occurs through aqueous pores between the epithelia. Intercellular spaces make up approximately 0.01% of the total surface area of the GI epithelium, and in addition, there are tight junctions between the cells.17 These tight junctions consist of a network of transmembrane proteins that form pores of approximately 8 Å in diameter and impede paracellular diffusion of molecules.70 As a result, only small, hydrophilic molecules can traverse this route. However, permeabilization of the GI epithelial cells can promote paracellular absorption, whereby it has been proposed that both hydrostatic and osmotic forces can modify the dimensions of intercellular spaces.71 Additionally, some molecules such as chitosan can induce reversible weakening of tight junction opening that promote paracellular uptake of lipid,72,73 without increasing the absorption of common endotoxins in the GIT.68 These factors can be exploited to enhance paracellular transport of lipid NPs.
Particles with diameters of <10 μm, approximately 1 μm, and <200 nm can be absorbed through the Peyer’s patches, phagocytosis by intestinal macrophages, and endocytosis by enterocytes, respectively.74 This indicates that lipid NPs can potentially be taken up from the GIT via these mechanisms. The transport of lipid NPs across intestinal epithelium can be investigated using techniques such as confocal laser scanning microscopy, flow cytometry, total internal reflection fluorescence microscopy, and transmission electron microscopy.75
Lipid NPs enter lymphatic tissue following oral administration via the Peyer’s patches.76 Lipid NPs can enhance chylomicron formation by enterocytes, which can improve solubilization of a loaded hydrophobic drug into the lipophilic matrix and enhance lymphatic uptake.77 Moreover, it has been shown that lipid-based formulations can increase lymphatic uptake of orally administered drugs. Specifically, several in-depth investigations have been performed to evaluate lymphatic uptake of NLCs and SLNs following oral or intraduodenal administration to laboratory rats.78–83
SLNs were found to increase the lymphatic transport of lopinavir by approximately five-fold.79 In another study, the uptake of coumarin-6-loaded SLNs modified with hydroxypropyl trimethyl ammonium chloride chitosan (HACC-C6-SLNs) through Peyer’s patches and small intestinal tissues near the patches was evaluated in male Sprague−Dawley rats. The results showed that the fluorescence intensity of HACC-C6-SLNs in the Peyer’s patches of the rats was higher than that in the normal small intestine, which was attributed to extensive phagocytosis by M cells in the Peyer’s patches.73 These findings clearly indicate that M cells, Peyer’s patches, and lymphatic transport are involved in the oral absorption of lipid NPs.
Baek et al also found that the paclitaxel concentration in the lymph nodes of male Sprague−Dawley rats was higher when the drug was orally delivered in hydroxypropyl-β-cyclodextrin-modified SLNs than when it was administered as a solution, clearly indicating a higher uptake of the surface-modified SLNs into the lymphatic vessels.84 In a subsequent study, the lymphatic uptake of curcumin-loaded SLNs and N-carboxymethyl chitosan (NCC)-coated curcumin-loaded SLNs was investigated in Sprague−Dawley rats following oral administration. The study was performed by measuring the amount of the drug in the mesenteric lymph nodes of the rats, and it was found that there were significantly higher amounts of curcumin in the lymph nodes of rats administered the SLNs compared to those administered curcumin solution. Additionally, the NCC-coated SLNs produced significantly higher amounts of curcumin in the lymph nodes compared to curcumin SLNs,85 indicating improved lymphatic uptake possibly due to surface modification of the SLNs.
Furthermore, NP bioadhesion, inhibition of P-glycoprotein mediated efflux, passive diffusion, and lipolysis (which improves the solubility and dissolution of poorly soluble drugs), are all important mechanisms by which the GI absorption of lipid NPs occurs or can be improved.86
Notwithstanding the proposed mechanisms of lipid NP uptake after oral delivery, and the fact that the small intestine is ideal for optimal drug absorption, it may be worthwhile investigating the absorption propensity of lipid NPs at specific sites in the gut. For instance, Li et al found that quercetin-loaded SLNs were absorbed at 6.20%, 13.00%, 17.57%, 30.44%, and 22.03% in the stomach, duodenum, jejunum, ileum, and colon of male Wister, respectively. Consequently, it was concluded that there are more Peyer’s patches and M cells in the ileum and colon compared to the other three segments.87 These findings indicate that the NPs will best be absorbed in the ileum and colon.
Transit of Dosage Forms in the GIT
Oral dosage forms are usually administered as solids, in the form of powders, granules, single-unit tablets, or multiparticulates, or as liquids, in the form of emulsions, solutions, or suspensions. Immediate-release solid dosage forms disintegrate and release the drug cargo immediately after dose intake, which means that such formulations are similar to solutions or suspensions in the GIT in respect of gastric emptying. Solid dosage forms can be fabricated to remain intact in the upper GIT or throughout their transit in the GIT.88 It is important to study the GI transit behavior of different oral dosage forms since their physical forms can influence their passage through the gut. NPs are usually prepared in the form of suspensions; as a result, they can be considered liquid dosage form. They can also be spray-dried or freeze-dried to obtain dry solids that can be administered as oral powders or further formulated as tablets or capsules. The removal of water from NP suspensions may be important in improving formulation stability.89
The transit of SLNs in the GIT of rats as well as the effect of food on the transit of the SLNs have been investigated.6,90 However, there are limited reports of such transit studies on lipid NPs in humans. This is not entirely surprising, as the transit behavior of liquids, multiparticulates, and tablets in the human GIT has been extensively studied, and it is expected that newer drug delivery systems will behave similarly. Notwithstanding the immense usefulness of available data on dosage form transit in the human GIT, we believe that specific transit studies on different drug delivery devices may be necessary to unravel any possible intricacies in drug absorption patterns. Estimated residence times of various dosage forms in the GIT that have been reported in literature are discussed below.
The transit of dosage forms within the human GIT is mainly affected by gastric emptying and colonic transit since small intestinal transit time (SITT) is relatively fixed. The transit of tablets in the esophagus is affected by size and shape while standing but not in the supine position. Esophageal transit time (ETT) of solid dosage forms is also affected by the volume of water administered with the formulations.91,92 It has been found that the orogastric transit time of size 2 hard gelatin capsules containing commonly used pharmaceutical excipients and microparticulate magnetite is in the range of 1.4−5.7 s in the upright position and 5.2−7.8 s in the supine position. The time decreases as the volume of the swallowed liquid is increased in either position. The study was conducted using a superconducting quantum interference device.92 ETT has also been estimated using gamma scintigraphy for single-unit tablets93 and video fluoroscopy for a film formulation.94 In the latter study, the ETT of the tested film was within 20 s for all participants, whether the formulation was taken with or without water.94 It has also been found that tablet coating, shape, and size can affect ETT. Specifically, film-coated, oval tablets (size, 5.7 × 11.5 mm; weight, 247 mg) were noted to transit faster in the esophagus compared to uncoated, round, convex tablets (diameter, 9.5 mm; weight, 200 mg).93
Gastric emptying time (GET) has been determined using methods such as alternate current biosusceptometry,95 magnetic moment imaging (MMI),96 gamma scintigraphy,96,97 real-time ultrasound,98 and the appearance of drug markers in plasma.99 It has been shown that GET is affected by food, being longer in the fed state and high calorie food.99 GET is also influenced by dosage form type, posture, and disease state.17 In a previous study, GET was estimated to be 9 min for a solution in the fasted state and 1.52 h and 3.87 h for pellets in the fasted and fed (following a heavy breakfast) states, respectively.97 GETs for multiparticulates have also been estimated to be 0.57 h and 2.08 h under fasted and fed states, respectively, in healthy volunteers.95 The GETs of an enteric-coated magnetic tablet and a non-disintegrating tablet have been found to be 1.59 h and 1.49 h, respectively, by MMI, and 1.62 h and 1.52 h, respectively, by scintigraphy.96 The double sampling technique has also been used to determine the GET of suspensions containing maltodextrins and medium-chain triglycerides in different amounts to be 23.2−35.8 min,100 whereas using that of a liquid meal (orange cordial) was estimated to be 22 min using real-time ultrasound.98
The small intestine is the main site of drug absorption because of its highly absorptive surface area. Consequently, the residence time of dosage forms in the small intestine can greatly affect the extent of drug absorption. Techniques that have been used to estimate SITT include direct methods such as gamma scintigraphy,101–103 MMI,104 radiotelemetry,105 X-ray imaging,106 and alternate current biosusceptometry,95 and indirect techniques such as hydrogen breath test107 and the use of marker compounds that are absorbed from specific sites.108 These techniques have their own merits and disadvantages and may yield different transit times; however, gamma scintigraphy surpasses the others because it provides real-time transit behavior of dosage forms.88 Formulations that have been evaluated in SITT determinations include pellets and tablets, and the estimated transit times vary between 2.6 and 8 h among different individuals in fasted and fed states. However, on average, SITT is proposed to be 3−4 h and is not generally affected by dosage form type or food. Thus, it is expected that NP formulations would transit the small intestines within this range as well. However, the type and amount of excipient used in a formulation can affect SITT, as has been shown for polyethylene glycol (PEG) 400109 and oleic acid.110
Drug absorption from the colon is incomplete and unpredictable because transit of dosage forms through this section of the GIT is highly variable and any unabsorbed drug from the upper GI regions may be embedded in fecal matter. However, the available data suggests that colonic transit of dosage forms is affected by feeding and the time of dosing relative to meal intake. Other factors that can affect colonic transit of drugs are posture, dosage form size, and patient-specific variabilities.111 It has been proposed that the mean mouth-to-cecum transit time of a 2 mm steel sphere is 13 h in the supine position, whereas total colonic transit time (CTT) is 43.5 h in healthy individuals.112 It has also been indicated that the CTT of pharmaceuticals can range from 2 h to 48 h, and that most people have mouth-to-anus transit times of 12−36 h, although this can go up to several days.17 These data are clear indications of the unpredictability of the residence times of pharmaceutical formulations in the large intestine.
The data presented above clearly show the depth of studies conducted on the GI transit of conventional dosage forms. It is therefore important that similar studies are conducted on NPs to estimate their ETT, GET, SITT, and CTT using the relevant techniques to appreciate how such particles travel through the GIT. This is particularly necessary as size has been shown to affect the GI transit of dosage forms. A good understanding of the impact of the nanosize dimensions of NPs on their movement in the GIT may help further clarify their ability to improve drug absorption among other findings. In the absence of extensive GI residence time data for NPs, we believe that the transit behavior of lipid NPs through the various segments of the GIT will be similar to that of a corresponding dosage form, whether a suspension or disintegrating or non-disintegrating tablet or capsule, with similar attendant effects of food as discussed above.
Surface Modification of Lipid NPs
One of the major challenges faced during the transit of drugs in the GIT following oral administration is rapid clearance. The GI mucus membrane is increasingly being recognized as a physiological feature that can be exploited to improve the pharmacokinetic profile of drugs.113 It has been shown that GI mucus, although protective and acts as a mechanical barrier for the GIT, can also be a barrier to effective drug absorption following oral delivery. Notwithstanding this limitation, it can be exploited as an anchoring material to improve the intestinal residence time of drug delivery systems such as NPs; this can be achieved by using materials that can interact with GI mucus.113
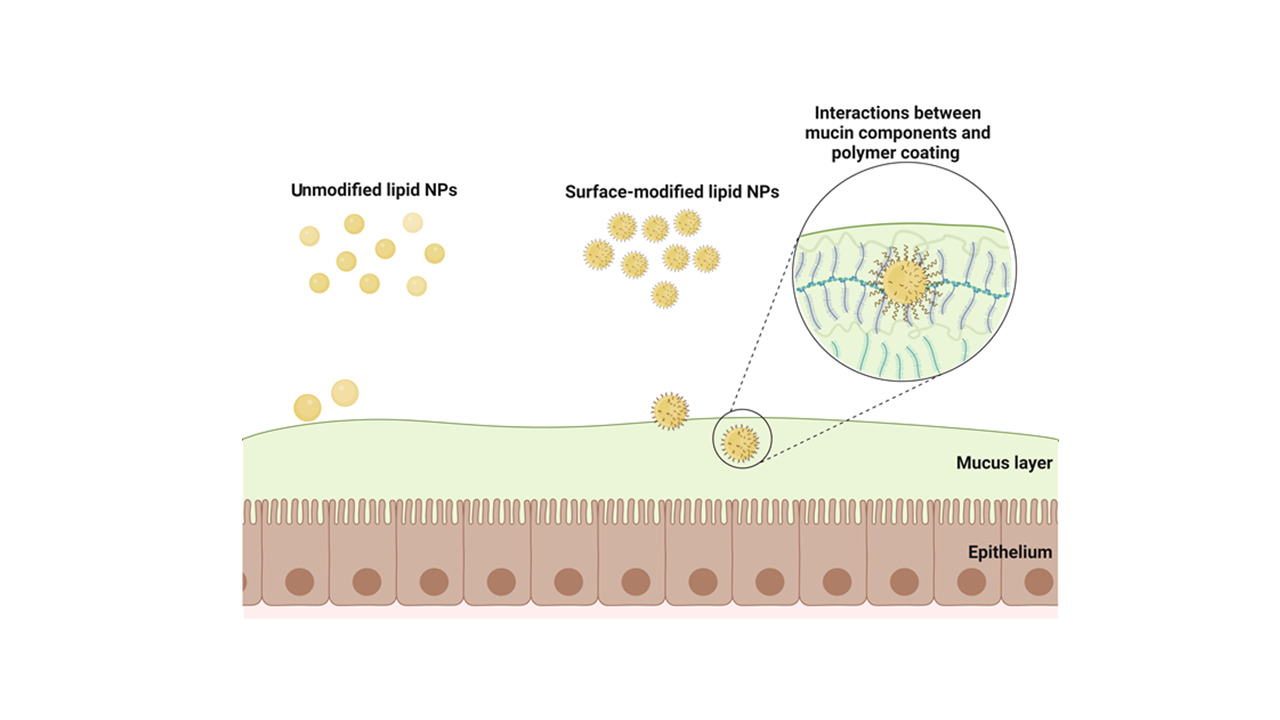
It has been shown that modifying the surface of oral lipid-based NPs, chiefly via coating, can result in improved specific distribution and targeting, prevention of particle aggregation, and increased circulation time, cellular uptake, lymphatic uptake, and bioavailability.85,114,115 These advantages can be realized through enhanced mucoadhesive and mucopenetrative properties of the particles that not only prolong their transit time in the GIT but also improve their propensity to traverse the epithelia (Figure 2). This means that the oral bioavailability of drugs encapsulated in lipid NPs can be further enhanced by increasing the GI transit time of the particles.116
 |
Figure 2 Mucoadhesion mechanisms underlying oral delivery of drug-loaded NPs. Created with BioRender.com. |
Mucoadhesion
It is often essential to maintain a fairly constant blood level in many disease conditions to ensure effective therapy. Unfortunately, following oral drug administration, unpredictable variations in plasma drug concentration can occur that lead to ineffective treatment due to subtherapeutic drug levels in the blood. There is therefore the need to formulate drugs with specific characteristics to achieve therapeutic objectives after oral delivery.117
One key strategy by which drug release from oral dosage forms can be optimized for improved absorption in the GIT is mucoadhesion, which is the attachment of the drug delivery system to a mucous membrane.118 The mucus layer that lines the GIT is a viscoelastic, translucent, aqueous gel. It serves as a protective layer that shields the GI epithelium from the effects of pathogens, toxins, and endogenous substances as hydrochloric acid, pepsin, and other digestive enzymes.119 Mucus is primarily composed of water and high molecular weight glycoproteins called mucins that form an entangled network. As a result of its composition, mucus can limit the free diffusion of materials within and through it and could, therefore, act as a barrier to drug delivery.120
Mucoadhesion is achieved by formulating preparations to adhere to the protective and sticky mucus lining the GI lumen, with the aim of extending the transit times of the dosage forms in the GIT. This objective can be achieved using positively charged materials that can adhere to the mucus layer via electrostatic binding, noting that the carbohydrate-bound ester sulfate residues and carboxyl groups of sialic acids in mucin impart a net negative charge to the mucus layer.121,122 Additionally, thiol-functionalized hydrogels can adhere to mucin glycoproteins in GI mucus through disulfide bonding.121,122
The mucus turnover encompasses synthesis, secretion, and degradation of mucus, which are regulated to preserve mucus function.123 It varies in different parts of the body and may range from a few minutes to several hours but is longer in the GIT compared to other physiological sites. In humans, the turnover time is approximately 4−6 h124 and 0.78−4.5 h125 in rats, clearly indicating a species dependency. Other characteristics of mucus, such as composition, mesh spacing, thickness, and viscosity, have not been exhaustively investigated, especially with respect to location, intersubject, and interspecies variation. Therefore, studies must be directed toward unravelling these parameters, in addition to the effects of nutrition, health, and other relevant factors on the integrity of the GI mucus.120
Notwithstanding these concerns and based on available data, it is possible to engineer mucoadhesive NPs to reside within the GI surface for up to 6 h, before mucus degradation. This invariably improves NP uptake. Furthermore, following mucus degradation, there is the possibility of re-adhesion/reattachment of loosened/freed NPs to mucus at other sites down the length of the GIT. Thus, re-attachment of NPs to GI mucus, coupled with normal transit, further increases the propensity of NP uptake of orally administered mucoadhesive NPs.
Mucoadhesive mechanisms include adsorption, diffusion, electronic interlocking, fracture, interpenetration of polymers, and wetting. The most widely accepted mechanisms are based on surface energy thermodynamics and interpenetration/diffusion theories (Figure 3).118 For mucoadhesion to occur, there must be intimate contact between a bioadhesive and a membrane, either via wetting or swelling, followed by penetration of the bioadhesive into the mucus membrane, which is termed interpenetration.126–128 Molecular interactions involved in mucoadhesion include hydrogen, hydrophobic, ionic, and van der Waals interactions.129
 |
Figure 3 Interaction of mucoadhesive and mucopenetrating NPs with the mucus layer for successful oral delivery. Created with BioRender.com. |
The main approach by which mucoadhesiveness can be imparted to lipid NPs is via surface modification, chiefly through coating with mucoadhesive polymers or attaching targeting ligands on the surface of the NPs.
Surface modification of lipid NPs with mucoadhesive polymers has led to an increase in residence time in the GIT, whereby contact of the NPs with the mucus increased the absorption of the API by passive diffusion.86 This coating approach relies on the use of bioadhesive materials, usually polymers, that interact with mucus. Furthermore, it has been shown that polymers that carry a positive charge density can be used as mucoadhesive agents.130,131 These polymers undergo electrostatic interactions with mucin, which is negatively charged, to increase the mucoadhesiveness of NPs. Thiolated polymers, commonly called thiomers, can also form covalent disulfide bonds with cysteine moieties in mucus as a result of their sulfhydryl groups.132 Additionally, some mucoadhesive polymers can improve drug absorption by inhibiting efflux pumps.133
Cellulose derivatives such as carboxymethylcellulose and hydroxypropyl cellulose and their grades may be mucoadhesive, depending on the amount of intestinal fluid. In limited intestinal fluids, these polymers appear to adhere to mucus (apparent mucoadhesion), whereby capillary attraction occurs between the dry water-absorbing polymer and the wet mucosal surface. This must be distinguished from the actual interaction between a mucoadhesive polymer and mucus in equilibrium with a third liquid phase.134
Typical polymers used for coating lipid NPs for mucoadhesive properties include chitosan and its derivatives N-trimethyl chitosan135 and N-carboxymethyl chitosan,136 alginate,130 PEG, and polyvinyl alcohol (PVA).137 Among these polymers, chitosan has arguably emerged as a frontrunner, largely because it is biocompatible. There are several polymers used to fabricate mucoadhesive NPs, with only a handful dedicated to imparting mucoadhesiveness to lipid NPs. In the following section, we review some of the key polymers studied on lipid NPs.
Materials Used to Enhance Mucoadhesion of Oral Lipid NPs
Chitosan
Chitosan is a polycationic, non-toxic, biocompatible polymer that has been widely investigated and reported to manifest excellent mucoadhesive properties.138 It is a biodegradable polysaccharide derived via partial deacetylation of chitin, which is the second most abundant polymer on earth after cellulose.139 Chitosan has superior qualities over other polymers because it is non-toxic, imparts high mucoadhesiveness, possesses antimicrobial property, and enhances oral drug absorption.135
The mucoadhesiveness of chitosan involves hydration, hydrogen bonding, and ionic interactions, although the primary mechanism at the molecular level is reported to be electrostatic interactions.130,140 These interactions are enhanced in slightly acidic to acidic media since the amine groups of chitosan are protonated in acidic pH.141 Additionally, high molecular weight chitosans have stronger mucoadhesive properties.134
The mucoadhesiveness of chitosan-coated amphotericin B−loaded NLCs has been studied in vitro and ex vivo by Tan et al, where a significant decrease in the zeta potential was observed after interaction with mucin.142 This decrease in mucoadhesion is a result of interaction between the amine groups of chitosan and mucin as previously reported.143 The ex vivo mucoadhesive study on excised intestine of Sprague−Dawley of uncoated and chitosan-coated amphotericin B−loaded NLCs showed that up to 30% more of the chitosan-coated NPs than the uncoated NPs remained adhered to the intestinal tissues. In another study, chitosan-coated amphotericin B-loaded NLCs were found to be more mucoadhesive than uncoated NLCs in an in vitro turbidimetric study.144 Fluconazole-loaded NLCs fabricated by Elkomy et al for the management of oral candidiasis were optimized by coating with chitosan. Mucoadhesion of the NLCs was assessed using buccal mucosal tissues isolated from rabbits. The investigators found that the mucoadhesive force of the NLCs increased about 1.6 folds due to the chitosan coating.145 Unfortunately, these studies did not include investigations on the uptake of the mucoadhesive NPs.
Fonte et al used confocal microscopy to study the mucoadhesive properties of oral chitosan-coated fluorescein isothiocyanate-insulin SLNs and found that the NPs were retained on the surface as well as within the intestinal epithelium of Wistar rats. This indicates the mucoadhesiveness of the coated SLNs. Additionally, API bioavailability from the coated SLNs was higher than that of the uncoated NPs, indicating an increase in systemic delivery.146 Luo et al also found that chitosan-coated SLNs have higher mucoadhesive property compared to uncoated SLNs in a quartz crystal microbalance test. The investigators posited that the coating strengthened hydrogen bonds between the NPs and mucin molecules due to the hydrophilicity of chitosan, thereby increasing mucoadhesion and uptake by Caco-2 cells.147 Similarly, Piazzini et al performed turbidimetric measurements via spectrophotometric evaluation at 500 nm and found that SLNs-coated chitosan increased the mucoadhesive property and in vitro cellular uptake of the SLNs.148 Chitosan-coated SLNs fabricated by Asfour et al149 and Parvez et al150 also showed more mucoadhesiveness than the respective uncoated SLNs. SLNs coated with thiolated chitosan derivatives showed improved mucoadhesion, being retained on porcine intestinal mucosa compared to uncoated SLNs.151 Cellular uptake and in vivo absorption were not investigated in these studies to assess the effects of the increased mucoadhesiveness of the respective lipid NPs on systemic delivery.
These findings clearly indicate that chitosan coating on lipid NPs improves mucoadhesive. However, assessment of the effect of the chitosan coating on in vivo uptake of the NPs was not conducted in most of the studies. Fonte et al investigated the fate of the SLNs administered to Wistar rats and found that the bioavailability of insulin from the chitosan-coated SLNs was twice as high as that from the uncoated SLNs after intragastric administration to the rats. The authors also explained that chitosan may be involved in transient opening of tight junctions between epithelial cells that facilitated paracellular insulin transport, further enhancing the bioavailability of insulin.146
PEG and PVA
It has been demonstrated that charged (anionic and cationic) polymers are more mucoadhesive compared to non-ionic polymers,152 because non-ionic polymers rely only on weak van der Waals type interactions but not electrostatic.
PEG is a non-ionic hydrophilic polymer commonly used to formulate drug delivery systems. With respect to its use as a surface-modifying polymer for lipid NPs, PEG is normally used to impart mucopenetrative capability to the NPs because of a neutral surface charge dispensation that prevents the particles from remaining attached to mucus, but rather penetrating the mucus network. PEG also enhances mucoadhesion of NPs through hydrophobic interactions, hydrogen bonding, and interactions with mucin via swelling, interpenetrating, and entangling with mucin fibers in mucus.153 Moreover, PEG can be thiolated to increase its mucoadhesion propensity. As indicated earlier, thiolated polymers can interact with the mucus layer in the GIT and thereby adhere to the mucus.132
The use of PEG as a mucoadhesive coating for lipid NPs is limited; however, mucoadhesiveness of docetaxel-loaded NLCs was increased by coating with an amphiphilic thiomer obtained by conjugating cysteine to PEG 2000 monostearate (Cy-PEG-MSA).154 Cysteine-modified (Cy-PEG-MSA-coated) drug-loaded NLCs showed significantly higher mucoadhesion (81.6%) compared to the unmodified (PEG-MSA-coated) drug-loaded NLCs (51.9%) in an in vitro assessment using porcine mucin. In a subsequent in vivo assessment using male Sprague−Dawley rats, the area under the plasma concentration–time curve (AUC) of docetaxel for the Cy-PEG-MSA-coated NLCs was 12.3-fold and 1.64-fold higher than that for docetaxel solution and the unconjugated NLCs, respectively, following oral administration of the formulations to the rats. These data clearly show an increase in systemic delivery of the mucoadhesive lipid NPs.154
PVA is a synthetic, uncharged, and hydrophilic polymer obtained from partial or full hydrolysis of polyvinyl acetate.155 In a study conducted by Chanburee and Tiyaboonchai, curcumin loaded NLCs coated with PEG or PVA were found to be more mucoadhesive to porcine mucus than uncoated NLCs. Interestingly, in the same study, the PEG400- and PVA-coated NLCs were more mucoadhesive than chitosan-coated NLCs. Furthermore, the extent of mucoadhesion from uncoated and chitosan-coated NLCs was comparable, albeit a slightly higher mucoadhesiveness from chitosan-coated NLCs in the first 2 h. The lower mucoadhesiveness from chitosan-coated NLCs was attributed to the larger size (1.9−2.1) than the PEG400- and PVA-coated NLCs. The investigation was conducted in pH 6.8 medium, where chitosan becomes unprotonated and therefore unable to electrostatically interact with mucin.137 We may conclude that NP size and pH contribute to the mucoadhesive behaviors of polymers.
Alginate
Alginate is a linear and water-soluble polysaccharide composed of alternating blocks of 1–4 linked α-L-guluronic and β-D-mannuronic acid residues, usually extracted from brown seaweed. It is a biocompatible and anionic with charged carboxyl end groups, which imparts the mucoadhesive characteristics.130 The extent of ionization of the charged groups is influenced by the ionic strength of the surrounding medium.156 Nonetheless, alginate has a very high mucoadhesive propensity compared to several polymers used in drug delivery.157 Unfortunately, there are limited reports on the use of alginate as a surface-modifying agent to impart mucoadhesiveness to lipid NPs.158
Mucopenetration
The thicknesses of the mucus lining the stomach, small intestine, ileum, and colon are 30–300, 150–400, 400–500, and 30–700 μm, respectively,159,160 which is clearly variable across the GIT. The variability in thickness across the gastrointestinal tract suggests that NPs of different sizes administered orally will reach epithelium at different times. Additionally, the spacing within the mesh-like structure of mucus as well as the brush-like structure of mucus cause it to filter off large particles.21,161,162 The mesh has average pore openings in the order of 200 nm; therefore, particles with size a size below 200 nm preferentially traverse.163 However, polymeric NPs up to 500 nm can rapidly traverse human mucus when coated with a muco-inert polymer since such polymers do not sufficiently interact with the mucus.164 Furthermore, there are concerns about mucus turnover, which can clear potentially mucoadhesive NPs within the mucosa. Thus, in addition to mucoadhesion, mucopenetration is equally important in ensuring that coated lipid NPs have the ability to traverse across the mucosa, and withstand turnover.124
Orally administered mucoadhesive adheres to the luminal mucus layer (LML) of the GIT, which is cleared more quickly than the adherent mucus layer (AML), underlining the LML and in direct contact with epithelium. In contrast, mucopenetrative NPs can readily traverse the LML into the AML, where they are retained even after clearance of the LML. Thus, a more enhanced absorption is observed from mucopenetrative compared to conventional mucoadhesive NPs.124
Although it has been shown that mucopenetration is size-dependent, surface characteristics are more consequential on mucopenetration.165 As previously indicated, mucoadhesion of lipid NPs is achieved mainly when the particles carry a positive charge and adhere to the mucus via electrostatic binding. This suggests that negatively charged or neutral NPs will undergo limited mucoadhesion to or entrapment in GI mucus.166 However, it has been noted that surface coatings comprising neutral or hydrophilic polymers confer increased mucopenetrative characteristics to particle formulations by overcoming the barrier properties of GI mucus.86
As previously stated, mucin is negatively charged, so oral lipid NPs with mucopenetrative properties can be formulated to have a net-neutral charge and thus undergo limited entrapment in GI mucus. Alternatively, the particles can be fabricated to have a charge-shifting ability to overcome endo/transcytosis of negatively charged particles.166,167 Net-neutral SLNs undergo faster and higher oral absorption in mice compared to similarly prepared anionic and cationic SLNs, attributable to mucopenetration in mucus-secreting HT29-MTX cells due to net-neutral charge on SLNs.168 Non-interacting polymers can also be used to evade electrostatic interactions with GI mucus. Mucolytic drug delivery systems disrupt the mucus meshwork and hence are able to penetrate GI mucus into the underlying epithelium;166,167 however, the disintegration of mucus can result in a break in the protective barrier and may therefore not be ideal.169
Materials Used to Enhance Mucopenetration of Oral Lipid NPs
PEGylation of NPs can produce smaller particles with a less negative surface charge that reduces hydrophobic or electrostatic interactions with mucus to encourage mucopenetration.170,171 Therefore, PEGs, low molecular weights (ie, 2–5 kDa) analogues, can be used to prepare mucopenetrative NPs because they impose a neutral surface charge on NPs and prevent static mucoadhesion.153 Doxorubicin-loaded PEGylated SLNs were found to have two-fold higher bioavailability compared to the unPEGylated SLNs, attributed to the above phenomenon. The SLNs permeated through mucus-secreting Caco-2/HT29 coculture cell monolayers better when they were PEGylated, indicating an enhanced mucus-penetrating ability. Additionally, PEGylated SLNs diffused through the mucus barriers of the gut sac models used, whereas unmodified SLNs were trapped. These findings indicate the ability of PEG to improve mucopenetration and perhaps systemic API delivery via lipid NPs.115
Other materials proposed as alternatives to PEG to enhance mucopenetration of NPs include poly(2-alkyl-2-oxazolines), polysarcosine, poly(vinyl alcohol), hydroxyl-containing non-ionic water-soluble polymers, and zwitterionic polymers (polybetaines).172 Unfortunately, there are limited published findings on the ability of these materials to actually exert a mucopenetrative effect on oral lipid NPs, which creates an avenue for more research to be conducted in this area.
Conclusions
Oral administration of therapeutics remains the mainstay option for systemic delivery of drugs. In the event of rate-limiting factors in GIT absorption of APIs, formulation interventions must be implemented aimed at exploiting biological features within the GIT or utilizing formulation approaches that surmount rate-limiting factors to absorption. Recent innovations aimed at improving the oral absorption of poorly absorbed APIs are almost exclusively limited to lipid NPs, whereby mucoadhesive and mucopenetrative properties of lipid NPs provide scope for enhanced systemic deployment of APIs. Moreover, oral administration of lipid NPs provides a conduit for bypassing the liver to reach systemic circulation, which further improves drug absorption. This review captures recent and key approaches in lipid NP formulation for oral delivery of APIs for researchers to align their efforts towards improved systemic delivery of APIs with low solubility. The impact of increased mucoadhesiveness or mucus-penetrative propensity of lipid NPs on cellular uptake requires much attention. Through this approach, there is potential for oral delivery of vaccines, peptides, and GIT labile APIs.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The publication fee for the manuscript was funded by Qatar University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wilding I. Site-specific drug delivery in the gastrointestinal tract. Crit Rev Ther Drug Carr Syst. 2000;17(6):557–620. doi:10.1615/CritRevTherDrugCarrierSyst.v17.i6.10
2. Osti C, Khadka M, Wosti D, Gurung G, Zhao Q. Knowledge and practice towards care and maintenance of peripheral intravenous cannula among nurses in Chitwan Medical College Teaching Hospital, Nepal. Nurs Open. 2019;6:1006–1012. doi:10.1002/nop2.288
3. Fairuz S, Nair RS, Billa N. Orally administered amphotericin B nanoformulations: physical properties of nanoparticle carriers on bioavailability and clinical relevance. Pharmaceutics. 2022;14(9):1823. doi:10.3390/pharmaceutics14091823
4. Sabra R, Billa N. Soliciting the oral route as a logical approach to managing colon cancer. Front Bioeng Biotechnol. 2021;9:645923. doi:10.3389/fbioe.2021.645923
5. Shakweh M, Ponchel G, Fattal E. Particle uptake by Peyer’s patches: a pathway for drug and vaccine delivery. Expert Opin Drug Deliv. 2004;1:141–163. doi:10.1517/17425247.1.1.141
6. Amekyeh H, Billa N, Yuen KH, Chin SLS. A gastrointestinal transit study on amphotericin B-loaded solid lipid nanoparticles in rats. AAPS Pharm Sci Tech. 2015;16:871–877. doi:10.1208/s12249-014-0279-4
7. Date AA, Hanes J, Ensign LM. Nanoparticles for oral delivery: design, evaluation and state-of-The-art. J Control Release. 2016;240:504–526. doi:10.1016/j.jconrel.2016.06.016
8. Lehr CM. Bioadhesion technologies for the delivery of peptide and protein drugs to the gastrointestinal tract. Crit Rev Ther Drug Carrier Syst. 1994;11:119–160.
9. De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomed. 2008;3(2):133–149. doi:10.2147/IJN.S596
10. Laniado-Laborín R, Cabrales-Vargas MN. Amphotericin B: side effects and toxicity. Rev Iberoam Micol. 2009;26(4):223–227. doi:10.1016/j.riam.2009.06.003
11. Jani P, Halbert GW, Langridge J, Florence AT. Nanoparticle uptake by the rat gastrointestinal mucosa: quantitation and particle size dependency. J Pharm Pharmacol. 1990;42(12):821–826. doi:10.1111/j.2042-7158.1990.tb07033.x
12. Du XJ, Wang JL, Iqbal S, et al. The effect of surface charge on oral absorption of polymeric nanoparticles. Biomater Sci. 2018;6(3):642–650. doi:10.1039/C7BM01096F
13. Asad S, Jacobsen A-C, Teleki A. Inorganic nanoparticles for oral drug delivery: opportunities, barriers, and future perspectives. Curr Opin Chem Eng. 2022;38:100869. doi:10.1016/j.coche.2022.100869
14. Hu Q, Katti PS, Gu Z. Enzyme-responsive nanomaterials for controlled drug delivery. Nanoscale. 2014;6:12273–12286. doi:10.1039/C4NR04249B
15. Crater JS, Carrier RL. Barrier properties of gastrointestinal mucus to nanoparticle transport. Macromol Biosci. 2010;10:1473–1483. doi:10.1002/mabi.201000137
16. Zandanel C, Ponchel G, Noiray M, Vauthier C. Nanoparticles facing the gut barrier: retention or mucosal absorption? Mechanisms and dependency to nanoparticle characteristics. Int J Pharm. 2021;609:121147. doi:10.1016/j.ijpharm.2021.121147
17. Ashford M. Biopharmaceutical principles of drug delivery. Introduction to biopharmaceutics. In: Aulton ME, Taylor KMG, editors. Aulton’s Pharmaceutics: The Design and Manufacture of Medicines.
18. Homayun B, Lin X, Choi H. Challenges and recent progress in oral drug delivery systems for biopharmaceuticals. Pharmaceutics. 2019;11(3):129. doi:10.3390/pharmaceutics11030129
19. Dressman JB, Berardi RR, Dermentzoglou LC, et al. Upper gastrointestinal (GI) pH in young, healthy men and women. Pharm Res. 1990;7(7):756–761. doi:10.1023/A:1015827908309
20. Rouge N, Buri P, Doelker E. Drug absorption sites in the gastrointestinal tract and dosage forms for site-specific delivery. Int J Pharm. 1996;136:117–139. doi:10.1016/0378-5173(96)85200-8
21. Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev. 2012;64:557–570. doi:10.1016/j.addr.2011.12.009
22. Gavaghan M. Anatomy and physiology of the esophagus. AORN J. 1999;69(2):370–386. doi:10.1016/S0001-2092(06)62494-0
23. Zhang L, Russell D, Conway BR, Batchelor H. Strategies and therapeutic opportunities for the delivery of drugs to the esophagus. Crit Rev Ther Drug Carrier Syst. 2008;25:259–304. doi:10.1615/CritRevTherDrugCarrierSyst.v25.i3.20
24. Soybel DI. Anatomy and physiology of the stomach. Surg Clin North Am. 2005;85:875–894. doi:10.1016/j.suc.2005.05.009
25. Koziolek M, Carrière F, Porter CJH. Lipids in the stomach – implications for the evaluation of food effects on oral drug absorption. Pharm Res. 2018;35(3):55. doi:10.1007/s11095-017-2289-x
26. Lemarié F, Cavalier JF, Garcia C, et al. Effect of preduodenal lipase inhibition in suckling rats on dietary octanoic acid (C8:0) gastric absorption and plasma octanoylated ghrelin concentration. Biochim Biophys Acta Mol Cell Biol Lipids. 2016;1861:1111–1120. doi:10.1016/j.bbalip.2016.06.009
27. Volk N, Lacy B. Anatomy and physiology of the small bowel. Gastrointest Endosc Clin N Am. 2017;27:1–13. doi:10.1016/j.giec.2016.08.001
28. Helander HF, Fändriks L. Surface area of the digestive tract-revisited. Scand J Gastroenterol. 2014;49:681–689. doi:10.3109/00365521.2014.898326
29. Kumar D, Phillips SF. The contribution of external ligamentous attachments to function of the ileocecal junction. Dis Colon Rectum. 1987;30:410–416. doi:10.1007/BF02556486
30. Lennernäs H. Human intestinal permeability. J Pharm Sci. 1998;87:403–410. doi:10.1021/js970332a
31. Rubin DC, Langer JC. Anatomy and development-small intestine: anatomy and structural anomalies. In: Podolsky DK, Camilleri M, Shanahan F, Fitz JG, Wang TC, Kalloo AN, editors. Yamada’s Atlas of Gastroenterology. Oxford, UK: Wiley Blackwell; 2016.
32. Stahl A, Gimeno RE, Tartaglia LA, Lodish HF. Fatty acid transport proteins: a current view of a growing family. Trends Endocrinol Metab. 2001;12:266–273. doi:10.1016/S1043-2760(01)00427-1
33. Cohn SM, Birnbaum EH, Friel CM. Colon: anatomy and structural anomalies. In: Yamada T, Alpers DH, Kalloo AN, Kaplowitz N, Owyang C, Powell DW, editors. Textbook of Gastroenterology.
34. Bardal SK, Waechter JE, Martin DS. Applied Pharmacology. St. Louis, MO: Elsevier Health Sciences; 2011.
35. Jannin V, Lemagnen G, Gueroult P, Larrouture D, Tuleu C. Rectal route in the 21st Century to treat children. Adv Drug Deliv Rev. 2014;73:34–49. doi:10.1016/j.addr.2014.05.012
36. Reinholz J, Landfester K, Mailänder V. The challenges of oral drug delivery via nanocarriers. Drug Deliv. 2018;25:1694–1705. doi:10.1080/10717544.2018.1501119
37. Schenk M, Mueller C. The mucosal immune system at the gastrointestinal barrier. Best Pract Res Clin Gastroenterol. 2008;22:391–409. doi:10.1016/j.bpg.2007.11.002
38. Leal J, Smyth HDC, Ghosh D. Physicochemical properties of mucus and their impact on transmucosal drug delivery. Int J Pharm. 2017;532:555–572. doi:10.1016/j.ijpharm.2017.09.018
39. Roger E, Lagarce F, Garcion E, Benoit JP. Biopharmaceutical parameters to consider in order to alter the fate of nanocarriers after oral delivery. Nanomedicine. 2010;5:287–306. doi:10.2217/nnm.09.110
40. Hunter AC, Elsom J, Wibroe PP, Moghimi SM. Polymeric particulate technologies for oral drug delivery and targeting: a pathophysiological perspective. Maturitas. 2012;73:5–18. doi:10.1016/j.maturitas.2012.05.014
41. Lundquist P, Artursson P. Oral absorption of peptides and nanoparticles across the human intestine: opportunities, limitations and studies in human tissues. Adv Drug Deliv Rev. 2016;106:256–276. doi:10.1016/j.addr.2016.07.007
42. Senapati PC, Sahoo SK, Sahu AN. Mixed surfactant based (SNEDDS) self-nanoemulsifying drug delivery system presenting efavirenz for enhancement of oral bioavailability. Biomed Pharmacother. 2016;80:42–51. doi:10.1016/j.biopha.2016.02.039
43. Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi:10.1023/a:1016212804288
44. Ganesan P, Narayanasamy D. Lipid nanoparticles: a challenging approach for oral delivery of BCS Class-II drugs. Futur J Pharm Sci. 2018;4:191–205. doi:10.1016/j.fjps.2018.04.001
45. U.S. Food and Drug Administration. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. Guidance for Industry; 2017. Available from: https://www.gmp-compliance.org/files/guidemgr/UCM070246.pdf.
46. Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/ elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi:10.1007/s11095-004-9004-4
47. Camenisch GP. Drug disposition classification systems in discovery and development: a comparative review of the BDDCS, ECCS and ECCCS concepts. Pharm Res. 2016;33:2583–2593. doi:10.1007/s11095-016-2001-6
48. Charalabidis A, Sfouni M, Bergström C, Macheras P. The biopharmaceutics classification system (BCS) and the biopharmaceutics drug disposition classification system (BDDCS): beyond guidelines. Int J Pharm. 2019;566:264–281. doi:10.1016/j.ijpharm.2019.05.041
49. Kawabata Y, Wada K, Nakatani M, Yamada S, Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. Int J Pharm. 2011;420:1–10. doi:10.1016/j.ijpharm.2011.08.032
50. Fasano A. Innovative strategies for the oral delivery of drugs and peptides. Trends Biotechnol. 1998;16:152–157. doi:10.1016/S0167-7799(97)01170-0
51. Thanou M, Verhoef JC, Junginger HE. Chitosan and its derivatives as intestinal absorption enhancers. Adv Drug Deliv Rev. 2001;50:91–101. doi:10.1016/S0169-409X(01)00180-6
52. Qiu Y, Chen Y, Zhang GGZ, Liu L, Porter W. Developing Solid Oral Dosage Forms: Pharmaceutical Theory and Practice. Elsevier Science; 2009.
53. Shargel L, Wu-Pong S, Yu AB. Applied Biopharmaceutics & Pharmacokinetics. McGraw-Hill; 2007.
54. Amidon GE, He X, Hageman MJ. Physicochemical Characterization and Principles of Oral Dosage Form Selection. In: Abraham DJ, editor. Burger’s Medicinal Chemistry and Drug Discovery. Wiley; 2003.
55. Wilczewska AZ, Niemirowicz K, Markiewicz KH, Car H. Nanoparticles as drug delivery systems. Pharmacol Rep. 2012;64:1020–1037. doi:10.1016/S1734-1140(12)70901-5
56. Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70(1–2):1–20. doi:10.1016/S0168-3659(00)00339-4
57. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. doi:10.1016/j.addr.2013.11.009
58. Gundogdu E, Yurdasiper A. Drug transport mechanism of oral antidiabetic nanomedicines. Int J Endocrinol Metab. 2014;12:1–5. doi:10.1016/j.addr.2013.11.009
59. Ganesan P, Narayanasamy D. Lipid nanoparticles: different preparation techniques, characterization, hurdles, and strategies for the production of solid lipid nanoparticles and nanostructured lipid carriers for oral drug delivery. Sustain Chem Pharm. 2017;6:37–56. doi:10.1016/j.scp.2017.07.002
60. Duan Y, Dhar A, Patel C, et al. A brief review on solid lipid nanoparticles: part and parcel of contemporary drug delivery systems. RSC Adv. 2020;10:26777–26791. doi:10.1039/D0RA03491F
61. Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery - A review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177. doi:10.1016/S0939-6411(00)00087-4
62. Wang G, Wang J, Wu W, Tony To SS, Zhao H, Wang J. Advances in lipid-based drug delivery: enhancing efficiency for hydrophobic drugs. Expert Opin Drug Deliv. 2015;12(9):1475–1499. doi:10.1517/17425247.2015.1021681
63. Kovacevic A, Savic S, Vuleta G, Müller RH, Keck CM. Polyhydroxy surfactants for the formulation of lipid nanoparticles (SLN and NLC): effects on size, physical stability and particle matrix structure. Int J Pharm. 2011;406:163–172. doi:10.1016/j.ijpharm.2010.12.036
64. Wissing SA, Kayser O, Müller RH. Solid lipid nanoparticles for parenteral drug delivery. Adv Drug Deliv Rev. 2004;56:1257–1272. doi:10.1016/j.addr.2003.12.002
65. Müller RH, Radtke M, Wissing SA. Nanostructured lipid matrices for improved microencapsulation of drugs. Int J Pharm. 2002;242:121–128. doi:10.1016/S0378-5173(02)00180-1
66. Üner M, Yener G. Importance of solid lipid nanoparticles (SLN) in various administration routes and future perspective. Int J Nanomed. 2007;2:289–300.
67. Feng L, Mumper RJ. A critical review of lipid-based nanoparticles for taxane delivery. Cancer Lett. 2013;334:157–175. doi:10.1016/j.canlet.2012.07.006
68. Salah E, Abouelfetouh MM, Pan Y, Chen D, Xie S. Solid lipid nanoparticles for enhanced oral absorption: a review. Colloids Surf B. 2020;196:111305. doi:10.1016/j.colsurfb.2020.111305
69. Muchow M, Maincent P, Müller RH. Lipid nanoparticles with a solid matrix (SLN®, NLC®, LDC®) for oral drug delivery. Drug Dev Ind Pharm. 2008;34:1394–1405. doi:10.1080/03639040802130061
70. Ménard S, Cerf-Bensussan N, Heyman M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol. 2010;3:247–259. doi:10.1038/mi.2010.5
71. Nellans HN. (B) Mechanisms of peptide and protein absorption. (1) Paracellular intestinal transport: modulation of absorption. Adv Drug Deliv Rev. 1991;7:339–364. doi:10.1016/0169-409X(91)90013-3
72. Yeh TH, Hsu LW, Tseng MT, et al. Mechanism and consequence of chitosan-mediated reversible epithelial tight junction opening. Biomaterials. 2011;32:6164–6173. doi:10.1016/j.biomaterials.2011.03.056
73. Shi LL, Xie H, Lu J, et al. Positively charged surface-modified solid lipid nanoparticles promote the intestinal transport of docetaxel through multifunctional mechanisms in rats. Mol Pharm. 2016;13:2667–2676. doi:10.1021/acs.molpharmaceut.6b00226
74. O’Hagan DT. The intestinal uptake of particles and the implications for drug and antigen delivery. J Anat. 1996;189(Pt 3):1.
75. Chai GH, Xu Y, Chen SQ, et al. transport mechanisms of solid lipid nanoparticles across Caco-2 cell monolayers and their related cytotoxicology. ACS Appl Mater Interfaces. 2016;8:5929–5940. doi:10.1021/acsami.6b00821
76. Lembo D, Cavalli R. Nanoparticulate delivery systems for antiviral drugs. Antivir Chem Chemother. 2010;21:53–70. doi:10.3851/IMP1684
77. Trevaskis NL, Charman WN, Porter CJH. Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev. 2008;60:702–716. doi:10.1016/j.addr.2007.09.007
78. Krishnan Y, Mukundan S, Akhil S, Gupta S, Viswanad V. Enhanced lymphatic uptake of leflunomide loaded nanolipid carrier via chylomicron formation for the treatment of rheumatoid arthritis. Adv Pharm Bull. 2018;8:257–265. doi:10.15171/apb.2018.030
79. Aji Alex MR, Chacko AJ, Jose S, Souto EB. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci. 2011;42:11–18. doi:10.1016/j.ejps.2010.10.002
80. Cavalli R, Bargoni A, Podio V, Muntoni E, Zara GP, Gasco MR. Duodenal administration of solid lipid nanoparticles loaded with different percentages of tobramycin. J Pharm Sci. 2003;92:1085–1094. doi:10.1002/jps.10368
81. Rangaraj N, Pailla SR, Shah S, Prajapati S, Sampathi S. QbD aided development of ibrutinib-loaded nanostructured lipid carriers aimed for lymphatic targeting: evaluation using chylomicron flow blocking approach. Drug Deliv Transl Res. 2020;10:1476–1494. doi:10.1007/s13346-020-00803-7
82. Cho HJ, Park JW, Yoon IS, Kim DD. Surface-modified solid lipid nanoparticles for oral delivery of docetaxel: enhanced intestinal absorption and lymphatic uptake. Int J Nanomed. 2014;9:495–504. doi:10.2147/IJN.S56648
83. Makwana V, Jain R, Patel K, Nivsarkar M, Joshi A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: elucidation of mechanism of uptake using chylomicron flow blocking approach. Int J Pharm. 2015;495:439–446. doi:10.1016/j.ijpharm.2015.09.014
84. Baek JS, So JW, Shin SC, Cho CW. Solid lipid nanoparticles of paclitaxel strengthened by hydroxypropyl-β-cyclodextrin as an oral delivery system. Int J Mol Med. 2012;30:953–959. doi:10.3892/ijmm.2012.1086
85. Baek JS, Cho CW. Surface modification of solid lipid nanoparticles for oral delivery of curcumin: improvement of bioavailability through enhanced cellular uptake, and lymphatic uptake. Eur J Pharm Biopharm. 2017;117:132–140. doi:10.1016/j.ejpb.2017.04.013
86. Talegaonkar S, Bhattacharyya A. Potential of lipid nanoparticles (SLNs and NLCs) in enhancing oral bioavailability of drugs with poor intestinal permeability. AAPS Pharm Sci Tech. 2019;20(3):121. doi:10.1208/s12249-019-1337-8
87. Li H, Zhao X, Ma Y, Zhai G, Li L, Lou H. Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. J Control Release. 2009;133:238–244. doi:10.1016/j.jconrel.2008.10.002
88. Yuen KH. The transit of dosage forms through the small intestine. Int J Pharm. 2010;395:9–16. doi:10.1016/j.ijpharm.2010.04.045
89. Li Y, Li M, Rantanen J, Yang M, Bohr A. Transformation of nanoparticles into compacts: a study on PLGA and celecoxib nanoparticles. Int J Pharm. 2022;611:121278. doi:10.1016/j.ijpharm.2021.121278
90. Amekyeh H, Billa N, Yuen KH, Lim SCS. Effect of food status on the gastrointestinal transit of amphotericin B-containing solid lipid nanoparticles in rats. AAPS Pharm Sci Tech. 2016;17:1060–1066. doi:10.1208/s12249-015-0438-2
91. Channer S, Virjee P. The effect of size and shape of tablets on their esophageal transit. J Clin Pharmacol. 1986;26(2):141–146. doi:10.1002/j.1552-4604.1986.tb02922.x
92. Osmanoglou E, Van Der Voort IR, Fach K, et al. Oesophageal transport of solid dosage forms depends on body position, swallowing volume and pharyngeal propulsion velocity. Neurogastroenterol Motil. 2004;16:547–556. doi:10.1111/j.1365-2982.2004.00541.x
93. Perkins AC, Wilson CG, Frier M, et al. The use of scintigraphy to demonstrate the rapid esophageal transit of the oval film-coated placebo risedronate tablet compared to a round uncoated placebo tablet when administered with minimal volumes of water. Int J Pharm. 2001;222:295–303. doi:10.1016/S0378-5173(01)00722-0
94. Okabe H, Suzuki E, Sugiura Y, et al. Development of an easily swallowed film formulation. Int J Pharm. 2008;355:62–66. doi:10.1016/j.ijpharm.2007.11.038
95. Miranda J, Corá L, Américo M, Romeiro F. AC biosusceptometry technique to evaluate the gastrointestinal transit of pellets under influence of prandial state. J Pharm Sci. 2010;99:317–324. doi:10.1002/jps.21794
96. Goodman K, Hodges LA, Band J, Stevens HNE, Weitschies W, Wilson CG. Assessing gastrointestinal motility and disintegration profiles of magnetic tablets by a novel magnetic imaging device and gamma scintigraphy. Eur J Pharm Biopharm. 2010;74:84–92. doi:10.1016/j.ejpb.2009.01.004
97. Wilding IR, Hardy JG, Maccari M, Ravelli V, Davis SS. Scintigraphic and pharmacokinetic assessment of a multiparticulate sustained release formulation of diltiazem. Int J Pharm. 1991;76:133–143. doi:10.1016/0378-5173(91)90351-N
98. Bateman DN, Whittingham TA. Measurement of gastric emptying by real-time ultrasound. Gut. 1982;23:524–527. doi:10.1136/gut.23.6.524
99. Choe SY, Neudeck BL, Welage LS, Amidon GE, Barnett JL, Amidon GL. Novel method to assess gastric emptying in humans: the pellet gastric emptying test. Eur J Pharm Sci. 2001;14:347–353. doi:10.1016/S0928-0987(01)00196-8
100. Beckers EJ, Jeukendrup AE, Brouns F, Wagenmakers AJM, Saris WHM. Gastric emptying of carbohydrate - medium chain triglyceride suspensions at rest. Int J Sports Med. 1992;13(08):581–584. doi:10.1055/s-2007-1024569
101. Billa N, Yuen KH, Khader MAA, Omar A. Gamma-scintigraphic study of the gastrointestinal transit and in vivo dissolution of a controlled release diclofenac sodium formulation in xanthan gum matrices. Int J Pharm. 2000;201(1):109–120. doi:10.1016/S0378-5173(00)00399-9
102. Coupe AJ, Davis SS, Wilding IR. Variation in gastrointestinal transit of pharmaceutical dosage forms in healthy subjects. Pharm Res. 1991;8(3):360–364. doi:10.1023/A:1015849700421
103. Yuen KH, Deshmukh AA, Newton JM, Short M, Melchor R. Gastrointestinal transit and absorption of theophylline from a multiparticulate controlled release formulation. Int J Pharm. 1993;97(1–3):61–77. doi:10.1016/0378-5173(93)90127-2
104. Weitschies W, Wedemeyer RS, Kosch O, et al. Impact of the intragastric location of extended release tablets on food interactions. J Control Release. 2005;108(2–3):375–385. doi:10.1016/j.jconrel.2005.08.018
105. Evans DF, Pye G, Bramley R, Clark AG, Dyson TJ, Hardcastle JD. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut. 1988;29(8):1035–1041. doi:10.1136/gut.29.8.1035
106. Fallingborg J, Christensen LA, Ingeman-Nielsen M, Jacobsen BA, Abildgaard K, Rasmussen HH. pH-Profile and regional transit times of the normal gut measured by a radiotelemetry device. Aliment Pharmacol Ther. 1989;3(6):605–614. doi:10.1111/j.1365-2036.1989.tb00254.x
107. Read NW, Cammack J, Edwards C, Holgate AM, Cann PA, Brown C. Is the transit time of a meal through the small intestine related to the rate at which it leaves the stomach? Gut. 1982;23(10):824–828. doi:10.1136/gut.23.10.824
108. Peh KK, Yuen KH. Indirect gastrointestinal transit monitoring and absorption of theophylline. Int J Pharm. 1996;139(1–2):95–103. doi:10.1016/0378-5173(96)04602-9
109. Schulze JDR, Waddington WA, Ell PJ, Parsons GE, Coffin MD, Basit AW. Concentration-dependent effects of polyethylene glycol 400 on gastrointestinal transit and drug absorption. Pharm Res. 2003;20(12):1984–1988. doi:10.1023/B:PHAM.0000008046.64409.bd
110. Dobson CL, Davis SS, Chauhan S, Sparrow RA, Wilding IR. The effect of oleic acid on the human ileal brake and its implications for small intestinal transit of tablet formulations. Pharm Res. 1999;16(1):92–96. doi:10.1023/a:1018827030210
111. Wilson CG. The transit of dosage forms through the colon. Int J Pharm. 2010;395(1–2):17–25. doi:10.1016/j.ijpharm.2010.04.044
112. Basile M, Neri M, Carriero A, et al. Measurement of segmental transit through the gut in man. A novel approach by the biomagnetic method. Dig Dis Sci. 1992;37(10):1537–1543. doi:10.1007/BF01296499
113. Subramanian DA, Langer R, Traverso G. Mucus interaction to improve gastrointestinal retention and pharmacokinetics of orally administered nano-drug delivery systems. J Nanobiotechnology. 2022;20(1):1–23. doi:10.1186/s12951-022-01539-x
114. Zhang C, Ma Y, Zhang J, et al. modification of lipid-based nanoparticles: an efficient delivery system for nucleic acid-based immunotherapy. Molecules. 2022;27:1–29. doi:10.3390/molecules27061943
115. Yuan H, Chen CY, Chai GH, Du YZ, Hu FQ. Improved transport and absorption through gastrointestinal tract by pegylated solid lipid nanoparticles. Mol Pharm. 2013;10(5):1865–1873. doi:10.1021/mp300649z
116. Tan SLJ, Billa N. Improved bioavailability of poorly soluble drugs through gastrointestinal muco-adhesion of lipid nanoparticles. Pharmaceutics. 2021;13(11):1817. doi:10.3390/pharmaceutics13111817
117. Thedrattanawong C, Manaspon C, Nasongkla N. Controlling the burst release of doxorubicin from polymeric depots via adjusting hydrophobic/hydrophilic properties. J Drug Deliv Sci Technol. 2018;46:446–451. doi:10.1016/j.jddst.2018.06.001
118. Mansuri S, Kesharwani P, Jain K, Tekade RK, Jain NK. Mucoadhesion: a promising approach in drug delivery system. React Funct Polym. 2016;100:151–172. doi:10.1016/j.reactfunctpolym.2016.01.011
119. Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev. 2009;61(2):75–85. doi:10.1016/j.addr.2008.09.008
120. Boegh M, Nielsen HM. Mucus as a barrier to drug delivery - understanding and mimicking the barrier properties. Basic Clin Pharmacol Toxicol. 2015;116(3):179–186. doi:10.1111/bcpt.12342
121. Li R, Deng L, Cai Z, et al. Liposomes coated with thiolated chitosan as drug carriers of curcumin. Mater Sci Eng C. 2017;80:156–164. doi:10.1016/j.msec.2017.05.136
122. Martins ALL, de Oliveira AC, Do Nascimento CMOL, et al. Mucoadhesive properties of thiolated pectin-based pellets prepared by extrusion-spheronization technique. J Pharm Sci. 2017;106:1363–1370. doi:10.1016/j.xphs.2017.01.028
123. Paone P, Cani PD. Mucus barrier, mucins and gut microbiota: the expected slimy partners? Gut. 2020;69:2232–2243. doi:10.1136/gutjnl-2020-322260
124. Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61:158–171. doi:10.1016/j.addr.2008.11.002
125. Lehr CM, Poelma FGJ, Junginger HE, Tukker JJ. An estimate of turnover time of intestinal mucus gel layer in the rat in situ loop. Int J Pharm. 1991;70:235–240. doi:10.1016/0378-5173(91)90287-X
126. Andrews GP, Laverty TP, Jones DS. Mucoadhesive polymeric platforms for controlled drug delivery. Eur J Pharm Biopharm. 2009;71:505–518. doi:10.1016/j.ejpb.2008.09.028
127. Chowdary KPR, Srinivas L. Mucoadhesive drug delivery systems: a review of current status. Indian Drugs. 2000;37:400–406.
128. Sankar R, Jain SK. Development and characterization of gastroretentive sustained-release formulation by combination of swelling and mucoadhesive approach: a mechanistic study. Drug Des Devel Ther. 2013;7:1455–1469. doi:10.2147/DDDT.S52890
129. Smart JD. The basics and underlying mechanisms of mucoadhesion. Adv Drug Deliv Rev. 2005;57:1556–1568. doi:10.1016/j.addr.2005.07.001
130. George M, Abraham TE. Polyionic hydrocolloids for the intestinal delivery of protein drugs: alginate and chitosan - A review. J Control Release. 2006;114:1–14. doi:10.1016/j.jconrel.2006.04.017
131. Ch’Ng HS, Park H, Kelly P, Robinson JR. Bioadhesive polymers as platforms for oral controlled drug delivery II: synthesis and evaluation of some swelling, water‐insoluble bioadhesive polymers. J Pharm Sci. 1985;74:399–405. doi:10.1002/jps.2600740407
132. Albrecht K, Bernkop-Schnürch A. Thiomers: forms, functions and applications to nanomedicine. Nanomedicine. 2007;2:41–50. doi:10.2217/17435889.2.1.41
133. Gradauer K, Dünnhaupt S, Vonach C, et al. Thiomer-coated liposomes harbor permeation enhancing and efflux pump inhibitory properties. J Control Release. 2013;165:207–215. doi:10.1016/j.jconrel.2012.12.001
134. Lehr C, Bouwstra JA, Schacht EH, Junginger HE. In vitro evaluation of mucoadhesive properties of chitosan and some other natural polymers. Int J Pharm. 1992;78:43–48. doi:10.1016/0378-5173(92)90353-4
135. Ramalingam P, Ko YT. Enhanced oral delivery of curcumin from N-trimethyl chitosan surface-modified solid lipid nanoparticles: pharmacokinetic and brain distribution evaluations. Pharm Res. 2015;32:389–402. doi:10.1007/s11095-014-1469-1
136. Venishetty VK, Chede R, Komuravelli R, Adepu L, Sistla R, Diwan PV. Design and evaluation of polymer coated carvedilol loaded solid lipid nanoparticles to improve the oral bioavailability: a novel strategy to avoid intraduodenal administration. Colloids Surf B. 2012;95:1–9. doi:10.1016/j.colsurfb.2012.01.001
137. Chanburee S, Tiyaboonchai W. Mucoadhesive nanostructured lipid carriers (NLCs) as potential carriers for improving oral delivery of curcumin. Drug Dev Ind Pharm. 2017;43:432–440. doi:10.1080/03639045.2016.1257020
138. Anselmo AC, Mitragotri S. Nanoparticles in the clinic: an update. Bioeng Transl Med. 2019;4:1–16. doi:10.1002/btm2.10143
139. Wang JP, Chen YZ, Ge XW, Yu HQ. Gamma radiation-induced grafting of a cationic monomer onto chitosan as a flocculant. Chemosphere. 2007;66:1752–1757. doi:10.1016/j.chemosphere.2006.06.072
140. Deacon MP, McGurk S, Roberts CJ, et al. Atomic force microscopy of gastric mucin and chitosan mucoadhesive systems. Biochem J. 2000;563:557–563. doi:10.1042/bj3480557
141. He P, Davis SS, Illum L. In vitro evaluation of the mucoadhesive properties of chitosan microspheres. Int J Pharm. 1998;166:75–88. doi:10.1016/S0378-5173(98)00027-1
142. Tan JSL, Roberts CJ, Billa N. Mucoadhesive chitosan-coated nanostructured lipid carriers for oral delivery of amphotericin B. Pharm Dev Technol. 2019;24:504–512. doi:10.1080/10837450.2018.1515225
143. Bonferoni MC, Sandri G, Ferrari F, et al. Comparison of different in vitro and ex vivo methods to evaluate mucoadhesion of glycol-palmitoyl chitosan micelles. J Drug Deliv Sci Technol. 2010;20:419–424. doi:10.1016/S1773-2247(10)50073-X
144. Ling JTS, Roberts CJ, Billa N. Antifungal and mucoadhesive properties of an orally administered chitosan-coated amphotericin B nanostructured lipid carrier (NLC). AAPS Pharm Sci Tech. 2019;20:1–11. doi:10.1208/s12249-019-1346-7
145. Elkomy MH, Elmowafy M, Shalaby K, et al. Development and machine-learning optimization of mucoadhesive nanostructured lipid carriers loaded with fluconazole for treatment of oral candidiasis. Drug Dev Ind Pharm. 2021;47:246–258. doi:10.1080/03639045.2020.1871005
146. Fonte P, Nogueira T, Gehm C, Ferreira D, Sarmento B. Chitosan-coated solid lipid nanoparticles enhance the oral absorption of insulin. Drug Deliv Transl Res. 2011;1:299–308. doi:10.1007/s13346-011-0023-5
147. Luo Y, Teng Z, Li Y, Wang Q. Solid lipid nanoparticles for oral drug delivery: chitosan coating improves stability, controlled delivery, mucoadhesion and cellular uptake. Carbohydr Polym. 2015;122:221–229. doi:10.1016/j.carbpol.2014.12.084
148. Piazzini V, Cinci L, D’Ambrosio M, Luceri C, Bilia AR, Bergonzi MC. Solid lipid nanoparticles and chitosan-coated solid lipid nanoparticles as promising tool for silybin delivery: formulation, characterization, and in vitro evaluation. Curr Drug Deliv. 2018;16:142–152. doi:10.2174/1567201815666181008153602
149. Asfour M, Salama A, Mohsen A. Fabrication of all-trans retinoic acid loaded chitosan/tripolyphosphate lipid hybrid nanoparticles as a novel oral delivery approach for management of diabetic nephropathy in rats. J Pharm Sci. 2021;110:3208–3220. doi:10.1016/j.xphs.2021.05.007
150. Parvez S, Yadagiri G, Karole A, et al. Recuperating biopharmaceutical aspects of amphotericin b and paromomycin using a chitosan functionalized nanocarrier via oral route for enhanced anti-leishmanial activity. Front Cell Infect Microbiol. 2020;10:1–12. doi:10.3389/fcimb.2020.570573
151. Wibel R, Braun DE, Hämmerle L, et al. In vitro investigation of thiolated chitosan derivatives as mucoadhesive coating materials for solid lipid nanoparticles. Biomacromolecules. 2021;22:3980–3991. doi:10.1021/acs.biomac.1c00776
152. Ludwig A. The use of mucoadhesive polymers in ocular drug delivery. Adv Drug Deliv Rev. 2005;57:1595–1639. doi:10.1016/j.addr.2005.07.005
153. Huckaby JT, Lai SK. PEGylation for enhancing nanoparticle diffusion in mucus. Adv Drug Deliv Rev. 2018;124:125–139. doi:10.1016/j.addr.2017.08.010
154. Fang G, Tang B, Chao Y, et al. Cysteine-functionalized nanostructured lipid carriers for oral delivery of docetaxel: a permeability and pharmacokinetic study. Mol Pharm. 2015;12:2384–2395. doi:10.1021/acs.molpharmaceut.5b00081
155. Suchaoin W, Pereira De Sousa I, Netsomboon K, et al. Mucoadhesive polymers: synthesis and in vitro characterization of thiolated poly(vinyl alcohol). Int J Pharm. 2016;503:141–149. doi:10.1016/j.ijpharm.2016.03.006
156. Muppalaneni S, Mastropietro D, Omidian H. Mucoadhesive drug delivery systems. In: Bader RA, Putnam DA, editors. Engineering Polymer Systems for Improved Drug Delivery. Wiley; 2013:319–342.
157. Chickering DE, Mathiowitz E. Bioadhesive microspheres: I. A novel electrobalance-based method to study adhesive interactions between individual microspheres and intestinal mucosa. J Control Release. 1995;34:251–262. doi:10.1016/0168-3659(95)00011-V
158. Fangueiro JF, Andreani T, Egea MA, Garcia ML, Souto SB, Souto EB. Experimental factorial design applied to mucoadhesive lipid nanoparticles via multiple emulsion process. Colloids Surf B Biointerfaces. 2012;100:84–89. doi:10.1016/j.colsurfb.2012.04.014
159. Strugala V, Allen A, Dettmar PW, Pearson JP. Colonic mucin: methods of measuring mucus thickness. Proc Nutr Soc. 2003;62:237–243. doi:10.1079/PNS2002205
160. Netsomboon K, Bernkop-Schnürch A. Mucoadhesive vs. mucopenetrating particulate drug delivery. Eur J Pharm Biopharm. 2016;98:76–89. doi:10.1016/j.ejpb.2015.11.003
161. Boegh M, García-Díaz M, Müllertz A, Nielsen HM. Steric and interactive barrier properties of intestinal mucus elucidated by particle diffusion and peptide permeation. Eur J Pharm Biopharm. 2015;95:136–143. doi:10.1016/j.ejpb.2015.01.014
162. Lieleg O, Ribbeck K. Biological hydrogels as selective diffusion barriers. Trends Cell Biol. 2011;21:543–551. doi:10.1016/j.tcb.2011.06.002
163. Florek J, Caillard R, Kleitz F. Evaluation of mesoporous silica nanoparticles for oral drug delivery-current status and perspective of MSNs drug carriers. Nanoscale. 2017;9:15252–15277. doi:10.1039/C7NR05762H
164. Lai SK, O’Hanlon DE, Harrold S, et al. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc Natl Acad Sci U S A. 2007;104:1482–1487. doi:10.1073/pnas.0608611104
165. Suk JS, Lai SK, Boylan NJ, Dawson MR, Boyle MP, Hanes J. Rapid transport of muco-inert nanoparticles in cystic fibrosis sputum treated with N-acetyl cysteine. Nanomedicine. 2011;6:365–375. doi:10.2217/nnm.10.123
166. Taipaleenmäki E, Städler B. Recent advancements in using polymers for intestinal mucoadhesion and mucopenetration. Macromol Biosci. 2020;20(3):1900342. doi:10.1002/mabi.201900342
167. Rohrer J, Partenhauser A, Hauptstein S, et al. Mucus permeating thiolated self-emulsifying drug delivery systems. Eur J Pharm Biopharm. 2016;98:90–97. doi:10.1016/j.ejpb.2015.11.004
168. Yu Z, Fan W, Wang L, Qi J, Lu Y, Wu W. Effect of surface charges on oral absorption of intact solid lipid nanoparticles. Mol Pharm. 2019;5013–5024. doi:10.1021/acs.molpharmaceut.9b00861
169. Menzel C, Bernkop-Schnürch A. Enzyme decorated drug carriers: targeted swords to cleave and overcome the mucus barrier. Adv Drug Deliv Rev. 2018;124:164–174. doi:10.1016/j.addr.2017.10.004
170. Cu Y, Saltzman WM. Controlled surface modification with poly(ethylene)glycol enhances diffusion of PLGA nanoparticles in human cervical Mucus. Mol Pharm. 2009;6:173–181. doi:10.1021/mp8001254
171. Wang YY, Lai SK, Suk JS, Pace A, Cone R, Hanes J. Addressing the PEG mucoadhesivity paradox to engineer nanoparticles that “slip” through the human mucus barrier. Angew Chem Int Ed Engl. 2008;47:9726–9729. doi:10.1002/anie.200803526
172. Khutoryanskiy VV. Beyond PEGylation: alternative surface-modification of nanoparticles with mucus-inert biomaterials. Adv Drug Deliv Rev. 2018;124:140–149. doi:10.1016/j.addr.2017.07.015
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.