")
Back to Journals » Drug Design, Development and Therapy » Volume 9
A pharmacokinetic and pharmacodynamic drug interaction between rosuvastatin and valsartan in healthy subjects
Authors Jung JA, Lee S, Kim J , Ko J , Jang SB, Nam SY, Huh W
Received 3 November 2014
Accepted for publication 27 November 2014
Published 2 March 2015 Volume 2015:9 Pages 745—752
DOI https://doi.org/10.2147/DDDT.S76942
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Wei Duan
Jin Ah Jung,1 Soo-Yun Lee,2 Jung-Ryul Kim,1 Jae-Wook Ko,1,2 Seong Bok Jang,3 Su Youn Nam,3 Wooseong Huh1,4
1Department of Clinical Pharmacology and Therapeutics, Samsung Medical Center, 2Department of Health Sciences and Technology, Samsung Advanced Institute for Health Sciences and Technology, Sungkyunkwan University, 3Yuhan Research Institute, Yuhan Corporation, 4Department of Internal Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
Purpose: Valsartan, an angiotensin-receptor blocker, and rosuvastatin, a competitive inhibitor of the 3-hydroxy-3-methylglutaryl coenzyme A reductase, are frequently coadministered to treat patients with hypertension and dyslipidemia. The study reported here sought to evaluate the pharmacokinetic and pharmacodynamic interactions between rosuvastatin and valsartan in healthy Korean subjects.
Subjects and methods: Thirty healthy male Korean subjects were administered with rosuvastatin (20 mg/day), valsartan (160 mg/day), and both drugs concomitantly for 4 days in a randomized, open-label, multiple-dose, three-treatment, three-period crossover study. Plasma concentrations of rosuvastatin, N-desmethyl rosuvastatin, and valsartan were determined using validated high-performance liquid chromatography with tandem mass spectrometry. Lipid profiles and vital signs (systolic and diastolic blood pressure and pulse rate) were measured for the pharmacodynamic assessment.
Results: For rosuvastatin, the geometric mean ratios (90% confidence intervals [CIs]) of coadministration to mono-administration were 0.8809 (0.7873-0.9857) for maximum plasma concentration at steady state and 0.9151 (0.8632-0.9701) for area under the concentration–time curve (AUC) over a dosing interval at steady state. For valsartan, the geometric mean ratios (90% CIs) of those were 0.9300 (0.7946-1.0884) and 1.0072 (0.8893-1.1406), respectively. There were no significant differences in the metabolic ratio of N-desmethyl rosuvastatin AUC to rosuvastatin AUC between coadministration and rosuvastatin alone. No interaction was found in terms of systolic or diastolic blood pressure or lipid profiles. Combined treatment with valsartan and rosuvastatin was generally well tolerated without serious adverse events.
Conclusion: The pharmacokinetic profiles of rosuvastatin and valsartan in combination were comparable with those of rosuvastatin and valsartan administered individually, suggesting that their individual pharmacokinetics were not affected by their coadministration. No dose adjustment was required and the results are supportive of a study in a larger patient population.
Keywords: pharmacokinetics, pharmacodynamics, hypertension, dyslipidemia
Introduction
Valsartan is one of the most commonly prescribed angiotensin II type 1 receptor blockers (ARBs) for the management of hypertension. Valsartan lowers blood pressure (BP) by antagonizing the renin–angiotensin–aldosterone system and competes with angiotensin II for interaction with the angiotensin II type 1 receptor, lowering BP by increasing the effects of angiotensin II. Valsartan is administered at a starting dose of 80 mg or 160 mg once daily and is titrated to a higher dose of 320 mg daily as required.1 Following oral administration, peak plasma concentrations of valsartan are reached in 2–4 hours and mean absolute oral bioavailability of valsartan is 23%.2 Valsartan is primarily eliminated largely unchanged by biliary excretion in feces and renally in the urine, and exhibits dose-linear pharmacokinetics within the clinical dosing range. Valsartan exhibits little accumulation in plasma following repeated administrations (accumulation ratio of 1.2 in healthy subjects),2 and its pharmacokinetics do not to differ significantly between males and females and dosage adjustments based on age are not necessary.3
Rosuvastatin is a competitive inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, a rate-limiting enzyme that converts HMG-CoA to mevalonate (a precursor for cholesterol), and is used in the treatment of hyperlipidemia and mixed dyslipidemia.4 The usual starting dose of rosuvastatin is 10–20 mg once daily, adjusted to 40 mg in accordance with lipid levels.5 Peak plasma concentrations of rosuvastatin are reached approximately 3–5 hours after oral administration and the elimination half-life ranges from approximately 13 to 21 hours.6 Rosuvastatin is not metabolized extensively and is primarily excreted in the feces (90%). Little accumulation of rosuvastatin was observed after repeated, once-daily dosing; the accumulation ratio was 1.05 in healthy subjects and 1.37 in patients with end-stage renal disease on dialysis.7 No effect of age, sex, or administration time on rosuvastatin pharmacokinetics has been observed.8,9
Due to the fact that hypertension and hyperlipidemia frequently coexist in the same patients and are responsible for an increased risk of cardiovascular events, the coadministration of valsartan and rosuvastatin is desirable in such patients. However, to our knowledge, there has been no report of any study on the interaction between valsartan and rosuvastatin. These two drugs share the same transporter proteins that are responsible for the hepatic uptake (organic anion-transporting polypeptide 1B1 and 1B3) and biliary excretion (multidrug resistance-associated protein 2).10,11 Through the inhibition of transporters, combination of rosuvastatin and telmisartan increased the peak plasma concentration (Cmax) of rosuvastatin and telmisartan by 2.01- and 1.35-fold, respectively.12 Hence, evaluation of drug–drug interactions is needed to ensure drug efficacy and the safety of patients for the combination therapy. Therefore, the aim of the study reported here (NCT01609907) was to evaluate potential interactions between valsartan and rosuvastatin after coadministration in a clinically relevant multiple-dose setting.
Subjects and methods
The Institutional Review Board of the Samsung Medical Center, Seoul, Korea, approved the protocol, and the study was conducted in accordance with the principles outlined in the Good Clinical Practice standard and the Declaration of Helsinki.13 All subjects provided written informed consent before their enrollment in the study.
Subjects
Thirty healthy male volunteers were enrolled between November 2011 and February 2012. Screening assessments included medical history, physical examination, 12-lead electrocardiogram (ECG), and standard laboratory tests including hematology, blood chemistry, urinalysis, and urine drug screening. Healthy male volunteers aged between 20 and 50 years with a body mass index of 19–27 kg/m2 were included in the trial. Volunteers who experienced hypersensitivity or allergy to any medication including statins or ARBs, had any other disease or abnormality of clinical relevance, or were positive in serology test (hepatitis C virus, hepatitis B virus, or HIV) were excluded from the trial. Volunteers were required to abstain from smoking, drinking alcohol, consuming caffeine-containing drinks, and undertaking strenuous activity from midnight before dosing until 72 hours after dosing.
Study design
This study was a randomized, open-label, six-sequence, three-period, three-treatment, multiple-dose crossover study in healthy male subjects. The number of subjects used in this study allowed for the detection of a 20% difference in area under the concentration–time curve (AUC)between the two drugs at a significance level of 0.05 with 80% power. Subjects received a different treatment in each period: 20 mg rosuvastatin (Crestor®, AstraZeneca plc, London, UK) alone once daily, 160 mg valsartan (Diovan®, Novartis International AG, Basel, Switzerland) alone once daily, and coadministration of 20 mg rosuvastatin or 160 mg valsartan once daily in an assigned order, with a 1-week washout period (Figure 1). To ensure compliance, study drugs were administered by the investigator at the Clinical Trial Center, Samsung Medical Center (Seoul, Korea).
 | Figure 1 Subjects (n=30) were randomized to one of the six sequence groups (five in each sequence in period 1). Subjects were administered the study drugs with a 1-week washout between treatments. |
Subjects in a fasted state received study drugs with 240 mL of water every morning for 4 days to reach steady state, determined by the known pharmacokinetic characteristics of each drug. Subjects were admitted to the Clinical Trial Center 12 hours before the last dosing in each treatment period and were confined until 24 hours after dosing. Additional 48- and 72-hour visits were made for pharmacokinetic sampling. The same procedure was repeated during each period for pharmacokinetic, pharmacodynamic, and safety-profile assessments.
Determination of plasma rosuvastatin, N-desmethyl rosuvastatin, and valsartan
To evaluate drug–drug interactions between rosuvastatin and valsartan, plasma concentrations of rosuvastatin and valsartan were analyzed. Valsartan is metabolized to a pharmacologically inactive metabolite, albeit slowly, by the action of cytochrome P450 2C9 (CYP2C9).14–16 N-desmethyl rosuvastatin, a pharmacologically active metabolite, is formed primarily by CYP2C9.17 Thus, the N-desmethyl metabolite was also measured.
The plasma concentrations were measured by BioCore (Seoul, South Korea), which is certified to the Good Laboratory Practice standard by the Ministry of Food and Drug Safety, Seoul, South Korea. The analytes were measured using high-performance liquid chromatography with tandem mass-spectrometric detection. For rosuvastatin and N-desmethyl rosuvastatin, the extracted upper organic phase of samples was filtered with a 0.2 μm filter and evaporated. Valsartan and the corresponding internal standard (valsartan-d3) were extracted from human plasma by protein precipitation. The analytes were chromatographically separated with a Unison UK-C18 column (2.0×75.0 mm, 3 μm; Imtakt Corporation, Tokyo, Japan) with a flow rate of 200 μL/min. The calibration curves for the three analytes were linear (r>0.990) over the concentration range of 0.15–150.00 μg/L for rosuvastatin, 0.1–50.00 μg/L for N-desmethyl rosuvastatin, and 40.00–20,000.00 μg/L for valsartan. Intra- and inter-assay accuracy and precision for the three analytes were within 15% of the theoretical values and stability was confirmed according to standard operating procedures and Ministry of Food and Drug Safety guidelines for bioanalytical method validation.18
Pharmacokinetic assessments
The primary pharmacokinetic endpoints were the AUC over a dosing interval τ at steady state (AUCτ) and maximum plasma concentration at steady state (Cmax,ss). The secondary pharmacokinetic endpoints were time from last dosing to maximum plasma concentration at steady state (Tmax,ss), terminal elimination half-life at steady state (t½,ss), and minimum plasma concentration at steady state (Cmin,ss). Additionally, metabolic ratio was calculated as N-desmethyl rosuvastatin AUC/rosuvastatin AUC. All pharmacokinetic variables were calculated using Phoenix WinNonlin (v 6.1; Pharsight Corporation, Cary, CA, USA).
Pharmacodynamic assessments
Lipid profiles and BP were measured to evaluate the pharmacodynamic effect of the drugs. To determine lipid profiles, fasting blood samples were drawn before the first dosing and 24 hours after the last dosing in each period. All lipid parameters, including serum total cholesterol, triglycerides, high- and low-density lipoprotein cholesterol (HDL-C and LDL-C, respectively), apolipoprotein A1 (ApoA1), and apolipoprotein B (ApoB) were measured using a Roche Modular P chemistry analyzer (Roche Diagnostics, Basel, Switzerland). Systolic blood pressure (SBP), diastolic blood pressure (DBP), and pulse rate (PR) were measured using a YM1000 Vital Signs Monitor (Mediana Technologies, San Antonio, TX, USA) after at least a 5-minute rest. Measurements were made from the same upper arm of each subject before the administration of daily dosing, then at 1 hour and 2, 4, 6, 12, 24, and 48 hours after the last dosing.
Safety assessments
Safety was evaluated based on physical examination, monitoring of vital signs, 12-lead ECGs, adverse events (AEs), and clinical laboratory tests.
AEs were monitored throughout the study. Their severity (mild, moderate, or severe) and relationship to the study drug (definite, probable, possible, probably not, definitely not, or nonassessable/unclassifiable) was assessed and recorded by the investigator. AEs were coded by using the Medical Dictionary for Drug Regulatory Activities (MedDRA; v 12.0).
Statistical analyses
All statistical analyses were performed using SAS software (v 9.1; SAS Institute, Cary, NC, USA) and the level of significance was defined as P<0.05. The effect of coadministration of rosuvastatin and valsartan on the steady-state pharmacokinetics of each agent alone was determined by calculation of the 90% confidence interval (CI) around the geometric mean ratios (GMRs) for Cmax,ss and AUCτ of rosuvastatin and valsartan.
Change in lipid profiles (total cholesterol, LDL-C, HDL-C, triglyceride, and ApoA1/ApoB) and change in SBP, DBP, and PR on day 4 from baseline (day 1) were compared between coadministration and administration of the drugs alone using a general linear mixed model.
Results
Study participants
Thirty healthy male subjects were enrolled and six subjects dropped out: two due to the occurrence of AEs and four due to consent withdrawal. All subjects were adult Korean men with a mean (standard deviation) age of 32 (7) years and body mass index of 23.0 (2.0) kg/m2. Demographic characteristics were comparable across the six sequence groups.
Pharmacokinetics
The trough plasma concentrations of these two agents confirmed that a steady state was reached after administration of rosuvastatin and valsartan for 4 consecutive days. The mean plasma concentration–time curves after coadministration of rosuvastatin and valsartan, and administration of each drug alone are shown in Figures 2 and 3. Pharmacokinetic variables for rosuvastatin, N-desmethyl rosuvastatin, and valsartan are summarized in Table 1. The Tmax,ss, Cmin,ss, and t½ of rosuvastatin and valsartan were not affected by their coadministration. Peak exposure to rosuvastatin and valsartan was 12% and 7% lower, respectively, when the drugs were coadministered, than when they were administered alone. However, this decrease was not clinically significant. For rosuvastatin, the 90% CI for the GMR was 0.7873–0.9857 for Cmax,ss and 0.8632–0.9701 for the AUCτ (Table 2). For valsartan, the 90% CI for the GMR was 0.7946–1.0884 for Cmax,ss and 0.8893–1.1406 for the AUCτ (Table 2).
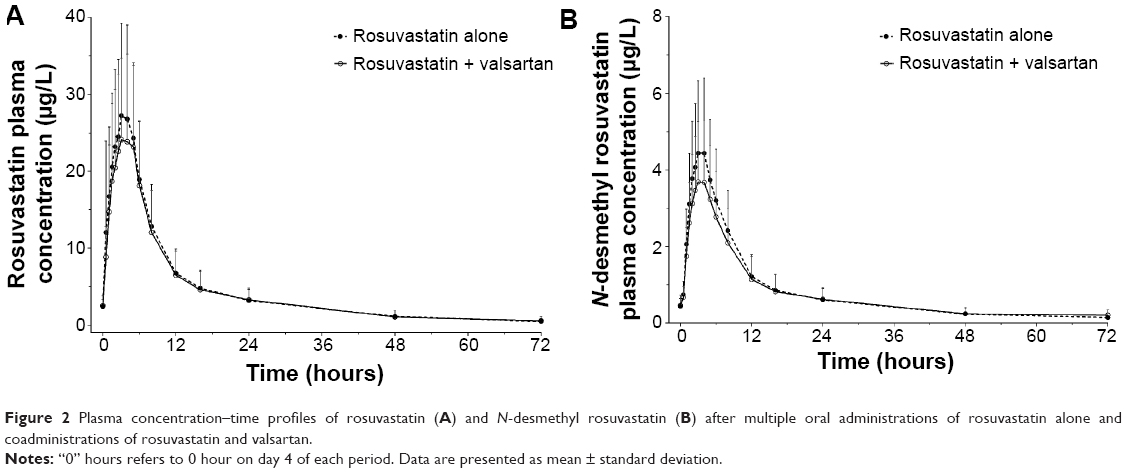 | Figure 2 Plasma concentration–time profiles of rosuvastatin (A) and N-desmethyl rosuvastatin (B) after multiple oral administrations of rosuvastatin alone and coadministrations of rosuvastatin and valsartan. |
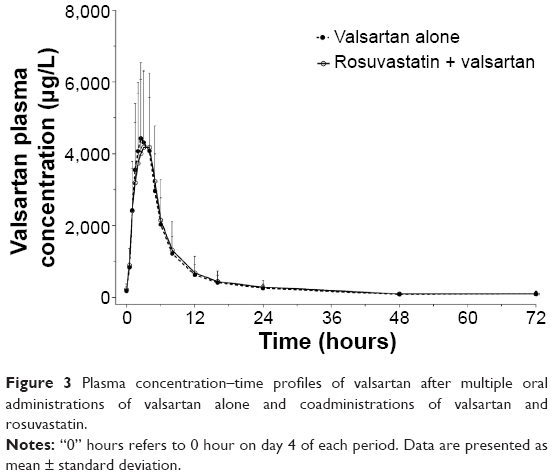 | Figure 3 Plasma concentration–time profiles of valsartan after multiple oral administrations of valsartan alone and coadministrations of valsartan and rosuvastatin. |
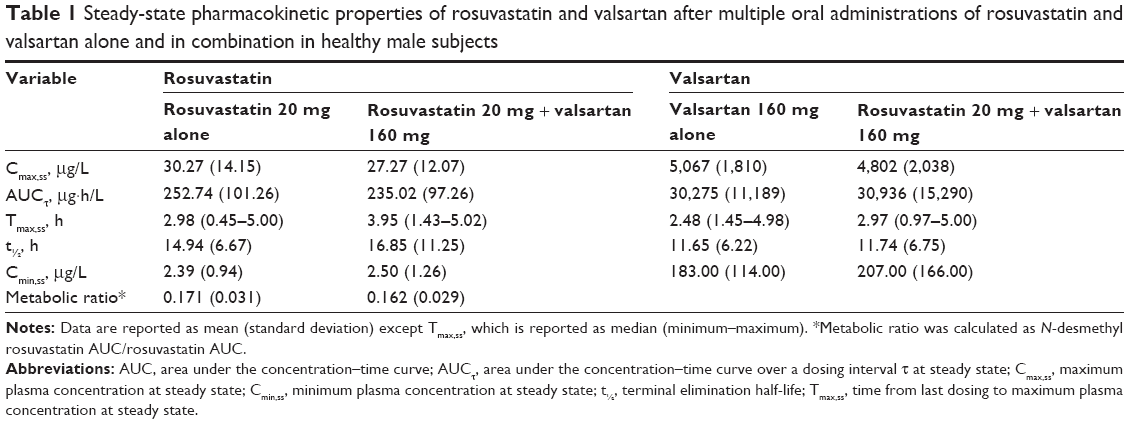 | Table 1 Steady-state pharmacokinetic properties of rosuvastatin and valsartan after multiple oral administrations of rosuvastatin and valsartan alone and in combination in healthy male subjects |
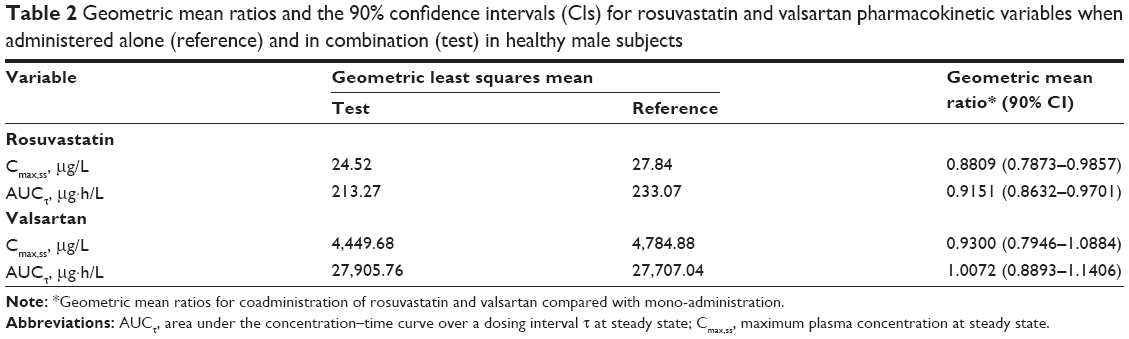 | Table 2 Geometric mean ratios and the 90% confidence intervals (CIs) for rosuvastatin and valsartan pharmacokinetic variables when administered alone (reference) and in combination (test) in healthy male subjects |
Pharmacodynamics
Mean changes in SBP, DBP, and PR from baseline following oral administrations of rosuvastatin and valsartan alone and in combination are presented in Table 3. SBP, DBP, and PR were significantly decreased following multiple oral administration of valsartan compared with rosuvastatin alone. There was no significant difference in changes of SBP, DBP, and PR between administration of valsartan alone and valsartan with rosuvastatin. Changes in BP recovered to baseline levels 24–48 hours after the last dosing. Maximum changes in BP were observed on average 6 hours after dosing with a compensatory PR increase. However, maximum changes of SBP, DBP, and PR were not altered by coadministration of valsartan and rosuvastatin (Table 3).
 | Table 3 Changes from baseline in systolic blood pressure (SBP), diastolic blood pressure (DBP), and pulse rate (PR) after multiple oral administrations of rosuvastatin and valsartan alone and in combination in healthy male subjects |
Mean changes from baseline in lipid profiles are summarized in Table 4. Total cholesterol, LDL-C, and ApoB/ApoA1 decreased significantly following coadministration of valsartan and rosuvastatin compared with valsartan alone (P<0.001). However, differences between rosuvastatin alone and in combination with valsartan were not significant.
 | Table 4 Change from baseline in lipid profiles following multiple oral administrations of rosuvastatin and valsartan alone and in combination in healthy male subjects |
Safety
There were no serious AEs during the study. Two subjects dropped out of the study because their blood creatine phosphokinase (CPK) levels increased (>10 times the upper limit of normal) accompanied by hepatic enzyme level increases without any symptoms. These AEs occurred 10 days after the most recent administration of valsartan, and these subjects reported a history of intense exercise.
Thirty-five AEs were reported in 18 subjects (nine [29.6%] with rosuvastatin, twelve [32.1%] with valsartan, and 14 [32.1%] with coadministration). There were five cases of drug-related AEs after rosuvastatin administration (14.8%), six cases after valsartan administration (21.4%), and six cases after coadministration of both drugs (24.1%). The frequency of drug-related AEs between treatments was not statistically different (Table 5). Common drug-related AEs were hemoglobin decreased (16.7%), asthenia (10.0%), and hypotension (10.0%). Other AEs (fatigue, nasopharyngitis, blood bilirubin increase, blood CPK increase, headache, and pruritus) were reported in one or two subjects (<10%). All drug-related AEs were mild in severity and subjects recovered without any medication. There were no clinically significant findings on physical examination or 12-lead ECGs.
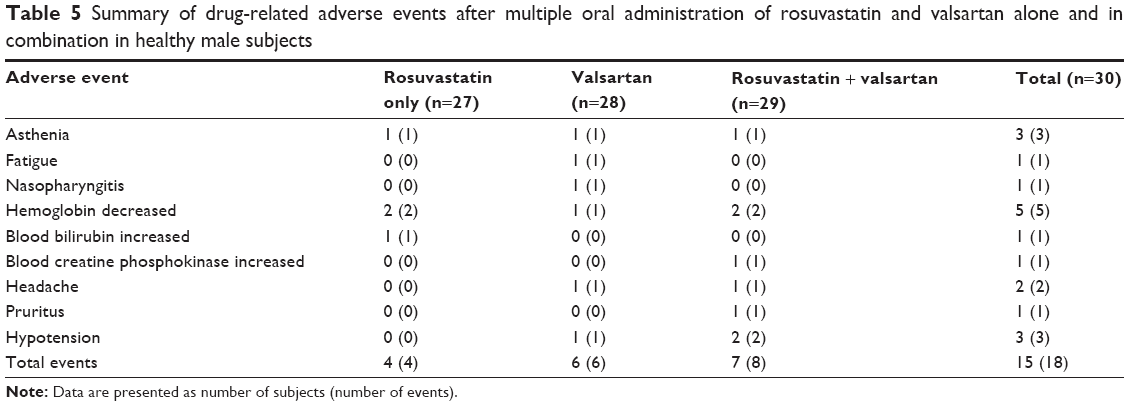 | Table 5 Summary of drug-related adverse events after multiple oral administration of rosuvastatin and valsartan alone and in combination in healthy male subjects |
Discussion
In the present study, we explored the pharmacokinetic interaction between rosuvastatin and valsartan in healthy male subjects. Peak and systemic exposure of rosuvastatin and valsartan at steady state was compared between administration of each drug alone and coadministration of both drugs. Rosuvastatin can be administered with or without a meal because food does not affect the AUC of rosuvastatin, whereas, food has been reported to decrease valsartan’s AUC by approximately 40% and its Cmax by approximately 50%.1,4,19 Therefore, this study was conducted under fasting conditions to avoid the effect of food on metabolizing enzymes and transporters.20
Observed pharmacokinetic parameters for valsartan were in agreement with those reported in the literature.21 As shown in a previous study, the Cmax,ss of rosuvastatin is increased in Asian subjects compared with in Caucasian subjects.22 In this study, the 90% CI for coadministered/individually administered AUCτ ratios indicated that no interaction criteria were fulfilled.20 The 90% CI in the Cmax,ss of valsartan and rosuvastatin fell slightly outside of the range of 0.80–1.25. However, this study was not sufficiently powered to demonstrate drug interaction based on Cmax using the strict criteria of 90% CIs falling in the range 0.80–1.25. Considering high intra-subject variability of Cmax and the wide therapeutic range of valsartan and rosuvastatin, a CI range from 0.75 to 1.34 is acceptable for Cmax.23–25 Therefore, the observed <12% decreases in Cmax,ss ratios are not clinically relevant. The median Tmax of valsartan and rosuvastatin was not changed after coadministration of valsartan and rosuvastatin.
Valsartan is excreted largely unchanged as the parent compound into the feces via bile and into the urine.16 An inhibitory or induction effect of valsartan on cytochrome P450 enzymes has not been observed. Rosuvastatin is also mainly excreted unchanged into the feces via bile and into the urine and only about 10% of rosuvastatin is metabolized by CYP2C9.4 In the present study, the Cmax of two drugs was only influenced by the combination, although the two drugs share the sample transporter proteins. This result is similar to those found with the of combination of rosuvastatin and olmesartan.26 Also, no clinically significant pharmacokinetic interactions have been reported between valsartan and other HMG-CoA reductase inhibitors such as simvastatin and pitavastatin.23,27
Multiple doses of valsartan and rosuvastatin, whether administered alone or coadministered, were generally well tolerated in this study. The frequency of AEs was similar whether the drugs were administered alone or coadministered. The observed AEs following administration of valsartan and rosuvastatin were not unexpected. No safety issues were reported, except in two subjects who dropped out because of an asymptomatic CPK increase related to intense exercise. The most important AEs associated with statins are myopathy and an asymptomatic increase in hepatic transaminases, both of which occur infrequently.28 Thus, frequency of these AEs with combination therapy should be evaluated on a long-term basis.
Comparable beneficial effects on both lipids (decrease in total cholesterol and LDL-C) and BP (decrease of SBP and DBP) were observed with combined drug treatment. It is conceivable that the lack of pharmacokinetic interaction enabled each drug to maintain its own pharmacodynamic effect. The results of the present study support the combined use of rosuvastatin and valsartan for the treatment of hypertension and hyperlipidemia. However, this study was conducted in a small cohort of healthy male Korean subjects, which limits its application, and the statistical analyses were powered for pharmacokinetic interactions only, not pharmacodynamic interactions. To generalize the result clinically, studies in hypertensive patients with hyperlipidemia or mixed dyslipidemia are necessary.
Conclusion
The pharmacokinetic profiles of rosuvastatin and valsartan when coadministered were comparable with those when the drugs were administered alone, suggesting that the pharmacokinetic interaction between valsartan and rosuvastatin was not significant in the healthy volunteers used in this study.
Acknowledgments
The authors are grateful to all study participants and volunteer subjects. This study was supported by a grant from the Yuhan Corporation, Seoul, Korea.
Disclosure
SY Nam and SB Jang disclose that they are employees of Yuhan Corporation. These two authors participated in the manuscript review. All other authors declare no conflicts of interest in this work.
References
Diovan (valsartan) tablets [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021283s044lbl.pdf. Accessed November 21, 2014. | ||
Flesch G, Müller P, Lloyd P. Absolute bioavailability and pharmacokinetics of valsartan, an angiotensin II receptor antagonist, in man. Eur J Clin Pharmacol. 1997;52(2):115–120. | ||
Sioufi A, Marfil F, Jaouen A, et al. The effect of age on the pharmacokinetics of valsartan. Biopharm Drug Dispo. 1998;19(4):237–244. | ||
McTaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of Rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol. 2001;87(5A):28B–32B. | ||
Crestor (rosuvastatin calcium) tablets [prescribing information]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021366s030lbl.pdf. Accessed November 21, 2014. | ||
Martin PD, Warwick MJ, Dane AL, et al. Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin Ther. 2003;25(11):2822–2835. | ||
Birmingham BK, Swan SK, Puchalski T, et al. Pharmacokinetic and pharmacodynamic profile of rosuvastatin in patients with end-stage renal disease on chronic haemodialysis. Clin Drug Investig. 2013;33(4):233–241. | ||
Martin PD, Mitchell PD, Schneck DW. Pharmacodynamic effects and pharmacokinetics of a new HMG-CoA reductase inhibitor, rosuvastatin, after morning or evening administration in healthy volunteers. Br J Clin Pharmacol. 2002;54(5):472–477. | ||
Martin PD, Dane AL, Nwose OM, Schneck DW, Warwick MJ. No effect of age or gender on the pharmacokinetics of rosuvastatin: a new HMG-CoA reductase inhibitor. J Clin Pharmacol. 2002;42(10):1116–1121. | ||
Yamashiro W, Maeda K, Hirouchi M, Adachi Y, Hu Z, Sugiyama Y. Involvement of transporters in the hepatic uptake and biliary excretion of valsartan, a selective antagonist of the angiotensin II AT1-receptor, in humans. Drug Metab Dispos. 2006;34(7):1247–1254. | ||
Hua WJ, Hua WX, Fang HJ. The role of OATP1B1 and BCRP in pharmacokinetics and DDI of novel statins. Cardiovasc Ther. 2012;30(5):e234–e241. | ||
Son M, Kim Y, Lee D, et al. Pharmacokinetic interaction between rosuvastatin and telmisartan in healthy Korean male volunteers: a randomized, open-label, two-period, crossover, multiple-dose study. Clin Ther. 2014;36(8):1147–1158. | ||
World Medical Association (WMA). Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. 7th revision, 64th WMA General Assembly, Fortaleza, Brazil. Ferney-Voltaire: WMA; 2013. Available from: www.wma.net/en/30publications/10policies/b3/. Accessed December 12, 2014. | ||
Schmidt B, Schieffer B. Angiotensin II AT1 receptor antagonists. Clinical implications of active metabolites. J Med Chem. 2003;46(12):2261–2270. | ||
Nakashima A, Kawashita H, Masuda N, et al. Identification of cytochrome P450 forms involved in the 4-hydroxylation of valsartan, a potent and specific angiotensin II receptor antagonist, in human liver microsomes. Xenobiotica. 2005;35(6):589–602. | ||
Waldmeier F, Flesch G, Müller P, et al. Pharmacokinetics, disposition and biotransformation of [14C]-radiolabelled valsartan in healthy male volunteers after a single oral dose. Xenobiotica. 1997;27(1):59–71. | ||
Lee HK, Ho CS, Hu M, Tomlinson B, Wong CK. Development and validation of a sensitive method for simultaneous determination of rosuvastatin and N-desmethyl rosuvastatin in human plasma using liquid chromatography/negative electrospray ionization/tandem mass spectrometry. Biomed Chromatogr. 2013;27(11):1369–1374. | ||
Ministry of Food and Drug Safety. Guideline on Bioanalytical Method Validation. Korea: Ministry of Food and Drug Safety; 2013. Available from: https://eirb2.ajoumc.or.kr/board/file/Notice/Notice_1055.pdf. Accessed January 13, 2015. | ||
Israili ZH. Clinical pharmacokinetics of angiotensin II (AT1) receptor blockers in hypertension. J Hum Hypertens. 2000;14 Suppl 1:S73–S86. | ||
US Food and Drug Administration (FDA) Center for Drug Evaluation and Research (CDER). Guidance for Industry: drug interaction studies – study design, data analysis, implications for dosing, and labeling recommendations [draft guidance]. Silver Spring, MD: FDA CDER; 2012. Available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf. Accessed November 22, 2014. | ||
Müller P, Flesch G, de Gasparo M, Gasparini M, Howald H. Pharmacokinetics and pharmacodynamic effects of the angiotensin II antagonist valsartan at steady state in healthy, normotensive subjects. Eur J Clin Pharmacol. 1997;52(6):441–449. | ||
Lee E, Ryan S, Birmingham B, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin Pharmacol Ther. 2005;78(4):330–341. | ||
Sunkara G, Reynolds CV, Pommier F, Humbert H, Yeh C, Prasad P. Evaluation of a pharmacokinetic interaction between valsartan and simvastatin in healthy subjects. Curr Med Res Opin. 2007;23(3):631–640. | ||
Martin PD, Schneck DW, Dane AL, Warwick MJ. The effect of a combination antacid preparation containing aluminium hydroxide and magnesium hydroxide on rosuvastatin pharmacokinetics. Curr Med Res Opin. 2008;24(4):1231–1235. | ||
Cooper KJ, Martin PD, Dane AL, Warwick MJ, Raza A, Schneck DW. The effect of erythromycin on the pharmacokinetics of rosuvastatin. Eur J Clin Pharmacol. 2003;59(1):51–56. | ||
Roh H, Son H, Lee D, Chang H, Yun C, Park K. Pharmacokinetic interaction between rosuvastatin and olmesartan: a randomized, open-label, 3-period, multiple-dose crossover study in healthy Korean male subjects. Clin Ther. 2014;36(8):1159–1170. | ||
Jung JA, Noh YH, Jin S, et al. Pharmacokinetic interaction between pitavastatin and valsartan: a randomized, open-labeled crossover study in healthy male Korean volunteers. Clin Ther. 2012;34(4):958–965. | ||
Bellosta S, Paoletti R, Corsini A. Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation. 2004;109(23 Suppl 1):III50–III57. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.