")
Back to Journals » The Application of Clinical Genetics » Volume 16
A New Inherited Syndrome Causing Sudden Cardiac Death with Distinct ST-Segment Depression and Ankyrin-2-Mutation
Authors von Korn H, Basso C, Pilichou K, Stefan V, Swojanowsky P
Received 11 October 2023
Accepted for publication 4 December 2023
Published 21 December 2023 Volume 2023:16 Pages 233—239
DOI https://doi.org/10.2147/TACG.S438957
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Hubertus von Korn,1 Cristina Basso,2 Kalliopi Pilichou,2 Victor Stefan,1 Patrick Swojanowsky1
1Department of Cardiology, Marienhaus Klinikum Hetzelstift, Neustadt, Weinstraße, 67434, Germany; 2Department of Cardiac, Thoracic and Vascular Sciences and Public Health, University of Padua Medical School, Padova, 35128, Italy
Correspondence: Hubertus von Korn, Department of Cardiology, Stiftstr. 10, Neustadt, 67434, Germany, Tel +496321 859 400, Fax +496321 859 4009, Email [email protected]
Introduction: Sudden cardiac death (SCD) is a serious threat. In individuals under the age of 35 years sudden arrhythmic death is the most frequent cause. In younger persons, genetically determined cardiac diseases (eg, cardiomyopathies and ion-channel diseases) account for an important proportion of these cases.
Methods: We investigated the case of a 23-year-old male with SCD, specific ECG changes and left ventricular hypertrophy. Family history was significant for SCD in the paternal line. A precise analysis was performed by an international multidisciplinary expert panel including autopsy of the index patient’s heart, molecular autopsy, whole-exome sequencing, analysis of the pedigree and examination of available family members.
Results: Three cases of SCD were reported in paternal relatives. The index patient exhibited specific ECG changes (ST-depression), which were also found in five paternal relatives and the brother of the index patient. Post-mortem analysis of the heart yielded mild idiopathic concentric hypertrophy without myocardial disarray. The genetic analysis of the index patient showed two nucleotide variations in two different genes (ANK2: c.11791G>A, MYO18B: c.3761G>A), which were also expressed in five relatives. Two family members had showed all indicators of the inherited syndrome including distinct ECG changes and genetic changes.
Conclusion: We describe a distinct inheritable syndrome causing SCD, characterized by specific ECG changes and mutations of ANK2 and MYO18. As far as we know this is the first description of this syndrome.
Keywords: sudden cardiac death, inherited syndrome, ion channel disorder, ankyrin 2, ST-segment depression, MYO18
Introduction
Sudden death of a young person is a tragic event. The incidence of sudden cardiac death (SCD) ranges from 50 to 100 per 100.000 inhabitants annually and increases with age.1 For younger individuals under the age of 40 years, the incidence of sudden cardiac death has been described as lower, between 0.7 and 6.2 per 100.000 person-years. In about 70% of cases the cause is cardiac. In individuals under the age of 35 years sudden arrhythmic death is the most frequent cause. In younger persons, genetically determined cardiac diseases (eg, cardiomyopathies and ion-channel diseases) account for an important proportion of these cases.2–8
Case Description
A previously healthy 23-year-old male was admitted following cardiac arrest. Manual chest compression was initiated immediately. Upon arrival of emergency medical services he was found with ventricular fibrillation and received defibrillation. On admission to our emergency department, the patient was still hemodynamically unstable and undergoing cardiopulmonary resuscitation. Bedside echocardiogram during resuscitation showed diffuse severe hypokinesis of the left ventricle. We performed emergent cardiac catheterization to rule out acute coronary syndrome. Coronary angiography showed regular smooth coronary arteries.9
Due to low cardiac output, an intra-aortic balloon pump was placed. Subsequently, a computed tomography of the chest and the brain ruled out relevant pathologic findings. Urine drug screening was negative. Laboratory results ruled out metabolic causes. Serology was negative for IgM to adenovirus, coxsackie and parvovirus.
Unfortunately, the patient expired 3 hr after admission due to refractory ventricular fibrillation and cardiogenic shock despite optimal treatment.
Since the family was concerned because of a high incidence of SCD in the paternal line, a more precise analysis of the case was initiated.
Methods
We have established a multidisciplinary multicenter international team of primary care physicians, cardiologists, pathologists, cardio-pathologists and geneticists with high expertise in the field of sudden cardiac death.
We thoroughly analyzed the past medical history and all available ECG records of the index patient.
Macroscopic and histologic examination of internal organs was performed, and the intact heart was then sent to the Department of Cardiac, Thoracic, Vascular Sciences and Public Health at the University of Padua (Padua, Italy) for specific analysis.
All available family members were seen and examined with our cardiology department and underwent resting and exercise ECGs, 24-hr Holter ECG monitoring and a standard transthoracic echocardiogram. A comprehensive family pedigree focused on identifying a history of cardiac disease, premature sudden death or ECG abnormalities was done.
Genetic testing of the index patients blood sample and in parts of the family was done. It was performed by sequencing in parallel 174 genes associated with inherited cardiac diseases on a MiSeq platform.10
The index patient and two clinically affected relatives (father and uncle [survivor of SCD]) underwent whole-exome sequencing. Libraries were constructed following Agilent recommendations present in SureSelect Clinical Research Exome V2 kit manual. DNA was randomly fragmented by tagmentation and adapter sequences were added to the ends. Tagmented DNA was purified using sample purification beads. Purified tagmented DNA was amplified through standard PCR reaction. After creating the library it was loaded on a HiSeq platform (Illumina). Variant classification was carried out according to American College of Medical Genetics (ACMG) revised criteria.11
Ethical Background
The study complied with the Declaration of Helsinki regarding research on humans. The parents of the patient (for the index patient and for themselves) and all affected family members provided written informed consent for research for this case and the case details to be published.
Results
Past Medical History of the Index Patient
Before the event, the patient was in good general health without known cardiac abnormalities or risk factors. No emotionally stressful event was recalled before cardiac arrest. However, prior to the event he had seen a general practitioner, who referred to outpatient cardiology for shortness of breath, fatigue, inability to exercise and sharp chest pain. At that time, echocardiogram showed a normal left ventricular function without pathologic findings. ST-changes had been noted, but no further testing had been done. The patient used no prescription or over-the-counter medications, remedies or illicit substances.
ECG Analysis of the Index Patient
Retrospective analysis of the ECG 12 weeks prior to SCD showed sinus rhythm, normal QT intervals, and no criteria indicating Brugada syndrome or early repolarization syndrome. A delta wave was not present.
Retrospective ECG analysis of the ECG 6 weeks prior to SCD showed sinus rhythm, normal QT intervals but new deep lowering of the ST-segment in lead III and aVF (the proximal part of the T-wave was involved) and a deep T inversion in aVR (Figure 1).
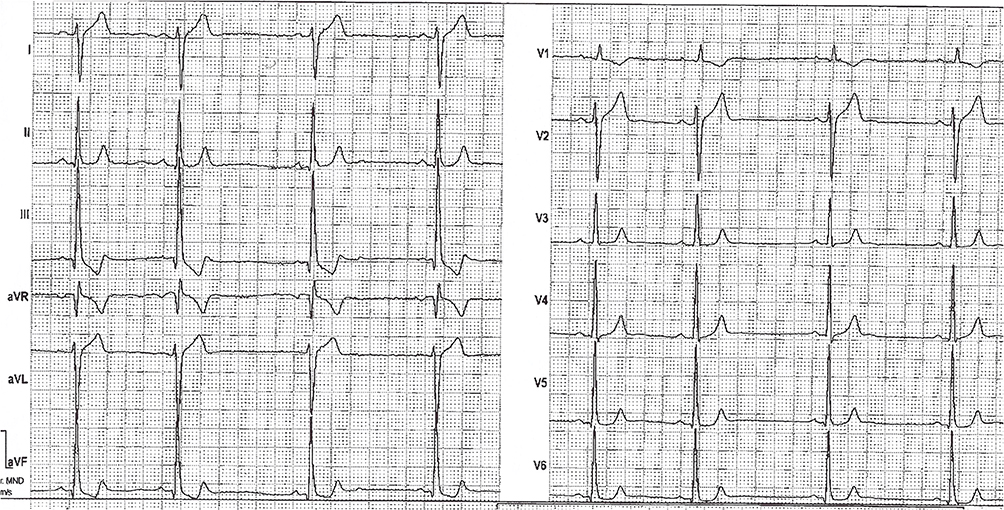 |
Figure 1 ECG of the index patient 6 weeks prior to SCD. |
It is important to note the dynamic pattern of the ECG changes.
General Autopsy of the Index Patient
Complete autopsy including dissection of internal organs in all body cavities showed no explanation for the sudden death.
Cardiac Autopsy of the Index Patient
The heart valves were normal. The course and origin of the coronary arteries were regular in a right dominant pattern. The LV thickness was 13 mm, septal thickness was 13 mm and the RV wall was 3 mm. The weight was 550 g.
The histology of myocardium showed a diffuse interstitial edema, a diffuse contraction band necrosis and neutrophilic infiltrates. The cardiac myocytes had a preserved size (mean diameter 15 µm, range 12–17 µm). The intramural vessels revealed moderate periadventitial fibrosis and medial hypertrophy. The inferior parts of the myocardium showed no additional features except for segmental gross hypertrophy (up to 15 mm thickness).
The final diagnosis was idiopathic left ventricular hypertrophy. Secondary forms such as pheochromocytoma and hypertensive heart disease were excluded.
Results of the Cardiac Evaluation of the Family Members
Comprehensive third-generation family pedigree focused on identifying a history of cardiac disease or premature sudden death was done (Figure 2).
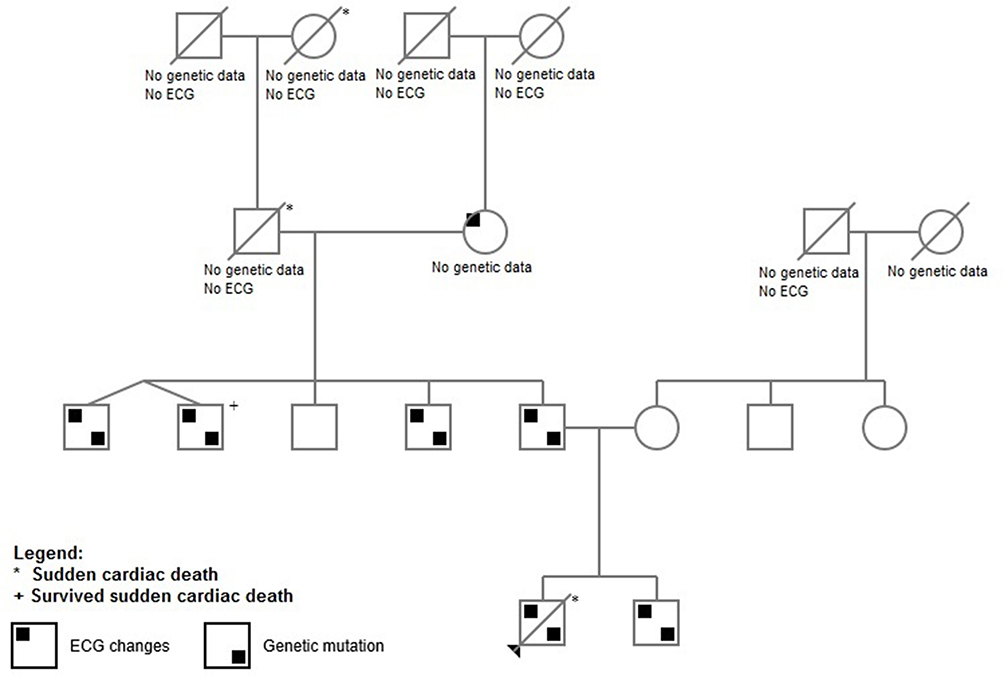 |
Figure 2 Pedigree. |
Results of the Cardiac Evaluation of the Family Members – Maternal Line
We followed the family line three generations up to the patient’s great-grandmother. No reports of SCD or syncope were remembered. ECG analyses of these relatives were unremarkable as well.
Results of the Cardiac Evaluation of the Family Members – Paternal Line
The brother of the index patient denied previous syncopal episodes and had no significant past medical history. Holter monitoring and echocardiography were normal. During Holter monitoring and treadmill test he remained asymptomatic but a transient change in the ECG pattern similar to the abnormalities of the index patient was observed. Cardiac MRI revealed a slightly reduced left ventricular function and a dilatation of the right ventricle and the right atrium.
The father had no significant past medical history and also denied previous syncopal episodes. The ECG showed the same transient pattern of abnormalities like the index patient and the echocardiography showed mild hypertrophy. Cardiac MRI confirmed mild hypertrophy of the left and right ventricles without late enhancement and normal left ventricular function.
We observed four uncles of the index patient. Two uncles were identical twins. One of them had survived a cardiac arrest due to ventricular fibrillation, and an ICD was implanted. We review the initial echocardiography, which showed a severely reduced left ventricular ejection fraction. Upon examination 3 years later, this was normalized. The angiography directly after resuscitation showed a high-grade stenosis of the LAD, a drug-eluting stent was implanted. We reviewed multiple ECGs obtained over the last 3 years, which showed the same pathologic pattern found in the index patient’s ECG (Figure 3).
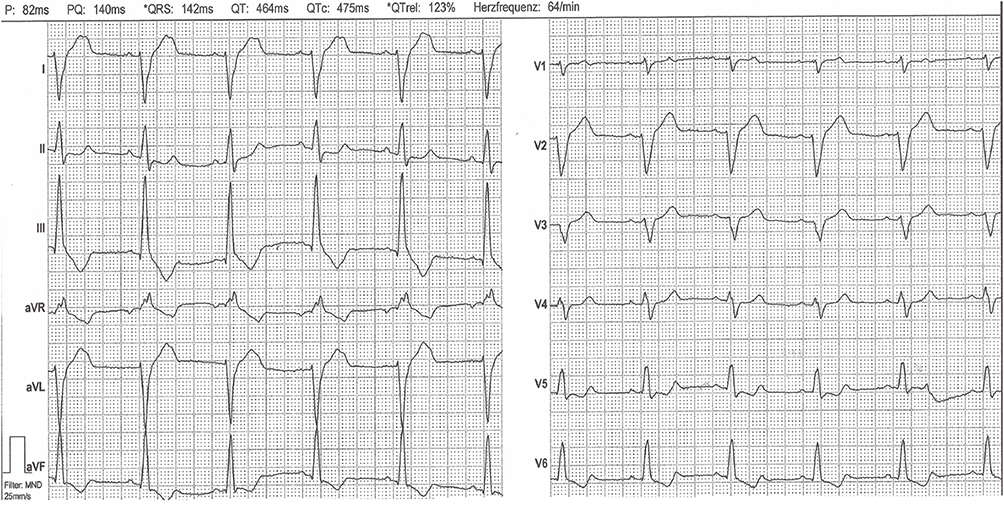 |
Figure 3 Representative ECG changes as also seen in the other family members (ECG from the uncle of the index patient). |
The other identical twin was healthy without significant past medical history or syncopal episodes. We observed the same pathologic ECG pattern. Echocardiography showed regular findings. A Single-Photon-Emission-Computed Tomography (SPECT) with Tc99m was done with reduced activity during exercise in the inferior parts of the myocardium, but coronary angiography was unremarkable.
Two other uncles were alive without episodes of sudden cardiac death. One of them had unclassified paroxysmal tachycardias with normal findings of echo, Holter monitoring and stress test. He had transient mild expression of the pathologic ECG pattern.
Another uncle had regular ECG records and treadmill testing revealed no pathologic ECG pattern.
Further analysis of the grandparents and great-grandparents showed two cases of sudden death (one male and one female) but a more detailed analysis including genetic analysis was not possible. His grandmother had a mild form of the pathologic ECG pattern.
Genetic Analysis
In the index patient, we found five nucleotide variations in four different genes (ANK2, CACNA2DS, HSPG2 and MYO18B). The cascade screening of the parents shows that two are coming from the father and three from the mother.
Additional genetic screening was performed of the brother, father and all four uncles in the paternal line. We found mutations of ANK2 (c.11791G>A) and MYO18 (c.3761G>A) in all of them except one uncle.
Based on all those informations we created a pedigree of the family containing informations about ECG changes and genetic mutations (Figure 2).
The gene ANK2 encodes Ankyrin-2 – a protein that is expressed in cardiomyocytes. Its main function is membrane stabilization of ion transporters and ion channels. It is known that mutations in Ankyrin-2 can cause cardiac arrhythmias (mainly long-QT syndrome) which is named “Ankyrin-B syndrome” and more infrequent LV hypertrophy.12
The preproprotein encoded by CACNA2D gene is cleaved into multiple chains that comprise the alpha-2 and delta subunits of the voltage-dependent calcium channel complex. Calcium channels mediate the influx of calcium ions into the cell upon membrane polarization. Mutations in this gene can cause cardiac deficiencies, including Brugada syndrome and short QT syndrome.13
The HSPG2 gene encodes the perlecan protein, which consists of a core protein to which three long chains of glycosaminoglycans (heparan sulfate or chondroitin sulfate) are attached. Perlecan is a key component of the vascular extracellular matrix, where it helps to maintain the endothelial barrier function. It is a major component of basement membranes, where it is involved in the stabilization of other molecules, as well as being involved with glomerular permeability to macromolecules and cell adhesion. Mutations in this gene cause Schwartz-Jampel syndrome type 1, Silverman-Handmaker type of dyssegmental dysplasia, and tardive dyskinesia.14
The MYO18B (Myosin XVIIIB) is a protein coding gene. Diseases associated with MYO18B include Klippel-Feil Syndrome 4. Among its related pathways are PAK pathway and actin nucleation by ARP-WASP complex. Gene ontology (GO) annotations related to this gene include nucleotide binding and cytoskeletal motor activity.15
Discussion
We describe a fatal case of sudden cardiac death due to ventricular fibrillation in a previously healthy 23 years old male with an uncommon and dynamic ECG pattern. Family history showed a high incidence of SCD in paternal relatives.
A detailed analysis of the descendent, his family and ancestors including genetic analysis with an international multidisciplinary team was initiated.
We recognized a distinct ECG pattern, to our knowledge not described before, which could be identified in the brother, the father, three uncles and the grandmother – all in the paternal line.
It should be noted in advance that a variety of different gene loci are involved in the development of sudden cardiac death, whereas the heart is macroscopically and microscopically normal. These include primary electric disorders caused by mutations in genes encoding ion channels and cardiomyopathies. For example, loss-of-function mutations in the KCNQ1 gene cause the Long QT syndrome, one of the most frequently causes of sudden cardiac death.16
A recently described mutation of the intermediate filament protein desmin, which is encoded by the gene DES, shows a new aspect of the genetic basis of SCD. This gene contributes to the mechanical stabilization of the striated muscle sarcomere. Mutations of the DES gene cause cardiac muscle diseases and have a high clinical arrhythmogenic potential.17
Therefore, we performed a genetic analysis of the index patient, which revealed five nucleotide variations in four different genes. Two of these gene mutations (ANK2 and MYO18) were present in the index patient, the brother of him, the father and three uncles. All affected male family members presenting with this specific ECG pattern had mutations of these two genes. One of them survived an episode of sudden cardiac death.
The gene ANK2 encodes Ankyrin-2 – a protein that is expressed in cardiomyocytes. Its main function is membrane stabilization of ion transporters and ion channels. It is known that mutations in Ankyrin-2 can cause cardiac arrhythmias (mainly long-QT syndrome) which is named “Ankyrin-B syndrome” and more infrequent LV hypertrophy.12 The mode of inheritance is autosomal dominant.
It could be hypothesized that this mutation is associated with an elevated risk of SCD due to an inherited ion channel disorder and causes the discovered (partially dynamic) abnormalities in baseline ECGs other than long-QT syndrome.
For these reasons, we conclude that at least ANK2 mutations are related to this specific and newly described ECG pattern associated with SCD. We further hypothesize that if all features (ECG-changes and described genetic mutations) are positive, the risk for SCD may be higher than in patients with none or only one of these markers.
We recommend the implantation of a subcutaneous ICD as a prophylactic measure in these few individuals.
Future Directions
A possible approach for further research into this form of sudden cardiac death is artificial intelligence. Artificial intelligence algorithms could process large volumes of patient and ECG data as in coronary artery disease and atrial fibrillation.18
We are planning to establish an international registry for further investigation of this syndrome and are accepting referral of cases for enrolment.
Conclusion
We describe an inheritable syndrome causing SCD, characterized by distinct ECG changes (specific ST-segment depression) and mutations of ANK2 and MYO18. Further expansion of testing in the population of SCD patients and their relatives is needed to gain more data.
Consent for Publication
We confirm that all details of our article can be published.
Acknowledgment
Abstract of this paper was presented at the EHRA 2021 Conference as a poster presentation with interim findings. In addition to the online presentations, all abstracts were published in the supplement of Europace: DOI https://academic.oup.com/europace/article/23/Supplement_3/euab116.326/6282943?login=false.
Funding
The family of the patient paid for the whole exome sequencing. No institutional approval was required to conduct this research and publish the case details. Our hospital is an academic teaching hospital.
Disclosure
There are no disclosures or competing interests interacting with our study.
References
1. Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation. 2014;125(4):620–637. doi:10.1161/CIRCULATIONAHA.111.023838
2. Winkel BG, Holst AG, Theilade J, et al. Nationwide study of sudden cardiac death in person aged 1-35 years. Eur Heart J. 2011;32(8):983–990. doi:10.1093/eurheartj/ehq428
3. van der Werf C, van Langen IM, Wilde AA. Sudden cardiac death in the young: what do we know about it and how to prevent? Circ Arrhythm Electrophysiol. 2010;3(1):96–104. doi:10.1161/CIRCEP.109.877142
4. Priori SG, Blomström-Lundquist C, Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36(41):2793–2867. doi:10.1093/eurheartj/ehv316
5. Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace. 2013;15(10):1389–1406. doi:10.1093/europace/eut272
6. Charron P, Arad M, Arbustini E, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on myocardial and pericardial diseases. Eur Heart J. 2010;31(22):2715–2726. doi:10.1093/eurheartj/ehq271
7. Risgaard B, Winkel BG, Jabbari R, et al. Burden of sudden cardiac death in persons aged 1 to 49: nationwide study in Denmark. Circ Arrhythm Electro. 2014;7(2):205–211. doi:10.1161/CIRCEP.113.001421
8. Basso C, Aguilera B, Banner J, et al. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows Arch. 2017;471(6):691–705. doi:10.1007/s00428-017-2221-0
9. von Korn H, Stefan V, van Ewijk R, et al. A Systematic Diagnostic And Therapeutic Approach For The Treatment Of Patients After Cardio-pulmonary Resuscitation: a Prospective Evaluation Of 212 Patients Over 5 Years. Intern Emerg Med. 2016;12(4):503–511. doi:10.1007/s11739-016-1480-0
10. Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm. 2011;8(8):1308–1339. doi:10.1016/j.hrthm.2011.05.020
11. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
12. Mohler PJ, Gramolini AO, Bennett V. Ankyrins. J Cell Sci. 2002;115(Pt8):1565–1566. doi:10.1242/jcs.115.8.1565
13. National Library of Medicine. Available from: https://www.ncbi.nlm.nih.gov/gene/781.
14. Gene cards. Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=HSPG2.
15. Gene cards. Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=MYO18B.
16. Bezzina CR, Lahrouchi N, Priori SG. Genetics of Sudden Cardiac Death. Circ Res. 2015;116(12):1919–1936. doi:10.1161/CIRCRESAHA.116.304030
17. Brodehl A, Dieding M, Klauke B, et al. The Novel Desmin Mutant p.A120D Impairs Filament Formation, Prevents Intercalated Disk Localization, and Causes Sudden Cardiac Death. Circ Cardiovasc Genet. 2013;6(6):615–623. doi:10.1161/CIRCGENETICS.113.000103
18. Hayiroglu MI, Altay S. The Role of Artificial Intelligence in Coronary Artery Disease and Atrial Fibrillation. Balkan Med J. 2023;40(3):151–152. doi:10.4274/balkanmedj.galenos.2023.06042023
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.