")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 17
Unraveling Potential Glyoxalase-I Inhibitors Utilizing Structure-Based Drug Design Techniques
Authors Fetian MH, Al-Balas QA
Received 17 October 2023
Accepted for publication 1 February 2024
Published 5 February 2024 Volume 2024:17 Pages 21—32
DOI https://doi.org/10.2147/AABC.S441074
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Maria Miteva
Mohammad H Fetian, Qosay A Al-Balas
Department of Medicinal Chemistry and Pharmacognosy, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan
Correspondence: Qosay A Al-Balas, Department of Medicinal Chemistry & Pharmacognosy, Faculty of Pharmacy, Jordan University of Science and Technology, P.O. Box 3030, Irbid, 22110, Jordan, Tel +962 2 720 1000, Fax +962 2 720 1071, Email [email protected]
Background: Glyoxalase system detoxifies methylglyoxal and other ketoaldehydes to produce innocuous metabolites that allow the cells to function normally. Its inhibition in cancer cells causes these toxic metabolites to accumulate, and the cancer cells enter the apoptotic stage.
Methods: The techniques of Computer-Aided Drug Design (CADD) were used, and the compounds possessing a zinc-binding group from commercial databases were extracted, using the pharmacophore search protocol. These compounds were subjected to robust docking using the CDOCKER protocol within the Discovery Studio. Docking was performed on both Glo-I twin active sites. The biological activities of candidate hits were assessed using an in vitro assay against Glo-I.
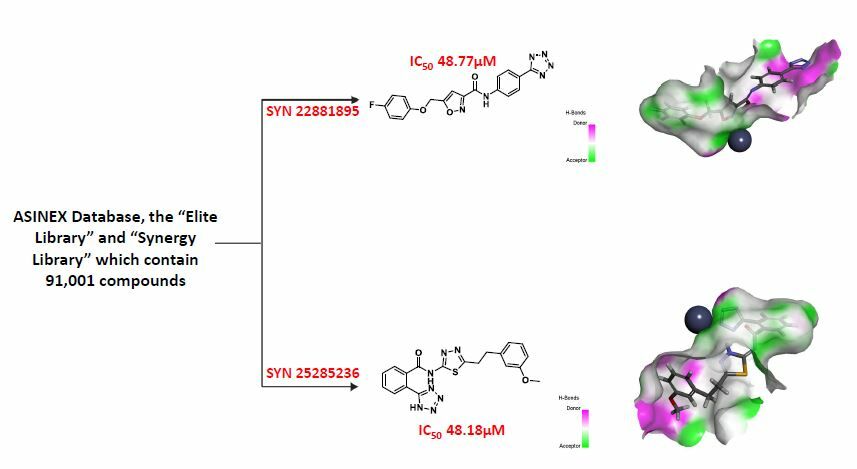
Results: Compounds containing zinc-binding groups were extracted from ASINEX® commercial database, which contains (91,001 compounds). This step has helped to retrieve 1809 ligands, which then were prepared and docked at the two active sites of Glo-I. The fourteen compounds, which have showed the highest scores in docking and returned acceptable Total Binding Energy values, were purchased and tested against the enzyme in vitro. Two compounds out of the fourteen, which were selected in the final step, possess tetrazole ring as zinc chelating moiety, and have showed moderate activity with an IC50 of 48.18μM for SYN 25285236 and 48.77 μM for SYN 22881895.
Conclusion: Two hits with moderate activity are identified as the lead compounds against Glo-I. Both compounds possess a negatively ionized tetrazole ring as the zinc-binding moiety. These compounds will lead to the development of inhibitors with improved activities.
Keywords: Glo-I, CDOCKER, zinc-binding group, tetrazole ring, structure-based drug design
Graphical Abstract:
Introduction
By dividing into new cells, human cells develop and multiply as required by the body. When old cells pass away due to aging or damage, new cells take their place. Cancer develops when the host cells grow out of control and spread to other parts of the body.1 It is an out-of-control growth that is caused by the accumulation of genetic and epigenetic alterations in healthy cells.2
To allow normal cell survival, the glyoxalase system detoxifies ketoaldehydes, such as methylglyoxal, to innocuous metabolites, such as D-lactic acid, in a GSH-catalyzing manner.3,4 The detailed detoxification process is shown in Figure 1. The isomerization mechanism of the glyoxalase system starts with proton abstraction from the C1 of hemithioacetal substrate by Glu172 that is produced non-enzymatically by the reaction of methylglyoxal with glutathione (GSH) to the produce enediol intermediate. Subsequently, Glu172 covey the proton to C2 of the substrate resulting in the formation of the S,D-lactoylglutathione (SLG), and this represents the substrate for Glo-II that converts the thioester into non-toxic D-lactic acid by hydrolysis.
 |
Figure 1 Methylglyoxal detoxification pathway by glyoxalase system. (GSH; glutathione). |
Many metalloenzymes in living organisms have been considered as pivotal targets for cancer treatment. The histone deacetylase family (HDAC), carbonic anhydrase, bacterial methionine aminopeptidase and matrix metalloproteinase 3 (MMP-3) were subjected to extensive research to find potential inhibitors. The main strategy was to find a suitable zinc-binding moiety that is considered the main determinant for enzyme inhibition.5–8
A series of studies have reported Glo-I overexpression in several cancer types, including pancreatic cancerous tissues,9 breast cancer proteomics,10 prostate cancer cells,11 and melanoma,12 while these tumor cells have increased glycolytic activity, which increases the cellular levels of hazardous substances, such as methylglyoxal.13–15
Inhibition of this enzyme leads to the accumulation of toxic byproducts which cause tumor cell death.16,17 Glo-I is a potential target for the design and discovery of new Glo-I inhibitors with various chemical scaffolds, because it is a rate-limiting step in this system. Glo-I overexpression has been linked to multidrug resistance in various malignancies.18–20
Over the past decades, there had been extensive research on the design and development of Glo-I inhibitors with potential anticancer activity.21,22 The earliest inhibitors that emerged were either substrates or transition-state analogues, which are known for being metabolically deactivated by glutamyl transpeptidase (GGT). Due to the obstacles related to metabolic instability of GSH-based inhibitors, and to the violations of drug-like properties, some researchers have created non-GSH-based Glo-I inhibitors.23,24 Natural flavonoids and anthocyanidins were investigated by Takasawa et al, as potential Glo-I enzyme inhibitors.25 In order to identify crucial structural elements of flavonoids to serve as Glo-I inhibitors, computational and experimental approaches were performed by Al-Balas et al, where the structure-activity relationship (SAR) of a panel of 24 compounds belong to the flavonoid class of natural products, was explored. It was found that a C2/C3 double bond, along with a di- or tri-hydroxylation of the benzene rings, particularly ring A, are pivotal for enzyme inhibition (primarily as zinc-binding features). Additionally, the inhibitory effect of these compounds does not entirely depend on the ketol system. Scutellarein is one of the flavonoids that had been studied and have been found the most effective inhibitor.26,27
Structurally, Glo-I has a homodimeric structure with two similar active sites, each monomer weighing 42kDa and containing 184 amino acids. The active sites could be easily identified due to the presence of zinc atom as a catalytic cofactor. Each active site had three primary areas: a positively charged entrance (Arg37, Arg122, and Lys156), a structurally central zinc atom – crucial for stabilizing the enediolate intermediate, and a deep hydrophobic pocket (Phe62, Leu69, Leu92, and Met179) (Figure 2).28–31 Our research team has worked on this target over the past period.32–35 This work is a continuation of previous efforts, and it is mainly based on the investigation of potential inhibitors, hinged on structure-based (SBDD) techniques, by utilizing the ASINEX® commercial database and testing the potential candidates by in vitro assays.
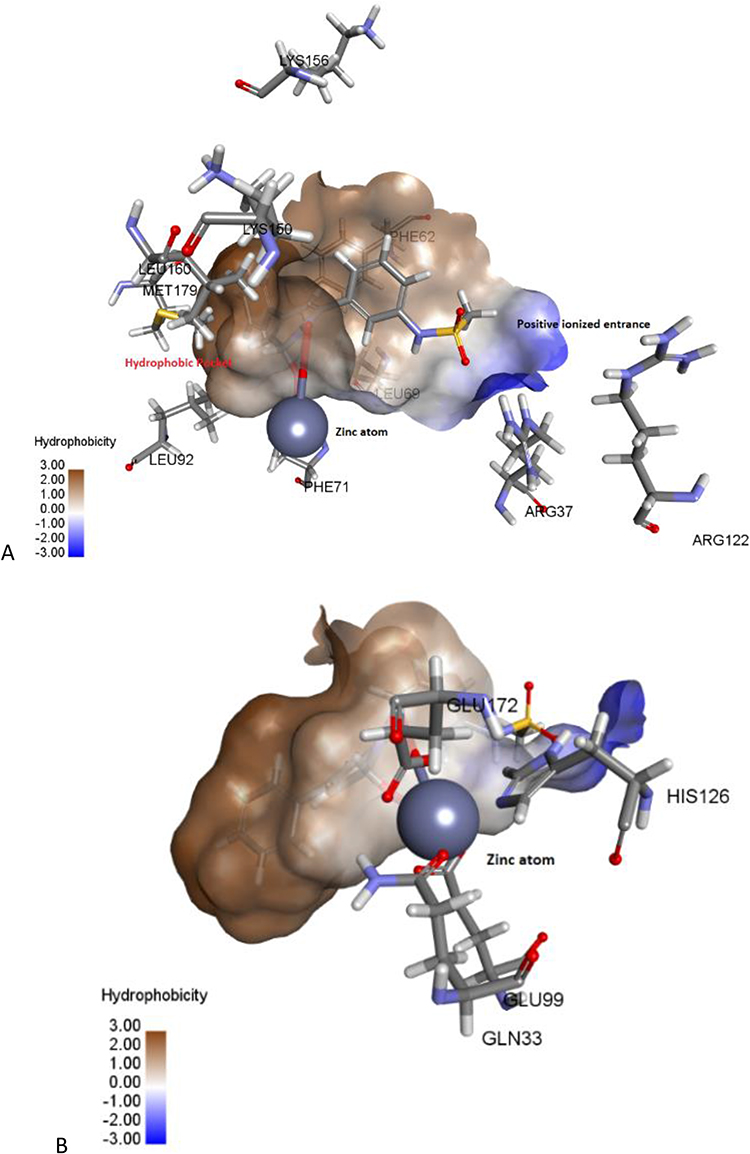 |
Figure 2 The active site of Glo-I enzyme; (A) Positively ionized mouth (blue), hydrophobic pocket (brown) amino acids, (B) Zinc atom (grey sphere) with major amino acids bound to it (Glu99, Gln33, his126, Glu172). |
Materials and Methods
The workflow of this effort is illustrated in Scheme 1.
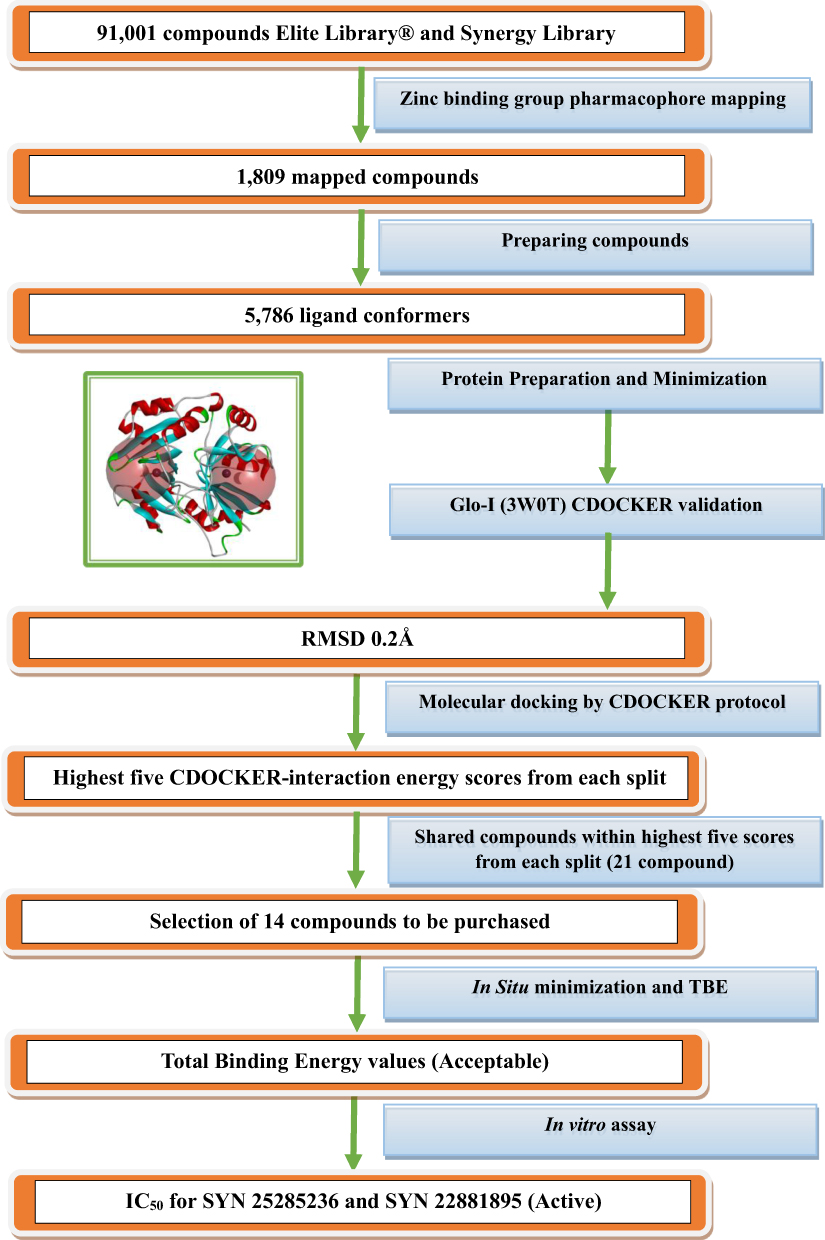 |
Scheme 1 Detailed scheme of the main steps that were conducted within this project. |
3D Ligand Pharmacophore Mapping
The first step was extracting ligands that possess zinc-binding groups, such as thiol; phosphate; carboxylic acid; five-membered aromatic ring possessing N-N atoms, such as (imidazole, tetrazole, and triazole); hydroxamic acid from ASINEX® database by using “3D Ligand Pharmacophore Mapping” protocol in Discovery Studio software from Biovia®. The ASINEX® database Elite Library and the Synergy Library contained 91,001 compounds, and these were screened in this process.
In silico Docking
The mapped compounds from the previous step were docked into the Glo-I active site using the CDOCKER protocol in the Discovery Studio software. This is a validated software, and it uses a CHARMm-based molecular dynamics (MD) scheme to dock ligands into a receptor binding site. Random ligand conformations are generated using high-temperature MD. The conformations are then translated into the binding site. Candidate poses are thereafter created using random rigid-body rotations, followed by simulated annealing. A final minimization is then employed to refine the ligand poses. Glo-I was retrieved from the Protein Databank PDB code 3W0T with a 1.35 Å resolution. The protein was prepared and minimized using default parameters before docking, and the zinc atom had a +2 charge, and was assigned an octahedral geometry. In addition, all mapped ligands were prepared using a preparation ligand protocol with default parameters, with the exception that the generated isomers were false (the geometry of the commercial compounds was conserved). The CDOCKER protocol was applied for the docking analysis. The parameters of the CDOCKER protocol were set as default; the number of top hits was 10, number of random conformations also 10, simulated annealing was set to true, the force field was CHARMm, the full potential was set to obtain more accurate results and Ligand Partial Charge Method was assigned to be Momany-Rone which uses a set of bond-charge-increment rules for assigning atom partial charges. Finally, this docking protocol was conducted on Glo-I enzyme active sites with a 10 Å sphere to ensure that all three major areas (a positively charged entrance, central zinc atom, and a deep hydrophobic pocket) were covered, and the docked compounds could be in contact with them. The second active site was assigned to a 9.95 Å sphere as a technical step to differentiate between the two active sites in the final stage of analysis.
Selection of Potential Inhibitors
The docking analysis was based on the CDOCKER-INTERACTION energy, and the best positions between the two active sites with the highest CDOCKER-INTERACTION energy were chosen. The next step was to select compounds with the highest CDOCKER-INTERACTION energy at the two active sites. This process yielded 21 poses that represent the highest pose from each compound was selected. Finally, 14 compounds were selected by avoiding repetition of the same functional groups and ensuring diversity in the zinc-binding functional groups.
In-Situ Minimization and Calculation of Binding Energy
The 14 selected compounds were checked for their suitability for purchase as they should have acceptable Total Binding Energy values (negative). In situ minimization was conducted as a preparatory step before calculating the Total Binding Energy. The poses that were elected for the in situ minimization were the top 14 poses from the previous docking step. Regarding the parameters, the, Adopted Basis NR was used as a minimization algorithm as it gives more accurate results in which better convergence to the local minima. Regarding RMSD gradient, it is a tolerance applied to the average gradient during a cycle of minimization (kcal/(mol-Å), it was set 109 to give more accurate results. Subsequently, the binding energy protocol was used to calculate the Total Binding Energy. With regard to parameters, Poisson Boltzmann with non-polar Surface Area (PBSA) was utilized as an implicit solvent model in which PB equation is fundamental for the most precise continuum models of solvation effects. In situ minimization was set False because this step was conducted previously as a separated step the Ligand Conformational Entropy set to True in order to determine the total binding energy. As the ligand flexibility is based on this method, the run time can be greatly extended. The negative total binding energy number means that the ligand bound spontaneously without using any energy, whereas a positive value means that the binding required energy to take place and could only do so if the energy was available.
Biological Assay
Once the selected compounds had been purchased, their Glo-I inhibitory activity was evaluated in vitro against human recombinant Glo-I, as described in other studies.32,33 All chemicals and bulk solvents were obtained from Acros (Thermo Fisher Scientific, New Jersey, US) and Sigma-Aldrich Co., respectively, and had the highest purity throughout the country. Following the manufacturer’s instructions (R&D Systems® Corporation, USA), the biological activity of the selected compounds was assessed based on their in vitro inhibitory activity against human recombinant Glo-I. The enzyme was reconstituted in our laboratory, kept at −70° C, which will be thawed prior to use on the day of the test. Regarding the assay buffer, it was prepared by utilizing 0.5 M sodium phosphate monobasic and 0.5 M sodium phosphate dibasic. DMSO was used to dissolve the test compounds to prepare a 10mM stock solution, and the absorbance of each well was measured for 200 s. at λmax 240 nm using a UV microplate reader at 25° C. The substrate solution was prepared by adding 25 mL buffer with 706 µL methylglyoxal and 706 µL glutathione, which was prepared previously, and then vortexed for 15s to incubate them in a water bath at 37° C for 15 min. A blank was prepared by mixing the substrate mixture with the assay buffer; the human recombinant Glo-I was then added. Three duplicates of each test were performed, and the average was calculated. To determine the top-hit compounds, IC50 was calculated using the Prism software. Myricetin was used as a positive control with an IC50 of 3.38 ± 0.41 µM.
Results and Discussion
3D Pharmacophore Mapping
The presence of zinc-binding groups is the main criterion for the discovery of potent Glo-I inhibitors. Therefore, the 3D pharmacophore mapping was conducted to extract the “Zinc Binding Groups” containing compounds, by building a zinc query file that contains the major groups; these were reported with good zinc-binding ability such as thiol, phosphate, carboxylic acid, and five-membered aromatic ring possessing N-N atoms, such as (imidazole, tetrazole, and triazole), and hydroxamic acid. “Elite and Synergy Libraries” that contain a panel of early ADMET filters have been used to screen these compounds and ensure that screening hits can be quickly optimized from hit to lead, and that they are free of ADMET issues. A total of 1809 zinc-binding group-containing compounds were obtained from the 91,001 compounds.
In silico Docking
Molecular docking was performed using the CDOCKER protocol, a grid-based molecular-docking algorithm. The two binding sites were defined using the Define and Edit Binding site tool within the DS, with a sphere of 10 Å and 9.95 Å radius, which is based on a co-crystallized ligand-protein complex. The CDOCKER protocols were employed with default parameters, with the exception of full potential to obtain more accurate results. The resulting pose ranking was based on the CDOCKER interaction energy, which included the interaction energy, plus ligand strain with the interaction energy alone (Table 1). This protocol gives 10 poses for each docked ligand with its score and in our work, the highest scored poses were adopted for the selection purposes.
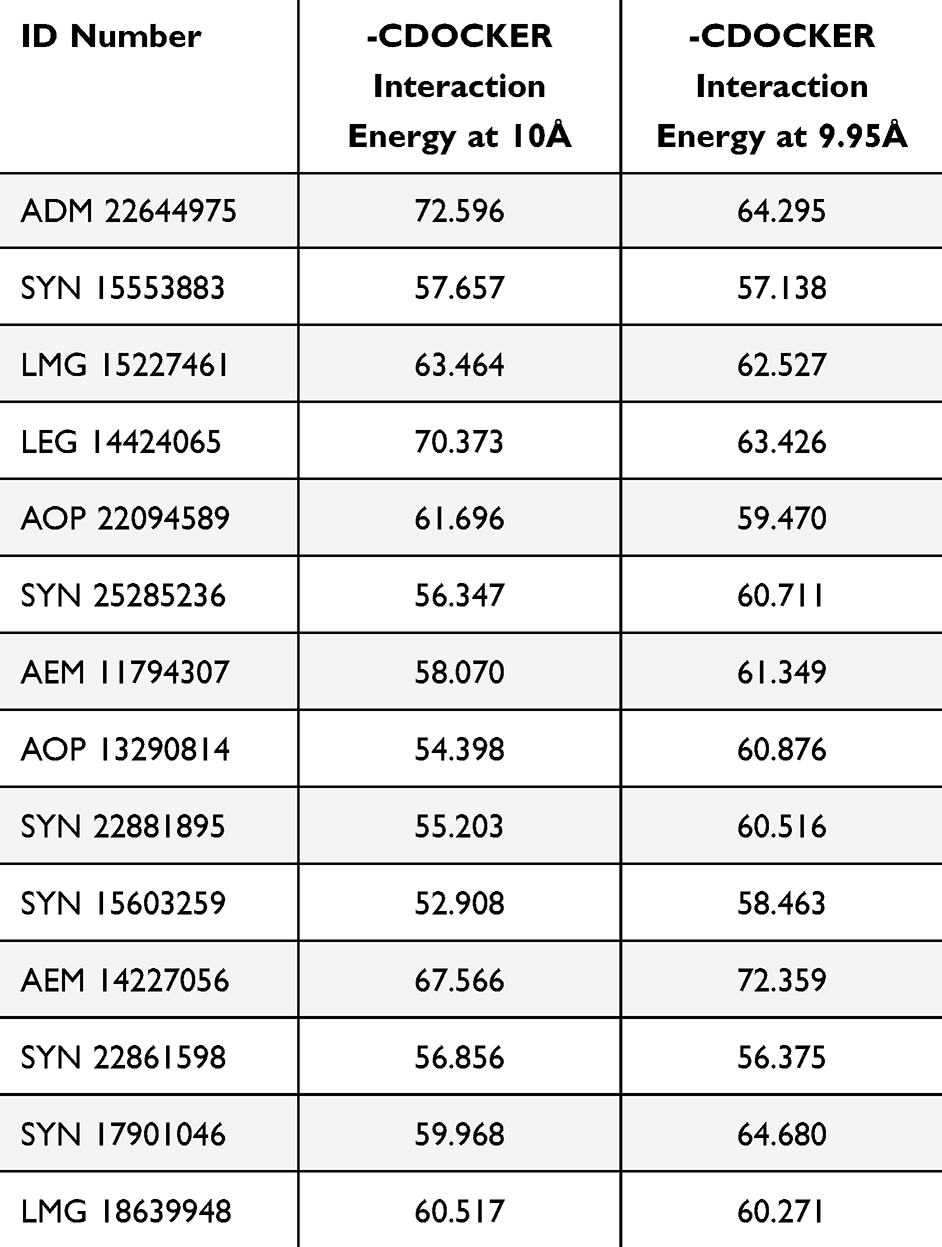 |
Table 1 The Values of -CDOCKER-Interaction at Two Active Sites |
Selection of Potential Inhibitors
The selection criteria were based on the highest CDOCKER interaction energy score, while their investigation manually is intended to avoid any inconsistencies that could happen during the docking process. The next step was to identify ligands with the highest CDOCKER interaction energy shared by the two active sites. ADM 22644975 scored the highest, with a value of 72.6 for one of the active sites, and 64.3 for the second. This has resulted in the identification of 21 common compounds that have high scores on both active sites (Table 1). Fourteen compounds were chosen for purchase, and particular attention was given to avoid the repetition of similar ligands (Figure 3).
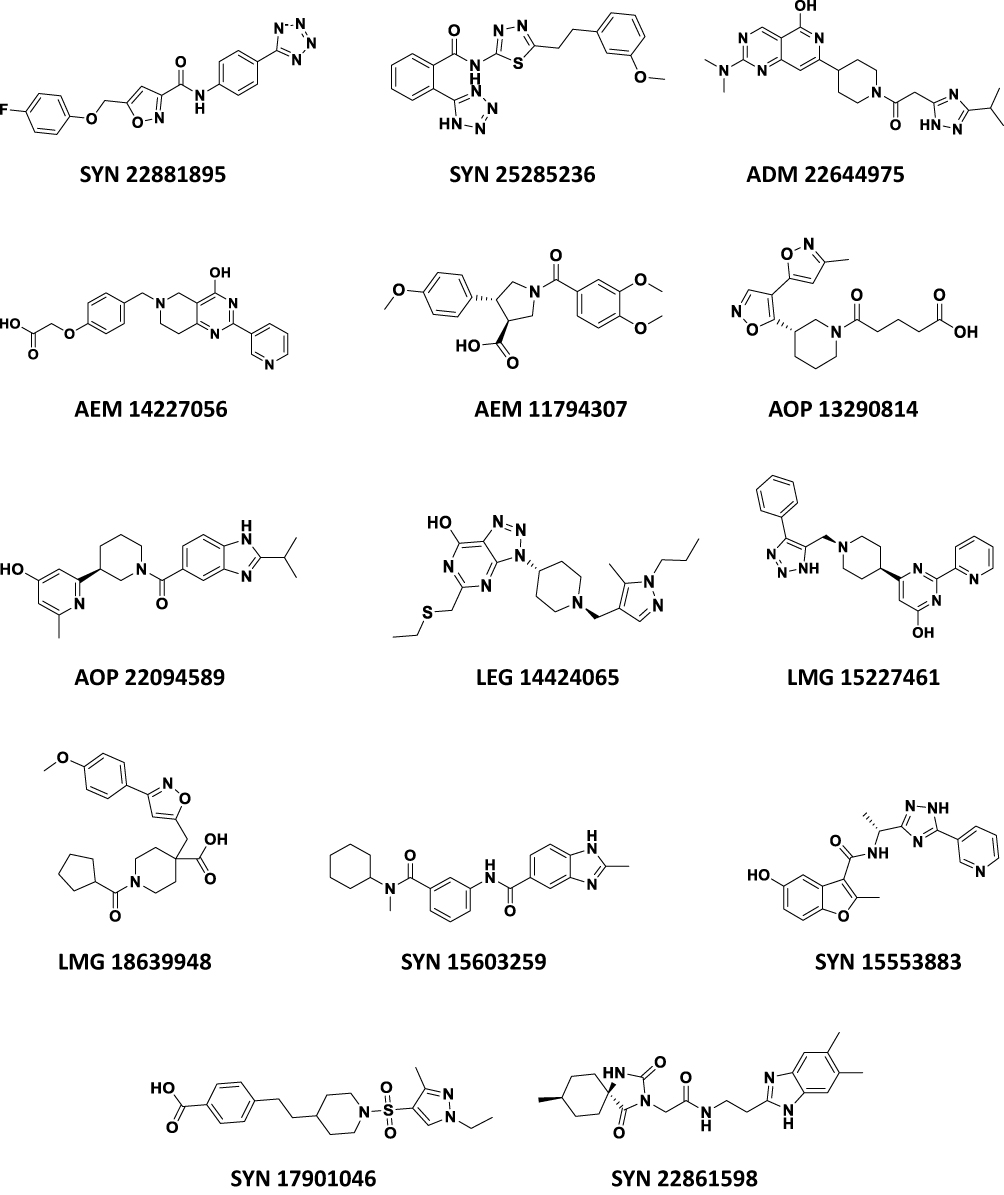 |
Figure 3 The chemical structures of the finally selected and purchased chemicals from the commercial database. |
In-Situ Minimization and Calculation of Binding Energy
For the chosen compounds, these two stages serve as confirmation steps prior to the purchase. Estimating the Total Binding Energy was conducted after the in situ minimization had taken place. The Adopted Basis Newton Raphson NR was used as a minimization algorithm, whereas the remaining parameters were left at their default settings. Given that we need to calculate the total binding energy rather than the binding energy, which is the sum of the binding energy and the conformational entropy of the ligand, the Poisson Boltzmann with non-polar Surface Area (PBSA) model was used as the implicit solvent model. All compounds showed total binding energy values within the range of −122.439 to −2.725 kcal/mol (Table 2). This has confirmed that the selected compounds were suitable for purchase.
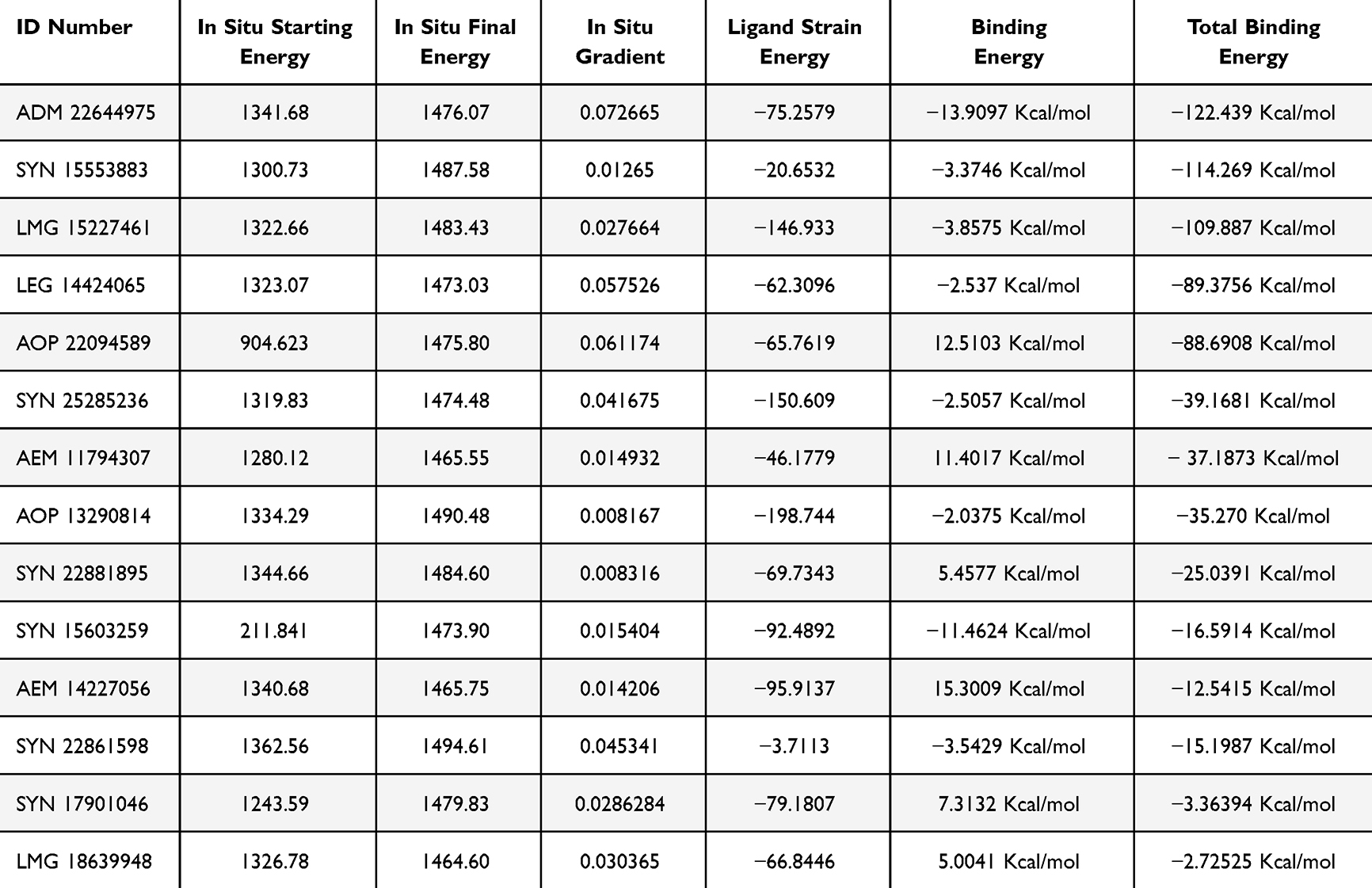 |
Table 2 The Values of in-situ Minimization, Binding Energy and Total Binding Energy |
Biological Evaluation
The inhibitory activities of the 14 compounds were conducted in vitro against human recombinant Glo-I. The slope for each compound was calculated by plotting absorbance versus time, and the inhibition percentage was computed as well. Myricetin was used as a positive control. Fourteen compounds were screened at a concentration of 50 µM, and the percentage of Glo-I inhibition was calculated and compared to inhibition of the positive control (myricetin).
Pursuant to the screening process, two compounds, SYN 22881895 and SYN 25285236, have showed inhibition percentages greater than 50%. The remaining compounds have showed a low percentage of inhibition. The average IC50 for SYN 22881895 and SYN 25285236 compounds was 48.77 µM and 48.18 µM, respectively (Table 3).
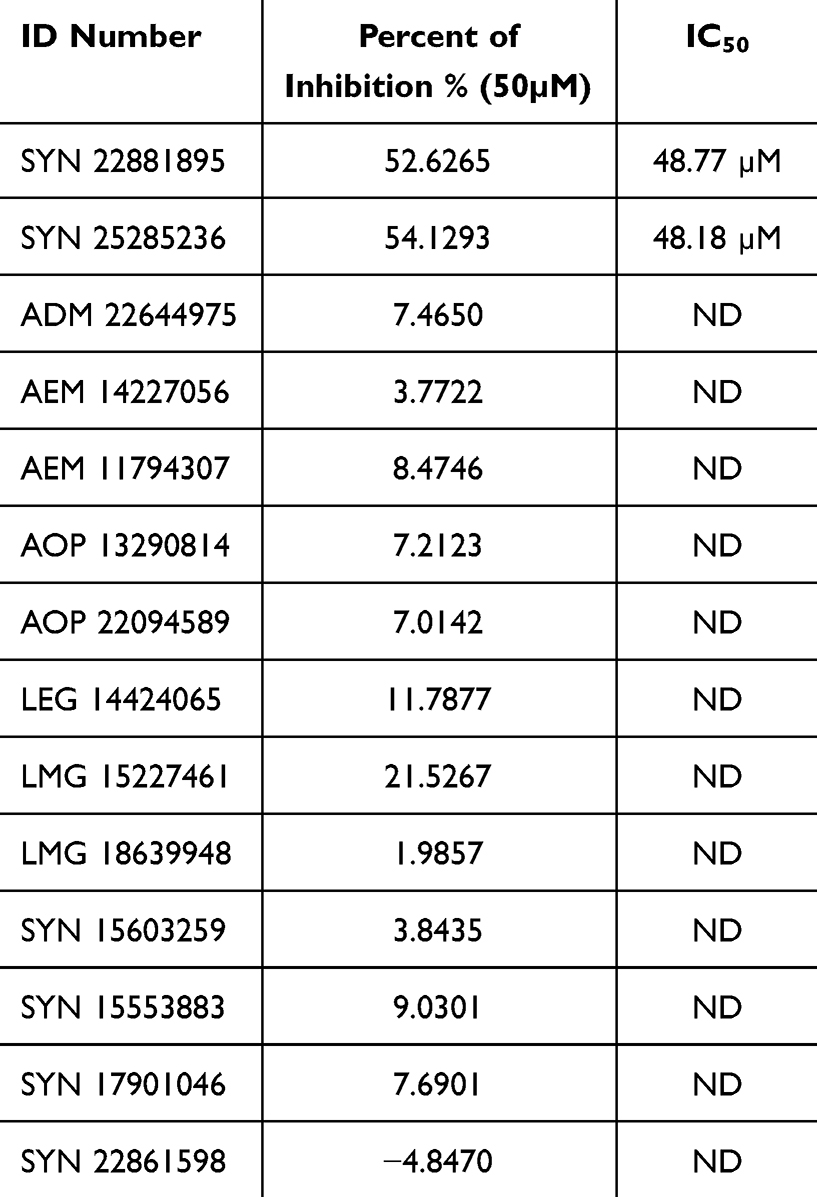 |
Table 3 The Average Percent of Inhibition and IC50 for the Tested Compounds |
Analysis of Docked Poses of the Active Compounds
Figure 4 shows the binding patterns of SYN22881895 and SYN25285236. Regarding compound SYN22881895, the benzene ring performed good hydrophobic interactions with amino acids Phe62 and Met179 in the hydrophobic pocket. The nitrogen atom of the isoxazole ring coordinates with the zinc atom via a metal-acceptor bond. The tetrazole ring showed ionic interactions with the positive mouth basic amino acids, Arg37 and Arg122. Regarding SYN25285236, the benzene ring showed hydrophobic interactions with the hydrophobic pocket of amino acids Met179, Leu92, and Phe62. These represent tetrazole-chelated zinc atoms via attractive charges. Additionally, the ether oxygen forms a hydrogen bond with the basic amino acids.
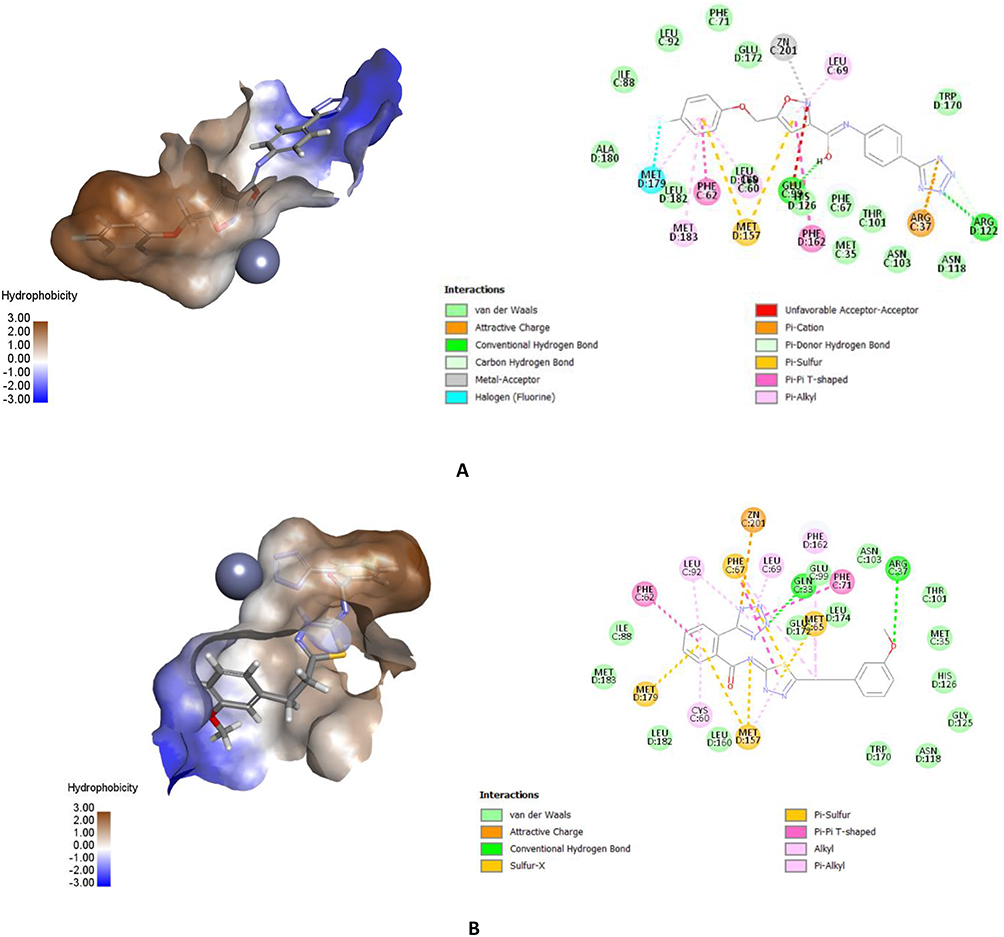 |
Figure 4 An interaction representation of the most active compounds depicted as 3D and 2D within the active site of Glo-I of (A) SYN22881895 with its most important interactions both 3D and 2D and (B) SYN25285236 with most important interactions both 3D and 2D. |
The tetrazole ring in the most active compound has performed optimal interaction with the zinc atom. This is due to the fact that the adjacent phenyl ring has positioned itself optimally inside the hydrophobic pocket next to the zinc atom. Meanwhile, the phenoxy group has showed ion dipole interaction with basic amino acid Arg37. On the other hand, in compound SYN 22881895, the zinc chelator was the isoxazole ring instead of the tetrazole ring. The para-fluoro methoxy moiety has perfect fit inside the hydrophobic pocket, while the negatively ionized tetrazole ring favored to have ionic interaction with the active site Arg 122 and Arg 37.
It is worth noting that the two active compounds were ranked as the top 21 of the entire database. This is a good indication of the ability of the software to choose actives. For example, the most active compound SYN25285236 ranked the eighth at one active site and the seventh at the other. SYN22881895 ranked the ninth at one active site and the eleventh at the other.
Conclusion
Two active lead hits were identified in this work, which possessed a tetrazole ring. The most active compound SYN25285236 offered the tetrazole ring as the zinc-chelating moiety. On the other hand, the SYN22881895 compound revealed that the tetrazole ring favored ionic interactions with positively ionized entrance, while the isoxazole was the zinc chelator. The most active compound had an IC50 of 48.18µM. This work is based on selecting the crystal structure of Glo-I with excellent resolution (1.35 Å), which was then prepared and minimized for docking purposes. The CDOCKER protocol was used as the main determinant to prioritize the compounds in the commercial ASINEX® database for the future purchase. The docking process was conducted on the two active sites of this protein to add more credit to the results retrieved from this step. The finally selected compounds were investigated as a confirmatory step by calculating their total binding energies which also returned satisfactory. An in vitro assay was conducted against this enzyme. Generally, acceptable correlation has been detected between the in silico and the in vitro results as those compounds were ranked as the top 21 of the in silico results and showed promising biological activity. In the future, these two compounds will be lead-guiding for deciphering more active compounds.
Acknowledgment
This study was funded by the Deanship of Research at the Jordan University of Science and Technology (Grant number 169-2023 & 131-2021).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Cronin KA, Scott S, Firth AU, et al. Annual report to the nation on the status of cancer, part 1: national cancer statistics. Cancer. 2022;128(24):4251–4284. doi:10.1002/cncr.34479
2. Prendergast GC, Jaffee EM. Cancer immunologists and cancer biologists: why we didn’t talk then but need to now. Cancer Res. 2007;67(8):3500–3504. doi:10.1158/0008-5472.CAN-06-4626
3. Al-Balas QA, Hassan MA, Al-Shar’i NA, et al. Novel glyoxalase-I inhibitors possessing a “zinc-binding feature” as potential anticancer agents. Drug Des Devel Ther. 2016;2623–2629. doi:10.2147/DDDT.S110997
4. Sukdeo N, Honek JF. Microbial glyoxalase enzymes: metalloenzymes controlling cellular levels of methylglyoxal. Drug Metab Drug Interact. 2008;23(1–2):29–50. doi:10.1515/DMDI.2008.23.1-2.29
5. Jackl D, K. M, Seo H, Karges J, Kalaj M, Cohen SM. Salicylate metal-binding isosteres as fragments for metalloenzyme inhibition. Chem Sci. 2022;13(7):2128–2136. doi:10.1039/D1SC06011B
6. Helgren C, R. T, Wangtrakuldee P, Staker BL, Hagen TJ. Advances in Bacterial Methionine Aminopeptidase Inhibition. Curr Top Med Chem. 2016;16(4):397–414. doi:10.2174/1568026615666150813145410
7. Lomelino B, L. C, Supuran CT, McKenna R. Non-Classical Inhibition of Carbonic Anhydrase. Int J Mol Sci. 2016;17(7):1150. doi:10.3390/ijms17071150
8. Sarkar A, Banerjee R, Amin S, Adhikari N, Jha T. Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: a review. Eur J Med Chem. 2020;192:112171.
9. Wang Y, Kuramitsu Y, Ueno T, et al. Glyoxalase I (GLO1) is up-regulated in pancreatic cancerous tissues compared with related non-cancerous tissues. Anticancer Res. 2012;32(8):3219–3222.
10. Fonseca-Sánchez MA, Rodríguez Cuevas S, Mendoza-Hernández G, et al. Breast cancer proteomics reveals a positive correlation between glyoxalase 1 expression and high tumor grade. Int j Oncol. 2012;41(2):670–680. doi:10.3892/ijo.2012.1478
11. Antognelli C, Mezzasoma L, Fettucciari K, Talesa VN. A novel mechanism of methylglyoxal cytotoxicity in prostate cancer cells. Int J Biochem Cell Biol. 2013;45(4):836–844. doi:10.1016/j.biocel.2013.01.003
12. Bair WB, Cabello CM, Uchida K, Bause AS, Wondrak GT. GLO1 overexpression in human malignant melanoma. Melanoma Res. 2010;20(2):
13. Audat SA, Al-Balas QA, Al-Oudat BA, Athamneh MJ. Design, Synthesis and Biological Evaluation of 1,4-Benzenesulfonamide Derivatives as Glyoxalase I Inhibitors. Drug Des Devel Ther. 2022;16:873–885. doi:10.2147/DDDT.S356621
14. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):
15. Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10(4):267–277. doi:10.1038/nrc2817
16. Rulli A, Carli L, Romani R, et al. Expression of glyoxalase I and II in normal and breast cancer tissues. Breast Cancer Res Treat. 2001;66(1):67–72. doi:10.1023/A:1010632919129
17. Thornalley PJ, Rabbani N. 2011. May. Glyoxalase in tumourigenesis and multidrug resistance. Seminars in Cell & Developmental Biology. Vol. 22. 3. 318–325. Academic Press..
18. Sakamoto H, Mashima T, Kizaki A, et al. Glyoxalase I is involved in resistance of human leukemia cells to antitumor agent-induced apoptosis. Blood J Am Soc Hematol. 2000;95(10):3214–3218.
19. Mearini E, Romani R, Mearini L, et al. Differing expression of enzymes of the glyoxalase system in superficial and invasive bladder carcinomas. Eur J Cancer. 2002;38(14):1946–1950. doi:10.1016/S0959-8049(02)00236-8
20. Antognelli C, Baldracchini F, Talesa VN, Costantini E, Zucchi A, Mearini E. Overexpression of glyoxalase system enzymes in human kidney tumor. Cancer J. 2006;12(3):222–228. doi:10.1097/00130404-200605000-00011
21. Zhang H, Huang Q, Zhai J, et al. Structural basis for 18-β-glycyrrhetinic acid as a novel non-GSH analog glyoxalase I inhibitor. Acta Pharmacol Sin. 2015;36(9):1145–1150. doi:10.1038/aps.2015.59
22. Zhang H, et al. In vitro inhibition of glyoxalase Ц# 134; by flavonoids: new insights from crystallographic analysis. Curr Top Med Chem. 2016;16(4):460–466. doi:10.2174/1568026615666150813150944
23. Jin T, Zhao L, Wang H-P, et al. Recent advances in the discovery and development of glyoxalase I inhibitors. Bioorg Med Chem. 2020;28(4):115243. doi:10.1016/j.bmc.2019.115243
24. Usami M, Ando K, Shibuya A, et al. Crystal structures of human glyoxalase I and its complex with TLSC702 reveal inhibitor binding mode and substrate preference. FEBS Lett. 2022;596(11):1458–1467. doi:10.1002/1873-3468.14344
25. Takasawa R, Takahashi S, Saeki K, et al. Structure–activity relationship of human GLO I inhibitory natural flavonoids and their growth inhibitory effects. Bioorg Med Chem. 2008;16(7):3969–3975. doi:10.1016/j.bmc.2008.01.031
26. Al‐Balas QA, Hassan MA, Al‐Shar’i NA, et al. Computational and experimental exploration of the structure–activity relationships of flavonoids as potent glyoxalase-I inhibitors. Drug Dev Res. 2018;79(2):58–69. doi:10.1002/ddr.21421
27. Al-Balas QA, Al-Sha’er MA, Hassan MA, et al. Identification of the First “Two Digit Nano-molar” Inhibitors of the Human Glyoxalase-I Enzyme as Potential Anticancer Agents. Med Chem. 2022;18(4):473–483. doi:10.2174/1573406417666210714170403
28. Takasawa R, Shimada N, Uchiro H, Takahashi S, Yoshimori A, Tanuma SI. TLSC702, a novel inhibitor of human glyoxalase I, induces apoptosis in tumor cells. Biol Pharm Bull. 2016;39(5):869–873. doi:10.1248/bpb.b15-00710
29. He Y, Zhou C, Huang M, et al. Glyoxalase system: a systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed Pharmacother. 2020;131:110663. doi:10.1016/j.biopha.2020.110663
30. Al-Balas Q, Hassan M, Al-Oudat B, Alzoubi H, Mhaidat N, Almaaytah A. Generation of the first structure-based pharmacophore model containing a selective “zinc binding group” feature to identify potential glyoxalase-1 inhibitors. Molecules. 2012;17(12):13740–13758. doi:10.3390/molecules171213740
31. Al-Balas QA, Hassan MA, Al-Shar’i NA, Al Jabal GA, Almaaytah AM. Recent advances in glyoxalase-I inhibition. Mini Rev Med Chem. 2019;19(4):
32. Al-Balasa A, Hassana QA, Al Jabala MA, Al-Shar NA, Almaaytahb AM, El-Elimata T. Novel thiazole carboxylic acid derivatives possessing a “zinc binding feature” as potential human glyoxalase-I inhibitors. Lett Drug Des Discovery. 2017;14(11):1324–1334. doi:10.2174/1570180814666170306120954
33. Al-Balas QA, Hassan MA, Al-Shar’i NA, et al. Novel glyoxalase-I inhibitors possessing a “zinc-binding feature” as potential anticancer agents. Drug Des Devel Ther. 2016;2623–2629.
34. Al-Sha’er MA, Al-Balas QA, Hassan MA, Al Jabal GA, Almaaytah AM. Combination of pharmacophore modeling and 3D-QSAR analysis of potential glyoxalase-I inhibitors as anticancer agents. Comput Biol Chem. 2019;80:102–110. doi:10.1016/j.compbiolchem.2019.03.011
35. Al-Oudat BA, Hana’a MJ, Al-Balas QA, Al-Shar’i NA, Bryant-Friedrich A, Bedi MF. Design, synthesis and biological evaluation of novel glyoxalase I inhibitors possessing diazenylbenzenesulfonamide moiety as potential anticancer agents. Bioorg Med Chem. 2020;28(16):
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.