")
Back to Journals » The Application of Clinical Genetics » Volume 12
Two novel variants in the TCF12 gene identified in cases with craniosynostosis
Authors Goumenos A, Tsoutsou E, Traeger-Synodinos J , Petychakis D, Gavra M, Kolialexi A, Frysira H
Received 14 October 2018
Accepted for publication 20 December 2018
Published 12 February 2019 Volume 2019:12 Pages 19—25
DOI https://doi.org/10.2147/TACG.S190855
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer

Athanasios Goumenos,1 Eirini Tsoutsou,1 Joanne Traeger-Synodinos,1 Dimitrios Petychakis,1,2 Maria Gavra,3 Aggeliki Kolialexi,1 Helena Frysira1
1Choremio Research Laboratory, Department of Medical Genetics, Faculty of Medicine, National and Kapodistrian University of Athens, Athens, Greece; 2Department of Pediatric Haematology-Oncology, Agia Sophia Children’s Hospital, Athens, Greece; 3CT, MRI & PET/CT Department, Agia Sophia Children’s Hospital, Athens, Greece
Abstract: Craniosynostosis (CS) is a condition where one or more of the cranial sutures fuse prematurely. It affects almost 1/2,000 newborns, and includes both syndromic and non-syndromic cases. To date, variants in over 70 different genes have been associated with the expression of CS. In this report, we describe two unrelated cases that presented with coronal CS. TCF12 sequencing analysis revealed novel frameshift nucleotide variants, which were evaluated as pathogenic according to the current guidelines for interpreting sequence variants. These findings expand the spectrum of TCF12 gene variants related with CS and support the importance of screening for such variants in patients with coronal synostosis.
Keywords: TCF12, craniosynostosis, clinical cases, frameshift, HEBβ
Introduction
The premature fusion of the calvarial sutures leads to a condition named craniosynostosis. Craniosynostosis is classified as “syndromic” when features such as facial asymmetry and limb deformities are present and as “non-syndromic” when the only feature is the premature fusion of the cranial sutures.1 Craniosynostosis affects ~1/2,000 newborns, and the cranial suture most commonly affected is the sagittal, followed by the coronal, the metopic and least frequently the lambdoid.1 Patients with a fused sagittal suture rarely have mutations in one of the craniosynostosis-related genes. However, it has been reported that chromosomal abnormalities may be present in some of these cases.2 On the other hand, patients with a fused coronal suture tend to have a specific genetic cause, most commonly located in one of the following genes: FGFR1 (MIM #136350), FGFR2 (MIM #176943), FGFR3 (MIM #134934), EFNB1 (MIM #300035), TWIST1 (MIM #601622) and TCF12 (MIM #600480).1,2,5 Of these genes, TCF12 has not been thoroughly researched due to its fairly recent association with craniosynostosis. TCF12 encodes a class-I E base–helix–loop–helix (bHLH) protein, which is expressed in multiple tissues and can form heterodimers with other bHLH proteins.3,6,7 Regarding its role in suture development, the Tcf12 protein forms heterodimers with the class-II bHLH transcription factor Twist1, helping to exert its action in lineage commitment and differentiation. It has been proven that this synergistic effect plays an important role during the formation of the coronal suture.2–4
In 2013, Sharma et al described 38 cases with mutations in the TCF12 gene and coronal craniosynostosis, all of which were toward the 3′ half of the gene (exons 10–19).3 Their phenotypic spectrum included bilateral or unilateral coronal synostosis accompanied by the clinical findings of dysmorphic face and ears, limb abnormalities, strabismus and mild to moderate learning difficulties.3,6 Clinically unaffected cases have also been reported.3,7 In this report, we describe two novel frameshift variants in two patients with coronal synostosis and phenotypes suggestive of TCF12-related craniosynostosis.
Case study
Patient 1 was a 9-month-old female with bilateral synostosis of the coronal suture. She was the first child of two non-consanguineous parents, born by Cesarean section due to maternal preeclampsia. Her birth weight was 3.130 g (50th centile) and the head circumference 31 cm (<3rd centile). Neonatal evaluation revealed wide anterior fontanel, flat forehead and dilation of the sagittal suture. At the age of 4 months, three-dimensional computed tomography (3D-CT) scan of the cranium revealed synostosis of both right and left coronal sutures (Figure 1A–D). At the age of 6 months, a surgical operation was performed in order to relieve the increased intracranial pressure (ICP) and reshape the cranial dome. The child’s psychomotor development was normal. Family history revealed that her mother, a 32-year-old woman, had a history of mild learning difficulties for which she had received professional support. She also exhibited a particular crus shape in both her ears. Her head circumference was between the normal range. Cranial or other facial morphological abnormalities or asymmetries were not observed. The child’s father was apparently healthy, with no clinical findings and with normal development.
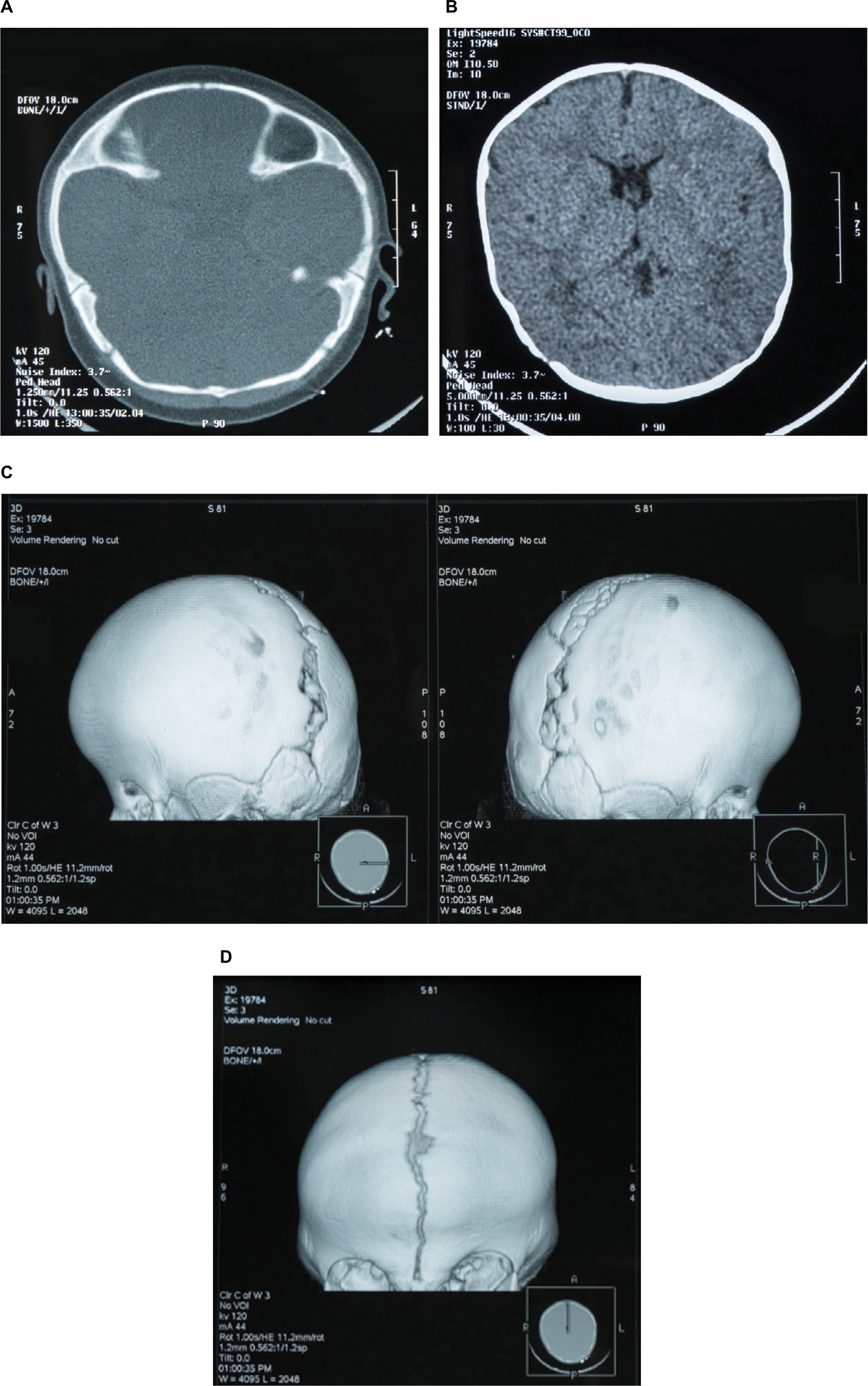 | Figure 1 (A–D) Radiological imaging of Patient 1. Three-dimensional reformatted CT scan, frontal and lateral views, demonstrate fusion of both the right and the left coronal sutures. Note: From the CT axial images, no brain parenchymal abnormalities were detected. Abbreviation: CT, computed tomography. |
Patient 2 was a 1-year-old female. She was born at term by Cesarean section due to increased body weight (birth weight of 4.490 g). The perinatal period was uneventful. At the age of 4 months, pediatric clinical evaluation revealed a closed anterior fontanel and morphological features such as plagiocephaly, facial asymmetry, a small nose with a broad base and a high-arched palate. A 3D-CT scan showed left coronal synostosis. Cardiologic, ophthalmologic and urinary tract examinations were normal, as well as auditory tests and psychomotor development. The child’s head circumference was in the 10th centile at the time of examination. At the age of 6 months, she underwent a surgical operation for cranial dome correction and ICP relief. The parents had no abnormal findings on physical examination.
Conventional karyotype and sequencing analysis of the three FGFR genes (FGFR1, FGFR2 and FGFR3) was performed in both patients initially. Due to negative results, we proceeded further to DNA analysis by sequencing the next four more commonly affected genes (TWIST1, TCF12, EFNB1 and ERF).
Two frameshift variants were identified. Patient 1 and her mother carried the same variant (c.1119delC/p.T373Tfs*23), whereas Patient 2 carried c.1836dupA/p.R613Tfs*5 (Figures S1 and S2). All variants were in heterozygous state. The consequences of the two frameshift variants identified were evaluated based on the American College of Medical Genetics and Genomics (ACMG) guidelines (see Table S1) and classified as pathogenic with the use of 1) MutationTaster (disease causing with a maximum probability score of 1.00), 2) the ACMG classification criteria which included evidence for six categories indicating pathogenicity (PVS1, PS4, PM1, PM2, PP3, PP4) and 3) previous reports of patients with TCF12 gene variants with frameshift mutations in various exons.3–5 The father of Patient 1 and the parents of Patient 2 did not carry any mutation in the TCF12 gene.
This study was approved by the bioethics committee of the Agia Sophia Children’s Hospital. Written informed consent was obtained from all four parents for the cases to be published.
Discussion
TCF12 consists of 21 exons and encodes three major transcripts: HEBβ (706 amino acids, with the inclusion of amino acids encoded by exon 15), HEBα (682 amino acids, with the exclusion of amino acids encoded by exon 15) and HEBalt (512 amino acids, alternative transcription starting from exon 9A). HEBβ the protein with 706 amino acids consists of two activation domains, one rep domain and a bHLH domain which is crucial for heterodimerization with other targets.3 The HEBα protein is identical to HEBβ, with the same structure, but lacks an analogous small site inside the second activation domain.3 It is suggested that HEBβ and HΕΒα play a far more significant role during suture development than other transcripts.3
Based on the evaluation of the variants identified in the patients, the c.1119delC (identified in Patient 1 and her mother), which is located in exon 14 of HEBβ (706 amino acids) protein transcript, creates a stop codon between exons 14 and 15, located 23 codons downstream to the single nucleotide deletion (Figure 2). More specifically, the new in-frame TGA stop triplet has TG located in exon 14 and A in exon 15. As inclusion or no inclusion of exon 15 creates two different proteins, in the case of ΗΕΒα, the stop codon is located again inside exon 15 (or the analogous exon 16 of HEBβ). As previously discussed, these two transcripts are quite similar and the effect is predicted to be the same in both of them; the mutant mRNAs are degraded by the nonsense-mediated decay (NMD) pathway. To the best of our knowledge, this is the first time that such a mutation, which is located inside that crucial location, has been described. Although incomplete penetrance in TCF12 is as high as 50% of the cases reported, it is interesting how two individuals, who are relatives (mother and daughter), have the same mutation, but the daughter exhibits the typical TCF12-related craniosynostosis phenotype, while the mother has only mild learning difficulties. Similar cases with varying phenotypes in different members of the family, including solely learning difficulties, have also been described in other reports, though they are rare.3,6,7 This supports the observation that clinical diagnosis in such cases might prove difficult without molecular testing.
 | Figure 2 Schematic representation of the TCF12 protein (HEBβ). The location of the mutations is indicated with a red stripe. The p.T373Tfs*23 (Patient 1 and mother) is located within AD2, whereas the p.R613Tfs*5 (Patient 2) is located within bHLH domain. Abbreviation: bHLH, base–helix–loop–helix. |
On the other hand, Patient 2 had c.1836dupA in exon 19 or p.R613Tfs*5 mutation. Evaluation of the altered nucleotide sequence showed a premature stop codon (TAA) inside exon 19. This mutation probably leads to an unstable transcript, which is degraded by NMD and by all means is responsible for the onset of craniosynostosis.
Previous reports indicate that all TCF12 mutations, located toward the 3′ half, affect crucial domains of the transcripts HEBβ and ΗEBα and are considered pathogenic, leading to coronal synostosis and/or other phenotypic features.3,4,6,7 As expected, both patients presented with coronal synostosis, as the null variants were unable to heterodimerize with TWIST1. Therefore, they could not mediate the formation of boundaries between the cephalic paraxial mesoderm and the neural crest, as well as control osteoblast differentiation via interactions with other key pathways and factors such as FGFRs, RUNX2 or BMPs.3,6,7 Both had undergone only one surgical operation (when 6 months old) in order to relieve ICP and reshape the cranial dome. No other complications have been reported since then (Table 1). Thus, we can confirm previous observations that patients suffering from TCF12 mutations have a good prognosis.
 | Table 1 Phenotypic features of the individuals described in this report Notes: 1: Patient 1 (daughter of Patient 2). 2: Mother of Patient 1. 3: Patient 2. |
Conclusion
Patients 1 and 2 exhibit a more severe phenotype than the mother of Patient 1, but overall, their development is satisfactory. Given the fact that a clinical diagnosis might prove difficult, especially in clinically unaffected cases, we suggest that TCF12 should be included in routine screening in patients with coronal synostosis and their parents.
Disclosure
The authors report no conflicts of interest in this work.
References
Slater BJ, Lenton KA, Kwan MD, Gupta DM, Wan DC, Longaker MT. Cranial sutures: a brief review. Plast Reconstr Surg. 2008;121(4):170e–178e. | ||
Wilkie AO, Byren JC, Hurst JA, et al. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics. 2010;126(2):e391–e400. | ||
Sharma VP, Fenwick AL, Brockop MS, et al. Mutations in TCF12, encoding a basic helix–loop–helix partner of Twist1, are a frequent cause of coronal craniosynostosis. Nat Genet. 2013;45(3):304–307. | ||
di Rocco F, Baujat G, Arnaud E, et al. Clinical spectrum and outcomes in families with coronal synostosis and TCF12 mutations. Eur J Hum Genet. 2014;22(12):1413–1416. | ||
Lee E, Le T, Zhu Y, et al. A craniosynostosis massively parallel sequencing panel study in 309 Australian and New Zealand patients: findings and recommendations. Genet Med. 2018;20(9):1061–1068. | ||
Goos JA, Fenwick AL, Swagemakers SM, et al. Identification of intragenic exon deletions and duplication of TCF12 by whole genome or targeted sequencing as a cause of TCF12-Related craniosynostosis. Hum Mutat. 2016;37(8):732–736. | ||
Paumard-Hernández B, Berges-Soria J, Barroso E, et al. Expanding the mutation spectrum in 182 Spanish probands with craniosynostosis: identification and characterization of novel TCF12 variants. Eur J Hum Genet. 2015;23(7):907–914. |
Supplementary materials
Methodology
Total DNA was extracted from the leukocytes of peripheral blood using the MagAttract DNA Blood M48 kit (Qiagen) on a BIOROBOT M48 (Qiagen). PCR amplification was performed using 0.5 μL primers (Bioanalytica), 6 μL HotStarTaq DNA Polymerase (Qiagen) and 4 μL H2O (RNase-free water; Qiagen) per 1 μL DNA/water mix. A Veriti® 96-well thermal cycler (Thermo Fisher Scientific) was used with the following conditions: 1) 5 minutes denaturation at 95°C; 2) 35 cycles of 30 seconds at 94°C, 1 minute annealing at 58°C and 2 minutes extension at 72°C and 3) 10 minutes extension at 72°C. The PCR product cleaning was performed with 0.75 μL ExoSAP-IT™ PCR Product Cleanup Reagent per 2.0 μL of PCR product using the conditions given by the manufacturer (Thermo Fisher Scientific). The sequencing reaction was performed on the Veriti 96-well thermal cycler using the following reagents: 0.25 μL BigDye term v3.1 mix (Thermo Fisher Scientific), 1.75 μL BigDye terminator 5x seq buffer (Thermo Fisher Scientific), 6.60 μL RNase-free water (Qiagen) and 0.4 μL primers (Bioanalytica) per 1.25 μL cleaned (sub-product free) PCR product. Sequencing products were run on an ΑBI-3500 Vs.1 (Thermo Fisher Scientific) instrument using the POP7 (Thermo Fisher Scientific), and the chromatograms were analyzed using the BioEdit sequence alignment editor v7.2.5 (Tom Hall).
 | Table S1 ACMG criteria Notes: Pathogenic criteria: PVS1, frameshift (very strong); PS4, the prevalence of the variant in affected individuals is significantly increased compared with that in controls (strong); PM1, located in a hotspot (moderate); PM2, absent from controls (moderate); PP3, computational evidence (supporting); PP4, patient’s phenotype is highly specific for a disease with a single genetic etiology (supporting). Abbreviation: ACMG, American College of Medical Genetics and Genomics. |
 | Figure S1 Represents DNA sequence analysis of exon 14. Notes: The transparent pink stripe in the wild-type sequence indicates the position of the deleted nucleotide (cytosine), whereas the transparent pink arrows in Patient 1 and her mother indicate the c.1119delC mutation. The father of Patient 1 carries the wild-type allele. |
 | Figure S2 Represents DNA sequence analysis of exon 19. Notes: The transparent blue stripe in the wild-type sequence indicates the position of the duplicated nucleotide (adenine), whereas the transparent blue arrow, in Patient 2, indicates the c.1836dupA mutation. Both parents of Patient 2 carry the wild-type allele. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.