")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 15
The Sphingosine Kinase 2 Inhibitor Opaganib Protects Against Acute Kidney Injury in Mice
Authors Maines LW, Green CL, Keller SN, Fitzpatrick LR, Smith CD
Received 18 August 2022
Accepted for publication 4 November 2022
Published 17 November 2022 Volume 2022:15 Pages 323—334
DOI https://doi.org/10.2147/IJNRD.S386396
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Lynn W Maines, Cecelia L Green, Staci N Keller, Leo R Fitzpatrick, Charles D Smith
Apogee Biotechnology Corporation, Hummelstown, PA, USA
Correspondence: Charles D Smith, Apogee Biotechnology Corporation, 1214 Research Blvd, Suite 2015, Hummelstown, PA, 17036, USA, Email [email protected]
Introduction: Acute kidney injury (AKI) is a common multifactorial adverse effect of surgery, circulatory obstruction, sepsis or drug/toxin exposure that often results in morbidity and mortality. Sphingolipid metabolism is a critical regulator of cell survival and pathologic inflammation processes involved in AKI. Opaganib (also known as ABC294640) is a first-in-class experimental drug targeting sphingolipid metabolism that reduces the production and activity of inflammatory cytokines and, therefore, may be effective to prevent and treat AKI.
Methods: Murine models of AKI were used to assess the in vivo efficacy of opaganib including ischemia-reperfusion (IR) injury induced by either transient bilateral occlusion of renal blood flow (a moderate model) or nephrectomy followed immediately by occlusion of the contralateral kidney (a severe model) and lipopolysaccharide (LPS)-induced sepsis. Biochemical and histologic assays were used to quantify the effects of oral opaganib treatment on renal damage in these models.
Results: Opaganib suppressed the elevations of creatinine and blood urea nitrogen (BUN), as well as granulocyte infiltration into the kidneys, of mice that experienced moderate IR from transient bilateral ligation. Opaganib also markedly decreased these parameters and completely prevented mortality in the severe renal IR model. Additionally, opaganib blunted the elevations of BUN, creatinine and inflammatory cytokines following exposure to LPS.
Conclusion: The data support the hypotheses that sphingolipid metabolism is a key mediator of renal inflammatory damage following IR injury and sepsis, and that this can be suppressed by opaganib. Because opaganib has already undergone clinical testing in other diseases (cancer and Covid-19), the present studies support conducting clinical trials with this drug with surgical or septic patients at risk for AKI.
Keywords: opaganib, sphingosine kinase, sphingolipid, acute kidney injury, ischemia-reperfusion injury, lipopolysaccharide, sepsis
Introduction
Acute kidney injury (AKI) is one of the most common and serious complications following major surgery or sepsis. For example, AKI occurs in up to 30% of patients who undergo cardiac surgery,1 of which there are more than 900,000 cardiac procedures performed in the United States alone each year. Mortality rates are over 20% for AKI patients following cardiac surgery2 and up to 50% in patients that require renal replacement therapy,1 with survivors needing extended stays in the intensive care unit and often continuing dialysis. The pathogenesis of AKI involves multiple pathways with overlapping hemodynamic, inflammatory, and nephrotoxic processes leading to kidney injury.1,3 AKI frequently follows ischemia-reperfusion (IR) of the kidney that occurs not only in cardiac surgery but also in other scenarios such as aortic aneurysm repair,4 trauma, organ transplantation5 and Covid-19.6 Additionally, sepsis which occurs in at least 1.7 million adults in the US per year frequently results in inflammatory damage to the kidneys resulting in more than 200,000 deaths annually. Other than supportive care medications, there are currently no drugs approved for AKI prevention or therapy. Consequently, there is a large unmet clinical need for drugs that can prevent and/or treat AKI resulting from surgery, Covid-19 or sepsis.
Inflammation is a critical component of AKI, and sphingolipids are known to regulate cellular signaling pathways in inflammation (reviewed in).7–9 Briefly, inflammatory cytokines, including TNFα and IL-6, activate sphingomyelinases that hydrolyze sphingomyelin to form ceramide which is then cleaved by ceramidases to yield sphingosine (Figure 1). Sphingosine kinases (SKs) phosphorylate the primary hydroxyl of sphingosine, forming sphingosine 1-phosphate (S1P) which promotes inflammation and cell proliferation. Two SK isozymes (SK1 and SK2) have different kinetic parameters and expression patterns in normal and diseased tissue and are therefore direct regulators of the equilibrium of ceramides, sphingosine, and S1P. Increasing attention is being paid to the roles of sphingolipid metabolism in renal pathologies (reviewed in).10–12 However, there are no approved products that modulate sphingolipid metabolism for the treatment and/or prevention of AKI.
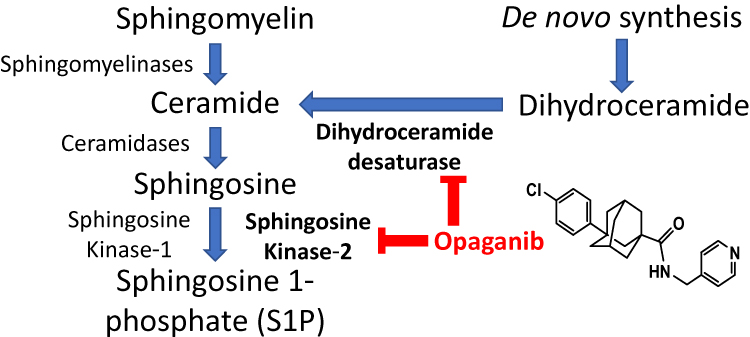 |
Figure 1 Inhibition of sphingolipid metabolism by opaganib. Opaganib (also known as ABC294640) inhibits sphingosine kinase-2 (SK2) resulting in reduced synthesis of sphingosine 1-phosphate (S1P), thereby suppressing pathologic inflammation. In parallel, opaganib also inhibits dihydroceramide desaturase (DES1) resulting in accumulation of dihydroceramides, which promotes autophagy. |
Opaganib (Figure 1) is an orally-active isozyme-selective inhibitor of SK2, and is competitive with respect to sphingosine.13,14 Opaganib depletes S1P and elevates ceramide in tumor cells, suppresses signaling through pERK, pAKT and NFκB, and promotes autophagy and/or apoptosis.13–17 Opaganib also down-regulates c-Myc in a variety of tumor cell lines.17–20 Because it acts as a sphingosine mimetic, opaganib also inhibits dihydroceramide desaturase (DES1), increasing levels of dihydroceramides18 and promoting autophagy in those cells. Opaganib has antitumor activity in a wide range of mouse models,13,17–19,21–25 as well as anti-inflammatory activity in several rodent models.26–30 Preclinical and certified Good Laboratory Procedure toxicology studies with opaganib in rodents and dogs demonstrated a lack of significant systemic toxicity, lack of hematologic suppression, and lack of organ-specific toxicity. This supported moving opaganib into clinical trials and the Investigational New Drug application was allowed by the FDA. A Phase I clinical trial with opaganib administered to patients with advanced solid tumors demonstrated that it is well-tolerated even with treatment of >40 weeks, and provided disease stabilization in most patients.31 Opaganib is currently in Phase II clinical testing in patients with cholangiocarcinoma (NCT03377179) or prostate cancer (NCT04207255). Because of its anti-inflammatory and antiviral properties, opaganib has been evaluated in hospitalized patients with Covid-19 pneumonia.32 A Phase 2a study that demonstrated that patients receiving oral opaganib required less supplemental oxygen and achieved earlier hospital discharge.33 Subsequently, a Phase 2/3 multinational randomized, placebo-controlled study enrolled 475 adult subjects hospitalized with severe Covid-19. This trial confirmed the safety of opaganib for these patients and demonstrated a clinical benefit to patients requiring lower oxygen supplementation (62% reduction in rate of ventilation and death).
Because opaganib has anti-inflammatory activity in several rodent models, we hypothesized that it will provide protection against AKI in vivo. In the present studies, we examine the ability of opaganib to suppress AKI using two well-established models in mice, specifically renal IR injury and lipopolysaccharide (LPS)-induced sepsis. These models were chosen to assess potential protective effects of opaganib against AKI induced by very different challenges representing a large number of patients with a need for improved therapies.
Materials and Methods
Chemicals and Animals
Opaganib (also known as ABC294640) was synthesized as previously described13 and dissolved in 0.375% Tween 80 in Phosphate-Buffered Saline (PBS). Lipopolysaccharide (L2030) and 3,3’,5,5’-tetramethylbenzidine (860336) were purchased from Sigma-Aldrich (St. Louis, MO, USA). C57BL/6 mice (Strain # 000664) were purchased from Jackson Labs (Bar Harbor, ME, USA), and were maintained at the Hershey Center for Applied Research (Hummelstown, PA, USA). Research was conducted under the supervision of the IACUC of the Penn State College of Medicine.
Renal Ischemia-Reperfusion Protocols
Multiple protocols for the induction of renal IR injury have been used to study AKI (reviewed in)34,35 and provide mild (unilateral renal IR), moderate (bilateral renal IR) or severe (unilateral IR with contralateral nephrectomy) pathologies. Variation among mice is highest in the mild and moderate IR models, but improves in the severe IR model.34,35 Therefore, we have used both a moderate and severe IR model in the present studies.
Moderate Model
Male C57/BL6 mice (approximately 24 g) were dosed with opaganib (50 mg/kg) or vehicle (0.1 mL in 0.375% Tween 80 in Phosphate-Buffered Saline (PBS)) by oral gavage (n = 4–6/group/experimental run), immediately followed by intraperitoneal injection of ketamine/xylazine for anesthesia. Surgery was performed on a heated surface using homeothermic pads to ensure the maintenance of animal body temperature. A midline incision was made and the two renal pedicles were located and clamped for 25 min. Total blockage of the renal pedicle and thus artery was confirmed after several minutes as the kidney was seen to be dark red to purple in color. After the scheduled time elapsed, the clamps were removed and the kidney was observed to ensure reperfusion as indicated by returning to its original color. Pre-warmed (37 °C) sterile saline (1 mL) was instilled into the peritoneum at the time of closing which used sutures for musculature and wound clips for the skin incision. Animals were maintained on the homeothermic pad until awakening from anesthesia and were carefully monitored post-operatively for morbidity according to the IACUC protocol. Control mice (“sham IR”) underwent surgery without clamping of the pedicles.
Severe Model
The severe IR model was performed in a similar manner to the moderate model with the exception that the right kidney pedicle was tied off and the kidney was removed. Immediately thereafter, the left kidney was clamped for 45 min before reperfusion was allowed. Animals were maintained on the homeothermic pad until awakening from anesthesia and were carefully monitored post-operatively for morbidity according to the IACUC protocol. Control mice (“sham IR”) underwent surgery without kidney removal or clamping of the pedicles.
Lipopolysaccharide Model of Sepsis
Male C57/BL6 mice (approximately 20 g) were dosed with opaganib (100 mg/kg) or vehicle by oral gavage 1 hr before (−1); 2 hr after (+2); or both before and after (−1, +2) intraperitoneal injection of LPS (5.0 mg/kg) (n = 4–6/group/experimental run). Animals were carefully monitored for morbidity according to the IACUC protocol. Control mice (“sham”) did not receive LPS injections.
Blood Chemistry and Cytokine Analyses
At the indicated times after completion of the IR surgery or LPS-treatment, mice were anesthetized with ketamine/xylazine and blood was harvested by cardiac puncture. The kidneys were then surgically removed and euthanasia was ensured by cervical dislocation. For the IR experiments serum chemistry values were obtained using a Roche Cobas Mira Plus automated chemistry analyzer on a fee-for-service basis from the Penn State Department of Comparative Medicine in Hershey, PA. Because of a change in instrumentation, plasma was analyzed using a VetScan VS2 Chemistry Analyzer (Zoetis, Boston, MA, USA) and Comprehensive Diagnostic Profile 2 rotors (10023220) for the LPS studies. Cytokine analyses of plasma were conducted on a Luminex 100 Multi-analyte System at the Cytokine Laboratory of the University of Maryland School of Medicine on a fee-for-service basis.
Myeloperoxidase (MPO) Assay
Kidney MPO activity was determined as a measure of granulocyte and monocytes infiltration into the kidney. At the time of collection, one half of each kidney was removed and snap-frozen in a methanol/dry ice bath and stored at −80 °C until the time of the MPO assay. The kidney samples were thawed and homogenized in 10 volumes (weight/volume) of cold PBS, and the homogenate was centrifuged at 22,000 × g for 15 min. The resulting pellet was resuspended and assayed for MPO by quantifying the metabolism of tetramethylbenzidine as described by Fitzpatrick et al.36
Kidney Histology
At the time of collection, one half of each kidney was placed in 10% formalin for at least 24 h and then dehydrated through graded alcohols. The kidney tissues were then embedded in paraffin wax and sections (5 μm) were stained with hematoxylin and eosin according to standard protocols. Kidney histology was quantitatively scored for 4–6 mice/treatment group and 4 areas/slide using a scale of 1 to 3 for each of five parameters (tubule cell swelling, tubular dilatation, edema, epithelial necrosis and tubular casts) that were added to generate a total Histology Score (range = 5–15).
Statistical Analyses
Using GraphPad (San Diego, CA, USA) Prism 5 software, survival rates were compared using the Kaplan–Meier approach with the Gehan-Breslow-Wilcoxon test. Other data shown in bar graphs were analyzed by one-way ANOVA using the Tukey post hoc test. Differences among treatment groups were considered to be statistically significant when p < 0.05.
Results
Opaganib Improves Kidney Function and Prevents Mortality Following Renal IR
Opaganib has efficacy in other inflammation models at oral doses of 50–100 mg/kg.26–30 To evaluate the effects of opaganib in the moderate IR model, mice were treated with 0 or 50 mg/kg opaganib and bilateral occlusion of the kidneys was maintained for 25 min. Ischemia-reperfusion resulted in significant increases in creatinine (Figure 2A) and BUN (Figure 2B) levels in vehicle-treated mice compared to non-IR sham animals (p < 0.01 for each). Mice treated with opaganib had substantially lower creatinine and BUN levels at 24 hr after reperfusion than did IR mice that received the vehicle. Similarly, IR promoted significant elevation of renal MPO activity (p < 0.01) reflecting granulocyte infiltration into the kidneys, and this was attenuated in IR mice that received opaganib (Figure 2C). However, because of the inherent variability of the moderate IR model,34,35 these differences between the vehicle- and opaganib-treatment groups did not reach the target level for designation as significant for creatinine (p = 0.12), BUN (p = 0.07) and MPO (p = 0.09).
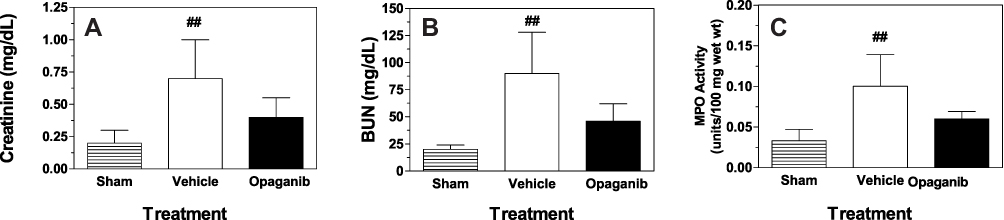 |
Figure 2 Opaganib improves renal function after moderate ischemia-reperfusion. Mice were treated with vehicle or opaganib (50 mg/kg) by oral gavage immediately prior to bilateral clamping of the kidney pedicles for 25 min. No surgery was performed in the Sham treatment group. Blood was drawn 24 h after reperfusion and assayed for serum creatinine (A) and BUN (B) levels. Additionally, kidney lysates were assayed for myeloperoxidase activity (C). Bars show the mean ± SD values for Sham-IR (n=5), vehicle-treated (n=5) and opaganib-treated (n=4) groups. ##Indicates p < 0.01 compared with the Sham treatment group. |
To reduce inter-animal variability, the effects of opaganib were evaluated using the severe IR model involving nephrectomy and immediate occlusion of the contralateral kidney for 45 min. Mice were treated with opaganib (50 mg/kg) or vehicle by oral gavage prior to severe renal IR and were carefully monitored. In this model, vehicle-treated IR mice consistently required sacrifice because of morbidity on post-surgery Day 2 (Figure 3). In marked contrast, mice that received 50 mg/kg opaganib preoperatively all appeared robust and maintained body weight until they were sacrificed on post-surgery Day 10 (p < 0.001). Blood was drawn from the mice at sacrifice (Day 2 or Day 10 for opaganib-treated mice) for BUN and creatinine analyses. Both creatinine (Figure 4A) and BUN (Figure 4B) levels were highly elevated in the vehicle-treated IR animals compared with sham IR animals (p < 0.01), indicating post-surgical renal failure. Creatinine and BUN concentrations were substantially higher, but less variable, than in the moderate IR model. In the severe IR model, animals treated with opaganib had significantly reduced serum creatinine and BUN levels at 48 hr post-IR (p < 0.05 compared with vehicle) with values returning to normal by Day 10 (p < 0.01 compared with Vehicle on Day 2). Similarly, MPO activity was elevated in vehicle-treated IR animals at 48 hr compared to sham IR controls (Figure 4C), and the increase in MPO activity was completely blocked in mice treated with opaganib (p < 0.001 compared with Vehicle).
 |
Figure 3 Opaganib promotes survival after severe ischemia-reperfusion. Mice were treated with vehicle (□, n=6) or 50 mg/kg opaganib (●, n=4) by oral gavage immediately prior to induction of severe kidney ischemia-reperfusion by ligating and removing the right kidney and then clamping the left kidney for 45 min. Mice were monitored for morbidity daily, and the percentages of surviving animals are shown. **Indicates p < 0.01 compared with the Vehicle treatment group. |
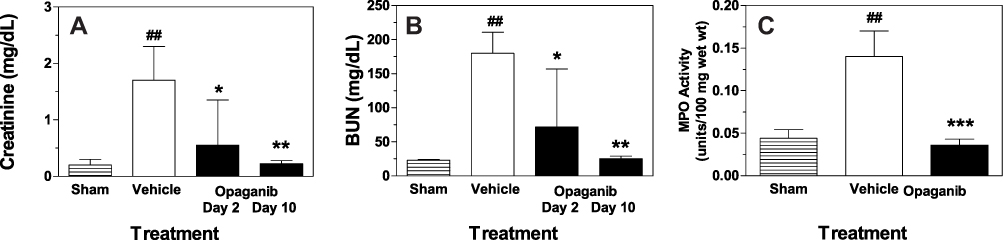 |
Figure 4 Opaganib improves renal function after severe ischemia-reperfusion. Mice were treated with vehicle or 50 mg/kg opaganib by oral gavage immediately prior to induction of severe kidney ischemia-reperfusion. No surgery was performed in the Sham treatment group. Serum levels of creatinine (A) and BUN (B) were measured on Day 2 or Day 10 (opaganib treatment group only). Kidneys were harvested on Day 2 for myeloperoxidase (MPO) assays (C). ##Indicates p < 0.01 compared with the Sham treatment group. *Indicates p < 0.05, **Indicates p < 0.01, and ***Indicates p < 0.001 compared with Vehicle treatment group. |
Renal thin sections were stained with hematoxylin/eosin and quantitatively scored on a scale of 1 to 3 for five parameters (tubule cell swelling, tubular dilatation, edema, epithelial necrosis and tubular casts) that were added to generate a total Histology Score. Kidneys from animals treated with opaganib had less morphologic damage (Figure 5A) and lower Histology Scores (Figure 5B, p < 0.001) than kidneys from vehicle-treated IR mice at 48 hr after reperfusion. An excellent correlation was observed between individual mouse serum creatinine level and Histology Score (r = 0.7556) for this relationship, suggesting a significant association between the two parameters (p <0.01) (Figure 5C). Overall, opaganib significantly improved kidney function and animal survival after IR surgery in this severe model of AKI.
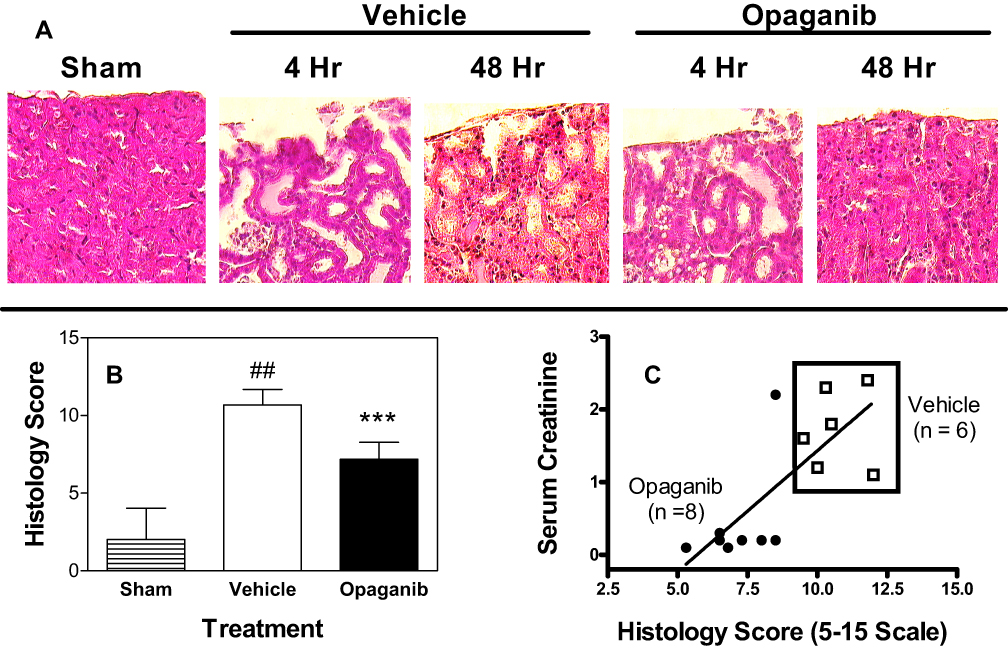 |
Figure 5 Opaganib attenuates histologic renal damage from severe ischemia-reperfusion. Mice were treated with vehicle or 50 mg/kg opaganib by oral gavage immediately prior to induction of severe kidney ischemia-reperfusion. No surgery was performed in the Sham treatment group. At 4 or 48 hr after reperfusion, mice were sacrificed and kidneys were harvested, fixed, sectioned and stained with hematoxylin and eosin. (A) Representative sections are shown. (B) Histology Scores were calculated as described in the Materials and Methods section. Bars show the mean ± SD values for Sham (n=5), vehicle (n=6) and opaganib (n=8) treatment groups. ##Indicates p < 0.01 compared with the Sham treatment group. ***Indicates p < 0.001 compared with the Vehicle treatment group. (C) To assess the consistency of the data, serum creatinine and Histology Score values are shown for individual mice from the vehicle (□) and opaganib (●) treatment groups. |
Opaganib Improves Kidney Function and Suppresses Inflammatory Cytokines in the LPS-Induced Sepsis Model
Lipopolysaccharide injection is widely used as a preclinical model for sepsis, resulting in transient AKI that is mediated by TLR-4-induced systemic expression of inflammatory cytokines including TNFα and IL-6. To evaluate the effects of opaganib in a sepsis model, mice were treated orally with 0 or 100 mg/kg opaganib before and/or after the administration of LPS (5 mg/kg). As shown in Figure 6A, LPS-treatment resulted in a significant increase in BUN levels at 24 hr in vehicle-treated mice compared with sham (non-LPS) animals of this model (p < 0.01). Pretreatment of mice with opaganib before LPS administration significantly suppressed the increase in BUN levels (p < 0.05). Treatment with opaganib after LPS-treatment substantially lowered BUN levels but the decrease did not reach the threshold of significance (p = 0.11). Creatinine levels were not significantly elevated after LPS-treatment; however, IL-1β (Figure 6B), IL-6 (Figure 6C) and TNFα (Figure 6D) were all significantly induced by LPS (p < 0.01). Increased expression of these inflammatory proteins was generally reduced in mice treated with opaganib, particularly when opaganib was administered both before and after LPS-treatment.
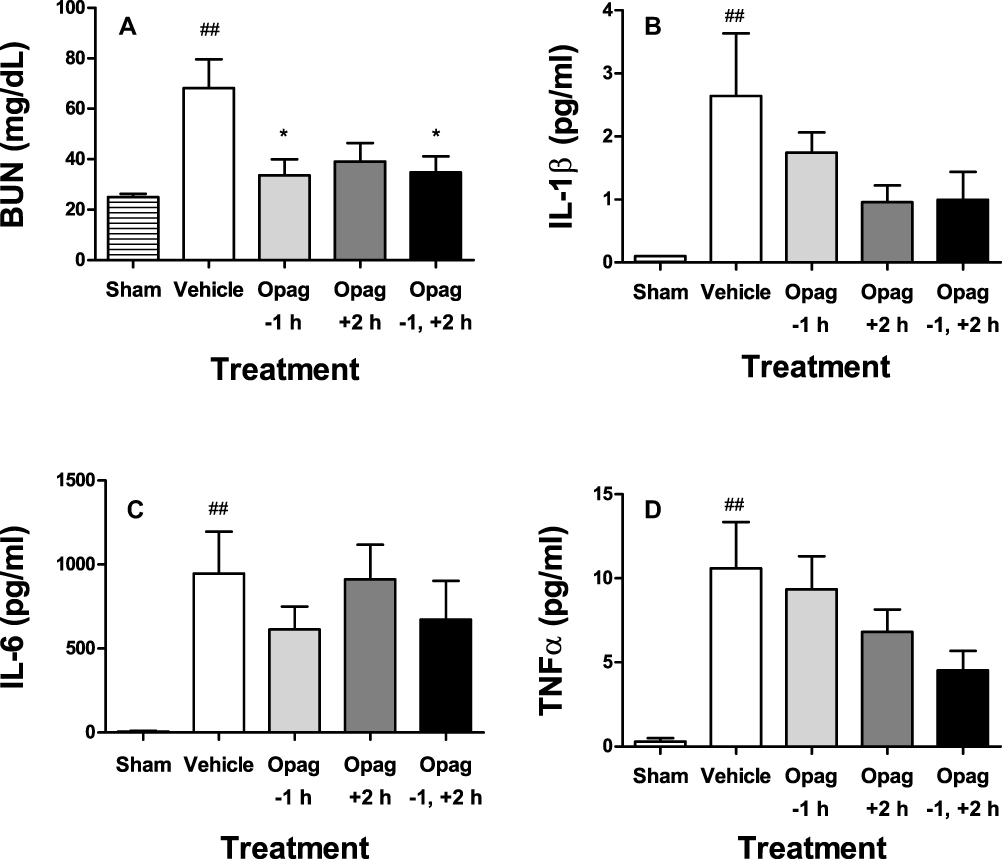 |
Figure 6 Opaganib preserves renal function and suppresses inflammation in lipopolysaccharide-induced sepsis. Mice were treated with opaganib (100 mg/kg) or vehicle by oral gavage at 1 hr before and/or 2 hr after lipopolysaccharide (LPS) injection. No LPS was administered in the Sham treatment group. Blood was drawn 24 hr after LPS administration and assayed for BUN (A), IL-1β (B), IL-6 (C) and TNFα (D). Bars show the mean ± SD values for n=4–6 mice per treatment group. ##Indicates p < 0.01 compared with the Sham treatment group. *Indicates p < 0.05 compared with the Vehicle treatment group. |
Discussion
It is well established that sphingolipid metabolism regulates pathologic inflammation underlying several disease states, and in particular, many studies have shown that SK activity regulates inflammation.7,12,37 Sphingosine 1-phosphate induces NFκB,38 which in turn can increase the proinflammatory enzymes nitric oxide synthase and cyclooxygenase-239–41 generating prostaglandins and oxidative and nitrative stress mediated that exacerbate inflammation.42 Inflammatory cytokines induce adhesion molecule expression via activation of SK and NFκB, and these proteins facilitate infiltration of neutrophils and macrophages into inflamed tissue. Sphingosine 1-phosphate promotes granulocyte activation leading to production of reactive oxygen species43,44 and activates S1P receptors further promoting inflammatory cascades at the site of tissue damage.45–47 Overall, a strong body of evidence demonstrates that activation of SKs alters sphingolipid metabolism in favor of S1P formation, resulting in pro-inflammatory responses.
More specific to the current studies, the roles of sphingolipids in IR injury to the kidneys have received attention,10–12,48 largely because of their direct regulation of inflammation and ROS generation.11,49–51 Altered sphingolipid metabolism in response to renal ischemia was first demonstrated in 1997.52 Plasma creatinine levels53 and pulmonary permeability and injury54 following renal IR were significantly lower in S1P receptor 3−/− mice compared with control animals, suggesting the importance of excessive S1P in IR injury. Supporting this, the S1P receptor superagonist FTY720 prevents lymphocyte egress from lymph nodes and thereby suppresses T cell influx into the kidney following IR.55,56 In parallel, roles for sphingolipid metabolism in sepsis have been studied since 19979,57–62 and generally focus on regulation of NFκB-induced cytokine production.
The anti-inflammatory activity of opaganib has been shown in several in vivo models26–28,30,63 and extensive preclinical toxicology studies demonstrated it does not have significant systemic, organ-specific or hematologic toxicity. In models of inflammatory bowel disease, opaganib decreased colonic levels of TNFα, IL-1β, IFN-γ and IL-6, and reduced S1P levels in colon tissue.28 In liver IR injury, opaganib reduced TLR4 expression, NFκB activation, TNFα, IL-1β and CXCL-10 production, adhesion molecule expression, and liver infiltration of granulocytes.64 Opaganib nearly completely ameliorated Pseudomonas aeruginosa (PA)-induced lung injury in mice, associated with decreased inflammatory cell and protein accumulation in the bronchoalveolar lavage fluid, and suppression of PA-induced elevation of IL-6, TNFα and H2O2.65
In studies described herein, we tested the ability of opaganib to suppress AKI in two well-established mouse models of renal IR injury and in LPS-induced sepsis. In the moderate IR model (bilateral renal IR), opaganib attenuated increases in creatinine, BUN and renal granulocyte infiltration; however, the greater variability in this AKI model prevented the effects from reaching a significance level of >95% (p values were 0.07–0.12). Therefore, we switched to a severe model consisting of contralateral nephrectomy immediately prior to renal IR for 45 min. As a severe model of disease, this protocol is expected to require a high level of efficacy for a drug to mitigate the renal damage, and so provides a rigorous evaluation of its potential clinical usefulness. Orally-administered opaganib suppressed the elevation of clinical markers of AKI following severe renal IR in association with decreased neutrophil infiltration into the kidney and reduced histologic damage. In this model of severe AKI, untreated mice all died within 2 days of renal IR; whereas, all opaganib-treated mice survived the IR insult long-term. Similarly, elevations of BUN, IL-1β, IL-6 and TNFα were suppressed by opaganib in the LPS-induced sepsis model. These effects are consistent with previous studies demonstrating that activation of NFκB by TNFα in vitro is dose-dependently suppressed by opaganib,30 and TNFα-induction of adhesion proteins (VCAM-1 and ICAM) involved in leukocyte recruitment and production of PGE2 are strongly suppressed by opaganib.28 Opaganib also suppresses titanium particle-induced TNFα and IL-6 production in RAW264.7 macrophages,66 and inhibits extracellular matrix deposition in human kidney fibroblasts.67
Our focus in these initial studies was to determine if opaganib has an effect on clinically relevant endpoints indicative of AKI. With that confirmation in hand, we are pursuing additional biochemical studies that should reveal the mechanistic basis for the therapeutic effects of opaganib. Multi-organ studies characterizing the pharmacokinetics and pharmacodynamics of opaganib include pharmacokinetic and sphingolipid analyses of kidneys from opaganib-treated normal C57BL/6 mice. These analyses demonstrated that opaganib reaches pharmacologically active concentrations (24 μg/g tissue or ~63 μM) in the kidney at 2 hr after oral dosing with a half-time of elimination of approximately 16 hr. This exposure to opaganib was associated with transient decreases in renal levels of S1P and increases in sphingosine and dihydroceramide, indicating in situ inhibition of SK2 and DES1.
Mechanistically, several key biochemical pathways involved in AKI are modulated by sphingolipids, and in some cases demonstrated to be suppressed by opaganib. First, signaling through pAkt is required for the pathology of AKI,68–71 and Akt inhibitors attenuate the excessive inflammation, cytokine storm, fibrosis, and thrombosis in AKI patients.72 We and others have demonstrated that opaganib efficiently inhibits Akt phosphorylation in multiple cell types, and so inhibition of Akt may underlie the ability of opaganib to suppress AKI. Second, c-Myc expression and activity is pathologic following IR.73–75 Similarly, LPS induces c-Myc,76–78 which may underlie its pro-apoptotic actions.79 Opaganib decreases the expression of c-Myc,17–20,80,81 likely due to depletion of nuclear S1P destabilizing the c-Myc protein.82 Therefore, inhibition of c-Myc may underlie the ability of opaganib to suppress AKI. Finally, a large body of evidence demonstrates that activation of autophagy suppresses AKI, leading to the suggestion that autophagy-inducing agents may effectively prevent and/or treat AKI (reviewed in).83–86 It is well established that: dihydroceramides induce autophagy;87–93 opaganib elevates dihydroceramides by inhibition of DES1;18,94,95 and opaganib promotes autophagy.15,96,97 Therefore, inhibition of DES1 may be involved in the ability of opaganib to suppress AKI.
Overall, sphingolipid metabolism is an emerging target for the prevention and/or treatment of AKI, and the present studies provide the first demonstration that the investigational drug opaganib protects against renal damage in two complementary models of AKI. The completed and ongoing clinical studies of opaganib demonstrate that oral opaganib is well-tolerated even by severely compromised cancer and Covid-19 patients, with treatment periods as long as 8 months for some patients. We envision that opaganib could be administered to patients at risk for developing AKI, eg in patients undergoing cardiovascular or other major surgery where periods of renal IR are expected. Similarly, trauma patients or military personnel experiencing hypovolemia or at risk for sepsis from gastrointestinal injury may benefit from opaganib treatment. Of additional interest is the potential for opaganib to mitigate sepsis from bacterial or viral infection, including SARS-CoV-2 and/or influenza viruses.
Abbreviations
AKI, Acute Kidney Injury; DES1, dihydroceramide desaturase 1; IR, ischemia-reperfusion; LPS, lipopolysaccharide; MPO, myeloperoxidase; PBS, phosphate-buffered saline; SK, sphingosine kinase; S1P, sphingosine 1-phosphate.
Ethical Statements
All protocols in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of the Penn State University College of Medicine, in compliance with US Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research and Training, which complies with the PHS Policy on Humane Care and Use of Laboratory Animals, and the United States Department of Agriculture Animal Welfare Act guidelines.
Funding
All studies were funded internally by Apogee Biotechnology Corporation.
Disclosure
Lynn W. Maines, Staci N. Keller and Charles D. Smith are current employees and own stock in Apogee Biotechnology Corporation. Apogee Biotechnology Corporation owns patent rights to opaganib (United States Patent numbers 7,338,961, 8,063,248, 8,324,237 and 8,557,800, and multiple parallel patents in other countries) that have been licensed to RedHIll Biopharma LTD. The value of these rights may be affected by the research reported in this paper. The authors report no other conflicts of interest in this work.
References
1. Lagny MG, Jouret F, Koch JN, et al. Incidence and outcomes of acute kidney injury after cardiac surgery using either criteria of the RIFLE classification. BMC Nephrol. 2015;16:76. doi:10.1186/s12882-015-0066-9
2. Boldt J, Brenner T, Lehmann A, Suttner SW, Kumle B, Isgro F. Is kidney function altered by the duration of cardiopulmonary bypass? Ann Thorac Surg. 2003;75(3):906–912. doi:10.1016/S0003-4975(02)04559-9
3. Rosner MH, Okusa MD. Acute kidney injury associated with cardiac surgery. Clin J Am Soc Nephrol. 2006;1(1):19–32. doi:10.2215/CJN.00240605
4. Chin J, Palop JJ, Puolivali J, et al. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2005;25(42):9694–9703. doi:10.1523/JNEUROSCI.2980-05.2005
5. Koo DD, Welsh KI, Roake JA, Morris PJ, Fuggle SV. Ischemia/reperfusion injury in human kidney transplantation: an immunohistochemical analysis of changes after reperfusion. Am J Pathol. 1998;153(2):557–566. doi:10.1016/S0002-9440(10)65598-8
6. Menez S, Parikh CR. Overview of acute kidney manifestations and management of patients with COVID-19. Am J Physiol. 2021;321(4):F403–F410.
7. Gomez-Larrauri A, Presa N, Dominguez-Herrera A, Ouro A, Trueba M, Gomez-Munoz A. Role of bioactive sphingolipids in physiology and pathology. Essays Biochem. 2020;64(3):579–589. doi:10.1042/EBC20190091
8. Hu Y, Dai K. Sphingosine 1-phosphate metabolism and signaling. Adv Exp Med Biol. 2022;1372:67–76.
9. Ziegler AC, Muller T, Graler MH. Sphingosine 1-phosphate in sepsis and beyond: its role in disease tolerance and host defense and the impact of carrier molecules. Cell Signal. 2021;78:109849. doi:10.1016/j.cellsig.2020.109849
10. Abou Daher A, El Jalkh T, Eid AA, Fornoni A, Marples B, Zeidan YH. Translational aspects of sphingolipid metabolism in renal disorders. Int J Mol Sci. 2017;18(12):2528. doi:10.3390/ijms18122528
11. Dupre TV, Siskind LJ. The role of sphingolipids in acute kidney injury. Adv Biol Regul. 2018;70:31–39. doi:10.1016/j.jbior.2018.11.003
12. Ueda N. A rheostat of ceramide and sphingosine-1-phosphate as a determinant of oxidative stress-mediated kidney injury. Int J Mol Sci. 2022;23(7):4010. doi:10.3390/ijms23074010
13. French KJ, Zhuang Y, Maines LW, et al. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther. 2010;333(1):129–139. doi:10.1124/jpet.109.163444
14. Gao P, Peterson YK, Smith RA, Smith CD. Characterization of isoenzyme-selective inhibitors of human sphingosine kinases. PLoS One. 2012;7(9):e44543. doi:10.1371/journal.pone.0044543
15. Beljanski V, Knaak C, Smith CD. A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J Pharmacol Exp Ther. 2010;333(2):454–464. doi:10.1124/jpet.109.163337
16. Antoon JW, White MD, Slaughter EM, et al. Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol Ther. 2011;11(7):678–689. doi:10.4161/cbt.11.7.14903
17. Schrecengost RS, Keller SN, Schiewer MJ, Knudsen KE, Smith CD. Downregulation of critical oncogenes by the selective SK2 inhibitor ABC294640 hinders prostate cancer progression. Mol Cancer Res. 2015;13(12):1591–1601. doi:10.1158/1541-7786.MCR-14-0626
18. Venant H, Rahmaniyan M, Jones EE, et al. The sphingosine kinase 2 inhibitor ABC294640 reduces the growth of prostate cancer cells and results in accumulation of dihydroceramides in vitro and in vivo. Mol Cancer Ther. 2015;14(12):2744–2752. doi:10.1158/1535-7163.MCT-15-0279
19. Venkata JK, An N, Stuart R, et al. Inhibition of sphingosine kinase 2 downregulates the expression of c-Myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood. 2014;124(12):1915–1925. doi:10.1182/blood-2014-03-559385
20. Lewis CS, Voelkel-Johnson C, Smith CD. Suppression of c-Myc and RRM2 expression in pancreatic cancer cells by the sphingosine kinase-2 inhibitor ABC294640. Oncotarget. 2016;7(37):60181–60192. doi:10.18632/oncotarget.11112
21. Antoon JW, White MD, Meacham WD, et al. Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology. 2010;151(11):5124–5135. doi:10.1210/en.2010-0420
22. Beljanski V, Knaak C, Zhuang Y, Smith CD. Combined anticancer effects of sphingosine kinase inhibitors and sorafenib. Invest New Drugs. 2011;29(6):1132–1142. doi:10.1007/s10637-010-9452-0
23. Beljanski V, Lewis CS, Smith CD. Antitumor activity of sphingosine kinase 2 inhibitor ABC294640 and sorafenib in hepatocellular carcinoma xenografts. Cancer Biol Ther. 2011;11(5):524–534. doi:10.4161/cbt.11.5.14677
24. Qin Z, Dai L, Trillo-Tinoco J, et al. Targeting sphingosine kinase induces apoptosis and tumor regression for KSHV-associated primary effusion lymphoma. Mol Cancer Ther. 2014;13(1):154–164. doi:10.1158/1535-7163.MCT-13-0466
25. Xun C, Chen MB, Qi L, et al. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J Exp Clin Cancer Res. 2015;34:94. doi:10.1186/s13046-015-0205-y
26. Fitzpatrick LR, Green CL, Maines LW, Smith CD. Experimental osteoarthritis in rats is attenuated by ABC294640, a selective inhibitor of sphingosine kinase-2. Pharmacology. 2011;87:135–143. doi:10.1159/000323911
27. Fitzpatrick LR, Green CL, Frauenhoffer EE, et al. Attenuation of arthritis in rodents by a novel orally-available inhibitor of sphingosine kinase. Inflammopharmacology. 2010;19:75–87. doi:10.1007/s10787-010-0060-6
28. Maines LW, Fitzpatrick LR, French KJ, et al. Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig Dis Sci. 2008;53(4):997–1012. doi:10.1007/s10620-007-0133-6
29. Maines LW, Fitzpatrick LR, Green CL, Zhuang Y, Smith CD. Efficacy of a novel sphingosine kinase inhibitor in experimental Crohn’s disease. Inflammopharmacology. 2010;18(2):73–85. doi:10.1007/s10787-010-0032-x
30. Maines LW, French KJ, Wolpert EB, Antonetti DA, Smith CD. Pharmacologic manipulation of sphingosine kinase in retinal endothelial cells: implications for angiogenic ocular diseases. Invest Ophthalmol Vis Sci. 2006;47(11):5022–5031. doi:10.1167/iovs.05-1236
31. Britten CD, Garrett-Mayer E, Chin SH, et al. A phase I study of ABC294640, a first-in-class sphingosine kinase-2 inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2017;23(16):4642–4650. doi:10.1158/1078-0432.CCR-16-2363
32. Smith CD, Maines LW, Keller SN, et al. Recent progress in the development of opaganib for the treatment of Covid-19. Drug Des Devel Ther. 2022;16:2199–2211. doi:10.2147/DDDT.S367612
33. Winthrop KL, Skolnick AW, Rafiq AM, et al. Opaganib in COVID-19 pneumonia: results of a randomized, placebo-controlled Phase 2a trial. Open Forum Infect Dis. 2022;9:ofac232. doi:10.1093/ofid/ofac232
34. Wei Q, Dong Z. Mouse model of ischemic acute kidney injury: technical notes and tricks. Am J Physiol. 2012;303(11):F1487–1494. doi:10.1152/ajprenal.00352.2012
35. Pabla N, Scindia Y, Gigliotti J, Bajwa A. Mouse models of acute kidney injury. In: Purevjav E, editor. Preclinical Animal Modeling in Medicine. IntechOpen; 2022:1–19.
36. Fitzpatrick LR, Wang J, Le T. In vitro and in vivo effects of gliotoxin, a fungal metabolite: efficacy against dextran sodium sulfate-induced colitis in rats. Dig Dis Sci. 2000;45(12):2327–2336. doi:10.1023/A:1005630723111
37. Pyne NJ, Adams DR, Pyne S. Sphingosine kinase 2 in autoimmune/inflammatory disease and the development of sphingosine kinase 2 inhibitors. Trends Pharmacol Sci. 2017;38(7):581–591. doi:10.1016/j.tips.2017.04.003
38. Xia P, Gamble JR, Rye KA, et al. Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci U S A. 1998;95(24):14196–14201. doi:10.1073/pnas.95.24.14196
39. Igarashi J, Bernier SG, Michel T. Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase. differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. J Biol Chem. 2001;276(15):12420–12426. doi:10.1074/jbc.M008375200
40. Igarashi J, Michel T. Sphingosine 1-phosphate and isoform-specific activation of phosphoinositide 3-kinase beta. Evidence for divergence and convergence of receptor-regulated endothelial nitric-oxide synthase signaling pathways. J Biol Chem. 2001;276(39):36281–36288. doi:10.1074/jbc.M105628200
41. Pettus BJ, Bielawski J, Porcelli AM, et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-alpha. FASEB J. 2003;17(11):1411–1421. doi:10.1096/fj.02-1038com
42. Kruidenier L, Kuiper I, Lamers CB, Verspaget HW. Intestinal oxidative damage in inflammatory bowel disease: semi-quantification, localization, and association with mucosal antioxidants. J Pathol. 2003;201(1):28–36. doi:10.1002/path.1409
43. Itagaki K, Hauser CJ. Sphingosine 1-phosphate, a diffusible calcium influx factor mediating store-operated calcium entry. J Biol Chem. 2003;278(30):27540–27547. doi:10.1074/jbc.M301763200
44. MacKinnon AC, Buckley A, Chilvers ER, Rossi AG, Haslett C, Sethi T. Sphingosine kinase: a point of convergence in the action of diverse neutrophil priming agents. J Immunol. 2002;169(11):6394–6400. doi:10.4049/jimmunol.169.11.6394
45. Stepanovska B, Huwiler A. Targeting the S1P receptor signaling pathways as a promising approach for treatment of autoimmune and inflammatory diseases. Pharmacol Res. 2020;154:104170. doi:10.1016/j.phrs.2019.02.009
46. Sukocheva OA, Furuya H, Ng ML, et al. Sphingosine kinase and sphingosine-1-phosphate receptor signaling pathway in inflammatory gastrointestinal disease and cancers: a novel therapeutic target. Pharmacol Ther. 2020;207:107464. doi:10.1016/j.pharmthera.2019.107464
47. Wang J, Goren I, Yang B, et al. Review article: the sphingosine 1 phosphate/sphingosine 1 phosphate receptor axis - a unique therapeutic target in inflammatory bowel disease. Aliment Pharmacol Ther. 2022;55(3):277–291. doi:10.1111/apt.16741
48. Zhang X, Ritter JK, Li N. Sphingosine-1-phosphate pathway in renal fibrosis. Am J Physiol. 2018;315(4):F752–F756. doi:10.1152/ajprenal.00596.2017
49. Bajwa A, Rosin DL, Chroscicki P, et al. Sphingosine 1-phosphate receptor-1 enhances mitochondrial function and reduces cisplatin-induced tubule injury. J Am Soc Nephrol. 2015;26(4):908–925. doi:10.1681/ASN.2013121351
50. Dupre TV, Doll MA, Shah PP, et al. Inhibiting glucosylceramide synthase exacerbates cisplatin-induced acute kidney injury. J Lipid Res. 2017;58(7):1439–1452. doi:10.1194/jlr.M076745
51. Sears SM, Dupre TV, Shah PP, et al. Neutral ceramidase deficiency protects against cisplatin-induced acute kidney injury. J Lipid Res. 2022;63(3):100179. doi:10.1016/j.jlr.2022.100179
52. Zager RA, Iwata M, Conrad DS, Burkhart KM, Igarashi Y. Altered ceramide and sphingosine expression during the induction phase of ischemic acute renal failure. Kidney Int. 1997;52(1):60–70. doi:10.1038/ki.1997.304
53. Jo SK, Bajwa A, Ye H, et al. Divergent roles of sphingosine kinases in kidney ischemia-reperfusion injury. Kidney Int. 2008;75:167–175.
54. Gon Y, Wood MR, Kiosses WB, et al. S1P3 receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity and is potentiated by TNF. Proc Natl Acad Sci U S A. 2005;102(26):9270–9275. doi:10.1073/pnas.0501997102
55. Fuller TF, Hoff U, Kong L, et al. Cytoprotective actions of FTY720 modulate severe preservation reperfusion injury in RatRenal transplants. Transplantation. 2010;89(4):402–408. doi:10.1097/TP.0b013e3181caa499
56. Kaudel CP, Schmiddem U, Frink M, et al. FTY720 for treatment of ischemia-reperfusion injury following complete renal ischemia in C57/BL6 mice. Transplant Proc. 2006;38(3):679–681. doi:10.1016/j.transproceed.2006.01.033
57. Claus RA, Bunck AC, Bockmeyer CL, et al. Role of increased sphingomyelinase activity in apoptosis and organ failure of patients with severe sepsis. FASEB J. 2005;19(12):1719–1721. doi:10.1096/fj.04-2842fje
58. Drobnik W, Liebisch G, Audebert FX, et al. Plasma ceramide and lysophosphatidylcholine inversely correlate with mortality in sepsis patients. J Lipid Res. 2003;44(4):754–761. doi:10.1194/jlr.M200401-JLR200
59. Graler MH. The role of sphingosine 1-phosphate in immunity and sepsis. Am J Clin Exp Immunol. 2012;1(2):90–100.
60. Haimovitz-Friedman A, Cordon-Cardo C, Bayoumy S, et al. Lipopolysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J Exp Med. 1997;186(11):1831–1841. doi:10.1084/jem.186.11.1831
61. Lo CJ, Fu M, Lo FR, Cryer HG. Macrophage TNF mRNA expression induced by LPS is regulated by sphingomyelin metabolites. Shock. 1999;11(6):411–415.
62. Peng H, Li C, Kadow S, et al. Acid sphingomyelinase inhibition protects mice from lung edema and lethal Staphylococcus aureus sepsis. J Mol Med. 2015;93(6):675–689. doi:10.1007/s00109-014-1246-y
63. Smith CD, Berk SG, Brandl MT, Riley LW. Survival characteristics of diarrheagenic Escherichia coli pathotypes and Helicobacter pylori during passage through the free-living ciliate, Tetrahymena sp. FEMS Microbiol Ecol. 2012;82(3):574–583. doi:10.1111/j.1574-6941.2012.01428.x
64. Liu Q, Rehman H, Shi Y, et al. Inhibition of sphingosine kinase-2 suppresses inflammation and attenuates graft injury after liver transplantation in rats. PLoS One. 2012;7(7):e41834. doi:10.1371/journal.pone.0041834
65. Ebenezer DL, Berdyshev EV, Bronova IA, et al. Pseudomonas aeruginosa stimulates nuclear sphingosine-1-phosphate generation and epigenetic regulation of lung inflammatory injury. Thorax. 2019;74(6):579–591. doi:10.1136/thoraxjnl-2018-212378
66. Yang G, Gu M, Chen W, et al. SPHK-2 promotes the particle-induced inflammation of RAW264.7 by maintaining consistent expression of TNF-alpha and IL-6. Inflammation. 2018;41(4):1498–1507. doi:10.1007/s10753-018-0795-6
67. Zhu X, Shi D, Cao K, et al. Sphingosine kinase 2 cooperating with Fyn promotes kidney fibroblast activation and fibrosis via STAT3 and AKT. Biochim Biophys Acta. 2018;1864(11):3824–3836. doi:10.1016/j.bbadis.2018.09.007
68. Kim IY, Park YK, Song SH, et al. Akt1 is involved in tubular apoptosis and inflammatory response during renal ischemia-reperfusion injury. Mol Biol Rep. 2020;47(12):9511–9520. doi:10.1007/s11033-020-06021-1
69. Sang Z, Dong S, Zhang P, Wei Y. miR214 ameliorates sepsis-induced acute kidney injury via PTEN/AKT/mTORregulated autophagy. Mol Med Rep. 2021;24(4). doi:10.3892/mmr.2021.12322
70. Wang H, Wang Y, Wang X, et al. PTEN alleviates maladaptive repair of renal tubular epithelial cells by restoring CHMP2A-mediated phagosome closure. Cell Death Dis. 2021;12(12):1087. doi:10.1038/s41419-021-04372-6
71. Zhan Y, Zhu M, Liu S, et al. MicroRNA93 inhibits the apoptosis and inflammatory response of tubular epithelial cells via the PTEN/AKT/mTOR pathway in acute kidney injury. Mol Med Rep. 2021;24(3). doi:10.3892/mmr.2021.12305
72. Somanath PR. Is targeting Akt a viable option to treat advanced-stage COVID-19 patients? Am J Physiol. 2020;319(1):L45–L47. doi:10.1152/ajplung.00124.2020
73. Hultstrom M, Becirovic-Agic M, Jonsson S. Comparison of acute kidney injury of different etiology reveals in-common mechanisms of tissue damage. Physiol Genomics. 2018;50(3):127–141. doi:10.1152/physiolgenomics.00037.2017
74. Liu D, Liu Y, Zheng X, Liu N. c-MYC-induced long noncoding RNA MEG3 aggravates kidney ischemia-reperfusion injury through activating mitophagy by upregulation of RTKN to trigger the Wnt/beta-catenin pathway. Cell Death Dis. 2021;12(2):191. doi:10.1038/s41419-021-03466-5
75. Xu D, Wang B, Chen PP, et al. c-Myc promotes tubular cell apoptosis in ischemia-reperfusion-induced renal injury by negatively regulating c-FLIP and enhancing FasL/Fas-mediated apoptosis pathway. Acta Pharmacol Sin. 2019;40(8):1058–1066. doi:10.1038/s41401-018-0201-9
76. Introna M, Hamilton TA, Kaufman RE, Adams DO, Bast RC. Treatment of murine peritoneal macrophages with bacterial lipopolysaccharide alters expression of c-fos and c-myc oncogenes. J Immunol. 1986;137(8):2711–2715.
77. Yao D, Dong Q, Tian Y, Dai C, Wu S. Lipopolysaccharide stimulates endogenous beta-glucuronidase via PKC/NF-kappaB/c-myc signaling cascade: a possible factor in hepatolithiasis formation. Mol Cell Biochem. 2018;444(1–2):93–102. doi:10.1007/s11010-017-3234-3
78. Zhang Y, Huang T, Jiang L, et al. MCP-induced protein 1 attenuates sepsis-induced acute lung injury by modulating macrophage polarization via the JNK/c-Myc pathway. Int Immunopharmacol. 2019;75:105741. doi:10.1016/j.intimp.2019.105741
79. Li B, Zhao Y, Song M, et al. Role of c-Myc/chloride intracellular channel 4 pathway in lipopolysaccharide-induced neurodegenerative diseases. Toxicology. 2020;429:152312. doi:10.1016/j.tox.2019.152312
80. Song K, Dai L, Long X, Cui X, Liu Y, Di W. Sphingosine kinase 2 inhibitor ABC294640 displays anti-epithelial ovarian cancer activities in vitro and in vivo. Onco Targets Ther. 2019;12:4437–4449. doi:10.2147/OTT.S208519
81. Sundaramoorthy P, Gasparetto C, Kang Y. The combination of a sphingosine kinase 2 inhibitor (ABC294640) and a Bcl-2 inhibitor (ABT-199) displays synergistic anti-myeloma effects in myeloma cells without a t(11;14) translocation. Cancer Med. 2018;7:3257–3268. doi:10.1002/cam4.1543
82. Lewis CS, Voelkel-Johnson C, Smith CD. Targeting sphingosine kinases for the treatment of cancer. Adv Cancer Res. 2018;140:295–325.
83. Bhatia D, Choi ME. Autophagy in kidney disease: advances and therapeutic potential. Prog Mol Biol Transl Sci. 2020;172:107–133.
84. Cui J, Bai X, Chen X. Autophagy and acute kidney injury. Adv Exp Med Biol. 2020;1207:469–480.
85. Duann P, Lianos EA, Ma J, Lin PH. Autophagy, innate immunity and tissue repair in acute kidney injury. Int J Mol Sci. 2016;17(5):662. doi:10.3390/ijms17050662
86. Gong L, Pan Q, Yang N. Autophagy and inflammation regulation in acute kidney injury. Front Physiol. 2020;11:576463. doi:10.3389/fphys.2020.576463
87. Casasampere M, Ordonez YF, Casas J, Fabrias G. Dihydroceramide desaturase inhibitors induce autophagy via dihydroceramide-dependent and independent mechanisms. Biochim Biophys Acta. 2017;1861(2):264–275. doi:10.1016/j.bbagen.2016.11.033
88. Gagliostro V, Casas J, Caretti A, et al. Dihydroceramide delays cell cycle G1/S transition via activation of ER stress and induction of autophagy. Int J Biochem Cell Biol. 2012;44(12):2135–2143. doi:10.1016/j.biocel.2012.08.025
89. Hernandez-Tiedra S, Fabrias G, Davila D, et al. Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy. 2016;12(11):2213–2229. doi:10.1080/15548627.2016.1213927
90. Lee AY, Lee JW, Kim JE, et al. Dihydroceramide is a key metabolite that regulates autophagy and promotes fibrosis in hepatic steatosis model. Biochem Biophys Res Commun. 2017;494(3–4):460–469. doi:10.1016/j.bbrc.2017.10.110
91. Munoz-Guardiola P, Casas J, Megias-Roda E, et al. The anti-cancer drug ABTL0812 induces ER stress-mediated cytotoxic autophagy by increasing dihydroceramide levels in cancer cells. Autophagy. 2021;17(6):1349–1366. doi:10.1080/15548627.2020.1761651
92. Signorelli P, Munoz-Olaya JM, Gagliostro V, Casas J, Ghidoni R, Fabrias G. Dihydroceramide intracellular increase in response to resveratrol treatment mediates autophagy in gastric cancer cells. Cancer Lett. 2009;282(2):238–243. doi:10.1016/j.canlet.2009.03.020
93. Wu CY, Jhang JG, Lin WS, et al. Dihydroceramide desaturase promotes the formation of intraluminal vesicles and inhibits autophagy to increase exosome production. iScience. 2021;24(12):103437. doi:10.1016/j.isci.2021.103437
94. McNaughton M, Pitman M, Pitson SM, Pyne NJ, Pyne S. Proteasomal degradation of sphingosine kinase 1 and inhibition of dihydroceramide desaturase by the sphingosine kinase inhibitors, SKi or ABC294640, induces growth arrest in androgen-independent LNCaP-AI prostate cancer cells. Oncotarget. 2016;7(13):16663–16675. doi:10.18632/oncotarget.7693
95. Shin SH, Kim HY, Yoon HS, et al. A novel selective sphingosine kinase 2 inhibitor, HWG-35D, ameliorates the severity of imiquimod-induced psoriasis model by blocking Th17 differentiation of naive CD4 T lymphocytes. Int J Mol Sci. 2020;21(21):8371. doi:10.3390/ijms21218371
96. Dai L, Bai A, Smith CD, Rodriguez PC, Yu F, Qin Z. ABC294640, a novel sphingosine kinase 2 inhibitor, induces oncogenic virus-infected cell autophagic death and represses tumor growth. Mol Cancer Ther. 2017;16(12):2724–2734. doi:10.1158/1535-7163.MCT-17-0485
97. Ding X, Chaiteerakij R, Moser CD, et al. Antitumor effect of the novel sphingosine kinase 2 inhibitor ABC294640 is enhanced by inhibition of autophagy and by sorafenib in human cholangiocarcinoma cells. Oncotarget. 2016;7(15):20080–20092. doi:10.18632/oncotarget.7914
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.