")
Back to Journals » Stem Cells and Cloning: Advances and Applications » Volume 16
The Regenerative Power of Stem Cells: Treating Bleomycin-Induced Lung Fibrosis
Authors Vats A, Chaturvedi P
Received 1 May 2023
Accepted for publication 6 September 2023
Published 12 September 2023 Volume 2023:16 Pages 43—59
DOI https://doi.org/10.2147/SCCAA.S419474
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Bernard Binetruy
Amrita Vats,1 Pankaj Chaturvedi2
1Department of Pharmacology and Regenerative Medicine, University of Illinois, Chicago, IL, 60612, USA; 2Department of Cell and Developmental Biology, University of Illinois at Urbana-Champaign, Urbana, IL, 61801, USA
Correspondence: Pankaj Chaturvedi, Email [email protected]
Abstract: Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive lung disease with no known cure, characterized by the formation of scar tissue in the lungs, leading to respiratory failure. Although the exact cause of IPF remains unclear, the condition is thought to result from a combination of genetic and environmental factors. One of the most widely used animal models to study IPF is the bleomycin-induced lung injury model in mice. In this model, the administration of the chemotherapeutic agent bleomycin causes pulmonary inflammation and fibrosis, which closely mimics the pathological features of human IPF. Numerous recent investigations have explored the functions of various categories of stem cells in the healing process of lung injury induced by bleomycin in mice, documenting the beneficial effects and challenges of this approach. Differentiation of stem cells into various cell types and their ability to modulate tissue microenvironment is an emerging aspect of the regenerative therapies. This review article aims to provide a comprehensive overview of the role of stem cells in repairing bleomycin-induced lung injury. It delves into the mechanisms through which various types of stem cells, including mesenchymal stem cells, embryonic stem cells, induced pluripotent stem cells, and lung resident stem cells, exert their therapeutic effects in this specific model. We have also discussed the unique set of intermediate markers and signaling factors that can influence the proliferation and differentiation of alveolar epithelial cells both during lung repair and homeostasis. Finally, we highlight the challenges and opportunities associated with translating stem cell therapy to the clinic for IPF patients. The novelty and implications of this review extend beyond the understanding of the potential of stem cells in treating IPF to the broader field of regenerative medicine. We believe that the review paves the way for further advancements in stem cell therapies, offering hope for patients suffering from this debilitating and currently incurable disease.
Keywords: pulmonary fibrosis, regeneration, differentiation, bleomycin, alveoli, proliferation
Introduction
The lungs are complex organs responsible for oxygen-carbon dioxide exchange and are susceptible to damage that can destroy alveolar epithelial cells.1 The alveoli, trachea, bronchiole, and bronchus constitute the structural and functional unit of the lung. Alveoli, which are the primary component of lung tissue, are responsible for gas exchange within the small air sacs.2 These expandable sacs have a large surface area, and the inhaled oxygen is absorbed by a network of capillaries that surround these sacs. The carbon dioxide from the capillaries is released to the alveoli.
The pulmonary alveolar epithelium acts as a physical barrier, shielding the lungs from environmental insults and foreign substances. Recently, this protective function of alveolar epithelia has garnered attention as it is a primary target of SARS-CoV-2 infection that caused COVID-19.3 Two types of epithelial cells make up the majority of the pulmonary alveolar epithelium: alveolar type I (AT1) and alveolar type II (AT2) cells. AT2 cells constitute less than 5% of the alveolar surface and are small, cuboidal cells responsible for synthesizing and secreting pulmonary surfactant protein C (SFTPC). Additionally, the AT2 cells also serve as alveolar stem cells that can self-renew and differentiate into AT1 cells during alveolar homeostasis and post-injury repair.4,5 AT1 cells, on the other hand, are large, flat cells that cover 95% of the alveolar surface area and form the epithelial component of the thin-air blood barrier. They are considered to be terminally differentiated cells.5
Remarkably, the lungs can repair and regenerate damaged alveolar cells through various biological processes. During alveolar regeneration, AT1 cells can dedifferentiate and give rise to AT2 cells. Studies have confirmed that AT1 cells also possess a certain degree of plasticity and can contribute to repair and regeneration to a varying degree,6 indicating lineage interchangeability between AT1 and AT2 cells in both in vitro and in vivo systems. Therefore, we believe that the regenerative potential of lung epithelia is an ideal system to understand the paradigms of stem cell biology and apply this knowledge for regenerative therapies to diverse organ systems. The review aims to summarize the current knowledge in the field about the regenerative potential of lung epithelial cells, specifically alveolar type I (AT1) and alveolar type II (AT2) cells, in a pulmonary fibrosis model. Here, we summarize the signaling pathways involved in the dedifferentiation and proliferation of these stem cells and explore the role of extrinsic factors in the regenerative process. By studying the regenerative capabilities of lung epithelial cells, the review aims to provide valuable insights into stem cell biology and potentially contribute to the development of regenerative therapies for various organ systems.
Bleomycin-Induced Lung Injury as a Model System for Pulmonary Fibrosis
Pulmonary fibrosis is a chronic disease characterized the development of fibrotic scar tissue in the lung parenchyma, impairing its function and causing respiratory failure. Histopathological examination of lung tissues from IPF patients reveals a distinctive pattern known as usual interstitial pneumonia (UIP), characterized by fibroblastic foci, collagen deposition, and distorted lung architecture.7,8 Alveolar epithelial cell injury and aberrant repair processes play a key role in IPF pathogenesis, leading to the activation of fibroblasts and their transformation into myofibroblasts. These myofibroblasts contribute to the excessive production and deposition of extracellular matrix components, causing irreversible tissue remodeling. Additionally, an inflammatory microenvironment with activated immune cells further exacerbates the fibrotic process.9–11 Recent studies have also documented development of pulmonary fibrosis in about half of the COVID-19 survivors.12,13 Nevertheless, the fibrosis identified in individuals who have survived COVID-19 seems to exhibit a self-resolving nature.13 However, further longitudinal investigations are ongoing to comprehensively ascertain the trajectory and mechanisms of this observed self-resolution of COVID-19-induced fibrosis.
This pulmonary fibrosis can result from drug use, environmental exposure, or collagen vascular disorders, and is often idiopathic.14 Idiopathic pulmonary fibrosis (IPF) is a progressively deteriorating and fatal lung disease. Widespread changes in DNA methylation, chromatin accessibility, and gene expression have been identified in pulmonary fibrosis. These epigenetic changes correlate well with pulmonary fibrosis predisposing factors such as smoking, age, gender, exposure to etiological agents such as asbestos and silica, or certain cancer treatments.15 Therefore, understanding the pathogenesis of idiopathic pulmonary fibrosis (IPF) and COVID-19-related pulmonary fibrosis could providing valuable insights into novel treatment approaches for both conditions, allowing for early intervention and better management of lung complications.
Idiopathic pulmonary fibrosis has also been reported in domestic animals such as dogs, cats, horses, and donkeys. Each of these animal models presents clinical features resembling those of human IPF, although the progressive and irreversible symptoms are not fully recapitulated. Several approaches to induce pulmonary fibrosis have been established: Fibrous or particulate aerosols (eg silica, asbestos, arsenic, cadmium), overexpression of cytokines such as TGF-β, TNFα, or IL13 in lung epithelia, induced senescence of AT2 cells by loss of Sin3a or Cdc42, exposure to radiations, immunizing haptens (eg Fluorescein isothiocyanate, trinitrophenol).16 Among these approaches, inducing lung injury in mice through the intratracheal administration of bleomycin has become a widely accepted model system for studying the risk factors associated with pulmonary fibrosis (Table 1).
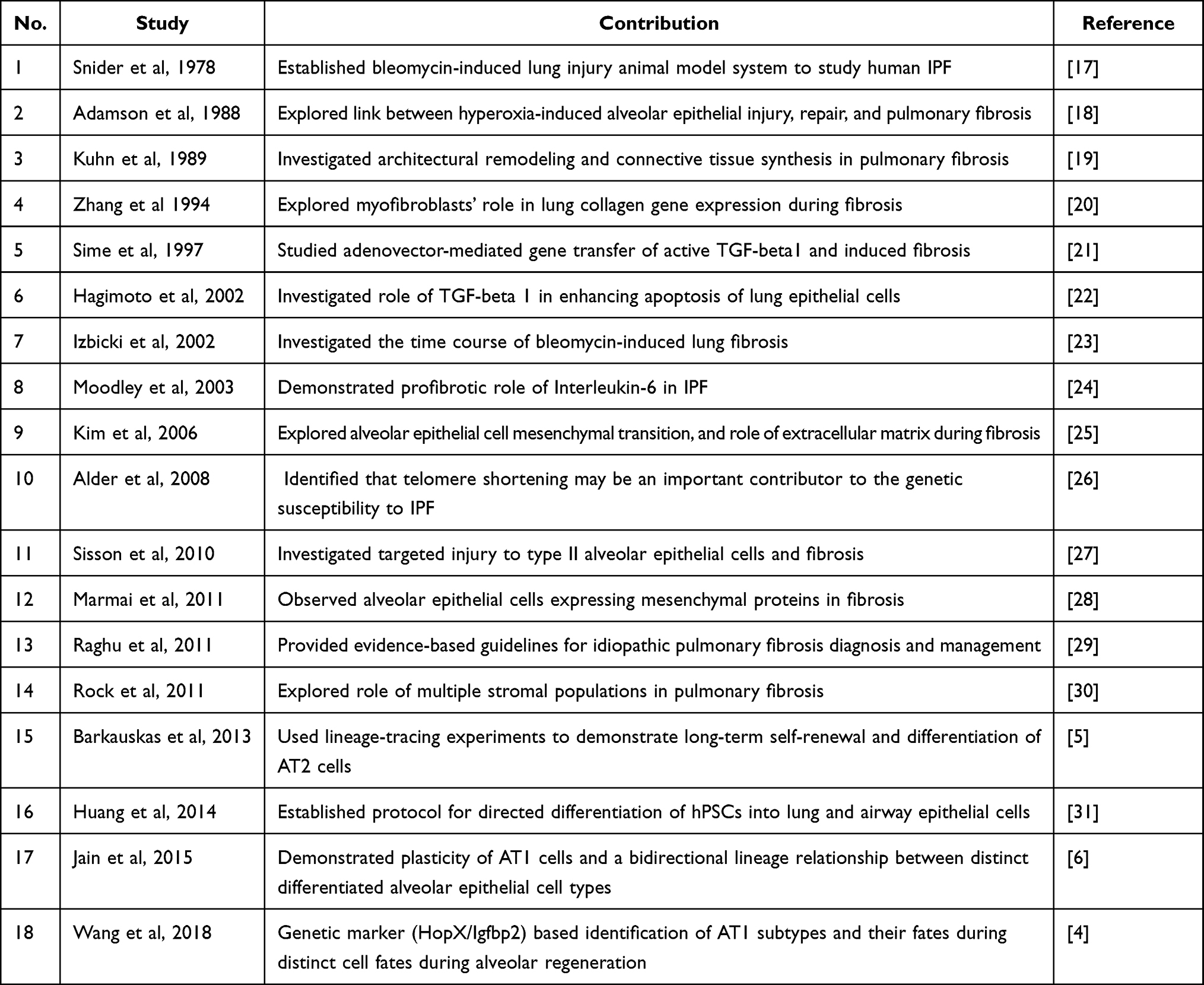 |
Table 1 Key Research Articles Contributing to the Field of Pulmonary Fibrosis |
Bleomycin is a glycopeptide antibiotic, used for the treatment of cervical, uterine, and testicular cancers, and lymphoma.32 Bleomycin causes cell cycle arrest by inducing DNA breaks and creating free radicals, leading to inflammation, necrosis, and apoptosis.33,34 Owing to the cytotoxic activity, a commonly observed adverse effect of bleomycin treatment is pulmonary toxicity in around 10% of patients undergoing bleomycin treatment.32 Even a single dose of bleomycin has been reported to induce lung fibrosis, but the extent of fibrotic damage in human lungs is at best achieved through repetitive intratracheal bleomycin administration. Such robust bleomycin-induced lung fibrosis in mice models has been achieved through multiple biweekly doses of 0.04 units of bleomycin.35 Immediate response to bleomycin administration is the production of inflammatory markers (interleukin-1, interleukin-6, interferon-γ, tumor necrosis factor-α). After about 9 days of bleomycin administration, the fibrotic response takes over and the pro-fibrotic factors (eg TGF-β, fibronectin, procollagen-1) are expressed.36 The resulting fibrosis is characterized by an increase in the production and deposition of collagen and other extracellular matrix components.37
Fibrosis typically appears four weeks after bleomycin administration, and more severe progression can be achieved by extending the duration of treatment. Although the fibrosis is self-limiting after 28 days, it becomes more difficult to resolve with age, possibly due to cellular senescence and telomere attrition.38,39 To create a more comprehensive model of persistent and progressive fibrosis, it is essential to understand the composition and presence of various cell types in the lung niche during fibrosis. These cell types include alveolar cuboidal type 2 epithelial cells (AT2s), Clara cells (CCSPs), and surfactant protein C (SPC) expressing cells.40 Several cell types, including AT1, AT2, fibroblasts, myofibroblasts, lymphocytes, neutrophils, endothelial cells, airway epithelial cells, and stem/progenitor cells are involved in bleomycin-induced lung fibrosis (Figure 1). Upon exposure to bleomycin, there is a temporary disruption of the alveolar structure before fibrosis occurs, leading to abnormal morphology and an increase in the number of myofibroblasts.41
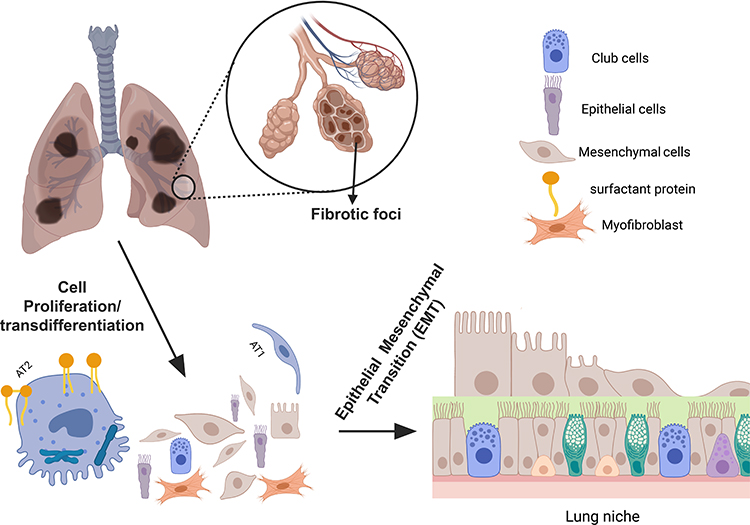 |
Figure 1 An illustration of AT2-AT1 cell transition during fibrotic lung injury. The figure represents the process of AT2-AT1 cell transition during fibrotic lung injury. AT2 cells are shown to proliferate after the lung injury, where a fraction of these cells differentiating into mature AT1 cells. Also included are the representative cell types involved in the repair process (top right). Created with BioRender.com. |
The severity of pulmonary fibrosis induced by bleomycin treatment in mice varies depending on the mouse strain, with C57BL/6 mice being more susceptible than BALB/c mice.42 This differential response to bleomycin treatment between the two strains is likely due to the lower expression of transforming growth factor-beta (TGF-β) in BALB/c mice.43 Similarly, higher susceptibility to fibrosis caused by intratracheal delivery of silica fibers was also observed in C57BL/6 mice compared to CBA/J mice.44 Such strain-specific differences may also be due to variations in immune response, pathogen susceptibility, or drug metabolism. The different immune response of mouse strains toward parasitic infections or tumors has been demonstrated elsewhere.45,46 Nevertheless, this strain-specific response to bleomycin treatment parallels human genetic predisposition to risk factors contributing to pulmonary fibrosis.
Mechanism of Lung Injury Induced by Bleomycin
Fibrosis is associated with excessive alveolar destruction, decreased lung capacity, and disrupted gas exchange function.47–50 The cause of pulmonary fibrosis is not fully understood, but it is believed to result from the infiltration of inflammatory cells and pro-inflammatory mediators in the lung parenchyma. Infiltration of T cells, B cells, and macrophages in the lung tissue in response to bleomycin treatment has been observed, and their activation can contribute to the progression of fibrosis.51–53 This infiltration leads to the release of pro-inflammatory mediators that eventually cause fibrosis, which is a major contributor to end-stage lung failure and death in many chronic cases. Release of cytokines such as TGF-β, IL-1, IL-6, and IL-13 has also been observed during the pro-fibrotic phenotype in damaged lungs.54 Further, increased expression of immune-responsive genes (eg Pteges, Orm1, and Zbp1) and antioxidative enzymes (eg Glrx, Prdx4, and Gstk1/2) has been shown in proliferating AT2 cells,55 indicating a role of inflammatory and oxidative-damage response in the repair process.
The chronic fibrotic response to bleomycin-induced lung injury is also linked to a loss of bleomycin hydrolase (BLMH) activity. Bleomycin hydrolase is a cytosolic aminopeptidase that presents antigens to the MHC-I complex,56 and is ubiquitously expressed in human tissues. In bleomycin-treated mice, the BLMH regulates the release of inflammatory cytokines and migration of activated effector cells in the lung. Enzymes such as cytosolic phospholipase A2 (cPLA2), transglutaminase (TG), and lysyl oxidases (LOX) have also been implicated in the progression of lung disorders and collagen deposition. cPLA2 is a key enzyme in the generation of pro-inflammatory eicosanoids, thromboxanes, and leukotrienes. Additionally, increased expression of TG and LOX has been suggested to contribute to pulmonary fibrosis by modifying the extracellular matrix (ECM).57 Both enzymes modulate ECM stiffness by mediating protein cross-linking.
TG2 promotes fibrosis by cross-linking extracellular collagen and fibronectin, while the LOX family of enzymes facilitates the covalent cross-linking of type I collagens. However, there is limited information available on the regulation of LOXL2 in lung fibrosis and its precise role in cross-linking fibrillar collagen.57 Evidently, more research is needed to understand the role of matrix stiffness and fibroblast contractility due to collagen deposition during bleomycin injury. Collagen and other ECM protein deposition lead to the production of pro-inflammatory cytokines that worsen the lung fibrotic process induced by bleomycin. Considering that CD44 is a hyaluronan receptor and an integral part of the ECM, understanding its role in the inflammatory and wound repair phases could provide deeper insights into the pathogenesis of lung injury.58
Epithelial-Mesenchymal Transition (EMT) in Bleomycin-Induced Lung Fibrosis
Epithelial-Mesenchymal Transition (EMT) is a fundamental cellular process in development, wound healing, and disease progression. During EMT, epithelial cells lose their tightly connected structure and polarity, acquiring mesenchymal characteristics like enhanced mobility and invasive abilities. Transcription factors like Snail, Twist, and ZEB act as central regulators of EMT by suppressing epithelial gene expression and promoting mesenchymal gene expression. These pathways converge to reorganize the cellular architecture, downregulate E-cadherin, upregulate N-cadherin and vimentin, and alter cytoskeletal dynamics.13,59 Although the origin and mechanism of lung fibrosis are not yet fully understood, current research has indicated the involvement of epithelial-mesenchymal transition (EMT) in idiopathic pulmonary fibrosis (IPF) (Table 1). The prevailing hypothesis suggests that defective alveolar epithelial type-II (AT2) cells initiate the fibrotic cascade. Their failure to differentiate to AT1 cell impairs the recovery of lung epithelia after injury, which is followed by induction of EMT and eventually fibrosis.9,60,61 The dysregulation of epithelial function, extracellular matrix deposition, fibroblast activation, and morphological changes in mesenchymal cells are all associated with IPF. The proper functioning of lung tissue depends on the dynamic interaction between the epithelium and the surrounding mesenchyme.62,63
Exposure to bleomycin can trigger the process of epithelial-mesenchymal transition (EMT), which transforms alveolar epithelial cells into myofibroblasts (Figure 1).64,65 These myofibroblasts overproduce collagen and other extracellular matrix proteins implicated in fibrosis. A common cell surface marker of these myofibroblasts, but not of healthy fibroblasts, is the fibroblast activation protein (FAP).59 While this process can damage pleural mesothelial cells, the exact mechanism behind EMT in these cells is not yet clear. The loss of alveolar epithelial cells and the activation of signaling pathways, such as TGF-β, and SMAD2-3, may play a role. The loss of regenerative potential in damaged epithelial cells results in sustained mechanical tension on AT2 cells. This mechanical tension triggers the activation of the TGF-β pathway, which propagates towards the center of the damaged tissue, leading to the spread of fibrosis in a periphery-to-center manner.66 The degree of fibrosis in the lungs can be accessed by the dynamic interactions of bronchoalveolar stem cells (BASCs), club cell secretory protein (CCSP), AT2 cells, and SPC2, which have similar functions to stem cells.67 Stem and progenitor cells are currently a growing field of research, with a focus on their use in regenerating damaged lungs. Tissue regeneration can occur through cell proliferation and differentiation of stem and progenitor cells.
Various epithelial cells express cytokeratin, which serves as an epithelial phenotypic marker. These markers form a cell-communication network with mesenchymal cells during lung fibrosis and repair. During bleomycin-induced EMT of pleural mesothelial cells, there is an increased expression of mesenchymal markers such as vimentin and α-smooth muscle actin, along with a reduction in expression of epithelial phenotypic markers (cytokeratin-8 and E-cadherin).68 Epithelia express keratins 5, 8, 14, and 18 and have a more rapid turnover rate.69 Thus, keratin expression likely plays a significant role in lung injury. Additionally, after influenza infection, there is a strong expression of keratin 5+ in the lung parenchyma, which creates a barrier to epithelial regeneration.70,71
After lung injury and tissue damage, epithelial cells can differentiate and undergo phenotypic switching or dedifferentiation. One effective way to study the role of different cells and their controlling mechanisms in the repair process is to use the CrER system (Figure 2). This bipartite system uses tissue or cell-specific promoters to drive Cre reporter expression at a safe-haven expression locus, such as the Gt(ROSA)26Sor locus. Cre-mediated recombination removes the intervening stop codon, producing robust tdTomato fluorescence. Tamoxifen, an estrogen receptor agonist, is a widely used inducer that activates the Cre-ERT2 fusion protein for lineage tracing experiments.72 CreER mouse studies have linked pre-alveolar type 1 transitional cell state (PATS) markers with AT1 cells, which emerge during lung injury and derive from AT2 cells (Figure 2).73 In the next section, we will discuss the role of alveolar epithelial cells, including AT2/AT1 transitional cells, in the development of pulmonary fibrosis.
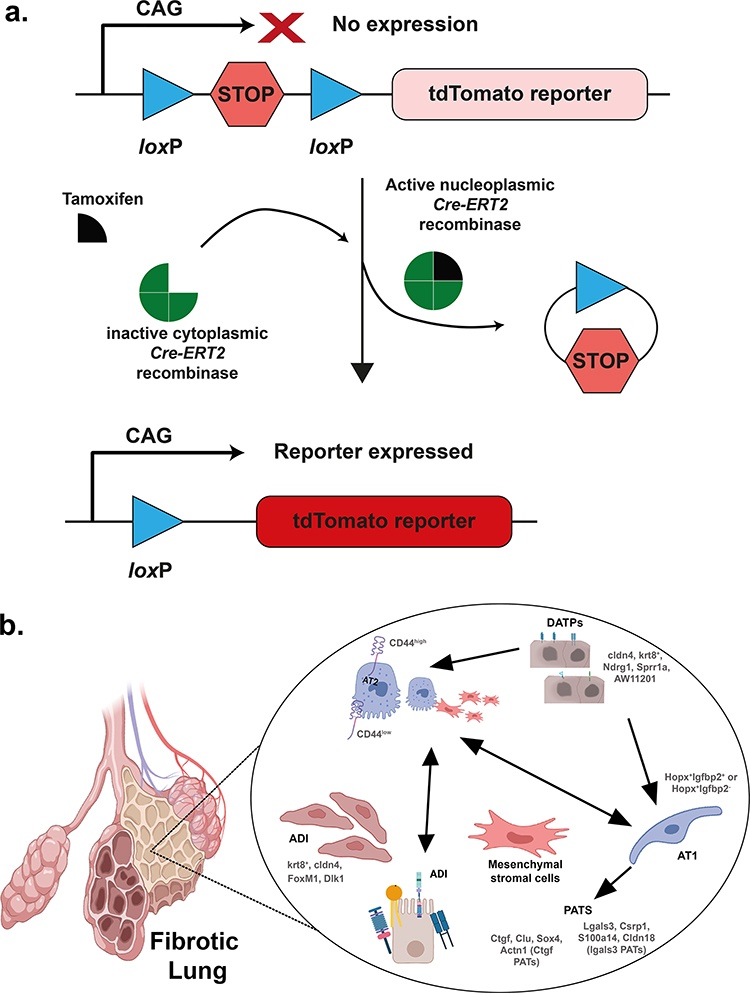 |
Figure 2 Lineage-tracing of cells during the repair phase of an injured lung by the Cre-ERT2 system. (a) Schematic representation of the Cre-ERT2 system. The presence of a stop codon flanked by loxP sites inhibits the expression of the reporter transgene (tdTomato). Upon tamoxifen treatment, the expressed Cre mediates site-specific recombination between loxP sites, which removes the intervening stop codon, and the reporter is expressed. (b) Lineage-tracing using Cre-ERT2 system enriches and identifies intermediate stages of transdifferentiated AT2 stem cells. Some of the identified intermediates are shown for representation. Similar enrichment can also be achieved using stem cell surface markers such as CD34, CD45, and Thy1. Created with BioRender.com. |
Acquisition of Stem Cell-Like Properties During the Transition from AT2 to AT1 Cells
One of the key areas of research in the development of pulmonary fibrosis is the understanding of the properties of stem cells and their origins during the AT2-AT1 transition. This process, known as alveolar epithelial cell plasticity, involves cellular differentiation, proliferation, and various phenotypic changes.74 During this transition, cells may acquire stem cell-like properties that help them regenerate damaged tissue. This AT2-AT1 transition state is characterized by the expression of Krt8+, Hbegf, and Areg intermediate stage markers (discussed in the next section).55,75 Another model of lung regeneration and alveolar repair is pneumonectomy, where one or multiple pulmonary lobes are surgically removed. In this system also AT2 cell proliferation has been observed during compensatory lung growth.76
Notably, the bone morphogenetic protein (BMP) signaling pathway has been identified as an important determinant of the AT2-AT1 transition. AT2 cell proliferation was inhibited by BMP4 in both in vitro (3D organoid culture) and in vivo (post-pneumonectomy) models, while the BMP4 agonists (eg noggin, follistatin) promoted the renewal of AT2 cells.76 Similarly, the local microenvironment comprising of the extracellular matrix (ECM), cytokines, growth factors, and cell-cell junctions also modulate stem cell proliferation and differentiation. With progressively increasing rigidity of the matrix the lineage outcome of mesenchymal stem cell differentiation shifts from neurogenic and myogenic to osteogenic lineage.77 Recently, it was demonstrated that the AT2 cells deficient in β1 integrins are also modulated by ECM.78 In the absence of β1 integrins, the AT2 cells proliferate but fail to differentiate into AT1 cells during the repair of alveolar damage. The study also suggests the existence of a feedback loop controlling the proliferation of AT2 cells during the repair process, where the proliferation of AT2 cells continues till the damaged epithelium is completely healed.78 The integrin-mediated mechano-transduction can remodel the actin cytoskeleton, thereby affecting cell-cell adhesion and cell polarity. This mechano-transduction pathway can also induce changes in nuclear architecture, function, and gene expression, and can be pathogenic.79–82 Aberrant nuclear morphology and organization have been implicated in the altered differentiation potential of stem cells,83 which can also hinder the repair process.
The AT2-AT1 transition is also accompanied by changes at the transcriptional levels. However, since the regenerating lungs are composed of tens of different cell types, the cell-specific changes in the gene expression can be better appreciated through single-cell RNA seq (scRNAseq) methods. The IPF Cell Atlas Data Mining portal (www.ipfcellatlas.com) is one such resource that permits transcriptional analysis and cell-type annotation based on RNAseq datasets from different research groups.84 Analysis of the global transcription profiles can resolve the distinct intermediate stages and the cell types contributing to the repair process. A demonstration of this approach was made recently by comparing different cell types in pulmonary fibrotic lungs to normal human lungs through scRNAseq. The study found that the SCGB3A2+ secretory cells can also differentiate into AT1 cells. These SCGB3A2+ cells may either differentiate first from AT2 cells or may share a common progenitor state with AT2 cells during this differentiation process.85 In a bleomycin-induced lung injury mice model scRNAseq analysis showed that the airway club cells converge to a common intermediate state during transdifferentiation to AT1 cells.86 These observations raise a pertinent question of whether the alveolar niche or the level of maturity of differentiated cells is a primary determinant of cellular identity during regeneration. Notably, these transdifferentiation mechanisms have also been observed to promote chemotherapy resistance and tumor diversification,87,88 ascribing a larger clinical relevance to cellular plasticity in regeneration and disease progression.
Stem cells are typically dormant during normal conditions, but in response to lung injury, they undergo proliferation.89 Although progenitor cells are believed to help maintain the epithelium’s integrity, their ability to regenerate the damaged lung epithelium is still uncertain, as studies have shown conflicting results.50 Following lung damage, resident and tissue-specific stem cells in the airways and structural units of the lung tissue undergo significant changes. In response to infection or injury, facultative stem and progenitor cell populations such as alveolar type II cells, Clara cells, basal cells (BCs), and Bronchioalveolar stem cells (BASCs) enter the cell cycle to activate progenitor cells.41 These progenitor cells are found in various regions of the lung, including the airways, alveoli, submucosal glands, and bronchoalveolar ductal junctions. They primarily participate in the local repair of lost cells in adjacent populations.90 AT2 (Alveolar type II) cells, which express surfactant protein C (SFTPC) genes, are also long-term stem cells. Recent studies indicate that AT2/AT1 transitional cells may play a role in the development of pulmonary fibrosis. Importantly, after bleomycin-induced injury to the alveolar region, the rate of differentiation of the lineage-labeled AT2 to AT1 is significantly higher.91,92
Although it is known AT2 cells transition to flattened AT1 cells, the exact mechanism of this transition and accompanying morphological change is still being investigated. Consistent with this hypothesis Chen et al identified a subpopulation of CD44 high and CD44 low on AT2 cells with the capability to differentiate into AT1 cells.93 CD44 is a receptor of hyaluronan and is a surface marker of various stem cells.94 In addition, distribution of Axin2 and Wnt proteins in AT2 subpopulation under homeostatic conditions and their essential role in inhibiting transdifferentiation was demonstrated.95,96 It is believed that upon Pseudomonas aeruginosa infection, a higher percentage of AT2 exit quiescence, differentiate, and start expressing Sca-1+ and FoxM1. These cells have a higher potential to differentiate into AT1 cells.97,98
Signal Transduction During the Transition from AT2 to AT1 Cells
During the repair of damaged pulmonary epithelium, the AT2 cells proliferate, undergo a cascade of intermediate transitional stages, and differentiate into AT1 cells (AT2-AT1 transition). Transcriptional analysis of cells derived from several in vivo murine or human repair models and organoids has revealed the presence of distinct intermediate progenitor cell or alveolar differentiation intermediate (ADI) states, each expressing a unique set of markers. These intermediate progenitors have been grouped into five distinct stages: ctgf+ pre-alveolar type 1 transitional cell state (ctgf PATs), Lgals3 PATs, Krt8+ expressing alveolar differentiation state (Krt8+ ADI), preAT1 state, and damage-associated transient progenitors (DATPs).55,75,92 Some of the intermediate markers expressed in ADI are shown in Figure 2. The ctgf+ PATs, first identified in organoids, specifically express marker genes Ctgf, Clu, Sox4, and Actn1, while the Lgals3 PATs are characterized by the expression of Lgals3, Csrp1, S100a14, and Cldn18 markers.75 DATPs are the cells expressing markers Cldn4, Krt8, Ndrg1, Sprr1a, and AW112010 and have been identified as an intermediate state during AT2 to AT1 transition.55 Interestingly, the DATPs were found to have the capacity to terminally differentiate into AT1 cells or revert to AT2 stem cells. It was also found that the IL-1β signaling indirectly promotes DATP formation, while HIF1α regulates their differentiation to the AT1 lineage.55 A similar population of cells has also been identified in the lungs of COVID-19 patients.55 The Krt8+ ADI state is transcriptionally similar to the DATPs. This state of cells is characterized by pro-fibrogenic SMAD signaling and high expression of markers such as Ctgf, Itgb6, Areg, Hbegf, Edn1, and Lgals3.86
Another transitional state known as alveolar type-0 (AT0) has also been identified in human lung tissues. AT0 cells can differentiate into alveolar type 1 (AT1) or respiratory bronchioles secretory cells (TRB-SCs) during alveolar type 2 (AT2) differentiation, like the previously mentioned ADI. These AT0 cells have been shown to express high levels of SCGB3A2, SFTPC, and SFTPB, which are secreted during AT2 injury and repaired during bleomycin injury.99 AT2 cells express STAT-3 (signal transducer and activator of transcription), which produces Brain-derived neurotrophic factor (BDNF) to improve the niche function of surrounding fibroblasts and play a role in alveolar repair through a non-autonomous mechanism.100 Studies have also shown that STATs can regulate the function of various tissue stem cells.101 Wnt, BMP, Notch, and NFκB signaling pathways also modulate the AT2 stem cell population to create different subsets of alveolar progenitor cells, which can alter the mechanical properties and structure of the tissue.73 Increased Notch signaling during the early repair phase of Pseudomonas aeruginosa induced acute lung injury in mice was found to promote AT2 cell proliferation. However, in a coordinated manner, non-canonical Notch ligand Dlk1 (delta-like homolog 1) attenuates this Notch signaling and permits the transition of proliferated cells to the AT1 lineage.102
The formation of abnormal epithelial cells and defective fibrotic foci, responsible for IPF, is controlled by complex signaling pathways that regulate the transition states of AT2-AT1 cells.103 Impaired alveolar regeneration can be caused by mechanical tension (Figure 3). AT2 cells deficient in Cdc42 are unable to differentiate into AT1 cells, resulting in the inability to generate new alveoli after lung injury.66 Cdc42 is a small GTPase Rho family member that regulates signaling pathways and controls various cellular functions, including differentiation, cell migration, and morphology. Additionally, Cdc42 is implicated in stem cell aging, leading to loss of polarity and altered epigenetic modification.104 As mice age, treating fibrosis after bleomycin administration becomes increasingly difficult, potentially due to the inability of damaged epithelial stem cells to return to normal function. The role of Cdc42 in regulating cytoskeletal organization, cell-cell matrix adhesion, and cell polarity in distinct cell types could be beneficial in resolving lung fibrosis by rejuvenating and regulating the phenotypic expression of stem cells.
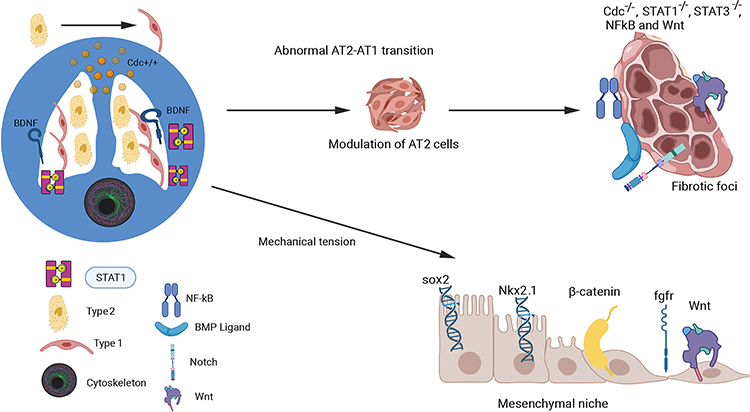 |
Figure 3 Mechanotransduction-Mediated Signaling in AT2-AT1 Transition. The figure illustrates the signaling factors and progenitor stem cells involved in the AT2-AT1 transition in fibrotic lungs, as well as the role of the mesenchymal niche microenvironment in this process. The complex mechanical signaling pathways that control this transition are depicted, with a focus on the involvement of the small Rho GTPase family member Cdc42 in regulating the STAT-3 and STAT-1 signaling pathways. The effects of mechanotransduction on Wnt, Sox2, EGF, β-catenin, and Nkx2 proteins in the lung mesenchyme, which can alter lung development and repair, have also been demonstrated (bottom right). Created with BioRender.com. |
During bleomycin-induced lung injury and repair in mice, diverse clusters and clonal cells with unique properties may develop. Lineage tracing experiments have revealed lineage-negative progenitor stem cells (LNEP) in normal epithelial lung cells that consist of p63+/krt5+. Accumulating evidence indicates that these LNEP cells can activate and migrate to heavily injured areas and proliferate after injury. These cells can differentiate into multipotent progenitor cells or mature AT2 cells, with other stem cells potentially contributing to lung fibrosis.105 Studies have revealed the presence of two distinct subsets of AT1 cells, Hopx+Igfbp2+ and Hopx+Igfbp2−, that have different fates during alveolar regeneration (Figure 2).4 The former subset represents the AT1 population which is terminally differentiated, while the latter subset constitutes ~5% of the total AT1 population and retains cellular plasticity.4 Igfbp2 (Insulin-like growth factor binding protein 2) is specifically expressed in postnatal AT1 cells.102 Hopx (Homeodomain-only protein X) plays a crucial role in lung development, alveolar damage, and maturation. Hopx also impedes surfactant protein expression, leading to defective AT1 cells and deformed alveoli.106 Interestingly, Hopx can also promote self-renewal and differentiation to AT2 cells, indicating bidirectional lineage differentiation in the alveolar epithelium (Figure 2).6 However, understanding the different subsets of the epithelial cell population and the immune signals during this phenotypic switch remains a challenge.
Recent studies have suggested that the rate of differentiation from AT2 to AT1 is more pronounced in bleomycin-induced injury than in other injury models such as Diphtheria Toxin A (DTA).5 Bleomycin injury may promote a distinct local signaling pathway which is not observed with other injury models. Comprehensive data analysis suggests that severe lung pathologies are associated with increased apoptosis, senescence, and mutations in surfactant proteins. Senescent cells have been found to hinder the regeneration of damaged cells by producing signaling proteins via senescent-associated secretory phenotype (SASP), which can cause fibrosis and block the function of neighboring cells.107 This can lead to stochastic profibrotic epigenetic reprogramming and premature epithelial cell senescence, preventing regeneration. It is unclear whether senescent cells express senescence-related genes that control differentiation towards AT2 or AT1 cells.
The mTOR complex which includes mTOR complexes 1 and 2, plays a crucial role in regulating cellular proliferation and biosynthetic processes. Activation of mTOR signaling can occur through various stimuli such as cytokines, growth factors, and mitogens.51 However, dysregulated mTOR signaling has been linked to a higher vulnerability to pulmonary and liver fibrosis.108,109 In a study by Hettiarachchi et al (2020), the targeted delivery of PI3K/mTOR agonist to myofibroblasts resulted in a significant reduction in the expression of fibrogenic mediators in BLM-injured mice and prolonged their survival,59 suggesting the potential of mTOR inhibitors in managing IPF. In a recent study, it was found that the constitutive activation of mTOR signaling in lung epithelial cells treated with bleomycin can potentially promote EMT. This effect might be attributed to the disruption of tight junctions in the lung epithelial cells.10
Stem Cells: A Potential Avenue for Treatment of Pulmonary Fibrosis
The current treatment options for IPF are limited and aim to slow the disease progression and manage symptoms. The standard pharmacological therapies include the use of antifibrotic agents, such as pirfenidone and nintedanib.110 These drugs have been shown to slow the decline in lung function and reduce the rate of acute exacerbations in patients with IPF. However, they do not cure the disease and have potential side effects. Other treatment options for IPF may include oxygen therapy, pulmonary rehabilitation, and lung transplantation in advanced cases. However, these treatments are not suitable for all patients and may have case-specific limitations.
In recent years, mesenchymal stem cells (MSCs) have emerged as a potential therapeutic option.8 MSCs are adult stem cells derived from various sources such as bone marrow, adipose tissue, umbilical cord tissue, menstrual blood, and placenta. These cells can differentiate into multiple cell types and secrete functional paracrine factors that possess anti-inflammatory, immunosuppressive, and angiogenic properties.111,112 Currently, several clinical trials are underway to evaluate MSCs for the treatment of IPF (ClinicalTrials.gov Identifiers: NCT05468502, NCT05016817, NCT01919827, NCT02013700, NCT01385644, NCT02594839, NCT02135380). However, at least for now, the therapeutic potential of MSCs is limited by their low survival rates in the intended organ and associated risks of embolism and tumorigenesis. An alternative approach is the use of extracellular vesicles (EVs) derived from MSCs. The use of EVs to deliver cargo has the advantages of target cell specificity, ability to cross biological barriers, low immunogenicity, and potential for chemical/biological modifications.113 The EVs are being clinically evaluated for health conditions such as Acute Respiratory Distress Syndrome, COVID-19, type-I diabetes, and cardiovascular disease.114 However, standardization of EV isolation and characterization methods is needed to ensure consistency and reproducibility of results. As IPF is caused by persistent immune reaction and deposition of extracellular matrix proteins the administration of MSCs or extracellular vesicles is a promising treatment option.115 This is evidenced by the use of MSCs in treating bleomycin-induced lung injury in mice, where improved wound healing and reduction in fibrosis were observed.115,116
Recently, human induced pluripotent stem cells (iPSCs) have also emerged as a promising therapeutic option for COVID-19 treatment. In this case, the stem cells infuse and migrate into damaged lung tissue, playing a role in reconstructing the damaged lung tissue and promoting regeneration.117 Studies using murine embryonic stem cells (ESCs) have demonstrated differentiation into alveolar epithelial cells and non-ciliated Clara cells, indicating their potential in treating lung injuries.118 In addition, transplantation of human ESC-derived AT2 cells was found to mitigate lung injury in mice by improving pulmonary function and prolonging survival (Table 1).119 CD166 is a glycoprotein found on the surface of lung epithelial cells that encodes the activated leukocyte cell adhesion molecule. Studies have demonstrated that CD166 derived from human ESCs and iPSCs can effectively mitigate bleomycin-induced acute lung injury (ALI) by enhancing pulmonary function and improving survival rates in mice.120 Additionally, both MSCs and iPSCs have the potential to differentiate into lung and airway progenitor cells with similar ultrastructural features and biological functions as normal AT2 cells. These cells can be produced from a variety of cell types, including basal cells, goblet cells, Clara cells, ciliated cells, and alveolar epithelial cells, both in vitro and in vivo.120 Human umbilical cord blood (HUCB) contains CD34+ cells that can differentiate into mesenchymal stem cells (MSCs) and potentially be used for cell therapies.121 CD44 is an early marker of differentiation for these immature CD34+ cells.122 Studies have shown that cells with higher CD44 expression levels have better proliferation and differentiation capabilities into AT1 cells.93 Therefore, the study of HSCs with a general expression of CD34 and CD44 markers could provide insight into the development of lung fibrosis.
Conclusion and Future Directions
The lung is a complex organ composed of various types of cells, including progenitor cells, endothelial cells, and myofibroblasts.40 While both mouse and human lungs harbor stem and progenitor cells in distinct niches, there are species-specific differences in their organization and challenges faced by these cells.8 For instance in mice, bronchoalveolar stem cells (BASC) are responsible for maintaining proximal and distal lung epithelia, whereas human lung BASC faces additional challenges from external factors such as environmental and behavioral influences.123 One contributing factor is the relative uniformity of the mouse lung niche, attributed in part to the controlled maintenance conditions in the laboratory. However, it is essential to consider that both wild and lab-raised mice, as well as human subjects, can experience alterations in lung niche development and function due to various environmental factors, including diet, microbiome, pollutants, and lifestyle choices. Several studies have demonstrated that exposure to environmental pollutants, such as particulate matter and cigarette smoke, can impair lung function and increase the risk of lung diseases in both humans and animals. Similarly, dietary factors and gut microbiota have been shown to play a critical role in lung health and disease.124 Despite these cross-species differences, the murine lung exhibits some complexity comparable to that of the human lung, making it a valuable model for investigating lung development and potential therapeutic approaches in the clinical setting.
Bleomycin-induced lung fibrosis is a model used to study the development of lung fibrosis, which is a condition characterized by the accumulation of excess fibrous tissue in the lungs. In this model, bleomycin, a chemotherapy drug, is administered to laboratory animals, typically mice, and its effects on the lung tissue are monitored. Bleomycin-induced lung fibrosis has also been used to study the therapeutic potential of stem cells in the treatment of lung fibrosis. The rationale behind this approach is that stem cells can differentiate into various cell types, including lung cells, and can be used to repair damaged tissue. Studies have shown that stem cells, including bone marrow-derived mesenchymal stem cells (BM-MSCs) and embryonic stem cells (ESCs), can reduce fibrosis and improve lung function when administered to animals with bleomycin-induced lung fibrosis. These cells appear to exert their therapeutic effects by reducing inflammation, suppressing fibroblast activation, and promoting the regeneration of lung tissue. These changes mimic those seen in human lung fibrosis, making the bleomycin-induced lung fibrosis model a valuable tool for studying the underlying mechanisms of this condition and for testing potential therapeutic approaches. In addition to changes in cellular morphology and proliferation rate, the local microenvironment of the damaged tissue also undergoes significant change. A complex network of signals and immune interactions may influence changes in stem cell behavior, quiescence, regeneration, and homeostasis. Exploring the functionality of resident stem cells, effector cells, and memory T cells may provide valuable insights into understanding the interactions between scarred epithelial tissue and stem cells.
Currently, researchers are employing lineage tracing experiments, cell labeling methods, and reporter systems to better understand the bleomycin-induced systemic injury (Table 1). However, there are several challenges in accurately identifying differentiating cells, characterizing transformed cells, and tracking transdifferentiation due to the spatial and temporal changes in the lineage trajectory. Additionally, the heterogeneity and existence of divergent subpopulations of fibroblasts present significant obstacles in comprehending lung fibrosis.125 Another challenge to the lineage tracing approach is the silencing of reporters during the transdifferentiation of cells, a problem acerbated when stem cells terminally differentiate. Bacterial artificial chromosome (BAC)-mediated stable gene expression is a powerful tool that enables the introduction of large genomic regions, including entire genes and their regulatory elements, into cells to achieve stable and physiologically relevant gene expression.126,127 This technique can be particularly useful in the context of lung fibrosis for introducing key reporters for lineage tracing, or through controlled expression of genes or signaling pathways regulating the differentiation and function of alveolar progenitor cells, including AT2 cells.
Importantly, there is a growing body of evidence supporting the use of human umbilical mesenchymal stem cells (HUMSCs) as a therapeutic approach for bleomycin-induced pulmonary fibrosis. HUMSCs regulate immune responses and synthesize hyaluronan to enhance lung regeneration.128 However, the use of stem cells in the treatment of lung fibrosis is still in its early stages and further research is needed to fully understand their mechanisms of action and to optimize their therapeutic potential. Despite these limitations, the use of stem cells in the treatment of bleomycin-induced lung fibrosis has provided valuable insights into the biological processes involved in lung fibrosis and has generated hope for the development of new therapeutic strategies for this debilitating disease. In the current review, we have summarized the various aspects of resident stem cells in lung tissue and their role in bleomycin-induced lung injury and the repair process. We believe that the key challenges in understanding the behavior of lung stem cell progenitors include:
- Characterizing newly identified AT2 stem cell subpopulations involved in post-injury repair: AT2 cells have been identified as alveolar stem cells that play an important role in lung repair and regeneration following injury.6 These cells possess the capacity for long-term self-renewal and differentiation to AT1 cells. Recent research has focused on characterizing newly identified subpopulations of AT2 cells that are involved in the post-injury repair. Approaches such as lineage tracing, 3D organoid culture, and single-cell RNA sequencing (scRNAseq) have been used to study these cells and identify key molecular pathways and signaling pathways involved in their activation and differentiation.
While 3D organoid culture has been effective in modeling the behavior of AT2 cells, it does not fully replicate the complex microenvironment of the human alveolar stem cell niche, which is critical for their differentiation into AT1 cells. To overcome these limitations, researchers are developing more sophisticated models that better recapitulate the niche microenvironment of AT2 cells.5,95 For example, microfluidic devices have been used to create dynamic environments that mimic the alveolar niche, allowing for the study of AT2 cells under more physiologically relevant conditions.129 Additionally, the use of decellularized lung scaffolds has shown promise in creating a more realistic microenvironment for studying AT2 cell behavior and differentiation.130 By improving our understanding of the complex microenvironment that supports the differentiation and maintenance of AT2 cells, researchers can develop more effective strategies for promoting lung repair and regeneration in patients with lung injury or disease.
Overall, a better understanding of the complex transcriptional regulatory networks that control AT2 stem cell properties will be critical for the development of novel therapeutic strategies for lung injury and disease. By identifying the specific transcription factors that control the proliferation and differentiation of these cells, researchers may be able to target these factors to promote lung repair and regeneration.
Summarily, the present review article holds significant importance in the field of regenerative medicine, specifically in the context of idiopathic pulmonary fibrosis (IPF). By using the bleomycin-induced lung injury model in mice, which mimics the pathological features of human IPF, researchers have demonstrated that stem cell therapy can reduce inflammation, prevent fibrosis progression, and promote tissue regeneration in the injured lung. The article delves into the mechanisms through which various types of stem cells, such as mesenchymal stem cells, embryonic stem cells, induced pluripotent stem cells, and lung resident stem cells, exert their therapeutic effects in this specific model. Understanding the regenerative capabilities of these stem cells could pave the way for the development of targeted and effective therapies for lung-related disorders in humans. Moreover, this review also highlights the challenges and opportunities associated with translating stem cell therapy to the clinic for IPF patients, shedding light on potential avenues for further advancements in regenerative medicine. In essence, this comprehensive review offers hope to IPF patients while also carrying broader implications for regenerative medicine, providing valuable insights into the regenerative potential of stem cells across various organ systems.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gonzales JN, Lucas R, Verin AD. The acute respiratory distress syndrome: mechanisms and perspective therapeutic approaches. Austin J Vascular Med. 2015;2(1):1009.
2. Eldridge L. Alveoli function, structure, and lung disorders that affect them. Lung Heal. 2022.
3. Carcaterra M, Caruso C. Alveolar epithelial cell type II as main target of SARS-CoV-2 virus and COVID-19 development via NF-Kb pathway deregulation: a physio-pathological theory. Med Hypotheses. 2021;146:110412. doi:10.1016/j.mehy.2020.110412
4. Wang Y, Tang Z, Huang H, et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc Natl Acad Sci. 2018;115(10):2407–2412. doi:10.1073/pnas.1719474115
5. Barkauskas CE, Cronce MJ, Rackley CR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123(7):3025–3036. doi:10.1172/JCI68782
6. Jain R, Barkauskas CE, Takeda N, et al. Plasticity of Hopx(+) type I alveolar cells to regenerate type II cells in the lung. Nat Commun. 2015;6:6727. doi:10.1038/ncomms7727
7. Adamson IY. Pulmonary toxicity of bleomycin. Environ Health Perspect. 1976;16:119–125. doi:10.1289/ehp.7616119
8. Álvarez D, Levine M, Rojas M. Regenerative medicine in the treatment of idiopathic pulmonary fibrosis: current position. Stem Cells Cloning. 2015;8:61–65. doi:10.2147/SCCAA.S49801
9. Confalonieri P, Volpe MC, Jacob J, et al. Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Cells. 2022;11(13). doi:10.3390/cells11132095
10. Saito M, Mitani A, Ishimori T, et al. Active mTOR in Lung epithelium promotes epithelial–mesenchymal transition and enhances lung fibrosis. Am J Respir Cell Mol Biol. 2020;62(6):699–708. doi:10.1165/rcmb.2019-0255OC
11. Yuan S, Zuo B, Zhou S-C, et al. Integrating Network pharmacology and experimental validation to explore the pharmacological mechanism of astragaloside iv in treating bleomycin-induced pulmonary fibrosis. Drug Des Devel Ther. 2023;17:1289–1302. doi:10.2147/DDDT.S404710
12. Hama Amin BJ, Kakamad FH, Ahmed GS, et al. Post COVID-19 pulmonary fibrosis; a meta-analysis study. Ann Med Surg. 2022;77:103590. doi:10.1016/j.amsu.2022.103590
13. Hirawat R, Jain N, Aslam Saifi M, Rachamalla M, Godugu C. Lung fibrosis: post-COVID-19 complications and evidences. Int Immunopharmacol. 2023;116:109418. doi:10.1016/j.intimp.2022.109418
14. Moore B, Lawson WE, Oury TD, Sisson TH, Raghavendran K, Hogaboam CM. Animal Models of Fibrotic Lung Disease. Am J Respir Cell Mol Biol. 2013;49(2):167–179. doi:10.1165/rcmb.2013-0094TR
15. Helling BA, Yang IV. Epigenetics in lung fibrosis: from pathobiology to treatment perspective. Curr Opin Pulm Med. 2015;21(5):454–462. doi:10.1097/MCP.0000000000000191
16. Tashiro J, Rubio GA, Limper AH, et al. Exploring Animal Models That Resemble Idiopathic Pulmonary Fibrosis. Front Med. 2017:4. doi:10.3389/fmed.2017.00118
17. Snider GL, Celli BR, Goldstein RH, O’Brien JJ, Lucey EC. Chronic interstitial pulmonary fibrosis produced in hamsters by endotracheal bleomycin. Lung volumes, volume-pressure relations, carbon monoxide uptake, and arterial blood gas studied. Am Rev Respir Dis. 1978;117(2):289–297. doi:10.1164/ARRD.1978.117.2.289
18. Adamson IYR, Young L, Bowden DH. Relationship of alveolar epithelial injury and repair to the induction of pulmonary fibrosis. Am J Pathol. 1988;130(2):377.
19. Kuhn C, Boldt J, King TE, Crouch E, Vartio T, McDonald JA. An Immunohistochemical study of architectural remodeling and connective tissue synthesis in pulmonary fibrosis. Am Rev Respir Dis. 2012;140(6):1693–1703. doi:10.1164/AJRCCM/140.6.1693
20. Zhang K, Rekhter MD, Gordon D, Phan SH. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am J Pathol. 1994;145(1):114.
21. Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100(4):768–776. doi:10.1172/JCI119590
22. Hagimoto N, Kuwano K, Inoshima I, et al. TGF-β1 as an Enhancer of Fas-Mediated Apoptosis of Lung Epithelial Cells. J Immunol. 2002;168(12):6470–6478. doi:10.4049/JIMMUNOL.168.12.6470
23. Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R. Time course of bleomycin-induced lung fibrosis. Int J Exp Pathol. 2002;83(3):111–119. doi:10.1046/J.1365-2613.2002.00220.X
24. Moodley YP, Misso NLA, Scaffidi AK, et al. Inverse Effects of Interleukin-6 on Apoptosis of Fibroblasts from Pulmonary Fibrosis and Normal Lungs. Am Rev Respir Dis. 2003;29(4):490–498. doi:10.1165/RCMB.2002-0262OC
25. Kim KK, Kugler MC, Wolters PJ, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103(35):13180–13185. doi:10.1073/PNAS.0605669103/SUPPL_FILE/05669FIG_9.JPG
26. Alder JK, Chen JJL, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13051–13056. doi:10.1073/PNAS.0804280105/SUPPL_FILE/0804280105SI.PDF
27. Sisson TH, Mendez M, Choi K, et al. Targeted injury of type ii alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181(3):254–263. doi:10.1164/rccm.200810-1615OC
28. Marmai C, Sutherland RE, Kim KK, et al. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am J Physiol - Lung Cell Mol Physiol. 2011;301(1):71–78. doi:10.1152/AJPLUNG.00212.2010/SUPPL_FILE/SUPPDATA.PDF
29. Raghu G, Collard HR, Egan JJ, et al. An Official ATS/ERS/JRS/ALAT Statement: idiopathic Pulmonary Fibrosis: evidence-based Guidelines for Diagnosis and Management. Am J Respir Crit Care Med. 2011;183(6):788. doi:10.1164/RCCM.2009-040GL
30. Rock JR, Barkauskas CE, Cronce MJ, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108(52):E1475–E1483. doi:10.1073/PNAS.1117988108/SUPPL_FILE/PNAS.201117988SI.PDF
31. Huang SXL, Islam MN, O’Neill J, et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol. 2013;32(1):84–91. doi:10.1038/nbt.2754
32. Brandt JP, Gerriets V. Bleomycin. xPharm Compr Pharmacol Ref. 2022;1–6. doi:10.1016/B978-008055232-3.61328-5
33. Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. 2008;40(3):362–382. doi:10.1016/j.biocel.2007.08.011
34. Schuster DP. Acute lung injury and predictors of mortality. Am J Physiol Lung Cell Mol Physiol. 2003;285(1):L18–9. doi:10.1152/ajplung.00052.2003
35. Degryse AL, Tanjore H, Xu XC, et al. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;299(4):L442–L452. doi:10.1152/ajplung.00026.2010
36. Chaudhary NI, Schnapp A, Park JE. Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am J Respir Crit Care Med. 2006;173(7):769–776. doi:10.1164/rccm.200505-717OC
37. Laurent GJ. Lung collagen: more than scaffolding. Thorax. 1986;41(6):418–428. doi:10.1136/thx.41.6.418
38. Tan W, Wang Y, Chen Y, Chen C. Cell tracing reveals the transdifferentiation fate of mouse lung epithelial cells during pulmonary fibrosis in vivo. Exp Ther Med. 2021;22(4):1188. doi:10.3892/etm.2021.10622
39. Redente EF, Black BP, Backos DS, et al. Persistent, progressive pulmonary fibrosis and epithelial remodeling in mice. Am J Respir Cell Mol Biol. 2021;64(6):669–676. doi:10.1165/rcmb.2020-0542MA
40. Habiel DM, Hogaboam CM. Heterogeneity of fibroblasts and myofibroblasts in pulmonary fibrosis. Curr Pathobiol Rep. 2017;5(2):101–110. doi:10.1007/s40139-017-0134-x
41. Hogan BL, Barkauskas CE, Chapman HA, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. 2014;15(2):123–138. doi:10.1016/j.stem.2014.07.012
42. Walkin L, Herrick SE, Summers A, et al. The role of mouse strain differences in the susceptibility to fibrosis: a systematic review. Fibrogenesis Tissue Repair. 2013;6(1):18. doi:10.1186/1755-1536-6-18
43. Hoyt DG, Lazo JS. Alterations in pulmonary mRNA encoding procollagens, fibronectin and transforming growth factor-beta precede bleomycin-induced pulmonary fibrosis in mice. J Pharmacol Exp Ther. 1988;246(2):765 LP–771.
44. Antonini JM, Hemenway DR, Davis GS. Quantitative image analysis of lung connective tissue in murine silicosis. Exp Lung Res. 2000;26(2):71–88. doi:10.1080/019021400269880
45. Hartmann W, Blankenhaus B, Brunn M-L, Meiners J, Breloer M. Elucidating different pattern of immunoregulation in BALB/c and C57BL/6 mice and their F1 progeny. Sci Rep. 2021;11(1):1536. doi:10.1038/s41598-020-79477-7
46. VanSeggelen H, Hammill JA, Dvorkin-Gheva A, et al. T Cells Engineered With Chimeric Antigen Receptors Targeting NKG2D Ligands Display Lethal Toxicity in Mice. Mol Ther. 2015;23(10):1600–1610. doi:10.1038/mt.2015.119
47. Mehta AJ, Guidot DM. Alcohol and the Lung. Alcohol Res. 2017;38(2):243–254.
48. Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345(7):517–525. doi:10.1056/NEJMra003200
49. Vyalov SL, Gabbiani G, Kapanci Y. Rat alveolar myofibroblasts acquire alpha-smooth muscle actin expression during bleomycin-induced pulmonary fibrosis. Am J Pathol. 1993;143(6):1754–1765.
50. Rock JR, Hogan BLM. Epithelial progenitor cells in lung development, maintenance, repair, and disease. Annu Rev Cell Dev Biol. 2011;27(1):493–512. doi:10.1146/annurev-cellbio-100109-104040
51. Huang E, Peng N, Xiao F, Hu D, Wang X, Lu L. The roles of immune cells in the pathogenesis of fibrosis. Int J Mol Sci. 2020;21(15):5203. doi:10.3390/ijms21155203
52. Moog MT, Hinze C, Bormann T, et al. B cells are not involved in the regulation of adenoviral tgf-β1– or bleomycin-induced lung fibrosis in mice. J Immunol. 2022;208(5):1259–1271. doi:10.4049/jimmunol.2100767
53. Pociask DA, Chen K, Choi SM, Oury TD, Steele C, Kolls JK. γδ T cells attenuate bleomycin-induced fibrosis through the production of CXCL10. Am J Pathol. 2011;178(3):1167–1176. doi:10.1016/j.ajpath.2010.11.055
54. Wilson MS, Madala SK, Ramalingam TR, et al. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med. 2010;207(3):535–552. doi:10.1084/jem.20092121
55. Choi J, Park JE, Tsagkogeorga G, et al. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell. 2020;27(3):366–382.e7. doi:10.1016/j.stem.2020.06.020
56. Riise R, Odqvist L, Mattsson J, et al. Bleomycin hydrolase regulates the release of chemokines important for inflammation and wound healing by keratinocytes. Sci Rep. 2019;9(1):20407. doi:10.1038/s41598-019-56667-6
57. Clarke DL, Carruthers AM, Mustelin T, Murray LA. Matrix regulation of idiopathic pulmonary fibrosis: the role of enzymes. Fibrogenesis Tissue Repair. 2013;6(1):20. doi:10.1186/1755-1536-6-20
58. Adair-Kirk TL, Senior RM. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol. 2008;40(6–7):1101–1110. doi:10.1016/j.biocel.2007.12.005
59. Hettiarachchi SU, Y-H L, Roy J, et al. Targeted inhibition of PI3 kinase/mTOR specifically in fibrotic lung fibroblasts suppresses pulmonary fibrosis in experimental models. Sci Transl Med. 2020;12(567):eaay3724. doi:10.1126/scitranslmed.aay3724
60. Salton F, Ruaro B, Confalonieri P, Confalonieri M. Epithelial–mesenchymal transition: a major pathogenic driver in idiopathic pulmonary fibrosis? Medicina. 2020;56(11). doi:10.3390/medicina56110608
61. Salton F, Volpe MC, Confalonieri M. Epithelial–mesenchymal transition in the pathogenesis of idiopathic pulmonary fibrosis. Medicina. 2019;55(4). doi:10.3390/medicina55040083
62. Dai H, Zhu M, Li W, Si G, Xing Y. Activation of PI3K/p110α in the Lung mesenchyme affects branching morphogenesis and club cell differentiation. Front Cell Dev Biol. 2022;10. doi:10.3389/fcell.2022.880206
63. Jolly MK, Ward C, Eapen MS, et al. Epithelial–mesenchymal transition, a spectrum of states: role in lung development, homeostasis, and disease. Dev Dyn. 2018;247(3):346–358. doi:10.1002/dvdy.24541
64. Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113(2):243–252. doi:10.1172/jci18847
65. Willis BC, Liebler JM, Luby-Phelps K, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166(5):1321–1332. doi:10.1016/s0002-9440(10)62351-6
66. Wu H, Yu Y, Huang H, et al. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells. Cell. 2020;180(1):107–121.e17. doi:10.1016/j.cell.2019.11.027
67. Tropea KA, Leder E, Aslam M, et al. Bronchioalveolar stem cells increase after mesenchymal stromal cell treatment in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L829–L837. doi:10.1152/ajplung.00347.2011
68. Chen L-J, Ye H, Zhang Q, et al. Bleomycin induced epithelial–mesenchymal transition (EMT) in pleural mesothelial cells. Toxicol Appl Pharmacol. 2015;283(2):75–82. doi:10.1016/j.taap.2015.01.004
69. Fuchs E. Scratching the surface of skin development. Nature. 2007;445(7130):834–842. doi:10.1038/nature05659
70. Vaughan AE, Brumwell AN, Xi Y, et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517(7536):621–625. doi:10.1038/nature14112
71. Kumar PA, Hu Y, Yamamoto Y, et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell. 2011;147(3):525–538. doi:10.1016/j.cell.2011.10.001
72. Ye R, Wang M, Wang QA, et al. Autonomous interconversion between adult pancreatic α-cells and β-cells after differential metabolic challenges. Mol Metab. 2016;5(7):437–448. doi:10.1016/j.molmet.2016.05.001
73. Tata PR, Mou H, Pardo-Saganta A, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503(7475):218–223. doi:10.1038/nature12777
74. Bergmann A, Steller H. Apoptosis, stem cells, and tissue regeneration. Sci Signal. 2010;3(145):re8. doi:10.1126/scisignal.3145re8
75. Kobayashi Y, Tata A, Konkimalla A, et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol. 2020;22(8):934–946. doi:10.1038/s41556-020-0542-8
76. Chung M-I, Bujnis M, Barkauskas CE, Kobayashi Y, Hogan BLM. Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development. 2018;145(9):dev163014. doi:10.1242/dev.163014
77. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–689. doi:10.1016/j.cell.2006.06.044
78. Sucre JMS, Bock F, Negretti NM, et al. Alveolar epithelial cell differentiation during lung repair requires cell-extracellular matrix interactions. bioRxiv. 2022. doi:10.1101/2022.08.05.502988
79. Parnaik VK, Chaturvedi P. Fluorescence recovery after photobleaching studies reveal complexity of nuclear architecture. Int J Chem. 2015;4(4):297–302.
80. Muralikrishna B, Chaturvedi P, Sinha K, Parnaik VK. Lamin misexpression upregulates three distinct ubiquitin ligase systems that degrade ATR kinase in HeLa cells. Mol Cell Biochem. 2012;365(1–2):323–332. doi:10.1007/s11010-012-1272-4
81. Chaturvedi P, Parnaik VK. Lamin A rod domain mutants target heterochromatin protein 1α and β for proteasomal degradation by activation of F-box protein, FBXW10. PLoS One. 2010;5(5):e10620. doi:10.1371/journal.pone.0010620
82. Thanumalayan S, Sehgal P, Muralikrishna B, et al. A rare mutation in lamin A gene is associated with dilated cardiomyopathy in Indian patients. Eur J Mol Biol Biochem. 2015;2(5):190–196.
83. Sehgal P, Chaturvedi P, Kumaran RI, Kumar S, Parnaik VK. Lamin A/C haploinsufficiency modulates the differentiation potential of mouse embryonic stem cells. PLoS One. 2013;8(2):e57891. doi:10.1371/journal.pone.0057891
84. Neumark N, Cosme C, Rose K-A, Kaminski N. The Idiopathic Pulmonary Fibrosis Cell Atlas. Am J Physiol Cell Mol Physiol. 2020;319(6):L887–L892. doi:10.1152/ajplung.00451.2020
85. Habermann AC, Gutierrez AJ, Bui LT, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv. 2020;6(28):eaba1972. doi:10.1126/sciadv.aba1972
86. Strunz M, Simon LM, Ansari M, et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun. 2020;11(1):3559. doi:10.1038/s41467-020-17358-3
87. Boumahdi S, de Sauvage FJ. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov. 2020;19(1):39–56. doi:10.1038/s41573-019-0044-1
88. Sehgal P, Chaturvedi P. Chromatin and cancer: implications of disrupted chromatin organization in tumorigenesis and its diversification. Cancers. 2023;15(2). doi:10.3390/cancers15020466
89. Potten CS, Morris RJ. Epithelial stem cells in vivo. J Cell Sci. 1988;1988(Supplement_10):45–62. doi:10.1242/jcs.1988.Supplement_10.4
90. Verheyden JM, Sun X. A transitional stem cell state in the lung. Nat Cell Biol. 2020;22(9):1025–1026. doi:10.1038/s41556-020-0561-5
91. Parimon T, Yao C, Stripp BR, Noble PW, Chen P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int J Mol Sci. 2020;21(7):2269. doi:10.3390/ijms21072269
92. Shen M, Luo Z, Zhou Y. Regeneration-Associated Transitional State Cells in Pulmonary Fibrosis. Int J Mol Sci. 2022;23(12). doi:10.3390/ijms23126757
93. Chen Q, Suresh Kumar V, Finn J, et al. CD44(high) alveolar type II cells show stem cell properties during steady-state alveolar homeostasis. Am J Physiol Lung Cell Mol Physiol. 2017;313(1):L41–L51. doi:10.1152/ajplung.00564.2016
94. Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11(4):254–267. doi:10.1038/nrc3023
95. Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. 2018;359(6380):1118–1123. doi:10.1126/science.aam6603
96. Zacharias WJ, Frank DB, Zepp JA, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. 2018;555(7695):251–255. doi:10.1038/nature25786
97. Liu Y, Kumar VS, Zhang W, Rehman J, Malik AB. Activation of type II cells into regenerative stem cell antigen-1(+) cells during alveolar repair. Am J Respir Cell Mol Biol. 2015;53(1):113–124. doi:10.1165/rcmb.2013-0497OC
98. Liu Y, Sadikot RT, Adami GR, et al. FoxM1 mediates the progenitor function of type II epithelial cells in repairing alveolar injury induced by Pseudomonas aeruginosa. J Exp Med. 2011;208(7):1473–1484. doi:10.1084/jem.20102041
99. Kadur Lakshminarasimha Murthy P, Sontake V, Tata A, et al. Human distal lung maps and lineage hierarchies reveal a bipotent progenitor. Nature. 2022;604(7904):111–119. doi:10.1038/s41586-022-04541-3
100. Paris AJ, Hayer KE, Oved JH, et al. STAT3–BDNF–TrkB signalling promotes alveolar epithelial regeneration after lung injury. Nat Cell Biol. 2020;22(10):1197–1210. doi:10.1038/s41556-020-0569-x
101. Levy DE. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651–662. doi:10.1038/nrm909
102. Finn J, Sottoriva K, Pajcini KV, et al. Dlk1-Mediated Temporal Regulation of Notch Signaling Is Required for Differentiation of Alveolar Type II to Type I Cells during Repair. Cell Rep. 2019;26(11):2942–2954.e5. doi:10.1016/j.celrep.2019.02.046
103. Olajuyin AM, Zhang X, Ji H-L. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discov. 2019;5(1):63. doi:10.1038/s41420-019-0147-9
104. Geiger H, Zheng Y. Cdc42 and aging of hematopoietic stem cells. Curr Opin Hematol. 2013;20(4):295–300. doi:10.1097/MOH.0b013e3283615aba
105. Xie T, Lynn H, Parks WC, et al. Abnormal respiratory progenitors in fibrotic lung injury. Stem Cell Res Ther. 2022;13(1):64. doi:10.1186/s13287-022-02737-y
106. Yin Z, Gonzales L, Kolla V, et al. Hop functions downstream of Nkx2.1 and GATA6 to mediate HDAC-dependent negative regulation of pulmonary gene expression. Am J Physiol Lung Cell Mol Physiol. 2006;291(2):L191–9. doi:10.1152/ajplung.00385.2005
107. Saul D, Kosinsky RL, Atkinson EJ, et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat Commun. 2022;13(1):4827. doi:10.1038/s41467-022-32552-1
108. Woodcock HV, Eley JD, Guillotin D, et al. The mTORC1/4E-BP1 axis represents a critical signaling node during fibrogenesis. Nat Commun. 2019;10(1):6. doi:10.1038/s41467-018-07858-8
109. Allen RJ, Guillen-Guio B, Oldham JM, et al. Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2019;201(5):564–574. doi:10.1164/rccm.201905-1017OC
110. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N Engl J Med. 2014;370(22):2071–2082. doi:10.1056/NEJMoa1402584
111. Miceli V, Pampalone M, Vella S, Carreca AP, Amico G, Conaldi PG. Comparison of Immunosuppressive and Angiogenic Properties of Human Amnion-Derived Mesenchymal Stem Cells between 2D and 3D Culture Systems. Stem Cells Int. 2019;2019:7486279. doi:10.1155/2019/7486279
112. Shu J, He X, Li H, et al. The Beneficial Effect of Human Amnion Mesenchymal Cells in Inhibition of Inflammation and Induction of Neuronal Repair in EAE Mice. J Immunol Res. 2018;2018:5083797. doi:10.1155/2018/5083797
113. Weng Z, Zhang B, Wu C, et al. Therapeutic roles of mesenchymal stem cell-derived extracellular vesicles in cancer. J Hematol Oncol. 2021;14(1):136. doi:10.1186/s13045-021-01141-y
114. Sang L, Guo X, Fan H, Shi J, Hou S, Lv Q. Mesenchymal stem cell-derived extracellular vesicles as idiopathic pulmonary fibrosis microenvironment targeted delivery. Cells. 2022;11(15):2322. doi:10.3390/cells11152322
115. Periera‐Simon S, Xia X, Catanuto P, et al. Anti-fibrotic effects of different sources of MSC in bleomycin-induced lung fibrosis in C57BL6 male mice. Respirology. 2021;26(2):161–170. doi:10.1111/resp.13928
116. Rubio GA, Elliot SJ, Wikramanayake TC, et al. Mesenchymal stromal cells prevent bleomycin-induced lung and skin fibrosis in aged mice and restore wound healing. J Cell Physiol. 2018;233(8):5503–5512. doi:10.1002/jcp.26418
117. Duan F, Guo L, Yang L, et al. Modeling COVID-19 with Human Pluripotent Stem Cell-Derived Cells Reveals Synergistic Effects of Anti-inflammatory Macrophages with ACE2 Inhibition Against SARS-CoV-2. Res Sq. 2020. doi:10.21203/rs.3.rs-62758/v1
118. Coraux C, Nawrocki-Raby B, Hinnrasky J, et al. Embryonic stem cells generate airway epithelial tissue. Am J Respir Cell Mol Biol. 2005;32(2):87–92. doi:10.1165/rcmb.2004-0079RC
119. Wang D, Morales JE, Calame DG, Alcorn JL, Wetsel RA. Transplantation of human embryonic stem cell-derived alveolar epithelial type II cells abrogates acute lung injury in mice. Mol Ther. 2010;18(3):625–634. doi:10.1038/mt.2009.317
120. Soh BS, Zheng D, Li Yeo JS, et al. CD166(pos) subpopulation from differentiated human ES and iPS cells support repair of acute lung injury. Mol Ther. 2012;20(12):2335–2346. doi:10.1038/mt.2012.182
121. Glassberg MK, Minkiewicz J, Toonkel RL, et al. Allogeneic Human Mesenchymal Stem Cells in Patients With Idiopathic Pulmonary Fibrosis via Intravenous Delivery (AETHER): a Phase I Safety Clinical Trial. Chest. 2017;151(5):971–981. doi:10.1016/j.chest.2016.10.061
122. Vats A, T-C H, Puc I, et al. Evidence that hematopoietic stem cells in human umbilical cord blood is infectable by dengue virus: proposing a vertical transmission candidate. Heliyon. 2021;7(4). doi:10.1016/j.heliyon.2021.e06785
123. Varghese B, Ling Z, Ren X. Reconstructing the pulmonary niche with stem cells: a lung story. Stem Cell Res Ther. 2022;13(1):161. doi:10.1186/s13287-022-02830-2
124. Charlson ES, Bittinger K, Haas AR, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184(8):957–963. doi:10.1164/rccm.201104-0655oc
125. Fries KM, Blieden T, Looney RJ, et al. Evidence of fibroblast heterogeneity and the role of fibroblast subpopulations in fibrosis. Clin Immunol Immunopathol. 1994;72(3):283–292. doi:10.1006/clin.1994.1144
126. Chaturvedi P, Zhao B, Zimmerman DL, Belmont AS. Stable and reproducible transgene expression independent of proliferative or differentiated state using BAC TG-EMBED. Gene Ther. 2018;25(5):376–391. doi:10.1038/s41434-018-0021-z
127. Zhao B, Chaturvedi P, Zimmerman DL, Belmont AS. Efficient and reproducible multigene expression after single-step transfection using improved bac transgenesis and engineering toolkit. ACS Synth Biol. 2020;9(5):1100–1116. doi:10.1021/acssynbio.9b00457
128. Moodley Y, Atienza D, Manuelpillai U, et al. Human umbilical cord mesenchymal stem cells reduce fibrosis of bleomycin-induced lung injury. Am J Pathol. 2009;175(1):303–313. doi:10.2353/ajpath.2009.080629
129. Hiemstra PS, Tetley TD, Janes SM. Airway and alveolar epithelial cells in culture. Eur Respir J. 2019;54(5):1900742. doi:10.1183/13993003.00742-2019
130. Gilpin SE, Charest JM, Ren X, et al. Regenerative potential of human airway stem cells in lung epithelial engineering. Biomaterials. 2016;108:111–119. doi:10.1016/j.biomaterials.2016.08.055
131. Sieber P, Schäfer A, Lieberherr R, et al. NF-κB drives epithelial-mesenchymal mechanisms of lung fibrosis in a translational lung cell model. JCI Insight. 2023;8(3). doi:10.1172/jci.insight.154719
132. Perona R, Montaner S, Saniger L, Sánchez-Pérez I, Bravo R, Lacal JC. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11(4):463–475. doi:10.1101/gad.11.4.463
133. Sehgal P, Kong X, Wu J, Sunyer R, Trepat X, Leckband D. Epidermal growth factor receptor and integrins control force-dependent vinculin recruitment to E-cadherin junctions. J Cell Sci. 2018;131(6):jcs206656. doi:10.1242/jcs.206656
134. Zepp JA, Zacharias WJ, Frank DB, et al. Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell. 2017;170(6):1134–1148.e10. doi:10.1016/j.cell.2017.07.034
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.