")
Back to Archived Journals » Gastrointestinal Cancer: Targets and Therapy » Volume 5
The potential of targeted antiangiogenesis therapies in the treatment of esophageal cancer
Received 12 February 2015
Accepted for publication 20 March 2015
Published 29 April 2015 Volume 2015:5 Pages 79—88
DOI https://doi.org/10.2147/GICTT.S49313
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Eileen O'Reilly
Wen Wen Xu,1,2 Bin Li,1,2,3 Annie L M Cheung1,2,3
1Department of Anatomy, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pokfulam, 2The University of Hong Kong-Shenzhen Institute of Research and Innovation (HKU-SIRI), 3Centre for Cancer Research, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pokfulam, Hong Kong SAR, People's Republic of China
Abstract: Esophageal cancer is one of the most lethal cancers worldwide and its incidence is increasing at an alarming rate. Despite advances in surgical techniques and combined multimodality therapy, the survival rate of esophageal cancer remains poor. Clearly, the time is ripe for introducing novel strategies such as targeted therapies to improve treatment outcome. The significance of angiogenesis and angiogenic factors in the progression and aggressiveness of esophageal cancer is well documented. However, although increasing numbers of antiangiogenic agents designed to inhibit angiogenesis through multiple mechanisms have been developed in the past few decades, and some of them have shown promising results in preclinical and clinical studies, there is as yet no antiangiogenic agent approved for esophageal cancer. This review provides a summary of angiogenesis and vasculogenesis, the molecular mechanisms that regulate these processes, and the strategies for targeting angiogenesis in tumors. We will also present the rationale and challenges of antiangiogenic therapy, and the antiangiogenic agents that have been tested in preclinical studies or clinical trials for esophageal cancer. With further research bringing a deeper understanding of the molecular mechanisms involved in angiogenesis, and as new angiogenesis-targeting agents continue to evolve, there are reasons to be optimistic that targeting angiogenesis may bring new opportunities to cure this highly lethal disease.
Keywords: esophageal cancer, angiogenesis, vascular endothelial growth factor, targeted therapies
Introduction
Esophageal cancer is the sixth most lethal cancer worldwide and its incidence is on the rise.1,2 Esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC) are the two predominant subtypes of esophageal cancer.3 ESCC develops from epithelial cells and can occur anywhere along the esophagus, whereas EAC arises from glandular cells in the lower third of the esophagus, which replace an area of squamous cells. For tumors that occur in the gastroesophageal junction (GEJ), those in which the epicenter is within 5 cm of the GEJ and also extends into the esophagus, are also classified by the International Union Against Cancer as esophageal cancer.4 Worldwide, ~90% of the new cases of esophageal cancer are ESCC and two-thirds of the cases occur in Asia. However, the past 30 years have witnessed a significant increase in the incidence of esophageal cancer in the United States and Europe. According to the latest data published by the American Cancer Society, there were 18,170 new cases and 15,450 deaths of esophageal cancer in the US in 2014. Smoking, alcohol consumption, hot drinks, and poor diet are regarded as the main risk factors for squamous cell-type esophageal cancer, whereas the most common causes of the adenocarcinoma-type esophageal cancer are gastroesophageal reflux disease, Barrett’s esophagus (BE), and obesity.3 The fact that most esophageal cancer patients have advanced disease at the time of diagnosis makes surgery futile in the majority of cases. For the patients deemed inoperable, the combination of chemotherapy and radiotherapy is becoming more and more important. Despite the great improvements in diagnosis, surgical resection, and neoadjuvant/adjuvant chemoradiotherapy in the past few decades, the 5-year survival rate of esophageal cancer rarely exceeds 30%.5 Therefore, new treatment strategies for this highly lethal disease are urgently needed.
Angiogenesis and vasculogenesis
The term “angiogenesis” was firstly proposed in 1935 by Hertig6 to describe the physiological process through which new blood vessels form from pre-existing vessels in the placenta of pregnant monkeys. Angiogenesis occurs during development, wound healing, pregnancy, and other physiological processes. However, angiogenesis is also an essential step in the conversion of a tumor from benign to malignant and metastatic phenotypes. Tumor growth is a complex and multistep process involving recruitment of the neighboring blood vessels or endothelial cells to deliver oxygen and nutrients into the tumor microenvironment to construct a favorable environment for tumor growth. The tumor microenvironment is a complex system.7 Compelling evidence has shown that besides cancer cells, endothelial progenitor cells, pericytes, and fibroblasts also significantly contribute to the development of tumors.8–10
The concept that tumor growth mimics embryonic development is not new.11 It has become increasingly evident that a tumor can effectively hijack and exploit various physiological functions and revert quiescent cell types to their embryonic state of proliferation and differentiation. In the 1971 issue of the New England Journal of Medicine, Judah Folkman launched the idea that all tumors are angiogenesis-dependent.12 In 1984, the first tumor angiogenic factor, basic fibroblast growth factor (bFGF or FGF-2), was characterized by Shing et al13 from a chondrosarcoma. This finding further supported the notion that tumors can induce neovascularization by secreting angiogenic factors. In subsequent decades, an increasing number of angiogenic factors with distinct functions in the regulation of vessel growth and vascular remodeling, including matrix metalloproteinase (MMPs)14 and angiopoietins,15 were identified. Another key concept termed “angiogenic switch” refers to the moment at which a tumor overwhelms the delicate balance between pro- and antiangiogenic factors, and begins to express high levels of proangiogenic factors, resulting in the transition from a dormant state to an outgrowing vascularized tumor and eventually to malignancies.16 Now, angiogenesis is gaining more and more recognition and is regarded as one of the important hallmarks of cancer.17
Distinct from “angiogenesis”, which refers to blood vessels sprouting from already established vasculature, the term “vasculogenesis” describes the formation of new blood vessels when there are no pre-existing ones. In 2001, Lyden et al18 demonstrated that impaired recruitment of bone marrow-derived endothelial and hematopoietic precursor cells could effectively block tumor angiogenesis and growth. For the first time, they proposed that tumor growth is not only angiogenesis-dependent, but also vasculogenesis-dependent.18 Furthermore, the study by Kioi et al19 documented the importance of blocking vasculogenesis in the prevention of tumor recurrence. The growth factors secreted by tumor cells, such as vascular endothelial growth factor (VEGF) and transforming growth factor beta (TGF-β), can augment mobilization and recruitment of bone marrow-derived cells (BMDCs) which, in turn, facilitate tumor neovascularization.20–22 Several subpopulations of BMDCs, for example, CD11b+/Gr1+ bone marrow-derived myeloid cells,23 Tie2+-expressing monocytes,24 and VEGF receptor 1-positive (VEGFR1+) bone marrow-derived hematopoietic progenitor cells,21 have been observed to significantly contribute to tumor growth. Pharmacological manipulation by using antiangiogenic drugs or genetic regulation can retard mobilization of the various BMDCs, and thus reduce the angiogenic response and inhibit tumor progression. Our recent preclinical study showed that simply targeting nontumor cells that express VEGFR1 and VEGFR2 using neutralizing antibodies is sufficient to significantly suppress tumor growth and angiogenesis in immunodeficient mice xenografted with ESCC cells.25 However, our understanding of the formation of tumor vessels by vasculogenesis is still in its infancy. The respective roles of selective subpopulations of BMDCs and how they are integrated into the growing tumor remain to be further elucidated.
VEGF and molecular mechanisms of angiogenesis
VEGF was discovered in 1983 and was first identified as an angiogenic factor in 1989.26 VEGF belongs to the platelet-derived growth factor (PDGF) family and it is a collective name for a broad range of proteins, including VEGF-A (also referred to as VEGF because it was the first VEGF isoform to be discovered), VEGF-B, VEGF-C, and VEGF-D. The normal function of VEGF is to create new blood vessels during embryonic development or after injury, whereas the overexpression of VEGF is often related to disease, such as cancer.27 Emerging evidence demonstrated the significant association between VEGF expression and cancer progression and prognosis in a variety of gastrointestinal malignancies, including esophageal,28,29 gastric,30 colon,31 and many other cancer types. VEGF can be secreted by tumor or stromal cells to exert functions in an autocrine or paracrine manner. VEGF plays a predominant role in angiogenesis by binding to VEGF receptors (VEGFR1, VEGFR2, and VEGFR3), which are kinase insert domain-containing receptors, or to neuropilins (such as NRP1 and NRP2) which are a class of nontyrosine kinase receptors.32 VEGF receptors (VEGFRs) are expressed in normal tissues as well as in tumor cells. VEGFRs have been reported to play important roles in the control of cell proliferation and differentiation. VEGFR1 is the only known receptor for placental growth factor (PlGF) and VEGF-B, and it can be expressed as a soluble variant. Increasing evidence shows that VEGFR1 may be a promising therapeutic target for the treatment of cancer.33,34 Although VEGFR1 binds with high affinity to VEGF, VEGFR2 seems to mediate almost all aspects of the cellular responses to VEGF.35 It was proposed that VEGFR1 may work as a decoy receptor to facilitate the binding of VEGFR2 to VEGF.36 VEGFR3 is widely expressed in malignant vascular tumors, but mostly in the capillaries and not in the solid tumors. Neuropilins have a high affinity to bind to certain isoforms of VEGF. VEGF can be regulated by many stimuli through a multitude of mechanisms, including various cytokines (eg, interleukin [IL]-4, IL-10, type I interferons), growth factors (eg, TGF-β; FGF), and hormones, as summarized by Xie et al.37 VEGF expression is also under the influence of various stresses such as oxidative stress,38 hypoxia,39 and glucose deficiency.40
The regulation of VEGF happens at the gene transcription, translation, and post-translation levels. In particular, regulation at the transcription level has been extensively studied, and hypoxia-inducible factor-1, which is a master transcription factor activated by hypoxia, and specific protein-1, which responds to various extracellular stimuli, have been demonstrated to play a fundamental role in the expression and secretion of VEGF.41
Architecturally different from the normal blood vessels, tumor blood vessels are a chaotic network of irregularly shaped, dilated, and tortuous vessels.16 The upregulation of VEGF, which results in endothelial cell sprouting, and the interaction between endothelial cells, pericytes, and stroma cells account for the main mechanisms of tumor angiogenesis.42–44 Blood vessel branching during angiogenesis is a complex process that involves completion of a complex succession of events: 1) the presence of angiogenic factors such as VEGF and FGF induces degradation of the basement membrane, pericyte detachment, and loosening of the cell–cell junctional contacts to favor the navigation of delta-like 4-expressing tip cells; 2) with the guidance of tip cells, stalk cells proliferate, elongate, and form a lumen; and 3) the establishment of new blood vessels, including vascular maintenance, basement membrane deposition, barrier formation, and pericyte maturation.45 In terms of molecular mechanisms, the VEGF/VEGFR pathway can activate a variety of signaling pathways, including the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K/AKT) and mitogen-activated protein kinases/extracellular signal-regulated kinases (MAPK/ERK, also known as Ras/Raf/MEK/ERK) pathways, to induce the proliferation, migration, and production of proteases, and vascular hyperpermeability.46
Angiogenesis is orchestrated by various activators and repressors. Besides the VEGF family, several other molecules or signaling pathways including PDGF, TGF-β, the FGF superfamily, epidermal growth factor (EGF), hepatocyte growth factor/scatter factor (HGF/SF), the tumor necrosis factor family (TNF), as well as the NOTCH/WNT signaling pathway, also significantly contribute to the development and maturation of vessels.45 Recent studies showed that vascular metabolism also plays an important role in regulating angiogenesis and is a viable target for manipulating the microvasculature.47 It remains to be further determined how these various components/signaling pathways functionally crosstalk to facilitate the tumor growth.
Strategies for targeting angiogenesis in tumors
The vital role of tumor angiogenesis in solid tumor holds much promise for designing efficacious treatments for cancers. Amongst the strategies for blocking angiogenesis, some drugs are designed to directly inhibit the proliferation or response of normal endothelial cells. Another important strategy aims to block the growth factors which mediate the cell signaling pathways that stimulate angiogenesis.48 Antiangiogenic agents that are/were on clinical trials are mainly divided into several categories based on their mechanisms: 1) drugs that directly inhibit endothelial cell proliferation and/or migration, such as thalidomide;49 2) agents that block angiogenesis signaling, such as bevacizumab;50 3) drugs that interfere with degradation of the extracellular matrix, such as several MMP inhibitors; and 4) drugs that have multiple functions.51
To date, two important antiangiogenic approaches have achieved clinical development for cancer therapy (Figure 1). One is the development of targeted small-molecule protein–tyrosine kinase inhibitors (TKIs), the other one is the generation of monoclonal antibodies. Both approaches have distinctive properties. Small-molecule agents can permeate through plasma membranes and disperse within the cells to reach their targets. In comparison, monoclonal antibodies are relatively large and generally cannot enter cells, so they are used to target unique cell-specific surface markers and redirect the targets to the immune system for resolution. Another major difference between TKIs and monoclonal antibodies is that the former can inhibit a wide range of tyrosine kinases and signaling pathways, but monoclonal antibodies are specific for one or two targets.52,53 Compared with the conventional cytotoxic chemotherapy, antiangiogenic therapy is proposed to have several advantages: 1) it directly targets endothelial cells instead of working on tumor/epithelial cells; 2) it is not restricted to a specific histologic tumor entity; and 3) it is less likely to acquire drug resistance due to a lower chance of endothelial cells acquiring mutations.54
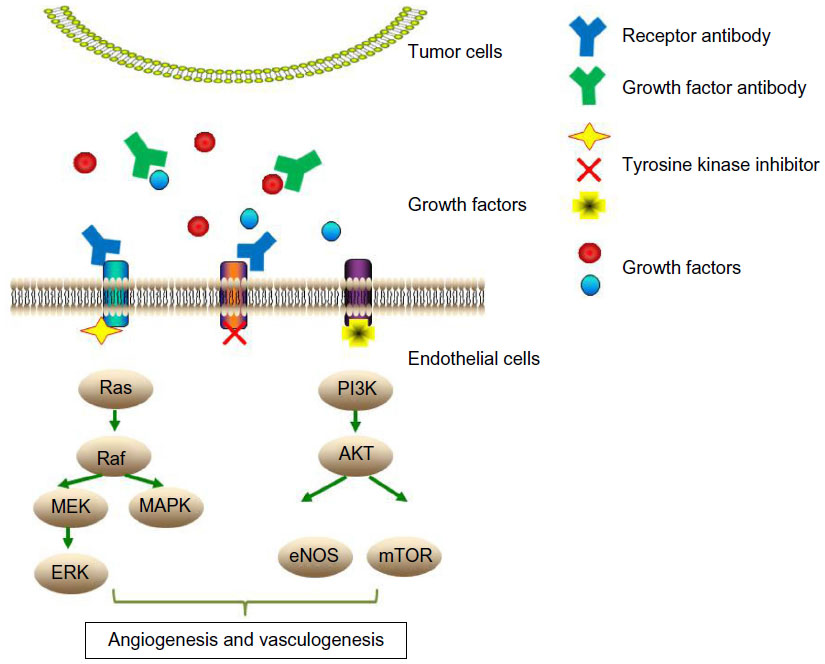 | Figure 1 Diagram depicting the key antiangiogenic approaches and the mechanisms of action of antiangiogenic agents. |
Rationale of targeting angiogenesis in esophageal cancer
The progression and aggressive behavior of esophageal cancer are associated with angiogenesis or VEGF/VEGFRs expression. BE is a precancerous condition for EAC, and patients with BE generally have a 30- to 50-fold greater risk of developing esophageal cancer.55 It had been reported that Barrett’s epithelium is strongly neovascularized.56 Another study based on 78 patients with Barrett’s adenocarcinoma found that VEGF is significantly correlated with vascularization in both metaplasia and tumor.57 These studies indicate that angiogenesis can happen as early as the transition from metaplasia to neoplasia in the development of esophageal malignancies. Several individual studies also found upregulation of VEGF in ESCC.58,59 Early lymphatic spread into local lymph nodes is a distinctive feature of esophageal cancer, and metastasis is one of the important reasons of high mortality.60 Kitadai et al61 evaluated the expression of VEGF-C in 12 preoperative biopsies and 48 ESCC surgical specimens, and found that VEGF-C expression has significant positive correlations with lymph node metastasis and distant metastasis. The positive association between VEGF expression and poor prognosis in patients with esophageal cancer was summarized in a meta-analysis covering 1,543 esophageal cancer patients with EAC or ESCC included in 19 studies published between 1997 and 2011.62 Another study comprising resected esophagus tissues in a cohort of 55 ESCC and 13 EAC patients showed that the high expression level of VEGFR1, one of the two dominant receptors for VEGF, is associated with poorer outcomes.63 Furthermore, our own recent study demonstrated a significant positive correlation between VEGFR2 expression and metastatic stage in a tissue array containing 86 cases of ESCC.25
Angiogenic factors circulating in the body fluid system, such as blood, can serve as useful biological markers. An analysis of serum VEGF-C and VEGF-D levels between esophageal cancer patients (110 with ESCC, 39 with EAC) and a healthy control group or patients with benign esophageal diseases showed that serum levels of VEGF-C and VEGF-D are associated with cancer stage and lymph node metastases.28 In particular, a high serum VEGF-C level was found to be a significant predictive marker for poor outcomes for patients undergoing potentially curative esophagectomy.28 In a more recent study, a comparison of serum levels between 89 patients with esophageal cancer (63 ESCC and 26 EAC) with a control group showed that high serum VEGF-A is associated with tumor progression and unfavorable prognosis in patients.64 This study showed that VEGF is not only an intratumoral biomarker, but also a circulating marker for esophageal cancer, which is in concordance with other cancer types.65–67
Besides VEGF, other angiogenic-related factors have been reported to have clinical relevance. For example, overexpression of FGF-2 messenger RNA in EAC and ESCC was observed to be associated with tumor recurrence and it reduced survival after the surgical resection of esophageal cancer.68 The expression of FGF-2, as well as EGF and HGF, was also evaluated in patients undergoing resection for GEJ adenocarcinoma, and the results confirmed that high levels of angiogenic factors are associated with unfavorable tumor characteristics and poorer overall survival.69 These studies collectively provide a rationale for antiangiogenic therapy in the treatment of esophageal cancer.
Antiangiogenic agents in the treatment of esophageal cancer
There is, as yet, no antiangiogenic agent approved for esophageal cancer. With recent advances in the development of antiangiogenic compounds, some of the promising antiangiogenic agents have been tested in preclinical studies or clinical trials (usually in combination with chemoradiotherapy or chemotherapy) (Tables 1 and 2). However, as will be described, most of the clinical studies involved subjects with advanced EAC or GEJ adenocarcinomas, so there is relatively little information of the efficacy of these drugs on ESCC.
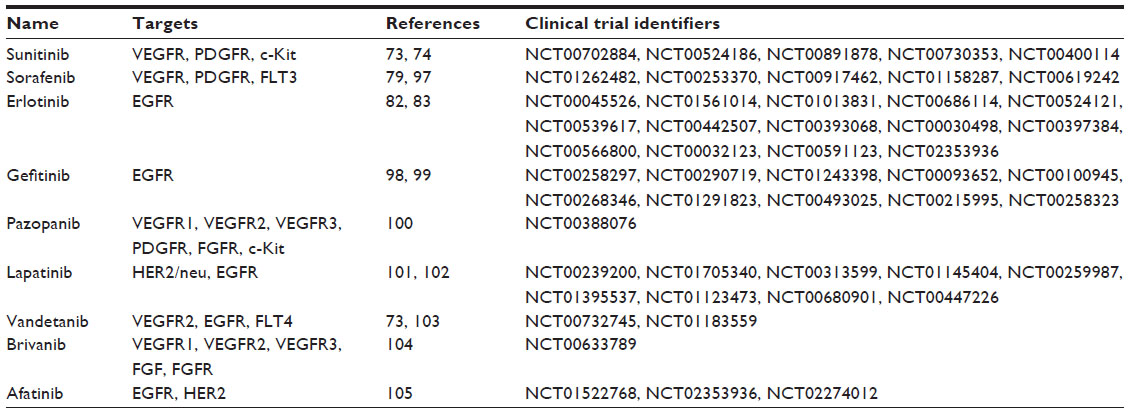 | Table 1 Summary of antiangiogenic small molecules used for the treatment of esophageal cancer |
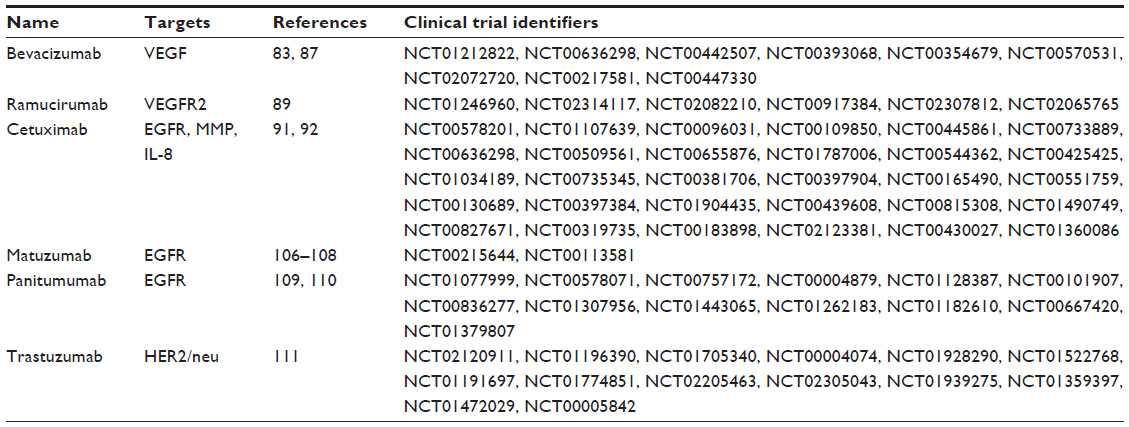 | Table 2 Summary of antiangiogenic monoclonal antibodies used for the treatment of esophageal cancer |
Small-molecule therapeutics
Sunitinib is an orally administered multitargeted TKI that blocks VEGFRs, PDGF alpha and beta receptors (PDGFR), stem cell-factor receptor (Kit), and other receptor tyrosine kinases.70 It was first approved by the US Food and Drug Administration (FDA) in 2006 for the treatment of gastrointestinal stromal tumor and metastatic renal cell cancer.71 With regard to esophageal cancer, a recent study showed that sunitinib inhibited autocrine VEGF signaling in Barrett’s neoplastic cells and reduced xenograft tumor growth in a mouse model.72 Another preclinical study showed that although sunitinib could inhibit the proliferation of an esophageal cancer cell line, there was no synergistic effect with cisplatin.73 In a Phase II study of patients with advanced esophageal cancer (mostly adenocarcinoma), the combination of sunitinib plus paclitaxel did not provide survival benefits for the patients and had serious toxic effects.74
Sorafenib is a kinase inhibitor drug approved for the treatment of advanced renal cell carcinoma75 and advanced primary liver cancer.76 In 2013, it was also approved for the treatment of locally recurrent or metastatic, progressive differentiated thyroid carcinomas, which are refractory to radioactive iodine treatment.77 Besides inhibiting the VEGFR pathway, it can also block signaling from the PDGFR and Fms-related tyrosine kinase 3 (FLT3) pathways.78 Sorafenib was demonstrated to inhibit acid or bile acid-stimulated proliferation of Barrett’s esophageal cancer cells via the regulation of the MAPK pathway.79 In a Phase II study, the combination of sorafenib, docetaxel, and cisplatin had an encouraging efficacy profile in the treatment of metastatic or advanced gastric and GEJ adenocarcinoma with tolerable toxicity.80
Erlotinib is an EGF receptor (EGFR) inhibitor and it was approved by the US FDA in 2013 for use as a first-line treatment in patients with metastatic nonsmall cell lung cancer, whose tumors have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations.81 A recent study showed that the combined regimen of radiation and erlotinib has a beneficial outcome for elderly patients aged >70 years with esophageal cancer.82 However, in another Phase II study, the addition of bevacizumab and erlotinib to neoadjuvant chemoradiation did not demonstrate survival benefits in patients with localized esophageal cancer.83
Other synthetic small-molecule drugs, including gefitinib and pazopanib, are also under close investigation. Some anti-inflammatory drugs with antiangiogenic potential also look promising. For example, aspirin was demonstrated to greatly improve the 5-year survival of patients with T2N0M0 ESCC.84
Large-molecule therapeutics
Bevacizumab, an anti-VEGF neutralizing antibody, was the first antiangiogenic agent to be approved by the US FDA in 2004 for the treatment of human colorectal cancer.85 Bevacizumab functions by blocking the binding of VEGF to VEGFRs, thus normalizing the disorganized vasculature of tumors. A Phase II trial of combination therapy consisting of docetaxel, cisplatin, and fluorouracil with bevacizumab was reported to be tolerable and was found to notably improve the outcome in patients with metastatic gastroesophageal adenocarcinoma.86 Another Phase II study combining bevacizumab, capecitabine, and oxaliplatin confirmed that bevacizumab can be given safely with chemotherapy in patients with metastatic gastroesophageal adenocarcinomas.87
Ramucirumab is a fully human immunoglobulin G1 monoclonal antibody to VEGFR2. It works as a receptor antagonist by binding to VEGFR2, thus preventing the binding of VEGF to VEGFR2. Ramucirumab was approved by the US FDA in 2014 to treat stomach cancer patients whose cancer could not be surgically removed or had metastasized.88 A recent Phase III study involving 355 patients with advanced gastric or GEJ adenocarcinoma progressing after first-line chemotherapy showed that ramucirumab significantly extended the median overall survival time from 3.8 months to 5.2 months.89
Cetuximab is the first chimeric monoclonal anti-EGFR antibody. It can suppress the expression of VEGF, MMP, or IL-8, thus inhibiting angiogenesis.90 Promising results from a recent Phase II clinical trial showed that the administration of cetuximab combined with chemoradiotherapy improved the clinical response rate of patients with nonresectable, locally advanced ESCC.91 Moreover, results from another Phase II study suggest that the combination of cetuximab and radiation therapy may be better tolerated than preoperative chemotherapy and radiation therapy in patients with resectable esophageal cancers.92
Clinical trials involving several other monoclonal antibodies, such as matuzumab and panitumumab, have also been conducted (Table 2). Besides monoclonal antibodies and receptor TKIs, a variety of other drugs have been shown to have one or more antiangiogenic properties. Aflibercept (VEGF-Trap), a soluble receptor chimeric protein, was engineered to have high affinity for VEGF and thus functions as a decoy VEGF receptor.93 It was approved by the US FDA in 2012 for the treatment of patients with metastatic colorectal cancer in combination with 5-fluorouracil, leucovorin, and irinotecan.94 Its efficacy in combination with FOLFOX in treating esophagogastric cancer is currently being investigated in a Phase II trial (NLM identifier: NCT00215644).
Antiangiogenic therapy is a double-edged sword in esophageal cancer therapy
Although increasing preclinical and clinical evidence shows that antiangiogenic therapies have promising effects, the clinical use of antiangiogenic therapy seemed more complex than anticipated. To date, evidence of successful antiangiogenic therapy in esophageal cancer is still lacking. The fact that a fraction of patients were refractory or developed resistance to anti-VEGF therapy through multiple mechanisms in different cancer types remains a critical challenge for antiangiogenic therapy. The mechanisms of refractoriness to anti-VEGF therapy may, in part, be attributable to the fact that several other key mediators, besides the VEGF family, can switch on angiogenesis so that blocking VEGF alone is not sufficient. Bergers and Hanahan94 referred to this phenomenon as “evasion of anti-angiogenic therapy” and proposed it as one of the important modes of resistance to antiangiogenic therapy. They also proposed “indifference to anti-angiogenic therapy” as another mode of resistance, which means that the angiogenic system supporting tumors in those patients resistant to antiangiogenic therapy may have already evolved before therapy.95 Another consideration is that in the dynamic tumor microenvironment, the persistent recruitment of BMDCs, such as CD11b+Gr1+ BMDCs, may revascularize tumors.23 Therefore, further studies are urgently called for to investigate the mechanisms of acquired resistance to antiangiogenic therapies, so as to facilitate the development of more advanced and effective strategies.
Conclusion and future perspectives
The incidence of esophageal cancer is increasing and the survival rate remains poor. As mentioned earlier, the progression of esophageal cancer is associated with angiogenesis. Although antiangiogenic therapies showed promising results from preclinical and clinical studies, many aspects of angiogenesis and antiangiogenic therapy remain to be further considered in order to overcome existing hurdles:
- Angiogenesis is a multicellular and multistep process. There is still a lack of validated biomarkers to predict which patients may benefit from antiangiogenic drugs. Until now, only a few candidate biomarkers have been identified, but most of the studies were based on small patient cohorts.
- Our understanding of the molecular mechanisms of the angiogenic switch is far from complete. Identification and deciphering of the roles/mechanisms of the molecules (including noncoding RNAs, which are currently under intensive investigation) involved in angiogenesis and vasculogenesis will facilitate the development of novel agents that can block the angiogenic switch.
- Resistance remains one of the critical challenges for antiangiogenic therapy. Therefore, novel strategies to diminish the resistance to antiangiogenic therapies should be identified. The fact that antiangiogenesis agents are often used together with a diverse combination of conventional chemotherapeutic drugs in the clinic needs to be taken into consideration when evaluating treatment response and the mechanism(s) of drug resistance.
- The same angiogenic signaling pathway that works in the tumors also applies to various normal physiological vascular processes, such as wound healing and pregnancy. Antiangiogenic therapy inevitably introduces undesirable or even serious side effects including fatigue, hypertension, and neurotoxicity.96 The side effects of antiangiogenic therapy should be minimized, and the drug delivery and dosing should be further optimized.
In summary, to combat esophageal cancer, concerted effort by bench scientists, clinicians, and the pharmaceutical industry is paramount in developing safe and effective antiangiogenic strategies that will ultimately benefit cancer patients.
Acknowledgments
This work was supported by General Research Funds from the Research Grants Council of the Hong Kong SAR, People’s Republic of China (project numbers HKU 763111M and HKU 17103814).
Disclosure
The authors report no conflicts of interest in this work.
References
Ulmar SB, Fleischer DE. Esophageal cancer: epidemiology, pathogenesis and prevention. Nat Clin Pract Gastroenterol Hepatol. 2008;5(9):517–526. | |
Zhang Y. Epidemiology of esophageal cancer. World J Gastroenterol. 2013;19(34):5598–5606. | |
Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349(23):2241–2252. | |
Sobin LH, Gospodarowicz MK, Wittekind C. The TNM Classification of Malignant Tumours. 7th ed. Hoboken, NJ: Wiley-Blackwell and UICC; 2009. | |
Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381(9864):400–412. | |
Hertig AT. Angiogenesis in the early human chorion and in the primary placenta of the macaque monkey. Contributions To Embryology. Washington DC: Carnegie Insitute of Washington; 1935;25(146). | |
Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. | |
Cheng L, Huang Z, Zhou W, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153(1):139–152. | |
Chiodoni C, Iezzi M, Guiducci C, et al. Triggering CD40 on endothelial cells contributes to tumor growth. J Exp Med. 2006;203(11):2441–2450. | |
Noma K, Smalley KS, Lioni M, et al. The essential role of fibroblasts in esophageal squamous cell carcinoma-induced angiogenesis. Gastroenterology. 2008;134(7):1981–1993. | |
Rafii S, Lyden D. Cancer. A few to flip the angiogenic switch. Science. 2008;319(5860):163–164. | |
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–1186. | |
Shing Y, Folkman J, Sullivan R, et al. Heparin affinity: purification of a tumor-derived capillary endothelial cell growth factor. Science. 1984;223(4642):1296–73, 10310499. | |
Haas TL, Milkiewicz M, Davis SJ, et al. Matrix metalloproteinase activity is required for activity-induced angiogenesis in rat skeletal muscle. Am J Physiol Heart Circ Physiol. 2000;279(4):H1540–H1547. | |
Thurston G. Role of Angiopoietins and Tie receptor tyrosine kinases in angiogenesis and lymphangiogenesis. Cell Tissue Res. 2003;314(1):61–68. | |
Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3(6):401–410. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Lyden D, Hattori K, Dias S, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7(11):1194–1201. | |
Kioi M, Vogel H, Schultz G, et al. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120(3):694–705. | |
Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8(12):1369–1375. | |
Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–827. | |
Yang L, Huang J, Ren X, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13(1):23–35. | |
Shojaei F, Wu X, Malik AK, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25(8):911–920. | |
De PM, Venneri MA, Galli R, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8(3):211–226. | |
Xu WW, Li B, Lam AK, et al. Targeting VEGFR1- and VEGFR2-expressing non-tumor cells is essential for esophageal cancer therapy. Oncotarget. 2015;6(3):1790–1805. | |
Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246(4935):1306–1309. | |
Ferrara N. Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: therapeutic implications. Semin Oncol. 2002;29(6 Suppl 16):10–14. | |
Kozlowski M, Kowalczuk O, Milewski R, et al. Serum vascular endothelial growth factors C and D in patients with oesophageal cancer. Eur J Cardiothorac Surg. 2010;38(3):260–267. | |
McDonnell CO, Harmey JH, Bouchier-Hayes DJ, Walsh TN. Effect of multimodality therapy on circulating vascular endothelial growth factor levels in patients with oesophageal cancer. Br J Surg. 2001;88(8):1105–1109. | |
Chen J, Zhou SJ, Zhang Y, et al. Clinicopathological and prognostic significance of galectin-1 and vascular endothelial growth factor expression in gastric cancer. World J Gastroenterol. 2013;19(13):2073–2079. | |
Cao D, Hou M, Guan YS, et al. Expression of HIF-1alpha and VEGF in colorectal cancer: association with clinical outcomes and prognostic implications. BMC Cancer. 2009;9:432. | |
Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25(4):581–611. | |
Schwartz JD, Rowinsky EK, Youssoufian H, Pytowski B, Wu Y. Vascular endothelial growth factor receptor-1 in human cancer: concise review and rationale for development of IMC-18F1 (Human antibody targeting vascular endothelial growth factor receptor-1). Cancer. 2010;116 (4 Suppl):1027–1032. | |
Wu Y, Zhu Z. Vascular endothelial growth factor receptor 1, a therapeutic target in cancer, inflammation and other disorders. Curr Med Chem. 2009;16(22):2890–2898. | |
Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19(10):2003–2012. | |
Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem. 1994;269(41):25646–25654. | |
Xie K, Wei D, Shi Q, Huang S. Constitutive and inducible expression and regulation of vascular endothelial growth factor. Cytokine Growth Factor Rev. 2004;15(5):297–324. | |
Xia C, Meng Q, Liu LZ, et al. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007;67(22):10823–10830. | |
Sanchez-Elsner T, Botella LM, Velasco B, et al. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J Biol Chem. 2001;276(42):38527–38535. | |
Shweiki D, Neeman M, Itin A, Keshet E. Induction of vascular endothelial growth factor expression by hypoxia and by glucose deficiency in multicell spheroids: implications for tumor angiogenesis. Proc Natl Acad Sci U S A. 1995;92(3):768–772. | |
Pages G, Pouyssegur J. Transcriptional regulation of the Vascular Endothelial Growth Factor gene--a concert of activating factors. Cardiovasc Res. 2005;65(3):564–573. | |
Baglole CJ, Ray DM, Bernstein SH, et al. More than structural cells, fibroblasts create and orchestrate the tumor microenvironment. Immunol Invest. 2006;35(3–4):297–325. | |
Chouaib S, Kieda C, Benlalam H, et al. Endothelial cells as key determinants of the tumor microenvironment: interaction with tumor cells, extracellular matrix and immune killer cells. Crit Rev Immunol. 2010;30(6):529–545. | |
Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18(6):884–901. | |
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. | |
Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling—in control of vascular function. Nat Rev Mol Cell Biol. 2006;7(5):359–371. | |
Stapor P, Wang X, Goveia J, Moens S, Carmeliet P. Angiogenesis revisited—role and therapeutic potential of targeting endothelial metabolism. J Cell Sci. 2014;127(Pt 20):4331–4341. | |
Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17(11):1359–1370. | |
D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91(9):4082–4085. | |
Mukherji SK. Bevacizumab (Avastin). AJNR Am J Neuroradiol. 2010;31(2):235–236. | |
Samant RS, Shevde LA. Recent advances in anti-angiogenic therapy of cancer. Oncotarget. 2011;2(3):122–134. | |
Dassonville O, Bozec A, Fischel JL, Milano G. EGFR targeting therapies: monoclonal antibodies versus tyrosine kinase inhibitors. Similarities and differences. Crit Rev Oncol Hematol. 2007;62(1):53–61. | |
Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6(9):714–727. | |
Boehm T, Folkman J, Browder T, O’Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390(6658):404–407. | |
Haggitt RC. Barrett’s esophagus, dysplasia, and adenocarcinoma. Hum Pathol. 1994;25(10):982–993. | |
Auvinen MI, Sihvo EI, Ruohtula T, et al. Incipient angiogenesis in Barrett’s epithelium and lymphangiogenesis in Barrett’s adenocarcinoma. J Clin Oncol. 2002;20(13):2971–2979. | |
Couvelard A, Paraf F, Gratio V, et al. Angiogenesis in the neoplastic sequence of Barrett’s oesophagus. Correlation with VEGF expression. J Pathol. 2000;192(1):14–18. | |
Mukherjee T, Kumar A, Mathur M, Chattopadhyay TK, Ralhan R. Ets-1 and VEGF expression correlates with tumor angiogenesis, lymph node metastasis, and patient survival in esophageal squamous cell carcinoma. J Cancer Res Clin Oncol. 2003;129(7):430–436. | |
Shimada Y, Imamura M, Watanabe G, et al. Prognostic factors of oesophageal squamous cell carcinoma from the perspective of molecular biology. Br J Cancer. 1999;80(8):1281–1288. | |
Nishimaki T, Tanaka O, Suzuki T, et al. Patterns of lymphatic spread in thoracic esophageal cancer. Cancer. 1994;74(1):4–11. | |
Kitadai Y, Amioka T, Haruma K, et al. Clinicopathological significance of vascular endothelial growth factor (VEGF)-C in human esophageal squamous cell carcinomas. Int J Cancer. 2001;93(5):662–666. | |
Peng J, Shao N, Peng H, Chen LQ. Prognostic significance of vascular endothelial growth factor expression in esophageal carcinoma: a meta-analysis. J BUON. 2013;18(2):398–406. | |
Kilic E, Schild SE, Thorns C, Bajrovic A, Rades D. Prognostic role of vascular endothelial growth factor and its receptor-1 in patients with esophageal cancer. Anticancer Res. 2014;34(9):5221–5226. | |
Kozlowski M, Laudanski W, Mroczko B, et al. Serum tissue inhibitor of metalloproteinase 1 (TIMP-1) and vascular endothelial growth factor A (VEGF-A) are associated with prognosis in esophageal cancer patients. Adv Med Sci. 2013;58(2):227–234. | |
Sharma BK, Srinivasan R, Kapil S, et al. Serum levels of angiogenic and anti-angiogenic factors: their prognostic relevance in locally advanced hepatocellular carcinoma. Mol Cell Biochem. 2013;383(1–2):103–112. | |
Bestas R, Kaplan MA, Isikdogan A. The correlation between serum VEGF levels and known prognostic risk factors in colorectal carcinoma. Hepatogastroenterology. 2014;61(130):267–271. | |
Fujita N, Okegawa T, Terado Y, et al. Serum level and immunohistochemical expression of vascular endothelial growth factor for the prediction of postoperative recurrence in renal cell carcinoma. BMC Res Notes. 2014;7:369. | |
Barclay C, Li AW, Geldenhuys L, et al. Basic fibroblast growth factor (FGF-2) overexpression is a risk factor for esophageal cancer recurrence and reduced survival, which is ameliorated by coexpression of the FGF-2 antisense gene. Clin Cancer Res. 2005;11(21):7683–7691. | |
Park dJ, Yoon C, Thomas N, et al. Prognostic significance of targetable angiogenic and growth factors in patients undergoing resection for gastric and gastroesophageal junction cancers. Ann Surg Oncol. 2014;21(4):1130–1137. | |
Matsui J, Wakabayashi T, Asada M, Yoshimatsu K, Okada M. Stem cell factor/c-kit signaling promotes the survival, migration, and capillary tube formation of human umbilical vein endothelial cells. J Biol Chem. 2004;279(18):18600–18607. | |
Cabebe E, Wakelee H. Sunitinib: a newly approved small-molecule inhibitor of angiogenesis. Drugs Today (Barc ). 2006;42(6):387–398. | |
Zhang Q, Yu C, Peng S, et al. Autocrine VEGF signaling promotes proliferation of neoplastic Barrett’s epithelial cells through a PLC-dependent pathway. Gastroenterology. 2014;146(2):461–472. | |
Lyros O, Mueller A, Heidel F, et al. Analysis of anti-proliferative and chemosensitizing effects of sunitinib on human esophagogastric cancer cells: Synergistic interaction with vandetanib via inhibition of multi-receptor tyrosine kinase pathways. Int J Cancer. 2010;127(5):1197–1208. | |
Schmitt JM, Sommers SR, Fisher W, et al. Sunitinib plus paclitaxel in patients with advanced esophageal cancer: a phase II study from the Hoosier Oncology Group. J Thorac Oncol. 2012;7(4):760–763. | |
Kane RC, Farrell AT, Saber H, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res. 2006;12(24):7271–7278. | |
Guan YS, He Q. Sorafenib: activity and clinical application in patients with hepatocellular carcinoma. Expert Opin Pharmacother. 2011;12(2):303–313. | |
Okamura R, Sugitani I. [Current progress and management in molecular targeted therapy for advanced thyroid cancer]. Gan To Kagaku Ryoho. 2014;41(2):148–152. Japanese. | |
Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. | |
Keswani RN, Chumsangsri A, Mustafi R, et al. Sorafenib inhibits MAPK-mediated proliferation in a Barrett’s esophageal adenocarcinoma cell line. Dis Esophagus. 2008;21(6):514–521. | |
Sun W, Powell M, O’Dwyer PJ, et al. Phase II study of sorafenib in combination with docetaxel and cisplatin in the treatment of metastatic or advanced gastric and gastroesophageal junction adenocarcinoma: ECOG 5203. J Clin Oncol. 2010;28(18):2947–2951. | |
Khozin S, Blumenthal GM, Jiang X, et al. U.S. Food and Drug Administration approval summary: Erlotinib for the first-line treatment of metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations. Oncologist. 2014;19(7):774–779. | |
Zhang XB, Xie CY, Li WF, Zhang P, Wu SX. [Phase II study of radiotherapy plus erlotinib for elder patients with esophageal carcinoma]. Zhonghua Yi Xue Za Zhi. 2012;92(23):1615–1617. Chinese. | |
Bendell JC, Meluch A, Peyton J, et al. A phase II trial of preoperative concurrent chemotherapy/radiation therapy plus bevacizumab/erlotinib in the treatment of localized esophageal cancer. Clin Adv Hematol Oncol. 2012;10(7):430–437. | |
Liu JF, Jamieson GG, Wu TC, Zhu GJ, Drew PA. A preliminary study on the postoperative survival of patients given aspirin after resection for squamous cell carcinoma of the esophagus or adenocarcinoma of the cardia. Ann Surg Oncol. 2009;16(5):1397–1402. | |
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–2342. | |
Shah MA, Jhawer M, Ilson DH, et al. Phase II study of modified docetaxel, cisplatin, and fluorouracil with bevacizumab in patients with metastatic gastroesophageal adenocarcinoma. J Clin Oncol. 2011;29(7):868–874. | |
Uronis HE, Bendell JC, Altomare I, et al. A phase II study of capecitabine, oxaliplatin, and bevacizumab in the treatment of metastatic esophagogastric adenocarcinomas. Oncologist. 2013;18(3):271–272. | |
Ramucirumab approved for gastric cancer. Cancer Discov. 2014;4(7):752–753. | |
Fuchs CS, Tomasek J, Yong CJ, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2014;383(9911):31–39. | |
Vincenzi B, Zoccoli A, Pantano F, Venditti O, Galluzzo S. Cetuximab: from bench to bedside. Curr Cancer Drug Targets. 2010;10(1):80–95. | |
Meng X, Wang J, Sun X, et al. Cetuximab in combination with chemoradiotherapy in Chinese patients with non-resectable, locally advanced esophageal squamous cell carcinoma: a prospective, multicenter phase II trail. Radiother Oncol. 2013;109(2):275–280. | |
Becerra CR, Hanna N, McCollum AD, et al. A phase II study with cetuximab and radiation therapy for patients with surgically resectable esophageal and GE junction carcinomas: Hoosier Oncology Group G05-92. J Thorac Oncol. 2013;8(11):1425–1429. | |
Ciombor KK, Berlin J. Aflibercept--a decoy VEGF receptor. Curr Oncol Rep. 2014;16(2):368. | |
FDA approves aflibercept (Zaltrap) for metastatic colorectal cancer. Oncology (Williston Park). 2012;26(9):842, 873. | |
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. | |
Verheul HM, Pinedo HM. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat Rev Cancer. 2007;7(6):475–485. | |
Delgado JS, Mustafi R, Yee J, et al. Sorafenib triggers antiproliferative and pro-apoptotic signals in human esophageal adenocarcinoma cells. Dig Dis Sci. 2008;53(12):3055–3064. | |
Adelstein DJ, Rodriguez CP, Rybicki LA, Ives DI, Rice TW. A phase II trial of gefitinib for recurrent or metastatic cancer of the esophagus or gastroesophageal junction. Invest New Drugs. 2012;30(4):1684–1689. | |
Dutton SJ, Ferry DR, Blazeby JM, et al. Gefitinib for oesophageal cancer progressing after chemotherapy (COG): a phase 3, multicentre, double-blind, placebo-controlled randomised trial. Lancet Oncol. 2014;15(8):894–904. | |
Burris HA, III, Dowlati A, Moss RA, et al. Phase I study of pazopanib in combination with paclitaxel and carboplatin given every 21 days in patients with advanced solid tumors. Mol Cancer Ther. 2012;11(8):1820–1828. | |
Guo XF, Zhu XF, Zhong GS, et al. Lapatinib, a dual inhibitor of EGFR and HER2, has synergistic effects with 5-fluorouracil on esophageal carcinoma. Oncol Rep. 2012;27(5):1639–1645. | |
Hou W, Qin X, Zhu X, et al. Lapatinib inhibits the growth of esophageal squamous cell carcinoma and synergistically interacts with 5-fluorouracil in patient-derived xenograft models. Oncol Rep. 2013;30(2):707–714. | |
Crescenzi M, Persano L, Esposito G, et al. Vandetanib improves anti-tumor effects of L19mTNFalpha in xenograft models of esophageal cancer. Clin Cancer Res. 2011;17(3):447–458. | |
Chou T, Finn RS. Brivanib: a review of development. Future Oncol. 2012;8(9):1083–1090. | |
Yap TA, Vidal L, Adam J, et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J Clin Oncol. 2010;28(25):3965–3972. | |
Rao S, Starling N, Cunningham D, et al. Phase I study of epirubicin, cisplatin and capecitabine plus matuzumab in previously untreated patients with advanced oesophagogastric cancer. Br J Cancer. 2008;99(6):868–874. | |
Rao S, Starling N, Cunningham D, et al. Matuzumab plus epirubicin, cisplatin and capecitabine (ECX) compared with epirubicin, cisplatin and capecitabine alone as first-line treatment in patients with advanced oesophago-gastric cancer: a randomised, multicentre open-label phase II study. Ann Oncol. 2010;21(11):2213–2219. | |
Trarbach T, Przyborek M, Schleucher N, et al. Phase I study of matuzumab in combination with 5-fluorouracil, leucovorin and cisplatin (PLF) in patients with advanced gastric and esophagogastric adenocarcinomas. Invest New Drugs. 2013;31(3):642–652. | |
Kordes S, van Berge Henegouwen MI, Hulshof MC, et al. Preoperative chemoradiation therapy in combination with panitumumab for patients with resectable esophageal cancer: the PACT study. Int J Radiat Oncol Biol Phys. 2014;90(1):190–196. | |
Lockhart AC, Reed CE, Decker PA, et al. Phase II study of neoadjuvant therapy with docetaxel, cisplatin, panitumumab, and radiation therapy followed by surgery in patients with locally advanced adenocarcinoma of the distal esophagus (ACOSOG Z4051). Ann Oncol. 2014;25(5):1039–1044. | |
Shen L, Xu JM, Feng FY, et al. [Trastuzumab in combination with chemotherapy versus chemotherapy alone for first-line treatment of HER2-positive advanced gastric or gastroesophageal junction cancer: a Phase III, multi-center, randomized controlled trial, Chinese subreport]. Zhonghua Zhong Liu Za Zhi. 2013;35(4):295–300. Chinese. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.