")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 16
The Molecular Mechanism of Renal Tubulointerstitial Inflammation Promoting Diabetic Nephropathy
Authors Xue R, Xiao H, Kumar V, Lan X, Malhotra A, Singhal PC, Chen J
Received 24 August 2023
Accepted for publication 30 November 2023
Published 5 December 2023 Volume 2023:16 Pages 241—252
DOI https://doi.org/10.2147/IJNRD.S436791
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Konstantinos Tziomalos
Rui Xue,1,* Haiting Xiao,2,* Vinod Kumar,3 Xiqian Lan,2 Ashwani Malhotra,4 Pravin C Singhal,4 Jianning Chen1
1Affiliated Mental Health Center & Hangzhou Seventh People’s Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, 310000, People’s Republic of China; 2Key Laboratory of Luzhou City for Aging Medicine, Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China; 3Department of Dermatology, Postgraduate Institute of Medical Education and Research, Chandigarh, 160012, India; 4Feinstein Institute for Medical Research and Donald and Barbara Zucker School of Medicine at Hofstra/Northwell, Manhasset, NY, 11030, USA
*These authors contributed equally to this work
Correspondence: Jianning Chen; Pravin C Singhal, Email [email protected]; [email protected]
Abstract: Diabetic nephropathy (DN) is a common complication affecting many diabetic patients, leading to end-stage renal disease. However, its pathogenesis still needs to be fully understood to enhance the effectiveness of treatment methods. Traditional theories are predominantly centered on glomerular injuries and need more explicit explanations of recent clinical observations suggesting that renal tubules equally contribute to renal function and that tubular lesions are early features of DN, even occurring before glomerular lesions. Although the conventional view is that DN is not an inflammatory disease, recent studies indicate that systemic and local inflammation, including tubulointerstitial inflammation, contributes to the development of DN. In patients with DN, intrinsic tubulointerstitial cells produce many proinflammatory factors, leading to medullary inflammatory cell infiltration and activation of inflammatory cells in the interstitial region. Therefore, understanding the molecular mechanism of renal tubulointerstitial inflammation contributing to DN injury is of great significance and will help further identify key factors regulating renal tubulointerstitial inflammation in the high glucose environment. This will aid in developing new targets for DN diagnosis and treatment and expanding new DN treatment methods.
Keywords: diabetic nephropathy, renal tubules, renal tubular injury, renal tubulointerstitial inflammation, DN injury
Introduction
Diabetic nephropathy (DN) is a common complication of diabetes mellitus (DM), affecting approximately 20–40% of diabetic patients. In recent years, the number of patients with DN has been increasing worldwide.1 According to statistics, China, India, and the United States have the highest number of DM patients globally, with China having the most significant number at 116 million.2 Most Chinese kidney inpatients are hospitalized due to renal function decline in the form of tubular injury in diabetic patients.2,3 This indicates that recurrent tubular injury in DN is likely to contribute to the progress of DN, which also affects their quality of life and survival.4,5
DN patients display varying kidney structural changes, including glomerular sclerosis, interstitial inflammatory infiltration and fibrosis, and tubular atrophy. Glomerular lesions are the most characteristic with different severity in DN.4,5 Therefore, studies on the pathogenesis of DN primarily focus on glomerular injury caused by hyperglycemia, and the clinical indicators used to evaluate DN are mainly based on changes in glomerular structure and function. However, recent clinical observations indicate that many DN patients’ renal function decreases despite showing no substantial glomerular lesions or albuminuria, the traditional indicator of kidney disease, which may not increase or even decrease at a time. Nonetheless, these patients display renal tubular lesions, which may have contributed to renal dysfunction.6,7 Interestingly, the degree of tubular injury often correlates more to reduced creatinine clearance than to the severity of glomerular injury.8 Studies confirm that tubulointerstitial fibrosis is associated with poor prognosis, renal impairment, and the intensity of tubulointerstitial inflammatory infiltration.9 Thus, the damage caused by tubulointerstitial inflammation may be playing a vital role in the development and progression of DN,10 and exploring the molecular mechanisms underlying the tubulointerstitial inflammation response is crucial for comprehending the widespread anti-inflammatory processes and informing the future development of novel immuno-targeted therapeutic drugs aimed at addressing tubulointerstitial inflammation.
Renal Tubular Injury in DN
Renal tubular cells are characterized by their high number of mitochondria, vigorous metabolism, and significant demand for ATP. Their primary function is reabsorption, which involves the transmembrane transport of beneficial substances and puts them under constant energy stress, making them vulnerable to damage due to insufficient energy supply. Moreover, in diabetic nephropathy (DN), these cells are exposed to high blood glucose, further exacerbating their susceptibility to injury.4,11 Renal tubule cells rely mainly on fatty acid oxidation (FAO) to fuel their intracellular physiological activities. However, in high glucose, the expression of critical enzymes and regulatory factors required for FAO decreases, leading to reduced energy production and ATP deficiency, which can cause lipid accumulation, epithelial-mesenchymal transition, and fibrosis in the tubulointerstitial tissue.12,13
Additionally, renal tubular epithelial cells are exposed to oxidative stress, which can further damage their mitochondria and compromise their energy supply.11 The excess glucose and secondary metabolites in the ultrafiltrate of the tubular lumen of DN patients exacerbate this stress, further increasing the demand for intracellular energy. This can lead to various functional injuries, such as electron transfer mechanism disorder, reactive oxygen species (ROS) overproduction, and abnormal mitochondrial morphology.14 Moreover, the production of advanced glycation end products (AGEs) and extracellular matrix increases, adding to cellular and tissue damage.15
The accumulation of ROS and AGEs activates the oxidative stress response, which has several consequences. Firstly, ROS can stimulate inflammatory cells, such as monocytes and macrophages, to release mediators that cause oxidative damage to cellular proteins. Secondly, ROS can directly reduce nitric oxide concentration in renal capillaries, leading to capillary contraction, reduced blood flow, hypoxia, and worsening tissue and cellular metabolic abnormalities, culminating in lactic acid accumulation and further tubular cell injury.15 Thirdly, AGEs produced by transforming growth factor (TGF) in renal tubular epithelial cells can increase and transmit signals, such as the direct stimulation of TGF-β mRNA production, which in turn activates cells to produce more ROS and expand the oxidative stress response of tubulointerstitial cells.14 Fourthly, extracellular AGEs can bind to AGE receptors, triggering the intracellular NF-κB signaling pathway, increasing the expression of inflammatory mediators, such as cytokine IL6 and chemokines, such as CCL2, and promoting the aggregation of inflammatory cells in renal interstitium, leading to renal tubular atrophy and interstitial fibrosis.16
Albuminuria is widely accepted as a biological marker of progression for diabetic and nondiabetic chronic kidney diseases (CKDs); it induces tubular cell expression of MCP1 TGF-beta1 and promotes tubular cell apoptosis.17,18 High glucose milieu induces podocyte apoptosis, leading to a breach in the glomerular filtration barrier, which contributes to albuminuria and, thus, plays a vital role in the progression of tubulointerstitial fibrosis.19–21 Moreover, Diabetic nephropathy impacts the interstitial and glomerular membranes, subjecting them to inflammatory cytokines and reactive oxygen species (ROS) damage. This damage results in the loss of proteins, such as albumin, and a decline in the glomerular filtration rate. Additionally, alterations in kidney hemodynamics may manifest through thinning of the basement membrane, expansion of renal mesangial cells, and hyperplasia of the extracellular matrix. Elevated ROS levels in the kidney contribute to vasoconstriction, endothelial dysfunction, and increased sodium reabsorption.22
It is crucial to acknowledge that albumin serves as an indicator of glomerular and renal tubular deterioration. The glomeruli play a vital role in blood filtration and typically act as a barrier to prevent the passage of large molecules like albumin. Any damage to segments of the glomerular filtration barrier (GFB), including endothelial cells, the glomerular basement membrane (GBM), and podocytes, can increase albumin’s permeability. Conversely, albumin is taken up by proximal tubule cells through the GFB. However, proteinuria can occur when reabsorption capacity is exceeded or when tubule cells are impaired. Albumin serves as a crucial indicator of both glomerular and tubular health, and its quantification is employed to categorize chronic kidney disease (CKD) and predict the prognosis of patients with kidney and cardiovascular issues. It is also utilized to monitor the effectiveness of treatments such as ACE inhibitors or ARBs, which reduce proteinuria. Clinicians can better interpret urinary albumin levels within the context of kidney health. For instance, microfiltered disease is characterized by podocyte damage without significant GBM changes, resulting in selective proteinuria, while diabetic nephropathy may involve various aspects of GFB and renal tubule dysfunction.23 Therefore, albumin is a valuable marker in assessing renal injury in diabetic nephropathy and can guide the selection of renal treatments, including anti-diabetic and anti-inflammatory medications.
A lack of glucose in cells can also lead to cellular starvation, stimulating starvation mechanisms and resulting in an increased food intake followed by hyperglycemia, polyuria, and dehydration.24 Hyperglycemia (high glucose levels in the blood) is often associated with hypervolemia, causing solute diuresis and polyuria, which contributes to dehydration and loss of electrolytes (sodium and potassium) and may invoke acute tubular injury. Recurrence of acute tubular injury and residual low function may contribute to the progression of DN without the classical occurrence of nephrotic range proteinuria.
Abnormal mitochondrial function and activation of the apoptosis pathway may be an important molecular mechanism for the increase of cellular oxidative stress induced by hyperglycemia.25 In another study, the proximal renal tubule cells under high glucose conditions showed higher caspase activation and apoptosis after ATP depletion and hypoxic injury; it was associated with Bax accumulation and cytochrome c release in mitochondria.24 In response to hypoglycemic injury, renal proximal tubule cells showed a significantly higher p53 induction, indicating that p53 and mitochondrial apoptosis pathways further aggravated tubular injury in high glucose milieu.26
From a pathological perspective, hyperglycemia is also a risk factor for endothelial dysfunction, and microvascular damage can significantly affect the kidneys in the short and long term. Short-term effects include endothelial cell expansion and apoptosis/necrosis, leading to microvascular obstruction. Therefore, post-ischemic reperfusion is inhibited, renal regeneration time is prolonged, and each episode of ischemic injury reduces the total vascular surface area in the kidney, followed by endothelial interstitial transdifferentiation, ultimately aggravating fibrosis and promoting DN injury.27
Inflammatory Response of Renal Tubulointerstitial Cells in DN
Although DN is not typically viewed as an inflammatory disease, recent clinical and experimental evidence has demonstrated that systemic and local renal inflammation, including glomerular and tubulointerstitial inflammation, is a significant factor in the initiation and progression of DN.28 Inflammatory cells from the circulatory system, such as monocytes and lymphocytes, are involved in addition to intrinsic kidney cells, including endothelial cells, mesangial cells, podocytes, tubular epithelial cells, and renal interstitial fibroblasts, which participate in the inflammatory response in DN.29 These cells can release proinflammatory cytokines, chemokines, adhesion molecules, and other substances that contribute to the onset and progression of renal inflammation and are vital to the development of DN.30
Macrophages play a significant role in the inflammatory response of DN by infiltrating the kidney. Various studies have established a positive correlation between the number of infiltrating macrophages and the progression of DN.31,32 Activated macrophages can secrete several proinflammatory factors like TNFα, IL1β, IL4, IL6, IL10, IL12, IL13, and IL23, which promote the onset and progression of inflammation, cause tissue inflammation, and exacerbate the inflammatory injury of kidney tissue.31–34 Besides macrophages, tubular epithelial cells contribute to kidney inflammation. Studies have demonstrated that in nephritis-related diseases, proximal renal tubule cells express and secrete some molecules related to antigen-presenting cells, such as T lymphocytes, which trigger the activation of T lymphocytes and associated inflammatory responses. These inflammatory molecules comprise histocompatibility complexes and anti-neutrophil antibodies.30–34
The renal cells and tissues of patients with diabetic nephropathy are exposed to a continuous and stimulating environment characterized by high levels of glucose, reactive oxygen species (ROS), and advanced glycation end products (AGEs). This environment not only facilitates the recruitment of monocytes and macrophages from the circulation to the renal tissue, which produces inflammatory mediators, but also promotes the production of inflammatory factors by intrinsic renal cells such as mesangial cells, renal tubular epithelial cells, and interstitial fibroblasts. Inflammatory chemotactic factors such as monocyte chemotactic protein-1 (MCP-1) and intercellular adhesion molecule-1 (ICAM-1) have been detected in the kidneys of DN patients, with an increased expression level of ICAM-1.35,36 Previous studies have shown that in vascular endothelial cells of diabetic mice, the expression of MCP-1 is negatively regulated by the transcription factor FOXO1.37 In the renal tissue, FOXO1 can alleviate oxidative stress in vivo and inhibit the activation of transforming growth factor (TGF), such as the TGF-β1 signal in diabetic rats, thus protecting the kidney and reducing renal damage.38 FOXO1 is a critical factor that can regulate cell cycle arrest, DNA damage repair, and inhibition of oxidative stress.39,40 These studies suggest that proinflammatory cytokines produced by intrinsic renal cells for various reasons can further recruit circulating inflammatory cells (such as macrophages) to infiltrate the renal tissue. The increase in these inflammatory factors and cells in the renal tissue perpetuates the inflammatory response of the kidney and exacerbates renal burden.38 For example, the phosphorylation and release of FOXO1 by MCP-1 in renal tissue activate the rapamycin target protein (mTOR), which in turn promotes FOXO1 phosphorylation, causing tubulointerstitial inflammation and ultimately leading to pathological renal dysfunction.41 Moreover, our recent study showed that the oxidative stress response in DN was significantly enhanced and that the TGF-β signaling pathway was activated considerably, with significantly increased levels of Smad2/3 and Smad1/5/8 proteins phosphorylation.42 This may enhance renal fibrosis and further compromise renal function in the diabetic milieu.
High glucose stimulates growth factors such as TGF, EGF, IGF, and platelet-derived growth factors (PDGF) by intrinsic kidney cells. The increase in these growth factors promotes the proliferation and hypertrophy of renal tubular epithelial cells, affecting their structure and function.29,43 Exposure of injured renal tubular cells to EGF and IGF-1 weakens their proliferation-promoting ability compared to normal levels, reducing the ability to recover renal tubular cells after injury.44
The intrinsic cells of the kidney in a high glucose environment also produce IL and CC chemokine ligands, known as common inflammatory agents. IL-6, IL-1β, CCL-2, and CCL-7 induce infiltration of inflammatory cells, triggering an inflammatory response in the tubulointerstitial region and contributing to the initiation and progression of kidney injury.38,45 IL-6 can produce secondary inflammatory reactions mediated by IL-1.46 Both TNF-α and IL-1β can upregulate the level of IL-6 in HK-2 (human embryonic kidney-2) cells, with IL-1β having a greater intensity of upregulating IL-6.47
Tumor necrosis factor (TNF) and its receptor (TNFR) are common inflammatory factors in DN and play a critical role in the pathogenesis of DN.48–50 Increased expression of TNF and TNFR is detected in various kidney cells, including tubular epithelial cells and mesangial cells, following prolonged exposure to high glucose, ROS, AGEs, and lipopolysaccharides. Inflammatory mediators, such as IL1α and IL6, can also stimulate the expression of TNF-α and its receptors in kidney cells.48,51 Studies have shown that inhibiting the expression of TNF and TNFR can reduce the mRNA levels of ICAM-1, VCAM-1, and MCP-1 in the kidney and decrease the number of F4/80-positive inflammatory cells (macrophages) compared to the control group. These findings suggest that TNF and TNFR inhibition can protect the kidney by exerting an anti-inflammatory effect, reducing kidney inflammation, and slowing down the progression of early DN.52 Furthermore, TNFR1 and TNFR2 have been identified as potential markers of DN, which can be used to diagnose the onset of DN in patients.28,53
Notably, renal tubular epithelial cells carry abundant mitochondria, and their dysfunction contributes to kidney inflammation.54,55 Mitochondrial dysfunction, which causes oxidative stress, cell death, metabolic dysfunction, and inflammation, plays a vital role in the initiation and progression of DN.56,57 Improved mitochondrial function has been shown to positively impact indices related to DN. One potential explanation for the protective effects of SGLT2 inhibitors on the kidneys is their ability to enhance mitochondrial function in the renal tubules.58 Additionally, repeated administration of mesenchymal stem cells has been shown to improve mitochondrial function and prevent albuminuria and renal tubular epithelial cell damage.59 Prolonged exposure to a high glucose milieu can activate the NLRP3 pathway, triggering the formation of NLRP3 complexes and activation of Caspase-1, contributing to ROS production, apoptosis, and pyroptosis followed by renal impairment and the progression of DN.60 Inhibition of the NLRP3 pathway has been shown to reduce renal fibrosis associated with DN and improve mitochondrial dysfunction,61,62 suggesting the potential value of inflammasome inhibition in treating DN.60
Studies have shown that dysfunctional renal tubular epithelial cells (TECs) can release CCL2 and CCL5 to attract monocytes and macrophages. These activated macrophages then transform into M1 proinflammatory macrophages, producing various molecules such as tumor necrosis factor-α, platelet-derived growth factor (PDGF), basic fibroblast growth factor (FGF-2), TGF-β, and ROS, ultimately leading to increased inflammation and fibrosis.63 Another study found that exposure of proximal renal tubular epithelial cells to a high-sugar medium for 6 hours significantly increased the secretion of pro-inflammatory factors like CCL2 and IL-6.64 Additionally, it has been reported that the activation of IL-6 in renal epithelial cells (HK2 cells and primary renal epithelial cells) is influenced by IL-1, IL-17, and TNF-α. The production of IL-6 induced by these factors was specifically hindered by a JNK inhibitor called SP600125, inhibiting c-jun NH2-terminal kinase activity. However, the selective inhibitor of p38 MAPK did not affect this process.
Furthermore, inhibiting the ERK1/2 pathway resulted in a moderate increase in IL-6 induced by these proinflammatory factors (IL-1, TNF-α, and IL-17). These findings suggest that CCL2 and IL-6 may serve as endogenous specific proinflammatory factors in tubulointerstitial cells responding to tubular inflammation associated with diabetic nephropathy. Notably, TECs are the most critical interstitial cells in the tubules, known for producing cytokines and chemokines that can induce inflammation. However, research indicates that glucocorticoids, commonly used to treat inflammatory diseases, are ineffective in suppressing the inflammatory mediators produced by TECs.65 This implies the need to focus on specific endogenous proinflammatory factors in renal tubulointerstitial cells and develop targeted treatments, offering a potential solution for treating diabetic nephropathy from an inflammatory standpoint.
Inflammation-Related Signaling Pathways in Renal Tubule Injury
In patients with DN, various signaling pathways play critical regulatory roles in renal tubulointerstitial inflammation. These pathways include nuclear factor kappa-B (NF-κB), mitogen-activated protein kinase (MAPK), Janus kinase/signal transducers and activators of transcription (JAK/STAT), diacylglycerol/protein kinase C (DAG/PKC) signaling pathways, among others.66
The NF-κB signaling pathway is critical in promoting inflammation by upregulating the production of proinflammatory factors such as IL-1β, IL-6, IL-8, and TNF-α.66 Without relevant signal stimulation, NF-κB dimer binds (in vivo) to the intracellular inhibitory protein IκB to form an inactive complex that dissociates in the cytoplasm. Upon receiving external stimulant signals, the inactive NF-κB complex induces related kinases to dissociate IκB from the complex, activate NF-κB, and promote its entry into the nucleus, leading to the transcription of proinflammatory cytokines (such as IL6 and IL10), chemokines (such as MCP-1 and CCL7), adhesion molecules (such as ICAM1), and mediating the occurrence and development of the inflammatory response.67,68 In DN patients, high levels of glucose, AGEs, ROS, cytokines such as IL6, and angiotensin II, etc., can activate the NF-κB signal transduction pathway in kidney cells, upregulate the expression of p65, and regulate the expression of glomerular mesangial nucleus-related inflammatory factors, exacerbating the inflammatory response in the kidney.67–69 Furthermore, the activated NF-κB signaling pathway promotes the polarization of macrophages in kidney tissues of DN patients, which secrete a large number of inflammatory cytokines, aggravate the inflammatory response, promote the secretion of extracellular matrix, and further worsen the fibrosis of the kidney, leading to the progression of DN.44 Inhibition of the NF-κB pathway in the kidney reduces the expression of downstream inflammatory factors, thus decreasing the inflammatory response and slowing down the progression of DN.66,70,71 Albumin is known to stimulate the NF-κB pathway, leading to increased production of chemokines such as MCP-1, and it can activate and regulate T cell expression and secretion.72 This association has been linked to inflammatory infiltration and tubular damage in diabetic nephropathy. Lefty-1 has demonstrated the ability to inhibit NF-κB-p65 nuclear translocation, I-κB-α degradation, and Smad7 degradation in the early stages of renal interstitial inflammation, consequently reducing renal fibrosis.73 Furthermore, MG53 (also known as TRIM72) has been discovered to bind to p65 and impede its nuclear translocation, thus inhibiting NF-κB activation and reducing renal fibrosis.74 It is suggested that Lefty-1 or MG53 could serve as potential novel therapeutic drugs for reducing tubulointerstitial inflammation and reversing the progression of diabetic nephropathy by targeting the NF-κB pathway.
The MAPK family regulates inflammation, cell proliferation and differentiation, and cell apoptosis. Among them, the p38-MAPK signaling pathway is highly active in renal tubular cells of DN patients, leading to the recruitment of inflammatory cells, production of inflammatory factors, and tubular injury.64,75,76 Studies have shown that in a high blood glucose model, the kidney tissue of rats treated with STZ (streptozotocin) to destroy islet B cells exhibits a different degree of increase in mRNA and protein levels related to the MAPK signaling pathway compared to normal circumstances.77 Downstream of the MAPK signaling pathway, the expression levels of inflammatory factors such as TNF-α, ICAM-1, and IL6 are increased. Inhibition of phosphorylated p38 MAPK activity in the MAPK signaling pathway can reduce the inflammatory response in the spontaneous diabetic mouse model, as evidenced by reduced mRNA content of the kidney inflammatory factor CCL2 at the transcription level in db/db diabetic mice.78,79 The role of MD2, a coreceptor of Toll-like receptor 4 (TLR4), in innate immunity has been elucidated in diabetic nephropathy (DN). Md2-deficient mice or the MD2 inhibitor L6H9 successfully eliminated the activation of MAPK signaling in proximal renal tubule cells and downstream expression of ACE, angiotensin receptors, and angiotensin II.80 This implies a positive role for MD2 in the anti-inflammatory response of renal tubules and underscores its potential as a novel target for drug development aimed at anti-inflammatory treatment for DN.
Under continuous exposure to high glucose, ROS, and AGEs, the JAK/STAT signaling pathway in the kidney can be activated by chemokines, TGF-β, and active ROS, inducing macrophage infiltration and proliferation, as well as further stimulating renal tubular epithelial cells to produce ROS and other harmful substances. The promotion of matrix protein synthesis can further accelerate cell apoptosis, thus exacerbating the pathophysiological process of DN.81–83 JAK/STAT signaling activation can also promote angiotensin II to mediate mesangial cell proliferation in the kidney. At the same time, TGF-β production can increase the expression of fibrosis-related proteins, ultimately aggravating renal fibrosis in DN patients.82 In a high glucose environment, inhibition of JAK/STAT signaling can reduce the content of caspase-9 and TGF-β, as well as significantly decrease the phagocytosis ability of macrophages, suggesting that inflammation in DN can be mitigated by blocking the JAK/STAT signaling pathway, thereby protecting the kidney.81 The cytokine signaling (SOCS) suppressor is a downstream response protein of the JAK/STAT pathway and negatively regulates its activation. In DN rats, vitamin D can phosphorylate JAK2, STAT1, and STAT3 in rat mesangial cells, inhibit the expression of the SOCS2 gene, further reduce the inflammatory response of renal tissue and fibrosis of the renal interstitium, and alleviate the oxidative stress state of the kidney; urinary albumin in DN rats is also decreased, thereby reducing the damage to the kidney.83–85
The over-activation of JAK-STAT in renal tubular epithelial cells (TEC) may play a pivotal role in escalating tubulointerstitial inflammation and advancing the progression of diabetic nephropathy (DN). Research indicates a significant elevation in STAT6 expression in TEC of mice induced with unilateral ureteral obstruction (UUO). Blocking the phosphorylation of STAT6 and reducing its stability has been shown to alleviate UUO-induced renal fibrosis.86 IL-6 binds to the IL-6 receptor (IL-6R) on the membrane, recruiting signal transduction glycoprotein 130 (gp130) to activate JAK and induce STAT phosphorylation. Recombinant gp130 linked to Fc can compete with the IL-6/IL-6R complex and endogenous gp130 for binding, thereby eliminating IL-6 signal transduction. Fc-gp130 has the potential to prevent renal fibrosis by inhibiting STAT3 activation.87,88 The kinase inhibitory region of SOCS1 exhibits a strong affinity for the substrate binding groove of JAK, acting as a pseudo-substrate for JAK kinase. Research has demonstrated that overexpression of SOCS1 or SOCS3 can lead to a decrease in interstitial fibrosis, macrophage infiltration, and proteinuria,89 highlighting the potential of the JAK-STAT signaling pathway in response to renal tubulointerstitial inflammation. Additionally, family proteins such as STATs and SOCSs show promise in suppressing inflammation, reducing fibrosis, and delaying the progression of DN.
The DAG/PKC signaling pathway is closely related to the occurrence and development of inflammation in DN. It mainly contributes to the microvascular complications of DM and promotes the onset and progression of DN by regulating vascular function. The activation of this pathway occurs mainly due to an increase in DAG on the cell membrane and cytoplasmic Ca2+ concentration, leading to the activation of PKC. This pathway regulates vascular endothelial permeability and causes vasoconstriction, initiates the polyol pathway, oxidative stress, and AGEs, ultimately promoting extracellular matrix synthesis and turnover and the expression of downstream inflammatory factors. Related growth factors secreted under the activation of this pathway encourage cell proliferation and angiogenesis and recruit inflammatory cells to adhere to renal tissues, thereby promoting the progression of DN.90,91 Inhibiting the DAG/PKC signaling pathway reduces macrophage infiltration in kidney tissue, diminishes PKC-β, TGF-β, and other factors, and lowers extracellular matrix synthesis, such as fibronectin. This mitigates the immune response and fibrosis of the kidney, thereby alleviating kidney damage caused by DN.12,90
Hyperglycemia can increase diacylglycerol (DAG) content, a critical factor in activating protein kinase C (PKC). This increase can be triggered by various mechanisms, including the synthesis of DAG from dihydroxyacetone phosphate, the inhibition of glyceraldehyde-3-phosphate dehydrogenase, and the oxidation and glycation of metabolites. All these processes contribute to elevated DAG levels and subsequent PKC activation.92 Research has highlighted the crucial role of DNA-dependent protein kinase (DNA-PKcs) catalytic subunits in controlling inflammation in renal tubule epithelial cells. Eliminating DNA-PKcs in these cells maintained the epithelial phenotype. It reduced fibroblast activation induced by transforming growth factor-β1, resulting in decreased renal fibrosis and lower expression of inflammatory cytokines such as interleukin-1β, interleukin-6, tumor necrosis factor-α, and MCP1 induced by unilateral ureteral obstruction (UUO).93 Furthermore, glucose-induced PKC activation has been found to impede insulin-stimulating chemokines and macrophage factors, such as interleukin-1 (IL-1). PKC-ε, a novel subtype of PKC, is activated in diabetic kidneys, and its gene deletion in diabetic mouse models has been associated with increased renal tubulointerstitial fibrosis, along with elevated expression of phosphorylated TGF-β1, Smad2, and p38 mitogen-activated protein kinases. It is hypothesized that the activation of PKC-ε in the diabetic state might represent a protective response to injury rather than a factor contributing to kidney damage, suggesting that PKC-ε overexpression could benefit diabetic nephropathy-related inflammatory responses.94
Conclusions and Future Perspective
It is well-established that diabetic nephropathy (DN) is more intricately linked to alterations in the tubulointerstitial region than the glomerular region. Hyperglycemia impacts the structure of tubules primarily through basolateral tubular cells. The increased filtration of glucose results in a heightened load and exposure to glucose within the tubules. Diabetic individuals exhibit elevated expression of SGLT2, a glucose transporter in the proximal renal tubules responsible for reabsorption. This upregulation leads to enhanced glucose reabsorption by the proximal tubules, activating the local angiotensin II system and inducing the expression of growth factors such as CTGF and TGF-β1. Consequently, this process results in tubular hypertrophy, heightened oxidative stress, apoptosis of tubular cells, inflammatory infiltration, and increased deposition of extracellular matrix. This sequence of events may elucidate how hyperglycemia-induced inflammation in DN can lead to renal tubulointerstitial fibrosis and exacerbate renal injury.95 Furthermore, the accumulation of reactive oxygen species (ROS) and advanced glycation end products (AGEs) resulting from linear body dysfunction due to renal tubular injury induced by hyperglycemia, along with the activation and induction of downstream pathways by PKC, serve as critical factors in the renal tubular inflammatory injury associated with DN.
In recent years, mounting clinical and experimental evidence has demonstrated the significance of renal tubulointerstitial inflammation in the onset and progression of DN. In addition to inflammatory cells derived from the circulatory system, such as monocytes and lymphocytes, intrinsic renal cells, including endothelial cells, mesangial cells, renal tubular epithelial cells, and fibroblasts, are also involved in the inflammatory response of the tubulointerstitium in DN.29 These cells secrete proinflammatory cytokines (TNF-α, IL1β, and IL6), chemokines (MCP-1, CXCL1, and CXCL8), adhesion molecules (such as ICAM-1 and VCAM-1) and participate in the inflammatory reaction process of renal tubules. These cytokines/chemokines promote the infiltration of myeloid inflammatory cells (lymphocytes, monocytes, and granulocytes), activate resident inflammatory cells in the interstitial region, produce more proinflammatory factors, aggravate the inflammatory response in the kidney, and enhance the secretion of extracellular matrix, thus promoting the process of DN, including worsening renal fibrosis and cellular apoptosis and necrosis, and leading to a decline in kidney function.30,31,33 In a high glucose environment, tubulointerstitial inflammation is regulated by NF-κB, p38-MAPK, JAK/STAT, and other signaling pathways.
Conversely, inhibiting the expression of proinflammatory factors (TNF and TNFR) or inhibiting related regulatory signaling pathways (NF-κB, p38-MAPK, JAK/STAT) can slow down the inflammatory response in the tubulointerstitial region, attenuate the production of inflammatory factors in the kidney, reduce the phagocytic ability of macrophages, and slow the progression of early DN.44,52,66,81,96–98 Based on the above database, a simple molecular regulatory mechanism diagram has been structured, displaying several crucial regulatory signaling pathways that are considered to affect tubular inflammation in DN. These pathways are important in influencing tubular inflammation, promoting renal fibrosis, and leading to loss of renal function (Figure 1).
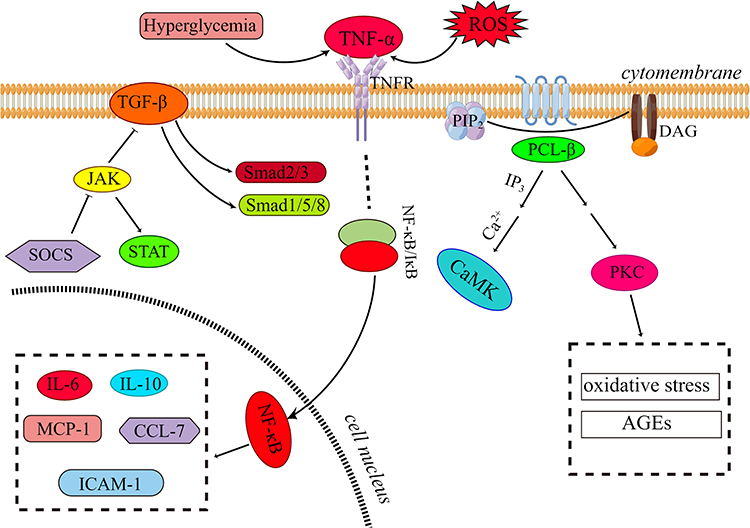 |
Figure 1 Diagram of regulatory signaling pathway mechanism that promotes renal tubule inflammation and renal fibrosis in DN. Activation of the TGF-β pathway increases downstream Smad2/3 and Smad1/5/8 protein phosphorylation levels. Inhibition of the JAK/STAT signaling pathway reduces the level of TGF-β, and SOCS is a negative regulator of the JAK/STAT signaling pathway. Hyperglycemia and ROS can activate TNF-α and then dissociate NF-κB from the NF-κB/IκB dimer complex through a series of reactions to activate NF-κB and then transfer it to cells and into cells. The activation of NF-κB induces the expression and transcription of proinflammatory factors, chemokines, and adhesion factors in the nucleus, thereby enhancing the damage of inflammatory response. PCL-β hydrolyzes PIP2 to generate IP3 and DAG and further activates calmodulin kinase and PKC; the activation of PKC can further induce oxidative stress and AGEs, contributing to severe renal fibrosis and decreased renal function. This figure was drawn by Figdraw. |
Renal tubulointerstitial inflammation is a crucial factor in developing DN, and inhibiting tubulointerstitial inflammation can slow DN’s progression and preserve renal cells’ function. Nonetheless, a non-specific anti-inflammatory therapy for DN may increase susceptibility to infection, making this regimen unsuitable for this disease.60 Drawing upon the compelling evidence outlined in this review, IL-6 and CCL2 emerge as crucial and specific targets for addressing renal tubulointerstitial inflammation in diabetic nephropathy (DN). Furthermore, Lefty-1, MG53, STATs, SOCSs, DNA-PKcs, and PKC-ε show substantial promise as anti-inflammatory targets against renal tubulointerstitial inflammation in DN. These factors demonstrate potential as novel diagnostic and therapeutic targets for managing diabetic nephropathy. However, a comprehensive understanding of their underlying molecular mechanisms requires further exploration. In conclusion, the process and mechanism of the renal inflammatory response are complex and involve a variety of chemical molecules, cells, and signaling pathways. The molecular mechanisms underlying renal tubulointerstitial inflammation in a high glucose environment require further investigation. In the future, identifying critical factors implicated in the renal tubular inflammatory response will enhance our understanding of widespread anti-inflammatory mechanisms and facilitate the development of new diagnostic and treatment targets for diabetic nephropathy.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the 2022 Health Sciences and Technology program of Hangzhou: General Project Category A (No. A20220084) and the Biomedical and Health Industry Development Support Science and Technology Project in Hangzhou (No.2022WJC064). This work was also supported by a grant RO1DK118017 (PCS) from the National Institutes of Health, Bethesda, MD, USA.
Disclosure
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
1. Bonner R, Albajrami O, Hudspeth J, Upadhyay A. Diabetic kidney disease. Primary Care. 2020;47(4):645–659. doi:10.1016/j.pop.2020.08.004
2. Saeedi P, Petersohn I, Salpea P, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the international diabetes federation diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019;157:107843. doi:10.1016/j.diabres.2019.107843
3. Parente EB, Harjutsalo V, Forsblom C, Groop PH. The impact of central obesity on the risk of hospitalization or death due to heart failure in type 1 diabetes: a 16-year cohort study. Cardiovasc Diabetol. 2021;20(1):153. doi:10.1186/s12933-021-01340-4
4. Zeni L, Norden A, Cancarini G, Unwin RJ. A more tubulocentric view of diabetic kidney disease. J Nephrol. 2017;30:701–717. doi:10.1007/s40620-017-0423-9
5. Chang DY, Li MR, Yu XJ, Wang SX, Chen M, Zhao MH. Clinical and pathological characteristics of patients with nonproteinuric diabetic nephropathy. Front Endocrinol. 2021;12:761386. doi:10.3389/fendo.2021.761386
6. Vallon V, Thomson SC. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat Rev Nephrol. 2020;16(6):317–336. doi:10.1038/s41581-020-0256-y
7. Gilbert RE. Proximal tubulopathy: prime mover and key therapeutic target in diabetic kidney disease. Diabetes. 2017;66(4):791–800. doi:10.2337/db16-0796
8. Risdon RA, Sloper JC, De Wardener HE. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet. 1968;2(7564):363–366. doi:10.1016/s0140-6736(68)90589-8
9. Bohle A, Mackensen-Haen S, von Gise H, et al. The consequences of tubulo-interstitial changes for renal function in glomerulopathies. A morphometric and cytological analysis. Pathol Res Pract. 1990;186(1):135–144. doi:10.1016/s0344-0338(11)81021-6
10. Rodríguez-Iturbe B, García García G. The role of tubulointerstitial inflammation in the progression of chronic renal failure. Nephron Clin Pract. 2010;116(2):c81–88. doi:10.1159/000314656
11. Habib SL. Kidney atrophy vs hypertrophy in diabetes: which cells are involved? Cell Cycle. 2018;17(14):1683–1687. doi:10.1080/15384101.2018.1496744
12. Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21(1):37–46. doi:10.1038/nm.3762
13. Yao L, Liang X, Qiao Y, Chen B, Wang P, Liu Z. Mitochondrial dysfunction in diabetic tubulopathy. Metabolism. 2022;131:155195. doi:10.1016/j.metabol.2022.155195
14. Yamagishi S, Inagaki Y, Okamoto T, Amano S, Koga K, Takeuchi M. Advanced glycation end products inhibit de novo protein synthesis and induce TGF-β overexpression in proximal tubular cells. Kidney Int. 2003;63(2):464–473. doi:10.1046/j.1523-1755.2003.00752.x
15. Bonventre JV. Can we target tubular damage to prevent renal function decline in diabetes? Semin Nephrol. 2012;32(5):452–462. doi:10.1016/j.semnephrol.2012.07.008
16. Youssef S, Nguyen DT, Soulis T, Panagiotopoulos S, Jerums G, Cooper ME. Effect of diabetes and aminoguanidine therapy on renal advanced glycation end-product binding. Kidney Int. 1999;55(3):907–916. doi:10.1046/j.1523-1755.1999.055003907.x
17. Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol. 2006;17(11):2974–2984. doi:10.1681/asn.2006040377
18. Wolf G, Ziyadeh FN. Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Neph Physiol. 2007;106(2):26–p31. doi:10.1159/000101797
19. Nagata M. Podocyte injury and its consequences. Kidney Int. 2016;89(6):1221–1230. doi:10.1016/j.kint.2016.01.012
20. Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol. 2002;13(12):3005–3015. doi:10.1097/01.asn.0000039661.06947.fd
21. Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225–233. doi:10.2337/diabetes.55.01.06.db05-0894
22. Caturano A, D’Angelo M, Mormone A, et al. Oxidative stress in type 2 diabetes: impacts from pathogenesis to lifestyle modifications. Curr Issues Mol Biol. 2023;45(8):6651–6666. doi:10.3390/cimb45080420
23. Pafundi PC, Garofalo C, Galiero R, et al. Role of albuminuria in detecting cardio-renal risk and outcome in diabetic subjects. Diagnostics. 2021;11(2):290. doi:10.3390/diagnostics11020290
24. Guthrie RA, Guthrie DW. Pathophysiology of diabetes mellitus. Crit Care Nurs Q. 2004;27(2):113–125. doi:10.1097/00002727-200404000-00003
25. Matsumoto N, Omagari D, Ushikoshi-Nakayama R, Yamazaki T, Inoue H, Saito I. Hyperglycemia induces generation of reactive oxygen species and accelerates apoptotic cell death in salivary gland cells. Pathobiology. 2021;88(3):234–241. doi:10.1159/000512639
26. Peng J, Li X, Zhang D, et al. Hyperglycemia, p53, and mitochondrial pathway of apoptosis are involved in the susceptibility of diabetic models to ischemic acute kidney injury. Kidney Int. 2015;87(1):137–150. doi:10.1038/ki.2014.226
27. Patschan D, Müller G. Acute kidney injury in diabetes mellitus. Int J Nephrol. 2016;2016:1–7. doi:10.1155/2016/6232909
28. Khanijou V, Zafari N, Coughlan MT, MacIsaac RJ, Ekinci EI. Review of potential biomarkers of inflammation and kidney injury in diabetic kidney disease. Diabetes Metabol Res Rev. 2022;38(6):e3556. doi:10.1002/dmrr.3556
29. Taniguchi K, Xia L, Goldberg HJ, et al. Inhibition of Src Kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes. 2013;62(11):3874–3886. doi:10.2337/db12-1010
30. Neumann K, Tiegs G. Immune regulation in renal inflammation. Cell Tissue Res. 2021;385(2):305–322. doi:10.1007/s00441-020-03351-1
31. Ma T, Li X, Zhu Y, et al. Excessive activation of notch signaling in macrophages promote kidney inflammation, fibrosis, and necroptosis. Front Immunol. 2022;13:835879. doi:10.3389/fimmu.2022.835879
32. Yonemoto S, Machiguchi T, Nomura K, Minakata T, Nanno M, Yoshida H. Correlations of tissue macrophages and cytoskeletal protein expression with renal fibrosis in patients with diabetes mellitus. Clin Exper Nephrol. 2006;10(3):186–192. doi:10.1007/s10157-006-0426-7
33. Tang PM, Nikolic-Paterson DJ, Lan HY. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. 2019;15(3):144–158. doi:10.1038/s41581-019-0110-2
34. Breda PC, Wiech T, Meyer-Schwesinger C, et al. Renal proximal tubular epithelial cells exert immunomodulatory function by driving inflammatory CD4(+) T cell responses. Am J Physiol Renal Physiol. 2019;317(1):F77–F89. doi:10.1152/ajprenal.00427.2018
35. Dehghanbanadaki H, Forouzanfar K, Kakaei A, et al. The role of CDH2 and MCP-1 mRNAs of blood extracellular vesicles in predicting early-stage diabetic nephropathy. PLoS One. 2022;17(4):e0265619. doi:10.1371/journal.pone.0265619
36. Li RX, Yiu WH, Wu HJ, et al. BMP7 reduces inflammation and oxidative stress in diabetic tubulopathy. Clin Sci. 2015;128(4):269–280. doi:10.1042/cs20140401
37. Lu X, Yin D, Zhou B, Li T. MiR-135a promotes inflammatory responses of vascular smooth muscle cells from db/db mice via downregulation of FOXO1. Int Heart J. 2018;59(1):170–179. doi:10.1536/ihj.17-040
38. Du M, Wang Q, Li W, et al. Overexpression of FOXO1 ameliorates the podocyte epithelial-mesenchymal transition induced by high glucose vitro and in vivo. Biochem Biophys Res. 2016;471(4):416–422. doi:10.1016/j.bbrc.2016.02.066
39. Puthanveetil P, Wan A, Rodrigues B. FoxO1 is crucial for sustaining cardiomyocyte metabolism and cell survival. Cardiovasc Res. 2013;97(3):393–403. doi:10.1093/cvr/cvs426
40. Xing YQ, Li A, Yang Y, Li XX, Zhang LN, Guo HC. The regulation of FOXO1 and its role in disease progression. Life Sci. 2018;193:124–131. doi:10.1016/j.lfs.2017.11.030
41. Sun H, Shao X, He J, Golos M, Shi B. Role of the mTOR‑FOXO1 pathway in obesity‑associated renal tubulointerstitial inflammation. Mol Med Rep. 2019;19(2):1284–1293. doi:10.3892/mmr.2018.9727
42. Chen J, Xiao H, Xue R, et al. Nicotine exacerbates diabetic nephropathy through upregulation of Grem1 expression. Mol Med. 2023;29(1):92. doi:10.1186/s10020-023-00692-9
43. Vallon V, Thomson SC. Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol. 2012;74:351–375. doi:10.1146/annurev-physiol-020911-153333
44. Weston CE, Feibelman MB, Wu K, Simon EE. Effects of EGF and IGF-1 on proliferation of cultured human proximal tubule cells after oxidant stress. Renal Failure. 2004;26(1):13–20. doi:10.1081/JDI-120028538
45. Jo HA, Kim JY, Yang SH, et al. The role of local IL6/JAK2/STAT3 signaling in high glucose-induced podocyte hypertrophy. Kidney Res Clin Pract. 2016;35(4):212–218. doi:10.1016/j.krcp.2016.09.003
46. Ridker PM. From C-reactive protein to interleukin-6 to interleukin-1: moving upstream to identify novel targets for atheroprotection. Circ Res. 2016;118(1):145–156. doi:10.1161/circresaha.115.306656
47. Li H, Wang J, Liu X, Cheng Q. MicroRNA-204-5p suppresses IL6-mediated inflammatory response and chemokine generation in HK-2 renal tubular epithelial cells by targeting IL6R. Biochem Cell Biol. 2019;97(2):109–117. doi:10.1139/bcb-2018-0141
48. Speeckaert MM, Speeckaert R, Laute M, Vanholder R, Delanghe JR. Tumor necrosis factor receptors: biology and therapeutic potential in kidney diseases. Am J Nephrol. 2012;36(3):261–270. doi:10.1159/000342333
49. Wang J, Feng Y, Zhang Y, et al. TNF-α and IL-1β promote renal podocyte injury in T2DM rats by decreasing glomerular VEGF/eNOS expression levels and altering hemodynamic parameters. J Inflamm Res. 2022;15:6657–6673. doi:10.2147/jir.S391473
50. Lampropoulou IT, Stangou Μ, Sarafidis P, et al. TNF-α pathway and T-cell immunity are activated early during the development of diabetic nephropathy in type II diabetes mellitus. Clin Immunol. 2020;215:108423. doi:10.1016/j.clim.2020.108423
51. Baud L, Ardaillou R. Tumor necrosis factor in renal injury. Mineral Electrol Metabol. 1995;21(4–5):336–341. doi:10.1016/S0344-0338(11)81021-6
52. Omote K, Gohda T, Murakoshi M, et al. Role of the TNF pathway in the progression of diabetic nephropathy in KK-A(y) mice. Am J Physiol Renal Physiol. 2014;306(11):F1335–F1347. doi:10.1152/ajprenal.00509.2013
53. Chen TK, Coca SG, Estrella MM, et al. Longitudinal TNFR1 and TNFR2 and kidney outcomes: results from AASK and VA NEPHRON-D. J Am Soc Nephrol. 2022;33(5):996–1010. doi:10.1681/asn.2021060735
54. Szeto HH. Pharmacologic approaches to improve mitochondrial function in AKI and CKD. J Am Soc Nephrol. 2017;28(10):2856–2865. doi:10.1681/asn.2017030247
55. Chung KW, Dhillon P, Huang S, et al. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. 2019;30(4):784–799.e785. doi:10.1016/j.cmet.2019.08.003
56. Yu EP, Bennett MR. The role of mitochondrial DNA damage in the development of atherosclerosis. Free Rad Biol Med. 2016;100:223–230. doi:10.1016/j.freeradbiomed.2016.06.011
57. Lindblom R, Higgins G, Coughlan M, de Haan JB. Targeting mitochondria and reactive oxygen species-driven pathogenesis in diabetic nephropathy. Rev Diabet Stud. 2015;12(1–2):134–156. doi:10.1900/rds.2015.12.134
58. Takagi S, Li J, Takagaki Y, et al. Ipragliflozin improves mitochondrial abnormalities in renal tubules induced by a high-fat diet. J Diabetes Investig. 2018;9(5):1025–1032. doi:10.1111/jdi.12802
59. Lee SE, Jang JE, Kim HS, et al. Mesenchymal stem cells prevent the progression of diabetic nephropathy by improving mitochondrial function in tubular epithelial cells. Exp. Mol. Med. 2019;51(7):1–14. doi:10.1038/s12276-019-0268-5
60. Tang SCW, Yiu WH. Innate immunity in diabetic kidney disease. Nat Rev Nephrol. 2020;16(4):206–222. doi:10.1038/s41581-019-0234-4
61. Zhu Y, Huang G, Yang Y, et al. Chinese herbal medicine Suyin detoxification granule inhibits pyroptosis and epithelial-mesenchymal transition by downregulating MAVS/NLRP3 to alleviate renal injury. J Inflamm Res. 2021;14:6601–6618. doi:10.2147/jir.S341598
62. Guo H, Bi X, Zhou P, Zhu S, Ding W. NLRP3 Deficiency attenuates renal fibrosis and ameliorates mitochondrial dysfunction in a mouse unilateral ureteral obstruction model of chronic kidney disease. Mediators Inflamm. 2017;2017:8316560. doi:10.1155/2017/8316560
63. Qi R, Yang C. Renal tubular epithelial cells: the neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. 2018;9(11):1126. doi:10.1038/s41419-018-1157-x
64. Tang SC, Chan LY, Leung JC, et al. Bradykinin and high glucose promote renal tubular inflammation. Nephrol Dial Transplant. 2010;25(3):698–710. doi:10.1093/ndt/gfp599
65. de Haij S, Adcock IM, Bakker AC, Gobin SJ, Daha MR, van Kooten C. Steroid responsiveness of renal epithelial cells. Dissociation of transrepression and transactivation. J Biol Chem. 2003;278(7):5091–5098. doi:10.1074/jbc.M209836200
66. Ren Q, Guo F, Tao S, Huang R, Ma L, Fu P. Flavonoid fisetin alleviates kidney inflammation and apoptosis via inhibiting Src-mediated NF-κB p65 and MAPK signaling pathways in septic AKI mice. Biomed. Pharmacother. 2020;122:109772. doi:10.1016/j.biopha.2019.109772
67. Zhang J, Gu C, Lawrence DA, Cheung AK, Huang Y. A plasminogen activator inhibitor type 1 mutant retards diabetic nephropathy in db/db mice by protecting podocytes. Exp Physiol. 2014;99(5):802–815. doi:10.1113/expphysiol.2013.077610
68. Lupachyk S, Watcho P, Stavniichuk R, Shevalye H, Obrosova IG. Endoplasmic reticulum stress plays a key role in the pathogenesis of diabetic peripheral neuropathy. Diabetes. 2013;62(3):944–952. doi:10.2337/db12-0716
69. Leng YP, Qiu N, Fang WJ, Zhang M, He ZM, Xiong Y. Involvement of increased endogenous asymmetric dimethylarginine in the hepatic endoplasmic reticulum stress of type 2 diabetic rats. PLoS One. 2014;9(2):e97125. doi:10.1371/journal.pone.0097125
70. Li F, Chen Y, Li Y, Huang M, Zhao W. Geniposide alleviates diabetic nephropathy of mice through AMPK/SIRT1/NF-κB pathway. Eur J Pharmacol. 2020;886:173449. doi:10.1016/j.ejphar.2020.173449
71. Qu X, Zhai B, Liu Y, et al. Pyrroloquinoline quinone ameliorates renal fibrosis in diabetic nephropathy by inhibiting the pyroptosis pathway in C57BL/6 mice and human kidney 2 cells. Biomed. Pharmacother. 2022;150:112998. doi:10.1016/j.biopha.2022.112998
72. Hodgkins KS, Schnaper HW. Tubulointerstitial injury and the progression of chronic kidney disease. Pediatr Nephrol. 2012;27(6):901–909. doi:10.1007/s00467-011-1992-9
73. Zhang L, Xu C, Hu W, Wu P, Qin C, Zhang J. Anti-inflammatory effects of Lefty-1 in renal tubulointerstitial inflammation via regulation of the NF-κB pathway. Int J Mol Med. 2018;41(3):1293–1304. doi:10.3892/ijmm.2017.3327
74. Li H, Duann P, Li Z, et al. The cell membrane repair protein MG53 modulates transcription factor NF-κB signaling to control kidney fibrosis. Kidney Int. 2022;101(1):119–130. doi:10.1016/j.kint.2021.09.027
75. Maik-Rachline G, Zehorai E, Hanoch T, Blenis J, Seger R. The nuclear translocation of the kinases p38 and JNK promotes inflammation-induced cancer. Sci Signal. 2018;11(525). doi:10.1126/scisignal.aao3428
76. Wu Q, Wu W, Fu B, Shi L, Wang X, Kuca K. JNK signaling in cancer cell survival. Med Res Rev. 2019;39(6):2082–2104. doi:10.1002/med.21574
77. Song Q, Xiang TY, Jian-Mei XU, Liu JG, Gao P. Effect of donglian capsules on protein expression of p38MAPK,CREB1 and TGFβ1 in diabetic nephropathy model rats induced by streptozotocin. Inform Trad Chin Med. 2016;33(1):1–5. doi:10.19656/j.cnki.1002-2406.2016.01.001
78. Wang SY, Gao K, Deng DL, et al. TLE4 promotes colorectal cancer progression through activation of JNK/c-Jun signaling pathway. Oncotarget. 2016;7(3):2878–2888. doi:10.18632/oncotarget.6694
79. Ma L, Wu F, Shao Q, Chen G, Xu L, Lu F. Baicalin alleviates oxidative stress and inflammation in diabetic nephropathy via Nrf2 and MAPK signaling pathway. Drug Des Devel Ther. 2021;15:3207–3221. doi:10.2147/dddt.S319260
80. Wang Y, Fang Q, Jin Y, et al. Blockade of myeloid differentiation 2 attenuates diabetic nephropathy by reducing activation of the renin-angiotensin system in mouse kidneys. Br J Pharmacol. 2019;176(14):2642–2657. doi:10.1111/bph.14687
81. Shen S, Huang J, Xu C, et al. ERK modulates macrophage polarization and alters exosome miRNA expression in diabetic nephropathy. Clin Lab. 2021;67(12). doi:10.7754/Clin.Lab.2021.210314
82. Wang K, Wu YG, Su J, Zhang JJ, Zhang P, Qi XM. Total glucosides of paeony regulates JAK2/STAT3 activation and macrophage proliferation in diabetic rat kidneys. Am J Chin Med. 2012;40(3):521–536. doi:10.1142/s0192415x12500401
83. Yang Y, Lei Y, Liang Y, et al. Vitamin D protects glomerular mesangial cells from high glucose-induced injury by repressing JAK/STAT signaling. Int Urol Nephrol. 2021;53(6):1247–1254. doi:10.1007/s11255-020-02728-z
84. Yang L, Xue J, Meng X, Wang Y, Bai Y. Effects of total flavonoids from Oxytropis falcata Bunge on the SOCS/JAK/STAT inflammatory signaling pathway in the kidneys of diabetic nephropathy model mice. Eur J Inflammation. 2019;17(2):205873921986187. doi:10.1177/2058739219861877
85. Hu J, Fan X, Meng X, Wang Y, Liang Q, Luo G. Evidence for the involvement of JAK/STAT/SOCS pathway in the mechanism of Tangshen formula-treated diabetic nephropathy. Planta Med. 2014;80(8–9):614–621. doi:10.1055/s-0034-1368454
86. Li J, Yang Y, Wei S, et al. Corrigendum: bixin protects against kidney interstitial fibrosis through promoting STAT6 degradation. Front Cell Dev Biol. 2020;8:643207. doi:10.3389/fcell.2020.643207
87. Johnson DE, Ra O, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15(4):234–248. doi:10.1038/nrclinonc.2018.8
88. Chen W, Yuan H, Cao W, et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics. 2019;9(14):3980–3991. doi:10.7150/thno.32352
89. Liau NPD, Laktyushin A, Lucet IS, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun. 2018;9(1):1558. doi:10.1038/s41467-018-04013-1
90. Zhang H, Zhang S, Wang L, Liu X, Wu Y. Chitooligosaccharide guanidine inhibits high glucose-induced activation of DAG/PKC pathway by regulating expression of GLUT2 in type 2 diabetic nephropathy rats. J Func Food. 2018;41:41–47. doi:10.1016/j.jff.2017.12.032
91. Suji G, Sivakami S. DNA damage by free radical production by aminoguanidine. Ann N Y Acad Sci. 2006;1067:191–199. doi:10.1196/annals.1354.023
92. Noh H, King GL. The role of protein kinase C activation in diabetic nephropathy. Kidney Int Suppl. 2007;(106):S49–53. doi:10.1038/sj.ki.5002386
93. Yang Y, Liu S, Wang P, et al. DNA-dependent protein kinase catalytic subunit (DNA-PKcs) drives chronic kidney disease progression in male mice. Nat Commun. 2023;14(1):1334. doi:10.1038/s41467-023-37043-5
94. Meier M, Menne J, Park JK, et al. Deletion of protein kinase C-epsilon signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J Am Soc Nephrol. 2007;18(4):1190–1198. doi:10.1681/asn.2005070694
95. Sagoo MK, Gnudi L. Diabetic nephropathy: is there a role for oxidative stress? Free Rad Biol Med. 2018;116:50–63. doi:10.1016/j.freeradbiomed.2017.12.040
96. Lim AK, Nikolic-Paterson DJ, Ma FY, et al. Role of MKK3-p38 MAPK signalling in the development of type 2 diabetes and renal injury in obese db/db mice. Diabetologia. 2009;52(2):347–358. doi:10.1007/s00125-008-1215-5
97. Ding W, Wang F, Fang Q, Zhang M, Chen J, Gu Y. Association between two genetic polymorphisms of the renin-angiotensin-aldosterone system and diabetic nephropathy: a meta-analysis. Mol Biol Rep. 2012;39(2):1293–1303. doi:10.1007/s11033-011-0862-7
98. Rodríguez-Iturbe B, Johnson RR, Herrera-Acosta J. Tubulointerstitial damage and progression of renal failure. Kidney Int Suppl. 2005;(99):S82–S86. doi:10.1111/j.1523-1755.2005.09915.x
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.